
Endogenous Stress Caused by Faulty Oxidation Reactions Fosters Evolution of 2,4-Dinitrotoluene-Degrading Bacteria
Environmental strain Burkholderia sp. DNT mineralizes the xenobiotic compound 2,4-dinitrotoluene (DNT) owing to the catabolic dnt genes borne by plasmid DNT, but the process fails to promote significant growth. To investigate this lack of physiological return of such an otherwise complete metabolic route, cells were exposed to DNT under various growth conditions and the endogenous formation of reactive oxygen species (ROS) monitored in single bacteria. These tests revealed the buildup of a strong oxidative stress in the population exposed to DNT. By either curing the DNT plasmid or by overproducing the second activity of the biodegradation route (DntB) we could trace a large share of ROS production to the first reaction of the route, which is executed by the multicomponent dioxygenase encoded by the dntA gene cluster. Naphthalene, the ancestral substrate of the dioxygenase from which DntA has evolved, also caused significant ROS formation. That both the old and the new substrate brought about a considerable cellular stress was indicative of a still-evolving DntA enzyme which is neither optimal any longer for naphthalene nor entirely advantageous yet for growth of the host strain on DNT. We could associate endogenous production of ROS with likely error-prone repair mechanisms of DNA damage, and the ensuing stress-induced mutagenesis in cells exposed to DNT. It is thus plausible that the evolutionary roadmap for biodegradation of xenobiotic compounds like DNT was largely elicited by mutagenic oxidative stress caused by faulty reactions of precursor enzymes with novel but structurally related substrates-to-be.
Published in the journal:
. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003764
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003764
Summary
Environmental strain Burkholderia sp. DNT mineralizes the xenobiotic compound 2,4-dinitrotoluene (DNT) owing to the catabolic dnt genes borne by plasmid DNT, but the process fails to promote significant growth. To investigate this lack of physiological return of such an otherwise complete metabolic route, cells were exposed to DNT under various growth conditions and the endogenous formation of reactive oxygen species (ROS) monitored in single bacteria. These tests revealed the buildup of a strong oxidative stress in the population exposed to DNT. By either curing the DNT plasmid or by overproducing the second activity of the biodegradation route (DntB) we could trace a large share of ROS production to the first reaction of the route, which is executed by the multicomponent dioxygenase encoded by the dntA gene cluster. Naphthalene, the ancestral substrate of the dioxygenase from which DntA has evolved, also caused significant ROS formation. That both the old and the new substrate brought about a considerable cellular stress was indicative of a still-evolving DntA enzyme which is neither optimal any longer for naphthalene nor entirely advantageous yet for growth of the host strain on DNT. We could associate endogenous production of ROS with likely error-prone repair mechanisms of DNA damage, and the ensuing stress-induced mutagenesis in cells exposed to DNT. It is thus plausible that the evolutionary roadmap for biodegradation of xenobiotic compounds like DNT was largely elicited by mutagenic oxidative stress caused by faulty reactions of precursor enzymes with novel but structurally related substrates-to-be.
Introduction
A diversity of xenobiotic compounds has made it to the environment in large amounts since the onset of synthetic chemistry due to urban and industrial activities [1]. Although many of such compounds bear bonds that are rare or non-existing in the natural realm, it is not infrequent to isolate bacteria able to use them as C, N and/or energy sources, especially in places with a history of chemical pollution [1]–[3]. This bear witness of the ability of existing metabolic networks to conquest new and diverse chemical landscapes. The process by which a given bacterium comes across a genetic and enzymatic solution to the challenge of degrading a new chemical is not trivial [4], [5]. Not only enzymes and complete pathways have to evolve new substrate specificities, but also they should be expressed at the right time and location. In addition, the resulting metabolic currency should be wired to the central biochemical network for eventual biomass buildup. This surely requires a mutual adaptation between the host and the pathway itself that involves multiple changes in both the catabolic genes and the rest of the cell transcriptome. On this basis, it comes as a surprise that many xenobiotic compounds containing strong chemical bonds and/or substituents can be degraded by environmental bacteria after being in the biosphere for only a relatively short period of time. One conspicuous case in this respect is that of nitroaromatic compounds. Although many chemical structures with C-NO2 bonds are found in natural products [6], [7], nitroaromatics could never become a significant selective pressure before their massive production and release in connection to the modern industry of explosives [1], [8]. Yet, a number of bacterial strains have been isolated that can mineralize many of such unusual chemical structures [1], [2], [9]. However, the mere genetic drift of pre-existing enzymes and operons that catabolize similar compounds towards new substrates can hardly explain the high rate at which new strains and catabolic operons appear [10]. The question thus arises on whether evolution of given catabolic properties benefit from some inherent accelerator of the process somehow encoded in the enzymes and genes involved.
Environmental bacteria capable of biodegradation of 2,4-dinitrotoluene (DNT) afford an exceptional opportunity to examine the factors at play in the emergence of new metabolic abilities. One of such isolates, Burkholderia sp. strain DNT, was isolated from contaminated surface water of an ammunition waste plant on the basis of utilizing DNT as C and N sources [11]. This capacity is due to the action of a route for catabolism of the nitroaromatic compound (Fig. 1A). The phylogeny of the dnt genes and the biochemical properties of the encoded products indicate that this cluster for DNT biodegradation has originated from a precursor pathway for catabolism of the naturally-occurring hydrocarbon naphthalene (Fig. 1B; [12], [13]). Still, the same data indicates that the pathway is even now evolving since the kinetic coupling of each of the biochemical steps is not well balanced [11], the regulation of the system keeps the same effector profile than the precursor operon [14], there are transposon remnants and vestigial genes in the genetic cluster [12] and the strain hardly grows on the substrate of interest [15]. These are all indicators that this strain has started its itinerary towards DNT catabolism but has not yet found an optimal solution to the multi-tiered problem of its viable degradation.

In this work we have examined possible physiological and biochemical drivers that frame the evolution of Burkholderia sp. DNT towards DNT catabolism and, by extension, many other environmental bacteria towards new chemical compounds. As shown below, reactive oxygen species (ROS) brought about by the faulty performance of the first enzyme of the pathway (DntA dioxygenase) on the xenobiotic substrate seem to be a key agent of the process. This is because ROS is translated into DNA mutagenesis and diversification of the host strain that foster the emergence of novel adaptive phenotypes. Moreover, the results provide an evolutionary logic to the abundance of enzymes containing Rieske-type Fe centers (as DntA) in pathways for biodegradation of xenobiotic compounds recently released to the environment [16]. This work showcases an evolutionary scenario in which the physiological stress caused by a metabolic problem triggers also the genetic diversification necessary for exploring the solution space to the same problem.
Results
Activity of the DNT pathway on its former and current substrate is detrimental for Burkholderia sp. DNT
The experimental system of choice for examining the transition of the DNT pathway from degrading naphthalene to metabolizing DNT involves only three players: strain Burkholderia sp. DNT and the two chemicals at stake. It should be noted that, according to the very high similarity between the leading ring oxygenases of the nag route in Ralstonia sp. U2 for naphthalene degradation and the dnt pathway of Burkholderia sp. DNT for DNT catabolism (Fig. 1B), the extant DntA enzyme retains the ability to act on the first substrate to produce 1,2-dihydroxy-1,2-dihydronaphthalene [13], [17]. Owing to its current catabolic role, this multi-component enzyme dioxygenates DNT in positions 4 and 5 to yield 4-methyl-5-nitrocatechol (4M5NC, Fig. 1A). In order to have a gross estimate of the physiological state of the cells when exposed to the former and current DntA substrates, we simply examined the survival of Burkholderia sp. DNT pre-grown on M9 minimal medium containing succinate and then exposed respectively to either chemical at a final concentration of 0.5 mM. In order to differentiate between the intrinsic effect of these compounds from their interplay with the dnt-encoded enzymes, we also employed a variant strain cured of the dnt genes (Burkholderia sp. DNT Δdnt), that has no chance of transforming the substrates at stake. While both naphthalene and DNT led to a very significant loss of viability of Burkholderia sp. DNT (Fig. 2A), which went down to <30% in the presence of DNT, addition of the same chemicals to the strain lacking the dnt genes had a considerably lesser toxicity (Fig. 2B). It should be noted that neither naphthalene nor DNT are inducers of the dnt pathway [14], which is expressed through its native promoter at a basal level under these experimental conditions. The detrimental effect of the former (naphthalene) and new (DNT) substrate can thus be both traced to the presence of the dnt genes and plausibly to the activity of their encoded products on each of the chemicals. In order to qualify the observed toxicity of the DntA substrates we measured the activity of the enzyme glucose-6-phosphate dehydrogenase (G6PDH) in cells exposed or not to DNT. The activity of this enzyme is a physiological stress marker in most bacteria [18], [19] and its increased activity reports a general response to metabolic hardship. When Burkholderia sp. DNT cells were exposed to DNT, the level of G6PDH activity more than doubled in the presence of DNT (Fig. 3). In contrast, the equivalent strain lacking the dnt genes had a G6PDH activity minimally affected by addition of the nitroaromatic compound. Taken together, these data suggested that activity of the dnt-encoded products on the two substrates tested caused physiological stress that was more pronounced in the case of DNT.


Exposure of Burkholderia sp. DNT to DNT results in massive production of ROS in individual cells
What could be the reason for the remarkable toxicity of DNT in cells expressing dnt genes? Since an increased activity of G6PDH would lead to enhanced NADPH turnover rates required to counteract oxidative stress [18]–[20], one plausible scenario was that the encounter of DNT with the dnt-encoded oxygenases could release ROS as a side product of the oxidative steps that shape the pathway (Fig. 1A). This is a frequent situation in evolving biodegradative routes of aerobic bacteria [21]–[24] and it often becomes a veritable bottleneck in the catabolism of some compounds [19], [25], [26]. On this background, we set out to measure ROS generation in Burkholderia sp. DNT cells treated with DNT along with their Δdnt counterparts. To quantify oxidative stress we resorted to staining cells with the ROS-activated green fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) followed by quantitative flow cytometry [19]. The minimum and maximum response levels of reference were fixed using succinate-grown cells added with either the H2DCF-DA solvent carrier (dimethyl sulfoxide, DMSO) or the same with 1.5 mM H2O2. Fig. 4 accredits the performance of the test and shows raw data obtained when cells were exposed to the toxicants at stake. Perusal of the results indicated that DNT caused an extraordinary and somewhat unexpected level of intracellular ROS, the intensity of which nearly equaled that produced by straight addition of H2O2 to the medium. In comparison, cells lacking the complement of dnt genes showed a much lower level of ROS in the presence of DNT, indicating that both the substrate and the genes were required for such a surprising synergy to cause oxidative stress. Addition of the ancestral substrate naphthalene also caused a lower, but still significant, level of ROS in Burkholderia sp. DNT (Fig. 5A and B). Even though permeability of the dye can be affected by the aromatic compounds tested [27], [28], pairwise comparisons of the different strains and substrates revealed the extension of the stress produced by each compound. Since the DntA originates in a precursor naphthalene dioxygenase [12], [13], we hypothesized that some of the ROS could be originated from an uncoupled, non-productive oxygen-delivery reaction between the evolved enzyme and the suboptimal substrate DNT, as reported for similar Rieske-type dioxygenases [29], [30]. This possibility placed the focus of the ensuing experiments on the first step of the DNT biodegradation route.


The bulk of ROS elicited by DNT in Burkholderia sp. DNT originates in the initial dioxygenation step
Since the most critical move in any biodegradative route of aromatics is the initial step that activates the ring for overcoming the resonance energy that stabilizes their structure [31], we concentrated on the leading reaction that converts DNT to 4M5NC (Fig. 1A). This compound, as is the case for other catechols, is highly toxic [32], [33]. Furthermore, it is known [11] that such yellow-colored intermediate transiently accumulates in the medium prior to be channeled towards the next step of the degradation route (Fig. 6A). The toxic effect of DNT on Burkholderia sp. DNT could thus originate in either faulty reactions of DntA on its substrate, in accumulation of 4M5NC or in both. Our strategy to distinguish these possibilities was to generate a strain that overproduced DntB (Fig. 1A). This enzyme is a monooxygenase that eliminates the remaining nitro substituent of 4M5NC to produce 2-hydroxy-5-methylquinone (2H5MQ; [34], [35]). Higher levels of DntB are thus predicted to drain 4M5NC faster towards 2H5MQ -thereby allowing us to separate the intrinsic physiological effect of DntA action from that of the toxic catechol that results from the reaction. To this end, we generated strain Burkholderia sp. DNT dntB↑, in which extra copies of the gene encoding the second oxygenase of the pathway were expressed from a constitutive promoter in a broad-host-range vector. Fig. 6B verifies such a prediction, as cultures of Burkholderia sp. DNT dntB↑ released a much lower amount (<20%) of 4M5NC to the medium. In the same setup, strain Burkholderia sp. DNT Δdnt did not accumulate any intermediate, while naphthalene would be biotransformed to 1,2-dihydroxy-1,2-dihydronaphthalene by any of the dntA+ strains. Once we had the three isogenic variants in hand, we set out to measure intracellular ROS production in wild-type, Δdnt and dntB↑ cells exposed to either DNT or naphthalene (Fig. 5A, B and C, respectively). These results provide an answer to the questions raised above. First, that DNT causes high ROS levels in both the wild-type and dntB↑ strains (Fig. 5A and C) clearly indicates that the bulk of oxidative stress can be traced to the very reaction of DntA with DNT, not to the product of the catalysis. Second, the effects of naphthalene were comparatively lower than those elicited by DNT, but they were still significant in respect to the untreated control conditions. ROS generated from the encounter of this substrate with the dnt-encoded enzymes must stem from uncoupled, faulty reactions with the leading DntA oxygenase, as there is no other enzyme that can recognize this aromatic compound as a potential but ultimately non-productive substrate. Since both the ancestral and the new substrate of the first dioxygenase release ROS when facing DntA, the cognate reaction has probably left behind a former biochemical optimum (i.e., when acting on naphthalene) but has not yet reached a new one with DNT.

DNT and naphthalene bring forth diversification of Burkholderia sp. DNT
In a further effort to identify the specific types of ROS that result from the above mentioned processes, we tested cells treated with the various substrates with nitro blue tetrazolium (NBT), a reagent that is specific for superoxide production. Consistently with the flow cytometry data above, the highest indications of superoxide presence were found in wild-type and dntB↑ cells exposed to DNT, while a lower level was detected in the Δdnt strain and in bacteria exposed to naphthalene (Fig. 7). Since superoxide originated in defective redox reactions promotes hydroxyl-radical formation and consequent DNA damage [36], [37] we wondered whether ROS stemming from the faulty oxygenations discussed above could eventually translate into an insult to the genome of Burkholderia sp. DNT. One direct way of quantifying such damage is the measurement of the 8-hydroxy-2′-deoxyguanosine (8-oxoG) content of genomic DNA as a coarse descriptor of HO• attack to purines [37], [38]. On this background we resorted to an immunoassay for quantifying 8-oxoG levels in cells exposed to DNT, using H2O2 as a positive control of oxidative stress. As shown in Fig. 8A, DNT indeed caused a 1.4-fold increase in the share of damaged purines in the Burkholderia sp. DNT genome (as opposed to a 2-fold increase elicited by H2O2). As such a chemical damage to DNA bases triggers the SOS response and eventually increases the mutation rate of the bacteria that undergo the insult [36], we next wondered whether the ultimate consequence of the uncoupling of the ring-hydroxylating reaction performed by DntA with their current and ancestral substrates was to increase genetic diversity by enhancing mutation rates. Since virtually nothing is known about the SOS response in Burkholderia sp. DNT, we directly measured such mutation rates as a descriptor of emerging genetic novelty. For this, we employed the standard test of appearance of rifampicin-resistant (RifR) clones under the various conditions assayed. As shown in Fig. 8B, both naphthalene and DNT triggered a considerable increase in the appearance of RifR colonies which was not noticeable in the strain deleted of the dnt genes The mutagenic effect of naphthalene in this context was slightly more pronounced than what could be expected from the sheer data on ROS production shown above. We speculate that ROS could react chemically with this bicyclic aromatic compound and generate additional DNA-intercalating agents (e.g. naphtoquinones and naphtodiols [39]) with a separate mutagenic action on DNA. In any case, it is worth noticing that maximum novelty (as measured with this procedure) is accompanied by an acute lethality (Fig. 2), so that the survivors to DNT exposure are more capable to explore the possible solution space to the next adaptive challenge (metabolic or otherwise) than those which had not been diversified because of the phenomena described here.


Discussion
Bacteria that inhabit environmental niches with a history of pollution by xenobiotic compounds offer a phenomenal experimental system for examining the expansion of the existing metabolic networks into new chemical spaces [10]. At least three bottlenecks must be overcome for the successful emergence of a novel catabolic pathway able to deal with a new-to-nature chemical structure. First, suitable enzymes must develop the right substrate specificity towards the new compound [40]. In most cases, such enzymes stem from precursors that act on structurally related chemicals but shift specificities through intermediate steps where the old and the new activities coexist in the same protein at no fitness cost [40], [41]. Second, the genes encoding the new enzymatic activities should be expressed only when required i.e. when the substrate-to-be is present in sufficient concentrations to grant a suitable return. More evolved catabolic systems are typically subject to a tight regulation by inducer substrates [42], while more recent counterparts are often expressed at low constitutive levels. Not infrequently, still evolving biodegradative operons carry along regulatory systems that respond to former substrates and not the new ones [43]. Finally, the metabolic action of the evolved pathways must be connected to some growth benefit that ultimately fosters its selective advantage [43]. This is brought about directly by entering metabolic currency from the biodegradation process into the central metabolism, or indirectly, by making cells more resistant to otherwise detrimental endogenous or exogenous stress. Even horizontal gene transfer granted, the number of mutations and genetic events that might be required for the emergence of a fully competent biodegradative strain able to deal with a novel xenobiotic compound can be considerable, far more than those necessary e.g. for new antibiotic resistances [44]. It thus comes as a surprise that a good number of xenobiotic compounds are indeed degraded by environmental strains not much after their production by the chemical industry [8], [10]. In view of all this, it could well be that evolution of biodegradative routes for xenobiotics is accelerated by factors inherent to the nature of the enzymatic reactions involved in the process.
The metabolic pathway of Burkholderia sp. DNT that acts on DNT has all what is biochemically needed for its complete biodegradation but it is clearly not optimal yet for coupling the process to efficient growth. Furthermore, we show above that the DNT dioxygenation reaction that constitutes the first step of the pathway exacerbates ROS production (Fig. 4 and Fig. 5). It is very likely that such reactive species result from the failure of the Fe-containing center of the α subunit of DntA to deliver active oxygen atoms to positions 4 and 5 of the DNT aromatic ring (Fig. 1A). Since the DntA enzyme complex originates in a precursor naphthalene dioxygenase (Fig. 1B), it is likely that such uncoupling of the reaction stems from a still poor geometry of the substrate-enzyme recognition. On the other hand, the enzyme still recognizes naphthalene as a substrate, but the reaction is not metabolically productive and also causes considerable ROS. These data not only pinpoints the DntA complex to be in the midst of a transition between the old and the new specificities, but also suggest key events in evolution of new xenobiotic-degrading strains. Since ROS mediate DNA damage, which in turn brings about a mutagenic SOS response [36], [37], [45], it seems that the generation of diversity associated to a faulty dioxygenation reaction could contribute to the exploration of the metabolic novelty space in a faster fashion that could be expected by a background, spontaneous mutagenesis. This evolutionary scenario is reminiscent - but not exactly identical, to that proposed for the rapid evolution of antibiotic-resistant bacterial strains, which is thought to be fostered as well by the boost of ROS and the ensuing mutagenesis and novelty generation that precedes death of bacteria treated with antimicrobials [46]–[49]. In the case of the DNT-degrading strain presented above, we could in fact trace the entire process to the master dioxygenation reaction that initiates the metabolic pathway. Because of its inherent enzymatic mechanism (e.g. Rieske-type active center) this reaction is prone to uncouple dioxygenations of suboptimal substrates and release ROS [29], [30]. In the case of strain DNT, the process might also be assisted by reactive nitrogen species connected to the nitrite [50] released as a by-product of the DntA and DntB-catalyzed reactions (Fig. 1). This issue deserves further studies, as nitrite has been claimed to be detrimental for in situ DNT degradation by environmental bacteria [51]. Additional mutagenic effects of biodegradation intermediates downstream of 4M5NC cannot be ruled out. However, since the only metabolite that is significantly accumulated during DNT turnover is precisely 4M5NC (the substrate of DntB, [11]), it is unlikely that the toxicity generated by such intermediates could be ultimately meaningful.
How general could be such a substrate-driven generation of diversity for evolution of other xenobiotic-biodegradation routes? A good number of results are found in the literature that can be interpreted under this light. For instance, the abundance of Rieske-containing enzymes in biodegradative operons [16] could reflect a key role in generation of novelty both in the catabolic genes themselves and in the host. This is an important detail, as changes that originate a better biodegradative strain involve genes other that those directly involved in the corresponding pathway. It is remarkable also that the transcriptomes and proteomes of strains degrading a suite of recalcitrant aromatics (e.g. polychlorobiphenyls and polyaromatic hydrocarbons) are overrepresented with functions that counteract oxidative stress [21]–[23]. Finally, it is not unusual to find oxidative pathways for xenobiotics in bacteria which are typical macrophage-resistant pathogens (e.g. Burkholderia and Mycobacterium) and thus adapted to endure a strongly oxidative milieu [52], [53]. Such routes might only develop in hosts able to endure the ROS-mediated lethality associated to the evolutionary roadmap from the enzymatic recognition of one substrate to another.
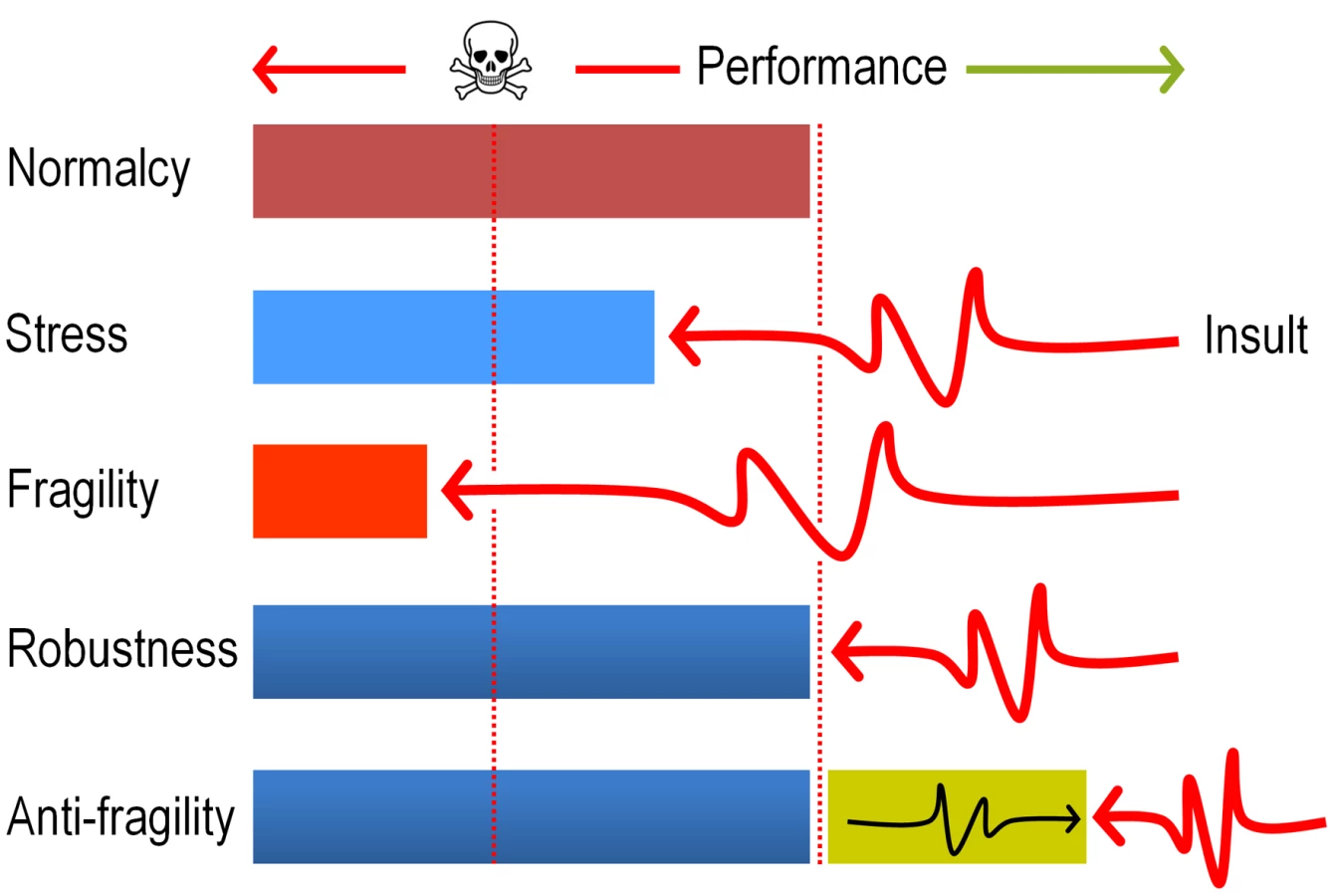
In sum, the data above strongly argue that evolution of DNT biodegradation pathways and possibly of many other catabolic systems, ultimately benefit from the mutagenic stress caused by faulty reactions of pre-existing enzymes on suboptimal substrates. The term anti-fragility (as opposed to robustness, [54], [55]) has been recently coined for describing such systems that reach a higher peak of efficacy after having nearly collapsed with stress and shocks (Fig. 9). The results discussed in this work seem not only to place the appearance of new xenobiotic-biodegradation strains within such an evolutionary scenario, but also suggest experimental approaches to accelerate their emergence in the Laboratory.
Materials and Methods
Bacterial strains, culture conditions and general methods
Burkholderia sp. DNT has been described before as a DNT degrading specimen [11]. The sequence, organization and function of each of the dnt genes, involved in mineralization of this aromatic compound, have been reported elsewhere [14]. The action of Burkholderia sp. DNT on the nitroaromatic substrate is very slow i.e. depletion of 1 mM DNT takes 3–5 days [15]. A spontaneous derivative of Burkholderia sp. DNT lacking the DNT-degradative phenotype was isolated by three consecutive cultivation rounds on LB medium, and the absence of key dnt genes (dntA, dntB and dntD) in this derivative was confirmed by PCR (data not shown). Accumulation of the yellow intermediate 4M5NC was spectrophotometrically quantified by absorbance readings at 420 nm [14]. The high segregational instability of the DNT plasmid, carrying the dnt genes, has been previously reported when cells are grown in rich media [56], [57]. Maintenance of the dnt+ phenotype (and thus of the DNT plasmid) was verified prior to each of the experiments by examining accumulation of 4M5NC (the first biodegradation intermediate; Fig. 1) in 200 clonal cultures added with 1 mM DNT. This control indicated that growth in minimal medium with succinate as the sole C source caused <1% loss of the catabolic ability of processing DNT. A strain overexpressing dntB, encoding the second enzyme of the pathway that removes 4M5NC, was constructed as follows. Oligonucleotides dntB-attB1-F (5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTG CTC TGT CGA TGA TTT GAG GA-3′) and dntB-attB2-R (5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTG TTG CGC ACC TGT CAT CG-3′), including the attB secuences required for Gateway-based cloning (shown in italics in the corresponding sequences), were used to amplify dntB from Burkholderia sp. DNT. The amplicon was cloned in the pDONR™/Zeo vector (Life Technologies Corp.) according to the manufacturer's instructions, and moved to the Gateway vector pBAV226 [58] (kindly provided by Jean T. Greenberg, University of Chicago, USA). This plasmid includes the nptII promoter to constitutively drive expression of dntB. The resulting construct was transformed into Burkholderia sp. DNT, giving rise to strain DNT dntB↑.
All strains were grown in 250-ml Erlenmeyer flasks containing 50 ml of M9 minimal medium [59] added with 2.5 ml/l of a trace elements solution [60] and 0.3% (w/v) sodium succinate as the sole C source. Overnight-grown Burkholderia cells were used as the inoculum by diluting them to a starting OD600 of ca. 0.1. Cells were grown in the same culture medium until they reached an OD600 of ca. 0.5, at which point the cell suspension was split into two 125-ml Erlenmeyer flasks, one culture served as a control experiment and was added with DMSO (the vehicle for both DNT and naphthalene), and the other one was amended with either DNT or naphthalene (from concentrated solutions in DMSO) to a final concentration of 0.5 mM. These conditions were used throughout the work. The solubility in H2O of DNT at 20°C is 1.7 mM, and that of naphthalene at 25°C is 0.2 mM; which results in dosages of 1.48 and 0.23 µmol/ml for DNT and naphthalene, respectively, assuming similar solubility values in water and in M9 minimal medium. Flasks were incubated at 170 rpm and 30°C, and samples collected at the times indicated in each case. Abiotic controls were run in parallel to estimate the possible disappearance of the aromatic compounds in the absence of cells. Non-inoculated flasks containing naphthalene (known to be a volatile compound) were incubated during 3 h as explained above, and the remaining substrate content was determined by gas chromatography. The determinations showed that ca. 88% of the initial naphthalene remained at the end of the experiment in these abiotic controls.
In vivo determination of ROS by flow cytometry
Fluorescence-activated cell sorter cytometry analysis was performed using the ROS-sensitive green fluorescent dye H2DCF-DA (Sigma-Aldrich Co.), essentially as described previously [19], [61], [62]. Cells were grown and treated with either DNT or naphthalene as explained above. In the cases indicated, 1.5 mM H2O2 was separately added as a positive control of oxidative stress conditions. After being incubated for 3 h, cells from 1.5–5.0 ml culture were pelleted by centrifugation at 5,000×g during 5 min, washed once with phosphate-buffered saline (PBS, pH = 7.5), and resuspended in PBS to adjust the OD600 to ca. 0.1. This cell suspension was added with 40 µM H2DCF-DA (added from a freshly-prepared 4 mM stock solution in DMSO), and then incubated in the dark for 15 min at room temperature. Flow cytometry analysis of fluorescence levels was performed in a Gallios™ flow cytometer (Beckman Coulter Inc.) equipped with an argon ion laser of 15 mW at 488 nm as the excitation source. The H2DCF-DA fluorescence emission at 525 nm was detected using a 530/30-nm band pass filter array. Size-related forward scatter signals gathered by the cytometer were used by the Cyflogic™ 1.2.1 software (CyFlo Ltd.) to gate fluorescence data from only bacteria in the stream, thus avoiding the mixing of data from bacteria with data from smaller, non-living particles in the suspension. Data for at least 25,000 cells per experimental condition were collected, and the Cyflogic™ 1.2.1 software was used to calculate the geometric mean of fluorescence per bacterial cell in each sample as well to generate histogram plots with a measure of -fluorescence intensity shown on the x-axis and the number of bacteria (i.e., events) counted at the specific fluorescence intensity used for H2DCF-DA detection shown on the y-axis.
Quantifying intracellular generation of hydrogen superoxide
Cell suspensions were treated with the compounds indicated before, and 1.5-ml culture aliquots were exposed to 350 µg/ml NBT during 30 min at 30°C in the dark. HCl was then added at 7.5 mM and the resulting mixture was centrifuged at 2,500×g for 15 min at room temperature. Pellets were treated twice with 0.35 ml DMSO to extract reduced NBT, and supernatants were pooled and mixed with 1 ml of 50 mM phosphate buffer (pH = 7.5). The NBT color change was monitored by absorbance at 575 nm and superoxide production was normalized to the protein concentration, measured by the Bradford method [63]. As the cultures had a similar OD600 value at the time of harvesting and processing, the amount of total protein used in these assays was considered to be roughly the same for each sample. Results are expressed as the fold-change in the ratio A575/mg protein among different strains and growth conditions as described elsewhere [64].
In vitro determination of enzymatic activity
G6PDH activity was analyzed by following the rate of NADP+ reduction at 340 nm at 30°C in a reaction mixture (1 ml final volume) containing 50 mM phosphate buffer (pH = 7.5), 10 mM MgSO4, 0.75 mM NADP+, 2 mM glucose-6-phosphate and 15–100 µl of the cell-free extract, obtained as previously described [19]. An extinction coefficient (εNADH) of 6.22 mM−1 cm−1 was used to calculate the specific enzymatic activity, and the protein concentration in cell-free extracts was measured by the Bradford method. One unit of enzymatic activity was defined as the quantity of enzyme that catalyzed the formation of 1 µmol product in 1 min at 30°C.
Determination of mutation frequencies
One hundred microliters of the 10−5, 10−6 and 10−7 dilutions of treated 24-h cultures were plated by quadruplicate onto LB agar, and 100 µl of undiluted cultures were spread (by quadruplicate also) onto LB-rifampicin plates (250 µg/ml). Colony counts were performed after 48 h of incubation. Mutation frequency values are reported as the dimensionless ratio between the number of RifR colonies and the total viable count [65].
DNA extraction and determination of 8-oxoG
A 25-ml aliquot of the cultures treated with either DNT or 1.5 mM H2O2 for 6 h was spun at 4°C at 4,500×g for 15 min. Cells were washed twice in cold 50 mM phosphate buffer (pH = 7.5) and genomic DNA was extracted using the UltraClean™ microbial DNA isolation kit (MoBio Labs Inc.). Genomic DNA was reconstituted in H2O, quantified by horizontal gel electrophoresis and using a NanoVue micro-volume UV/Vis spectrophotometer (GE Healthcare Bio-Sciences AB), and immediately hydrolyzed as follows. A suitable volume of the suspension, containing 1 µg DNA, was boiled at 97°C during 5 min, and immediately placed in an ice bath. Digestion reactions (1 ml) contained 1 µg denatured genomic DNA, 0.75 mM ZnCl2, 10 mM CH3COOK (pH = 5.5), 2.5 U/ml nuclease P1 from Penicillium citrinum, and 1.75 U/ml phosphatase acid from Ipomoea batatas (enzymes purchased from Sigma-Aldrich Co.). Digestions were carried out during 18 h at 37°C, after which the reaction was loaded into a 2-ml Centricon YM-10™ device (Millipore Corp.) and clarified by centrifugal ultrafiltration. Hydrolysis of genomic DNA was assessed by horizontal gel electrophoresis and spectrophotometry. Samples were processed within 6 h after the hydrolysis procedure. The content of 8-oxoG (in equilibrium with the keto form 8-oxo-2′-deoxyguanosine) was assessed by competitive ELISA using a commercial kit (Cayman Chemical Co.). The assay is based on the competition of free 8-oxoG and an 8-oxoG-acetylcholinesterase conjugate for a limited amount of 8-oxoG monoclonal antibody. The free acetylcholinesterase activity was evaluated with the Ellman's reagent [66]. The detection limit for 8-oxoG is 30 pg/ml, and calibration curves were run in parallel in each set of experiments using an authentic 8-oxoG standard.
Statistical analysis
All reported experiments were independently repeated at least twice (as specified in the figure legends), and the mean value of the corresponding parameter ± SD is presented. Statistical significance was assessed using analysis of variance (ANOVA). For flow cytometry experiments, the median value is reported in box plots along with the 1st and 3rd quartiles, and the statistical significance was evaluated with the Mann-Whitney U test. In all cases, data were considered statistically significant when P<0.05.
Zdroje
1. JuKS, ParalesRE (2010) Nitroaromatic compounds, from synthesis to biodegradation. Microbiol Mol Biol Rev 74 : 250–272.
2. SpainJC (1995) Biodegradation of nitroaromatic compounds. Annu Rev Microbiol 49 : 523–555.
3. KivisaarM (2011) Evolution of catabolic pathways and their regulatory systems in synthetic nitroaromatic compounds degrading bacteria. Mol Microbiol 82 : 265–268.
4. JohnsonGR, SpainJC (2003) Evolution of catabolic pathways for synthetic compounds: bacterial pathways for degradation of 2,4-dinitrotoluene and nitrobenzene. Appl Microbiol Biotechnol 62 : 110–123.
5. van der MeerJR (1997) Evolution of novel metabolic pathways for the degradation of chloroaromatic compounds. Antonie van Leeuwenhoek 71 : 159–178.
6. WinklerR, HertweckC (2007) Biosynthesis of nitro compounds. ChemBiochem 8 : 973–977.
7. ParryR, NishinoS, SpainJ (2011) Naturally-occurring nitro compounds. Nat Prod Rep 28 : 152–167.
8. SinghB, KaurJ, SinghK (2012) Microbial remediation of explosive waste. Crit Rev Microbiol 38 : 152–167.
9. SymonsZC, BruceNC (2006) Bacterial pathways for degradation of nitroaromatics. Nat Prod Rep 23 : 845–850.
10. van der MeerJR (2006) Environmental pollution promotes selection of microbial degradation pathways. Front Ecol Environ 4 : 35–42.
11. SpanggordRJ, SpainJC, NishinoSF, MortelmansKE (1991) Biodegradation of 2,4-dinitrotoluene by a Pseudomonas sp. Appl Environ Microbiol 57 : 3200–3205.
12. JohnsonGR, JainRK, SpainJC (2002) Origins of the 2,4-dinitrotoluene pathway. J Bacteriol 184 : 4219–4232.
13. SuenWC, HaiglerBE, SpainJC (1996) 2,4-Dinitrotoluene dioxygenase from Burkholderia sp. strain DNT: similarity to naphthalene dioxygenase. J Bacteriol 178 : 4926–4934.
14. de las HerasA, ChavarríaM, de LorenzoV (2011) Association of dnt genes of Burkholderia sp. DNT with the substrate-blind regulator DntR draws the evolutionary itinerary of 2,4-dinitrotoluene biodegradation. Mol Microbiol 82 : 287–299.
15. NishinoSF, PaoliGC, SpainJC (2000) Aerobic degradation of dinitrotoluenes and pathway for bacterial degradation of 2,6-dinitrotoluene. Appl Environ Microbiol 66 : 2139–2147.
16. GibsonDT, ParalesRE (2000) Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr Opin Biotechnol 11 : 236–243.
17. ParalesRE, EmigMD, LynchNA, GibsonDT (1998) Substrate specificities of hybrid naphthalene and 2,4-dinitrotoluene dioxygenase enzyme systems. J Bacteriol 180 : 2337–2344.
18. MaJF, HagerPW, HowellML, PhibbsPV, HassettDJ (1998) Cloning and characterization of the Pseudomonas aeruginosa zwf gene encoding glucose-6-phosphate dehydrogenase, an enzyme important in resistance to methyl viologen (paraquat). J Bacteriol 180 : 1741–1749.
19. NikelPI, Pérez-PantojaD, de LorenzoV (2013) Why are chlorinated pollutants so difficult to degrade aerobically? Redox stress limits 1,3-dichloprop-1-ene metabolism by Pseudomonas pavonaceae. Phil Trans R Soc B 368 : 20120377.
20. SinghR, MaillouxRJ, Puiseux-DaoS, AppannaVD (2007) Oxidative stress evokes a metabolic adaptation that favors increased NADPH synthesis and decreased NADH production in Pseudomonas fluorescens. J Bacteriol 189 : 6665–6675.
21. Bobadilla FazziniRA, PretoMJ, QuintasAC, BieleckaA, Martins dos SantosVAP (2010) Consortia modulation of the stress response: proteomic analysis of single strain versus mixed culture. Environ Microbiol 12 : 2436–2449.
22. ChávezFP, LünsdorfH, JerezCA (2004) Growth of polychlorinated-biphenyl-degrading bacteria in the presence of biphenyl and chlorobiphenyls generates oxidative stress and massive accumulation of inorganic polyphosphate. Appl Environ Microbiol 70 : 3064–3072.
23. PatrauchanMA, FlorizoneC, EapenS, Gomez-GilL, SethuramanB, et al. (2008) Roles of ring-hydroxylating dioxygenases in styrene and benzene catabolism in Rhodococcus jostii RHA1. J Bacteriol 190 : 37–47.
24. TamburroA, RobuffoI, HeipieperHJ, AllocatiN, RotilioD, et al. (2004) Expression of glutathione S-transferase and peptide methionine sulphoxide reductase in Ochrobactrum anthropi is correlated to the production of reactive oxygen species caused by aromatic substrates. FEMS Microbiol Lett 241 : 151–156.
25. KangYS, LeeY, JungH, JeonCO, MadsenEL, et al. (2007) Overexpressing antioxidant enzymes enhances naphthalene biodegradation in Pseudomonas sp. strain As1. Microbiology 153 : 3246–3254.
26. PonceBL, LatorreVK, GonzálezM, SeegerM (2011) Antioxidant compounds improved PCB-degradation by Burkholderia xenovorans strain LB400. Enzyme Microb Technol 49 : 509–516.
27. CzechowskaK, van der MeerJR (2012) Reversible and irreversible pollutant-induced bacterial cellular stress effects measured by ethidium bromide uptake and efflux. Environ Sci Technol 46 : 1201–1208.
28. SeguraA, MolinaL, FilletS, KrellT, BernalP, et al. (2012) Solvent tolerance in Gram-negative bacteria. Curr Opin Biotechnol 23 : 415–421.
29. ImbeaultNY, PowlowskiJB, ColbertCL, BolinJT, EltisLD (2000) Steady-state kinetic characterization and crystallization of a polychlorinated biphenyl-transforming dioxygenase. J Biol Chem 275 : 12430–12437.
30. LeeK (1999) Benzene-induced uncoupling of naphthalene dioxygenase activity and enzyme inactivation by production of hydrogen peroxide. J Bacteriol 181 : 2719–2725.
31. FuchsG, BollM, HeiderJ (2011) Microbial degradation of aromatic compounds - from one strategy to four. Nat Rev Microbiol 9 : 803–816.
32. SchweigertN, ZehnderAJ, EggenRI (2001) Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ Microbiol 3 : 81–91.
33. GeorgeKW, HayA (2012) Less is more: reduced catechol production permits Pseudomonas putida F1 to grow on styrene. Microbiology 158 : 2781–2788.
34. HaiglerBE, NishinoSF, SpainJC (1994) Biodegradation of 4-methyl-5-nitrocatechol by Pseudomonas sp. strain DNT. J Bacteriol 176 : 3433–3437.
35. HaiglerBE, SuenWC, SpainJC (1996) Purification and sequence analysis of 4-methyl-5-nitrocatechol oxygenase from Burkholderia sp. strain DNT. J Bacteriol 178 : 6019–6024.
36. BjellandS, SeebergE (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res 531 : 37–80.
37. CadetJ, DoukiT, GasparuttoD, RavanatJL (2003) Oxidative damage to DNA: formation, measurement and biochemical features. Mutat Res 531 : 5–23.
38. Le PageF, GentilA, SarasinA (1999) Repair and mutagenesis survey of 8-hydroxyguanine in bacteria and human cells. Biochimie 81 : 147–153.
39. TikkanenL, MatsushimaT, NatoriS, YoshihiraK (1983) Mutagenicity of natural naphthoquinones and benzoquinones in the Salmonella/microsome test. Mutat Res 124 : 25–34.
40. O'BrienPJ, HerschlagD (1999) Catalytic promiscuity and the evolution of new enzymatic activities. Chem Biol 6: R91–R105.
41. KhersonskyO, RoodveldtC, TawfikDS (2006) Enzyme promiscuity: evolutionary and mechanistic aspects. Curr Opin Chem Biol 10 : 498–508.
42. TropelD, van der MeerJR (2004) Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev 68 : 474–500.
43. CasesI, de LorenzoV (2001) The black cat/white cat principle of signal integration in bacterial promoters. EMBO J 20 : 1–11.
44. van der MeerJR (2006) Evolution of catabolic pathways in Pseudomonas through gene transfer. Pseudomonas 4 : 189–236.
45. TenaillonO, DenamurE, MaticI (2004) Evolutionary significance of stress induced mutagenesis in bacteria. Trends Microbiol 12 : 264–270.
46. KohanskiMA, DePristoMA, CollinsJJ (2010) Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37 : 311–320.
47. WongA, RodrigueN, KassenR (2012) Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet 8: e1002928.
48. MacLeanRC, BucklingA (2009) The distribution of fitness effects of beneficial mutations in Pseudomonas aeruginosa. PLoS Genet 5: e1000406.
49. CouceA, GuelfoJR, BlázquezJ (2013) Mutational spectrum drives the rise of mutator bacteria. PLoS Genet 9: e1003167.
50. BowmanLA, McLeanS, PooleRK, FukutoJM (2011) The diversity of microbial responses to nitric oxide and agents of nitrosative stress close cousins but not identical twins. Adv Microb Physiol 59 : 135–219.
51. FortnerJD, ZhangC, SpainJC, HughesJB (2003) Soil column evaluation of factors controlling biodegradation of DNT in the vadose zone. Environ Sci Technol 37 : 3382–3391.
52. KanalyRA, HarayamaS (2010) Advances in the field of high-molecular-weight polycyclic aromatic hydrocarbon biodegradation by bacteria. Microb Biotechnol 3 : 136–164.
53. VialL, ChapalainA, GroleauM-C, DézielE (2011) The various lifestyles of the Burkholderia cepacia complex species: a tribute to adaptation. Environ Microbiol 13 : 1–12.
54. DanchinA, BinderPM, NoriaS (2011) Antifragility and tinkering in biology (and in business) flexibility provides an efficient epigenetic way to manage risk. Genes 2 : 998–1016.
55. Taleb NN (2012) Antifragile: things that gain from disorder. New York: Random House.
56. JohnsonGR, JainRK, SpainJC (2000) Properties of the trihydroxytoluene oxygenase from Burkholderia cepacia R34: an extradiol dioxygenase from the 2,4-dinitrotoluene pathway. Arch Microbiol 173 : 86–90.
57. SuenWC, SpainJC (1993) Cloning and characterization of Pseudomonas sp. strain DNT genes for 2,4-dinitrotoluene degradation. J Bacteriol 175 : 1831–1837.
58. LeeJ, TeitzelGM, MunkvoldK, del PozoO, MartinGB, et al. (2012) Type III secretion and effectors shape the survival and growth pattern of Pseudomonas syringae on leaf surfaces. Plant Physiol 158 : 1803–1818.
59. Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory.
60. AbrilMA, MichanC, TimmisKN, RamosJL (1989) Regulator and enzyme specificities of the TOL plasmid-encoded upper pathway for degradation of aromatic hydrocarbons and expansion of the substrate range of the pathway. J Bacteriol 171 : 6782–6790.
61. NikelPI, de LorenzoV (2013) Implantation of unmarked regulatory and metabolic modules in Gram-negative bacteria with specialised mini-transposon delivery vectors. J Biotechnol 163 : 143–154.
62. HerreraG, MartínezA, O'ConnorJE, BlancoM (2003) Functional assays of oxidative stress using genetically engineered Escherichia coli strains. Curr Protoc Cytom 11 : 16.11–16.19.
63. BradfordMM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72 : 248–254.
64. LeeY, Peña-LlopisS, KangYS, ShinHD, DempleB, et al. (2006) Expression analysis of the fpr (ferredoxin-NADP+ reductase) gene in Pseudomonas putida KT2440. Biochem Biophys Res Commun 339 : 1246–1254.
65. RoscheWA, FosterPL (2000) Determining mutation rates in bacterial populations. Methods 20 : 4–17.
66. RiddlesPW, BlakeleyRL, ZernerB (1983) Reassessment of Ellman's reagent. Methods Enzymol 91 : 49–60.
67. MattsonMP (2008) Hormesis defined. Ageing Res Rev 7 : 1–7.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy