
Hard Selective Sweep and Ectopic Gene Conversion in a Gene Cluster Affording Environmental Adaptation
Among the rare colonizers of heavy-metal rich toxic soils, Arabidopsis halleri is a compelling model extremophile, physiologically distinct from its sister species A. lyrata, and A. thaliana. Naturally selected metal hypertolerance and extraordinarily high leaf metal accumulation in A. halleri both require Heavy Metal ATPase4 (HMA4) encoding a PIB-type ATPase that pumps Zn2+ and Cd2+ out of specific cell types. Strongly enhanced HMA4 expression results from a combination of gene copy number expansion and cis-regulatory modifications, when compared to A. thaliana. These findings were based on a single accession of A. halleri. Few studies have addressed nucleotide sequence polymorphism at loci known to govern adaptations. We thus sequenced 13 DNA segments across the HMA4 genomic region of multiple A. halleri individuals from diverse habitats. Compared to control loci flanking the three tandem HMA4 gene copies, a gradual depletion of nucleotide sequence diversity and an excess of low-frequency polymorphisms are hallmarks of positive selection in HMA4 promoter regions, culminating at HMA4-3. The accompanying hard selective sweep is segmentally eclipsed as a consequence of recurrent ectopic gene conversion among HMA4 protein-coding sequences, resulting in their concerted evolution. Thus, HMA4 coding sequences exhibit a network-like genealogy and locally enhanced nucleotide sequence diversity within each copy, accompanied by lowered sequence divergence between paralogs in any given individual. Quantitative PCR corroborated that, across A. halleri, three genomic HMA4 copies generate overall 20 - to 130-fold higher transcript levels than in A. thaliana. Together, our observations constitute an unexpectedly complex profile of polymorphism resulting from natural selection for increased gene product dosage. We propose that these findings are paradigmatic of a category of multi-copy genes from a broad range of organisms. Our results emphasize that enhanced gene product dosage, in addition to neo - and sub-functionalization, can account for the genomic maintenance of gene duplicates underlying environmental adaptation.
Published in the journal:
. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003707
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003707
Summary
Among the rare colonizers of heavy-metal rich toxic soils, Arabidopsis halleri is a compelling model extremophile, physiologically distinct from its sister species A. lyrata, and A. thaliana. Naturally selected metal hypertolerance and extraordinarily high leaf metal accumulation in A. halleri both require Heavy Metal ATPase4 (HMA4) encoding a PIB-type ATPase that pumps Zn2+ and Cd2+ out of specific cell types. Strongly enhanced HMA4 expression results from a combination of gene copy number expansion and cis-regulatory modifications, when compared to A. thaliana. These findings were based on a single accession of A. halleri. Few studies have addressed nucleotide sequence polymorphism at loci known to govern adaptations. We thus sequenced 13 DNA segments across the HMA4 genomic region of multiple A. halleri individuals from diverse habitats. Compared to control loci flanking the three tandem HMA4 gene copies, a gradual depletion of nucleotide sequence diversity and an excess of low-frequency polymorphisms are hallmarks of positive selection in HMA4 promoter regions, culminating at HMA4-3. The accompanying hard selective sweep is segmentally eclipsed as a consequence of recurrent ectopic gene conversion among HMA4 protein-coding sequences, resulting in their concerted evolution. Thus, HMA4 coding sequences exhibit a network-like genealogy and locally enhanced nucleotide sequence diversity within each copy, accompanied by lowered sequence divergence between paralogs in any given individual. Quantitative PCR corroborated that, across A. halleri, three genomic HMA4 copies generate overall 20 - to 130-fold higher transcript levels than in A. thaliana. Together, our observations constitute an unexpectedly complex profile of polymorphism resulting from natural selection for increased gene product dosage. We propose that these findings are paradigmatic of a category of multi-copy genes from a broad range of organisms. Our results emphasize that enhanced gene product dosage, in addition to neo - and sub-functionalization, can account for the genomic maintenance of gene duplicates underlying environmental adaptation.
Introduction
Analyses of nucleotide sequence variation bear great promise for advancing our understanding of evolutionary processes. However, such analyses have so far rarely targeted loci of experimentally established roles in naturally selected adaptive traits, and, instead, have mostly been conducted on candidate loci or even anonymous sequences [1]–[3]. Among the highest selection pressures known in ecology are those encountered by plants on metalliferous soils, which contain high, toxic levels of heavy metals from geological anomalies or anthropogenic contamination [4]. Examples of metalliferous soils are the widespread ultramafic (serpentine) soils rich in Ni, Co and Cr, and calamine soils containing high levels of Zn, Cd, and Pb. The extremophile species Arabidopsis halleri is one of the few plant taxa capable of colonizing calamine metalliferous soils [5]. In addition to its hypertolerance to Zn, Cd and likely Pb, A. halleri groups among approximately 500 known taxa of so-called hyperaccumulators of metals such as Ni, Co, Zn or Cd [6], [7]. Hyperaccumulators are characterized by leaf metal concentrations exceeding those of ordinary non-accumulator plants by more than two orders of magnitude. Metal hyperaccumulation contributes to metal hypertolerance and has been proposed to act as an elemental defense against biotic stress [8], [9].
A. halleri is closely related to Arabidopsis lyrata and to the genetic model plant Arabidopsis thaliana, both of which are non-hyperaccumulators and exhibit only basal metal tolerance common to all vascular plants [10]. Different from A. thaliana, A. halleri is an outcrossing, stoloniferous perennial, with a nuclear genome of 2 n = 16 chromosomes [6]. In an attempt to address the molecular basis of Zn and Cd hyperaccumulation and associated hypertolerance in A. halleri, cross-species transcriptomics approaches employing the accession Langelsheim (Germany) established dozens of candidate genes with potential functions in metal homeostasis, of which transcript levels were elevated in A. halleri when compared to A. thaliana [11]–[13]. Functional characterization through various molecular approaches supported a role for several of these genes including HEAVY METAL ATPASE4 (HMA4) [8], [12], HMA3 [11], METAL TRANSPORT PROTEIN1 (MTP1) [11], [14], NICOTIANAMINE SYNTHASE2 (NAS2) [13], [15], and IRON-REGULATED TRANSPORTER3 (IRT3) [6], [7], [16]. Transcript abundance of HMA4 was highest of all identified candidate genes, with more than 100-fold higher transcript levels in both roots and shoots of A. halleri than in A. thaliana or A. lyrata [12], [17]. The HMA4 protein is a plasma membrane transport protein acting in ATP-driven cellular export-mediated detoxification of Zn2+ and Cd2+, as well as root-to-shoot translocation of both metals [8], [18]. The strongly enhanced HMA4 transcript levels present in A. halleri were shown to be necessary not only for metal hypertolerance but also for metal hyperaccumulation, by employing RNA interference-mediated silencing in the A. halleri accession Langelsheim. The introduction into A. thaliana of an AhHMA4 promoter fused to an AhHMA4 cDNA suggested that AhHMA4 alone, however, is not sufficient to generate either metal hypertolerance or hyperaccumulation [8]. In agreement with these findings, genetic studies identified HMA4 and MTP1 to be located within rather large QTL regions for metal hypertolerance in a segregating back-cross 1 population of an inter-specific hybrid cross between A. halleri (accession Auby, France) and A. lyrata [17], [19]. Moreover, HMA4 co-localized with one out of several major QTL for leaf Zn and Cd hyperaccumulation, respectively, in a segregating F2 population [20], [21]. Among the candidate genes of A. halleri characterized in detail to date, HMA4 thus makes the largest contribution to both metal hyperaccumulation and metal hypertolerance.
High HMA4 transcript levels were shown to be attributable to a combination of tandem gene triplication and cis-activation in the Langelsheim accession of A. halleri [8]. Promoter-reporter fusions suggested approximately equivalent quantities and localizations of promoter activity for all three A. halleri HMA4 gene copies, in agreement with copy-specific transcript quantification through quantitative real-time RT-PCR [8]. Because of almost identical protein-coding sequences, the functions of the three HMA4 protein isoforms of A. halleri have not been individually characterized. All these findings supported a critical role of enhanced HMA4 gene product dosage in naturally selected metal hyperaccumulation and hypertolerance of A. halleri [8]. Interestingly, high HMA4 transcript levels, copy number expansion and cis-activation were also reported in Noccaea caerulescens [22], [23], another Zn/Cd hyperaccumulator in the Brassicaceae family, in which metal hyperaccumulation and associated hypertolerance must have evolved independently. Moreover, copy number expansion appears to be common among additional highly expressed metal hyperaccumulation/hypertolerance candidate genes of A. halleri, for example the ZINC-REGULATED TRANSPORTER, IRON-REGULATED TRANSPORTER-RELATED PROTEIN (ZIP) genes ZIP3, ZIP6 and ZIP9 [12], MTP1 [14], [24] and PLANT DEFENSIN (PDF) genes [25].
Gene duplication is known as a major driver of genome evolution over long timescales [26]. In eukaryotic genomes, gene duplications occur spontaneously at rates that are between 100 and 10,000 times higher per locus than those of base substitutions per site [27], [28], thus explaining the presence of substantial gene copy number variation polymorphism in genomes. For example, per haploid genome and generation, S. cerevisiae was estimated to spontaneously acquire about 0.002 non-synonymous base substitutions within coding regions and 0.02 gene duplications [28]. A number of genetic diseases of humans are caused by gene duplication events [29], [30]. Current theory predicts the rapid loss of recent duplicates unless they undergo neo - or sub-functionalization, with few exceptions [26], [31], [32]. However, the factual contribution of gene duplication to evolutionary adaptation as an outcome of natural selection remains poorly understood. Functional diversity and evolutionary dynamics of multigene families are of particular importance in plant and animal immunity, as exemplified by plant Resistance (R) and human Major Histocompatibility Complex (MHC) genes [33]. Natural selection for increased gene product dosage was implied to account for copy number expansion of the BOT1 boron tolerance locus of barley [34], the MATE1 aluminum tolerance locus of maize [35] and the human salivary amylase gene (AMY1) [36]. However, these reports were based merely on functional data encompassing genotype-phenotype relationships, without evidence for selection from an analysis of sequence polymorphism.
Here, we address two gaps in present knowledge, namely whether a signature of selection can indeed be identified at a locus known to functionally govern an adaptive trait and, more specifically, whether positive selection for increased gene product dosage can result in the fixation of gene duplications [37]. We detect positive selection at the copy-number expanded HMA4 metal hypertolerance locus of Arabidopsis halleri. Moreover, we show that the profile of polymorphism is unexpectedly complex as a result of ectopic gene conversion. This work can act as a guide for related studies on other duplicated genes, and warrants caution in targeted analyses as well as genome-wide scans of polymorphism when dealing with presently or historically copy-number expanded loci.
Results/Discussion
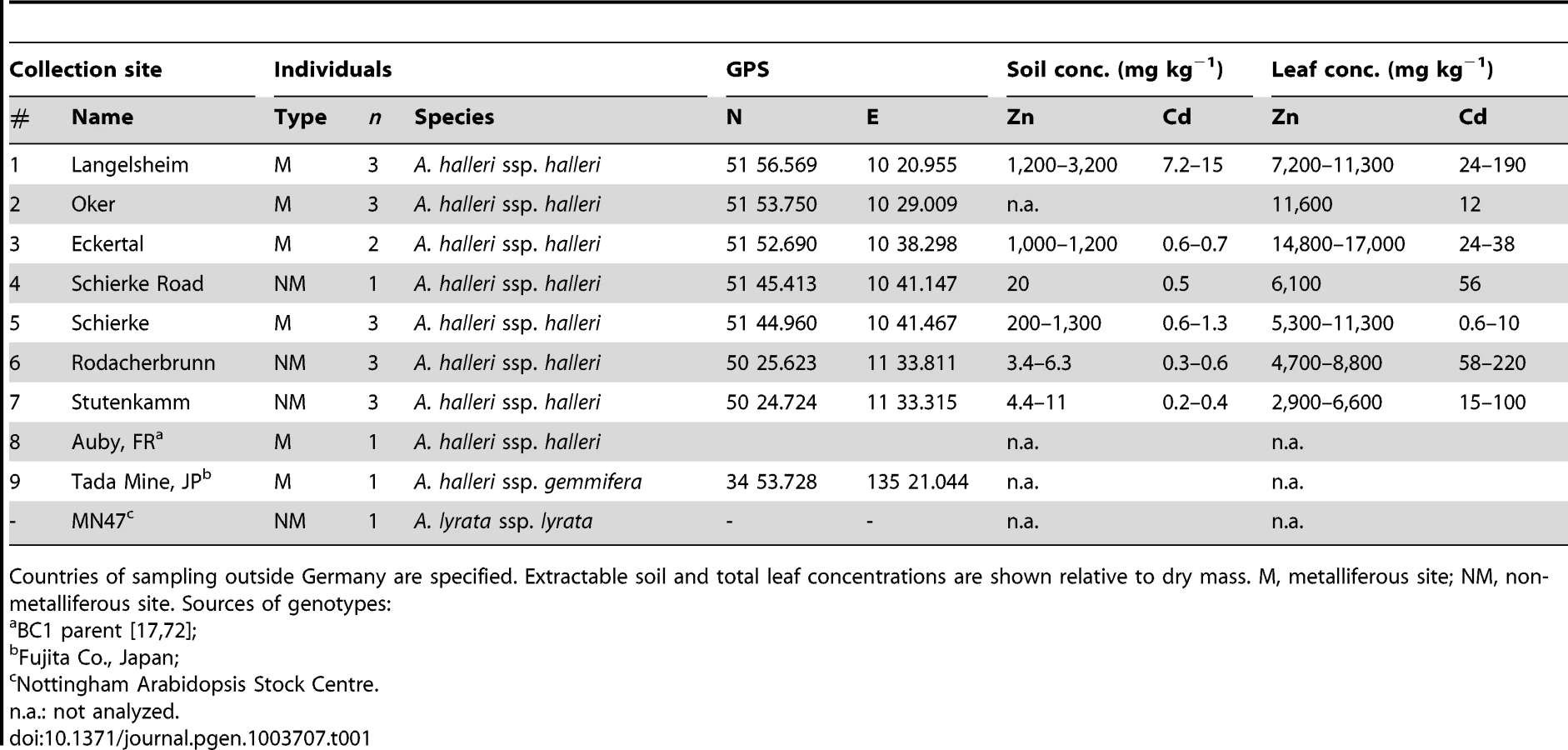
Evidence for Positive Selection in HMA4 Promoter Regions
For an analysis of intra-specific nucleotide sequence diversity across the triple HMA4 genes of A. halleri, we sequenced from multiple individuals (Table 1) series of 13 genomic DNA segments positioned consecutively along the 150-kb HMA4 region and in flanking regions (Figure 1, Figure S1A and Table S1). In more detail, amplicons of between 492 and 2245 bp in length (see Table S1) were designed based on published sequence data, and sequenced from between 15 and 20 individuals (see Table S2; http://www.ebi.ac.uk, accession nos. HE995813 to HE996227). The number of alleles observed per genotype never exceeded expectations of a maximum of two for any of the amplicons (see Table S2, lower section; see Materials and Methods section ‘Sequencing, Sequence Assembly and Assignment of Consensi’). We confirmed leaf metal accumulation in these same individuals by Inductively-Coupled Atomic Emission Spectrometry analysis of field-collected leaves. Maximal concentrations exceeded 10,000 µg Zn g−1 leaf dry biomass and 100 µg Cd g−1 leaf dry biomass in individuals from both non-metalliferous and metalliferous sites that are characterized by toxic levels of metals in the soil and a specialist vegetation (Table 1). For comparison, we also obtained nucleotide sequence data from single individuals of the Zn/Cd-hypertolerant and -hyperaccumulating subspecies A. halleri ssp. gemmifera [38] from East Asia and the closely related Zn/Cd-sensitive, non-hyperaccumulating Arabidopsis lyrata. The genome of A. lyrata contains a single functional HMA4 gene (Figure S1B) in a region that is overall syntenic to A. halleri (Figure 1 and S1A) and A. thaliana (Figure S1C) [39]. In addition, the A. lyrata genome uniquely contains a second, 5′-truncated HMA4-like pseudogene in a non-syntenic position.

If a novel mutation confers a strong selective advantage, the corresponding haplotype is likely to sweep through a population. This reduces or even eliminates pre-existing nucleotide sequence diversity at the affected locus and – proportionately to the extent of genetic linkage – at flanking loci through genetic hitchhiking [40]. In order to test for evidence of a selective sweep in the HMA4 genomic region of A. halleri, we calculated statistics of genetic diversity. At distant control loci (S1 and S13) and loci flanking the ∼150-kb HMA4 genomic region (S2 and S12), average pairwise nucleotide sequence diversity (π) was between 4.9 and 9.1‰ (Figure 1A and Table S2), and thus within the published range for random neutral loci in A. halleri [41]–[43]. Comparable studies on A. halleri ssp. halleri have reported a median π of 3.9‰ (between 0.3 and 37.7‰) for 24 randomly chosen loci [42] and a median π of 4.3‰ (between 1.8 and 32.7‰) for a total of 8 loci [41] (Figure S2). Indeed, this was in sharp contrast with much lower values for π of between 0.1 and 1.8‰ for segments comprising sequences in the promoter regions of the three paralogous HMA4 gene copies (S4, S6, S9; Figure 1A, Figure S1D and Table S2). Compared to the distant and flanking control loci, π decreased gradually towards and within the HMA4 region and reached a minimum of 0.1‰ at the HMA4-3 promoter (S9), yielding a profile as expected upon a hard selective sweep. This characteristic profile of nucleotide sequence diversity was found to be interrupted, however, by elevated π values of between 3.2 and 5.2‰ for segments positioned within the coding sequences of the three HMA4 gene copies (S5, S7, S10) and also for the additional segment S8, all comprising sequences that are present in two or more, almost identical copies in the HMA4 genomic region (Figure 1A, Tables S2 to S4). The overall profile of nucleotide sequence diversity across the HMA4 region was robust against error in sequence assignment to S5, S7 and S10 (see Materials and Methods, Table S4A), as well as towards a regionally separate analysis of individuals from the Harz Mountains and the Thuringian Forest (Table S4B).
To further substantiate the evidence for positive selection in the genomic HMA4 region of A. halleri, we conducted statistical tests of molecular population genetics by calculating Tajima's D, Fu and Li's D*, and Fu and Li's F* [44], [45]. For segments in the promoter regions of HMA4-1 (S4), HMA4-2 (S6) and HMA4-3 (S9), these three tests unanimously indicated an excess of rare polymorphisms resulting from a depletion of higher-frequency, ancestral polymorphisms. A statistically significant deviation from expectations under neutral evolution was detected at the promoter of HMA4-3 (S9; Figure 1B and Table S2), diagnostic of positive selection. Indeed, diversity statistics indicated a unique combination of a very low value for π with a highly negative Tajima's D for S9 (see Figure S2). In agreement with these results, there were fewer long and intermediate-length branches in the topologies of maximum likelihood phylogenetic trees for HMA4 (S4, S6, S9) than observed for control loci on either side of the HMA4 region (S1, S2, S12, S13; Figure 2, Figure S3) [44]. In the region of extremely low sequence diversity in the promoter region of HMA4-3 (S9) of A. halleri ssp. halleri (see Figure 1A), for example, all polymorphisms were unique to single observations (e.g., Figure 2B, 1.1-2, 1.3-2, 5.1-1, 7.2-1; see also Figure 1B). Taken together, statistical tests of sequence diversity, molecular population genetics and sequence phylogenies concordantly support a hard selective sweep centered on the promoter of HMA4-3, with genetic hitchhiking [40] covering a total of 250 kb. This is comparable to previously reported selective sweeps, which affect chromosomal regions of between 60 and 600 kb in length linked to domestication loci of crop plants [46].

HMA4 Transcript Levels
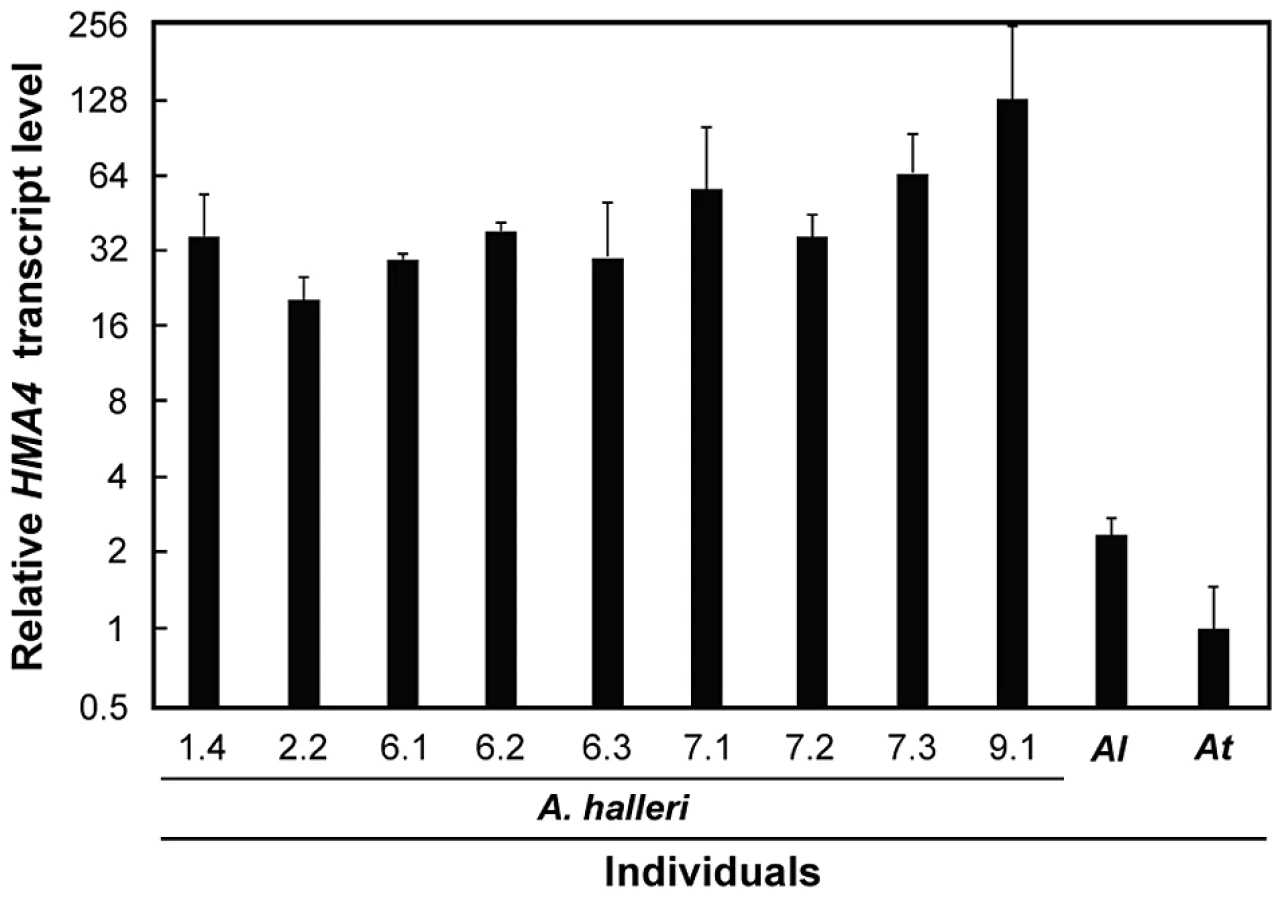
As demonstrated in a single individual of A. halleri [8], the combination of gene copy number expansion and cis-regulatory divergence results in strongly enhanced steady-state HMA4 transcript levels that are necessary for metal hyperaccumulation and hypertolerance. If this was selected for in the entire species A. halleri, as indicated by the diversity statistics (see Figure 1), then we would expect high HMA4 transcript levels in all A. halleri individuals. Indeed, we observed between 20 - and 130-fold higher HMA4 transcript levels across individuals of A. halleri from different collection sites, when compared to A. thaliana (Figure 3). This result supports a substantial increase in HMA4 gene product dosage in all A. halleri ssp. halleri and ssp. gemmifera individuals analyzed here, by comparison to A. lyrata and A. thaliana.
Ectopic Gene Conversion among Protein-Coding Sequences of HMA4 Paralogs
For segments located within coding sequences of the three HMA4 gene copies (S5, S7, S10), relationships among haplotypes differed from those for segments located in HMA4 promoters (S4, S6, S9). Phylogenetic reconstructions of S5, S7 and S10 did not recover three distinct groups of haplotypes as expected for three independently evolving paralogs (Figure S4). Instead, the genealogy resembled a network-like structure, with complex relationships between HMA4 haplotypes at different loci (Figure 4A). For example, out of a total of 25 haplotypes, three were found at two or more of the paralogous HMA4 genes (h13, h20, h25; Figure 4B). These results demonstrate a recurrent transfer of genetic information between the coding sequences of different HMA4 gene copies of A. halleri.

Segmental transfer of genetic information between paralogous sequences can arise in somatic cells during homologous recombination-based repair of double-strand breaks, addressed here as ectopic gene conversion (EGC, also termed interlocus or non-allelic gene conversion), or alternatively result from unequal crossing-over events during meiosis [29], [47], [48]. Quantitative PCR analysis of genomic DNA of A. halleri individuals from different collection sites was consistent with the species-wide presence of three HMA4 gene copies per haploid genome (Figure 5). Average gene copy number was estimated at 3.2±0.2 for A. halleri, compared to 1.8±0.2 and 1.0±0.1 for A. lyrata and A. thaliana, respectively (arithmetic means ± SD), whereby one of the two gene copies detected in A. lyrata is a truncated pseudogene in a non-syntenic position (see Figure S1B). A total of three HMA4 gene copies is in agreement with our observations of a maximum of six alleles observed per individual upon joint PCR amplification of all 3′ HMA4 coding sequences (S5/S7/S10; Table S2), and a maximum of two alleles observed in the promoter region of each HMA4 gene copy (S4, S6, S9; Table S2). The lack of evidence for HMA4 copy number variation among A. halleri individuals suggests that recurrent EGC events account for the segmental transfer of genetic information between paralogous HMA4 coding regions. EGC is known to be common among some genes, for example rRNA genes [29], [47], [49]–[51]. Paralogous genes of eukaryotes have been reported to exchange sequence information at per-locus frequencies even higher than those of spontaneous gene duplications [52], [53], thus contributing significantly to human disease [54]. The contribution of EGC to adaptation, however, is poorly understood.

EGC is predicted to transfer a newly arisen mutation from the site of its origin in one HMA4 paralog to the corresponding sites in the other two paralogs, thus cumulatively enriching species-wide sequence diversity in each individual HMA4 gene copy [55]. This explains the higher levels of nucleotide sequence diversity detected at S5, S7, S8 and S10, when compared to S4, S6 and S9 (see Figure 1) [29], [32]. Simultaneously, EGC suppresses between-copy sequence divergence and thus results in the concerted evolution of the affected loci [29]. Our findings imply that EGC accounts for the high extent of 99 to 99.3% inter-copy sequence identity among A. halleri HMA4-1 to -3 coding sequences (Table S3) [8], consistent with the prevalence of EGC among duplicates of >95% sequence identity known in other organisms [29], [47].
Hallmarks of EGC were also detected in the multi-copy portion of segment S8 outside the HMA4 coding sequence (Figure S5A, Table S3), again with a network-like genealogy (Figure S5B–D) and a comparably high π of 9.5‰ (as opposed to π of 1.7‰ for the single-copy 3′-portion of S8). As in A. halleri ssp. halleri, EGC was also evident among the coding sequences of HMA4 gene copies of A. halleri ssp. gemmifera (Figure S5D and S6), with an apparent additional EGC event between the promoters of HMA4-2 and -3 that was uniquely observed in this individual (S6, S9; see Figure S1D and compare Figure 2B and Figure S3C and S3D).
Population genetics theory and simulations have been developed for small multigene families undergoing concerted evolution [56]–[59]. Nucleotide substitution rates were predicted to be strongly enhanced with increasing gene copy number for beneficial mutations, whereas gene copy number had no effect on substitution rates for selectively neutral mutations [57]. Indeed, the AhHMA4 protein-coding sequences represented in S5/S7/S10, which correspond to the cytoplasmic C-terminal regulatory domain of the HMA4 protein [60], show an over-proportionately high nucleotide sequence divergence of 22% from A. thaliana [8]. By comparison, within coding regions in general, average divergence of both A. halleri and A. lyrata from A. thaliana is around 6%. In the corresponding region of HMA4, A. lyrata is 9% divergent from A. thaliana and 22% divergent from A. halleri. This suggests an enhanced rate of fixed sequence alterations in 3′ AhHMA4 coding sequence of S5/S7/S10, which – according to theoretical considerations – is likely to constitute evidence for positive selection [57]. Different from predictions, however, there is no prevalence of non-synonymous over synonymous nucleotide substitutions in this region, but a prevalence of indel polymorphisms instead. Nonetheless, these considerations suggest that HMA4 gene copy number expansion is not only a result of selection for enhanced gene product dosage, but – in combination with EGC – accommodates an enhanced evolutionary rate of HMA4 under positive directional selection.
Regions of the human genome hosting multigene families that undergo segmental exchange of sequence information have been addressed as hypermutable [8], [30]. Similarly, sequence exchange was proposed to contribute to the unusually high levels of sequence diversity among plant disease Resistance (R) genes, which typically belong to multigene families and are often present in the genome as tandem arrays of multiple paralogous genes [61]. Alongside unequal crossing over and illegitimate inter-allelic recombination, EGC was implicated in the generation of novel pathogen recognition specificities [33], [61]–[64]. The pervasiveness of sequence differences between paralogous R genes, despite sequence exchange, was attributed to small exchanged tracts of sequence of mostly <100 bp among multiple paralogs [61], [63], [64], to the suppression of unequal crossing over within R gene clusters of homozygotes [61], to the occurrence of inter-allelic rather than inter-locus gene conversion [33], or to the past discontinuation of sequence exchange [65]. By comparison to the high sequence diversification among paralogous R genes, the concerted evolution of A. halleri HMA4 paralogs is in stark contrast. This could be interpreted to indicate a prominent role for selection in determining the outcome of inter-locus sequence exchange, a process that appears to be common at least in some classes of multigene families [61], [62], [64].
Ancestry of Nucleotide Sequence Polymorphism in the HMA4 Genomic Region
The evolutionary events reflected in the profile of nucleotide sequence diversity across the HMA4 region of A. halleri occurred concurrent with or after the divergence from the A. lyrata lineage. Whereas nucleotide sequence diversity within A. halleri ssp. halleri was not positively correlated with the genetic divergence from A. lyrata across the HMA4 region (Figure S7A), we detected shared ancestry of nucleotide sequence diversity profiles in the two subspecies of A. halleri, ssp. halleri and ssp. gemmifera. This is supported by a positive correlation between inter-subspecies sequence divergence and sequence diversity π within ssp. halleri (Figure S7B), by the grouping of ssp. gemmifera alleles among ssp. halleri alleles in genealogies (Figures 2, S1D, S3, S4), and by shared polymorphisms among the two A. halleri subspecies in the coding sequences of HMA4 genes (Figure S6) as well as downstream of HMA4-2 (S8) (Figure S5D). These findings also indicate that our sampling captured a large proportion of sequence diversity within A. halleri, which was further confirmed by a larger genetic diversity of A. halleri within collection sites than between collection sites or between regional subgroups of collection sites according to analyses of molecular variance (AMOVA) (Table S5).
Our results support two consecutive duplications of HMA4 with or after the split of the lyrata and halleri lineages, which was estimated at between 2 mio. years ago according to sequence divergence [66] and around 0.34 mio. years ago according to approximate Bayesian computation [43]. Previous estimates of the timing of HMA4 duplication events 0.36 and 0.25 mio. years ago, respectively, are likely to require downward adjustment as they were based on single A. halleri sequences for each of the three gene copies and did not take into account EGC [43].
Conclusion
Enhanced HMA4 gene product dosage is known to functionally underlie the environmental adaptations of heavy metal hyperaccumulation and hypertolerance in the wild plant A. halleri [8]. Here, we detect positive selection in HMA4 promoter regions of A. halleri, incurred by either activating cis-regulatory mutations or gene copy number expansion of HMA4, and likely by both. Furthermore, we identify ectopic gene conversion to effect the concerted evolution of paralogous HMA4 coding sequences, a finding that adds unexpected complexity to the profile of sequence polymorphism. We expect that, together, our results coin a class of multi-copy genes associated not only with instances of environmental adaptation in plants [6], [51], [67], but also more generally with eukaryotic adaptation [29], [32], [36], [37], [68]. Thus, this work will stimulate the development of crop breeding strategies based on gene copy number variation [34], [69]. In the future, complex profiles of nucleotide sequence polymorphism, as exemplified by the HMA4 region of A. halleri, will deserve designated attention in advanced targeted studies as well as in large-scale genome scanning approaches [2], [3], [70]. Subsequent to gene duplication events [27], alongside neo - and sub-functionalization, selection for more of the same gene product is of higher evolutionary relevance than previously appreciated [26], [31], [32], [71].
Materials and Methods
Plant and Soil Sampling, Processing and Multi-Element Analyses
Leaf tissues and soil samples were collected in the field from 18 randomly selected A. halleri ssp. halleri individuals at 7 European sites (Table 1). A minimum distance of 2 m was kept between sampled individuals to avoid sampling clones because A. halleri is stoloniferous. From a subset of collected genotypes, clones were propagated vegetatively and maintained in a greenhouse. For element analysis by Inductively-Coupled Plasma Atomic Emission Spectrometry (ICP-AES), leaf material was washed with ultrapure water and dried at room temperature (RT) for >1 week, followed by processing of samples and measurements as described [11], [12]. For the determination of extractable soil metal concentrations, soil cores were taken down to 0.05 m depth within 0.1 m distance from each individual. Three g of air-dried, sieved soil (2 mm particle size) were extracted in 25 ml of 0.1 M HCl with rotary shaking at 150 rpm at RT for 0.5 h.
For DNA extraction, leaf tissues were frozen in liquid nitrogen immediately after harvest, kept on dry ice for up to 20 h, and stored at −80°C until further processing. Additionally, previously characterized greenhouse-cultivated, clonally propagated genotypes were included in some experiments: the BC1 parent individual from Auby (individual 8.1) [17], [72], individuals 1.1/Lan 3.1 [8], [12] and 1.4/W504 [13] from Langelsheim, and an individual (9.1) of A. halleri ssp. gemmifera [38] (Table 1).
Genomic DNA Preparation and DNA Cloning
Genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, Venlo, The Netherlands) from 100 mg of frozen leaf material of each genotype. The thirteen amplicons designed to analyze sequence diversity (S1 to S13) comprised either non-coding (i.e., promoter, UTR and intron) or both non-coding and coding sequences, and were positioned within all three of the HMA4 gene copies and at loci of increasing distances upstream and downstream of HMA4 (Figure 1, Figure S1A, Tables S1 and S2). No additional amplicons could be designed in the repeat - and transposon-rich genomic regions between HMA4 genes [8]. Primer sequences for amplicons S2 to S12 were designed based on available A. halleri BAC sequences (Genbank accession numbers EU382073.1 and EU382072.1) (Table S1) [8]. Primer design for S1 and S13 was based on the Arabidopsis thaliana and Arabidopsis lyrata ssp. lyrata genome sequences [39], [73]. In A. thaliana and A. lyrata, S1 is located 116 and 198 kb upstream of S2, and S13 is located 113 kb and 2.47 Mbp downstream of S12, respectively. Amplicons comprising the 3′-portions of AhHMA4-1 (S5), AhHMA4-2 (S7) and AhHMA4-3 (S10) were simultaneously amplified in each of three independent PCRs using primer pairs that were not copy-specific (Table S1). In contrast, primers for S8 amplified only the 3′-end of AhHMA4-2 and additional downstream intergenic sequence, taking advantage of copy-specific sequence polymorphisms in the design of the reverse primer (see Figure S5A).
For PCR amplification, 2 µl of genomic DNA were used with GoTaq DNA polymerase (S1, S2, S11 and S13, Promega, Leiden, The Netherlands), Bio-X-Act Long DNA polymerase (S4, S6, Bioline/Gentaur, Brussels, Belgium) or a mix of both enzymes (S3, S9, S12 and S5/S7/S10), the respective primer pairs (0.5 µM each) (Table S1) and dNTPs (200 µM each) (Fermentas, St. Leon-Rot, Germany) in a final volume of 25 µl, the latter enzyme allowing more efficient amplification. PCR reactions were carried out as follows: 3 min at 95°C, followed by 30–32 cycles of 30 s at 95°C, 30 s at 58°C, 1 min per kb at 70–72°C, and a final extension step of 7 min at 70–72°C. PCR products were gel-purified and cloned into the pGEM-T easy vector (Promega, Leiden, The Netherlands) before transformation of E. coli DH5α.
Sequencing, Sequence Assembly and Assignment of Consensi
In order to ensure with high probability that both alleles were sampled in heterozygous individuals through DNA sequencing, plasmid DNA was isolated from overnight cultures of at least eight independent bacterial colonies per amplicon and genotype, 20 clones for S6 and a total of 56 clones for S5/S7/S10, respectively, before sequencing of inserts by the Sanger method on an ABI 3730xl automated sequencer (Applied Biosystems, Darmstadt, Germany) using vector-specific and additional locus-specific primers when required (Table S1). For two individuals, 48 additional clones from two further independent PCRs were sequenced for S5/S7/S10 to resolve remaining sequence ambiguities.
For the S6 amplicon (corresponding to the promoter region of HMA4-2), a set of substantially divergent sequences was initially obtained, and, including these, a total of more than the expected maximum of two types of S6 sequences, corresponding to two alleles expected per individual at this single locus, were found in several A. halleri individuals. Using a combination of PCR, BAC end sequencing and DNA gel blot analyses of previously isolated A. halleri BACs harboring HMA4 and related sequences [8], the divergent set of sequences was unequivocally attributed to the promoter of AhHMA2, which was found to occur in tandem with AhHMA3 on a BAC clone, but this BAC did not contain any AhHMA4 coding sequence. AhHMA2 and AhHMA3 are orthologs of AtHMA2 and AtHMA3 that are located in tandem on chromosome 4 of A. thaliana whereas AtHMA4 is on chromosome 2. HMA4, HMA2 and HMA3 genes all encode divalent transition metal cation-transporting P1B-type ATPases [18], [74].
Sequence assembly was conducted with DNASTAR (DNASTAR Inc., Madison, USA). First, a consensus sequence was generated for each clone. Then, each consensus was compared to all other consensi from the same amplicon in a given individual and to all consensi of the same amplicon from all other individuals to i) correct Taq polymerase errors, ii) identify recombinant chimeras that resulted from template switches during PCR amplification [75] and iii) distinguish heterozygous from homozygous loci.
For the 3′-regions of the three HMA4 gene copies (S5, S7, S10, S8) more than 800 sequences were obtained in total. Among these, sequences were considered to be authentic when the same sequence was observed at least three times from one PCR reaction or in at least two independent PCRs of the same genotype. After removal of chimeras (which accounted for ca. 5% of the sequences), a total of 25 consensus sequences were retained for the 3′-regions of HMA4 gene copies. These consensi were assigned to the three HMA4 loci taking advantage of i) the copy-specific sequence information for AhHMA4-2 via the overlap between S7 and S8 for each individual (see Figure S5A), ii) position information available from two completely sequenced BACs [8], and iii) step-wise inference using a strictly parsimonious approach, similar to the strategy used to solve a SUDOKU in two times three double-blind independent replicates to ensure reproducibility.
Statistical and Phylogenetic Analyses
After sequence assembly and alignment, DnaSP v5 [76] was used to calculate sequence diversity (π), Tajima's D [45], Fu and Li's D* and F* [44], and to conduct other statistical tests of molecular population genetics. MEGA v5 was used for phylogenetic analyses [77]. The ML trees shown throughout were constructed using a general time-reversible model. Rates among sites were assumed to be gamma-distributed with invariant sites, and 5 discrete categories of gamma were used. All sites were used. To estimate bootstrap support for the nodes, 1000 replicates were calculated. Neighbor joining methods yielded essentially the same results for tree branching orders. Genome sequence information from A. lyrata ssp. lyrata was used as a reference [39]. Network analyses for HMA4 genes and for S8 were conducted with TCS v1.21 using a connection limit of 95% [78]. Alignment gaps were re-coded with nucleotides to reflect the exact number of mutational steps between sequences in the respective sequence portion. AMOVA (Analysis of Molecular Variance) was carried out with Arlequin 3.5 [79] to compare the contribution of three hierarchical levels to genetic variance: among the geographic regions of the Thuringian Forest (A. halleri ssp. halleri), the Harz Mountains (A. halleri ssp. halleri), and Japan (A. halleri ssp. gemmifera), among geographic collection sites in each of these three regions, and within single geographic collection sites. A total of 1000 permutations were carried out for each locus, with equal weights of 1 for transitions and transversions, and a deletion weight of 0.
Determination of HMA4 Gene Copy Number
Quantitative PCR reactions were performed on 5 ng of genomic DNA in 384-well plates with an ABI Prism 7900HT system (Applied Biosystems, Brussels, Belgium) using MESA GREEN qPCR MasterMix (Eurogentec, Liège, Belgium). Mean reaction efficiencies were determined from all reactions for each amplicon (>270 reactions, Table S6) [80] and used to calculate relative gene copy number by normalization with the qBase software [81] using (i) multiple single-copy reference amplicons and (ii) A. thaliana genomic DNA (Col-0) as a calibrator [82]. Three single-copy reference amplicons were selected and designed at the 5′ - and 3′-ends of the AhFRD3 gene [12] and in the S13 amplicon (this study), respectively. Their adequacy to normalize gene copy number in our experimental conditions was validated using the geNorm module in qBase (gene stability measure M = 0.309, pairwise variation CV = 0.121) [83].
Quantification of Relative HMA4 Transcript Levels
Fresh cuttings of greenhouse-grown A. halleri and A. lyrata genotypes were cultivated hydroponically in 0.1× Hoagland solution for about 2 weeks [13]. After rooting, plants were transferred to pots with soil and further grown in a greenhouse with temperature settings of 22°C (day)/20°C (night) and a photoperiod of 16 h light and 8 h dark. Leaf material was harvested twice independently from the same individuals at an interval of eight weeks, immediately frozen in liquid nitrogen and stored at −80°C. A. thaliana and A. lyrata plants were grown from seeds as described, with harvest of leaves from 6-week-old plants, alongside harvest of A. halleri tissues [12]. Total RNA was extracted with TRIzol Reagent (Invitrogen, Karlsruhe, Germany), cDNA was synthesized from 1 µg of DNaseI-treated (Invitrogen) total RNA using oligo-dT and the SuperScript First-Strand Synthesis System (Invitrogen). Quantitative PCR was conducted in 96-well plates with a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad, Munich, Germany) using SYBR Green qPCR Master Mix (Eurogentec, Cologne, Germany). A total of three technical repeats were run per cDNA and primer pair combination. Data were analyzed using iQ5 Optical System Software version 2.0 (Bio-Rad). Relative transcript levels of HMA4 were calculated by normalization to EF1α as a constitutively expressed reference gene [12]. Primers were as follows: AhHMA4 primers (5′ - GCTGCAGCGATGAAAAACAAAC-3′ and 5′-TCCATACAACATCCCGAGGAAC-3′; amplification efficiency: 1.88); AlHMA4 primers (5′ - TGAAGGTGGTGGTGATTGCA-3′ and 5′-CTCTCCACATTGACCAACTTTG-3′; amplification efficiency: 1.90). AtHMA4 and EF1α primers were described earlier [12].
Accession Numbers
Sequence data are available through EBI (http://www.ebi.ac.uk), accession nos. HE995813 to HE996227.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HoekstraHE, CoyneJA (2007) The locus of evolution: evo devo and the genetics of adaptation. Evolution 61 : 995–1016.
2. Mitchell-OldsT, WillisJH, GoldsteinDB (2007) Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat Rev Genet 8 : 845–856.
3. StinchcombeJR, HoekstraHE (2008) Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100 : 158–170.
4. AntonovicsJ, BradshawAD, TurnerRG (1971) Heavy metal tolerance in plants. Adv Ecol Res 7 : 1–85.
5. Ernst WHO (1974) Schwermetallvegetationen der Erde. Stuttgart, Germany: Gustav Fischer Verlag.
6. KrämerU (2010) Metal hyperaccumulation in plants. Annu Rev Plant Biol 61 : 517–534.
7. VerbruggenN, HermansC, SchatH (2009) Molecular mechanisms of metal hyperaccumulation in plants. New Phytol 181 : 759–776.
8. HanikenneM, TalkeIN, HaydonMJ, LanzC, NolteA, et al. (2008) Evolution of metal hyperaccumulation required cis-regulatory changes and triplication of HMA4. Nature 453 : 391–395.
9. BoydR (2010) Elemental defenses of plants by metals. Nature Education Knowledge 1 : 6.
10. ClemensS (2001) Molecular mechanisms of plant metal tolerance and homeostasis. Planta 212 : 475–486.
11. BecherM, TalkeIN, KrallL, KrämerU (2004) Cross-species microarray transcript profiling reveals high constitutive expression of metal homeostasis genes in shoots of the zinc hyperaccumulator Arabidopsis halleri. Plant J 37 : 251–268.
12. TalkeIN, HanikenneM, KrämerU (2006) Zinc-dependent global transcriptional control, transcriptional deregulation, and higher gene copy number for genes in metal homeostasis of the hyperaccumulator Arabidopsis halleri. Plant Physiol 142 : 148–167.
13. WeberM, HaradaE, VessC, Roepenack-LahayeEV, ClemensS (2004) Comparative microarray analysis of Arabidopsis thaliana and Arabidopsis halleri roots identifies nicotianamine synthase, a ZIP transporter and other genes as potential metal hyperaccumulation factors. Plant J 37 : 269–281.
14. DrägerDB, Desbrosses-FonrougeAG, KrachC, ChardonnensAN, MeyerRC, et al. (2004) Two genes encoding Arabidopsis halleri MTP1 metal transport proteins co-segregate with zinc tolerance and account for high MTP1 transcript levels. Plant J 39 : 425–439.
15. DeinleinU, WeberM, SchmidtH, RenschS, TrampczynskaA, et al. (2012) Elevated nicotianamine levels in Arabidopsis halleri roots play a key role in zinc hyperaccumulation. Plant Cell 24 : 708–723.
16. LinYF, LiangHM, YangSY, BochA, ClemensS, et al. (2009) Arabidopsis IRT3 is a zinc-regulated and plasma membrane localized zinc/iron transporter. New Phytol 182 : 392–404.
17. CourbotM, WillemsG, MotteP, ArvidssonS, RoosensN, et al. (2007) A major QTL for Cd tolerance in Arabidopsis halleri co-localizes with HMA4, a gene encoding a heavy metal ATPase. Plant Physiol 144 : 1052–1065.
18. HussainD, HaydonMJ, WangY, WongE, ShersonSM, et al. (2004) P-type ATPase heavy metal transporters with roles in essential zinc homeostasis in Arabidopsis. Plant Cell 16 : 1327–1339.
19. WillemsG, DrägerDB, CourbotM, GodeC, VerbruggenN, et al. (2007) The genetic basis of zinc tolerance in the metallophyte Arabidopsis halleri ssp. halleri (Brassicaceae): An analysis of quantitative trait loci. Genetics 176 : 659–674.
20. WillemsG, FrérotH, GennenJ, SalisP, Saumitou-LapradeP, et al. (2010) Quantitative trait loci analysis of mineral element concentrations in an Arabidopsis halleri×Arabidopsis lyrata petraea F2 progeny grown on cadmium-contaminated soil. New Phytol 187 : 368–379.
21. FrérotH, FauconMP, WillemsG, GodeC, CourseauxA, et al. (2010) Genetic architecture of zinc hyperaccumulation in Arabidopsis halleri: the essential role of QTL×environment interactions. New Phytol 187 : 355–367.
22. PapoyanA, KochianLV (2004) Identification of Thlaspi caerulescens genes that may be involved in heavy metal hyperaccumulation and tolerance. Characterization of a novel heavy metal transporting ATPase. Plant Physiol 136 : 3814–3823.
23. O'LochlainnS, BowenHC, FrayRG, HammondJP, KingGJ, et al. (2011) Tandem quadruplication of HMA4 in the zinc (Zn) and cadmium (Cd) hyperaccumulator Noccaea caerulescens. PloS One 6: e17814.
24. ShahzadZ, GostiF, FrérotH, LacombeE, RoosensN, et al. (2010) The five AhMTP1 zinc transporters undergo different evolutionary fates towards adaptive evolution to zinc tolerance in Arabidopsis halleri. PLoS Genet 6: e1000911.
25. MirouzeM, SelsJ, RichardO, CzernicP, LoubetS, et al. (2006) A putative novel role for plant defensins: a defensin from the zinc hyper-accumulating plant, Arabidopsis halleri, confers zinc tolerance. Plant J 47 : 329–342.
26. Ohno S (1970) Evolution by gene duplication. New York: Springer.
27. LipinskiKJ, FarslowJC, FitzpatrickKA, LynchM, KatjuV, et al. (2011) High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol 21 : 306–310.
28. LynchM, SungW, MorrisK, CoffeyN, LandryCR, et al. (2008) A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A 105 : 9272–9277.
29. ChenJM, CooperDN, ChuzhanovaN, FerecC, PatrinosGP (2007) Gene conversion: mechanisms, evolution and human disease. Nat Rev Genet 8 : 762–775.
30. MichaelsonJJ, ShiY, GujralM, ZhengH, MalhotraD, et al. (2012) Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151 : 1431–1442.
31. LynchM, ConeryJS (2000) The evolutionary fate and consequences of duplicate genes. Science 290 : 1151–1155.
32. InnanH, KondrashovF (2010) The evolution of gene duplications: classifying and distinguishing between models. Nat Rev Genet 11 : 97–108.
33. MichelmoreRW, MeyersBC (1998) Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8 : 1113–1130.
34. SuttonT, BaumannU, HayesJ, CollinsNC, ShiBJ, et al. (2007) Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science 318 : 1446–1449.
35. MaronLG, GuimaraesCT, KirstM, AlbertPS, BirchlerJA, et al. (2013) Aluminum tolerance in maize is associated with higher MATE1 gene copy number. Proc Natl Acad Sci U S A 110 : 5241–5246.
36. PerryGH, DominyNJ, ClawKG, LeeAS, FieglerH, et al. (2007) Diet and the evolution of human amylase gene copy number variation. Nat Genet 39 : 1256–1260.
37. KondrashovFA (2012) Gene duplication as a mechanism of genomic adaptation to a changing environment. Phil Trans R Soc B 279 : 5048–5057.
38. KubotaH, TakenakaC (2003) Arabis gemmifera is a hyperaccumulator of Cd and Zn. Int J Phytoremediation 5 : 197–201.
39. HuTT, PattynP, BakkerEG, CaoJ, ChengJF, et al. (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43 : 476–481.
40. BartonNH (2000) Genetic hitchhiking. Phil Trans R Soc B 355 : 1553–1562.
41. Ramos-OnsinsSE, StrangerBE, Mitchell-OldsT, AguadeM (2004) Multilocus analysis of variation and speciation in the closely related species Arabidopsis halleri and A. lyrata. Genetics 166 : 373–388.
42. HeidelAJ, Ramos-OnsinsSE, WangWK, ChiangTY, Mitchell-OldsT (2010) Population history in Arabidopsis halleri using multilocus analysis. Mol Ecol 19 : 3364–3379.
43. RouxC, CastricV, PauwelsM, WrightSI, Saumitou-LapradeP, et al. (2011) Does speciation between Arabidopsis halleri and Arabidopsis lyrata coincide with major changes in a molecular target of adaptation? PloS One 6: e26872.
44. FuYX, LiWH (1993) Statistical tests of neutrality of mutations. Genetics 133 : 693–709.
45. TajimaF (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 : 585–595.
46. PuruggananMD, FullerDQ (2009) The nature of selection during plant domestication. Nature 457 : 843–848.
47. BenovoyD, DrouinG (2009) Ectopic gene conversions in the human genome. Genomics 93 : 27–32.
48. PetesTD, HillCW (1988) Recombination Between Repeated Genes in Microorganisms. Ann Rev Genet 22 : 147–168.
49. CasolaC, ConantGC, HahnMW (2012) Very low rate of gene conversion in the yeast genome. Mol Biol Evol 89 : 3817–3826.
50. GaoLZ, InnanH (2004) Very low gene duplication rate in the yeast genome. Science 306 : 1367–1370.
51. KroymannJ, DonnerhackeS, SchnabelrauchD, Mitchell-OldsT (2003) Evolutionary dynamics of an Arabidopsis insect resistance quantitative trait locus. Proc Natl Acad Sci U S A 100 : 14587–14592.
52. BoschE, HurlesME, NavarroA, JoblingMA (2004) Dynamics of a human interparalog gene conversion hotspot. Genome Res 14 : 835–844.
53. AssisR, KondrashovAS (2012) A strong deletion bias in nonallelic gene conversion. PLoS Genet 8: e1002508.
54. CasolaC, ZekonyteU, PhillipsAD, CooperDN, HahnMW (2012) Interlocus gene conversion events introduce deleterious mutations into at least 1% of human genes associated with inherited disease. Genome Res 22 : 429–435.
55. InnanH (2002) A method for estimating the mutation, gene conversion and recombination parameters in small multigene families. Genetics 161 : 865–872.
56. InnanH (2003) The coalescent and infinite-site model of a small multigene family. Genetics 163 : 803–810.
57. ManoS, InnanH (2008) The evolutionary rate of duplicated genes under concerted evolution. Genetics 180 : 493–505.
58. TeshimaKM, InnanH (2012) The coalescent with selection on copy number variants. Genetics 190 : 1077–1086.
59. OhtaT (1983) On the evolution of multigene families. Theor Popul Biol 23 : 216–240.
60. BaekgaardL, MikkelsenMD, SorensenDM, HegelundJN, PerssonDP, et al. (2010) A combined zinc/cadmium sensor and zinc/cadmium export regulator in a heavy metal pump. J Biol Chem 285 : 31243–31252.
61. ParniskeM, Hammond-KosackKE, GolsteinC, ThomasCM, JonesDA, et al. (1997) Novel disease resistance specificities result from sequence exchange between tandemly repeated genes at the Cf-4/9 locus of tomato. Cell 91 : 821–832.
62. KuangH, CaldwellKS, MeyersBC, MichelmoreRW (2008) Frequent sequence exchanges between homologs of RPP8 in Arabidopsis are not necessarily associated with genomic proximity. Plant J 54 : 69–80.
63. KuangH, WooSS, MeyersBC, NevoE, MichelmoreRW (2004) Multiple genetic processes result in heterogeneous rates of evolution within the major cluster disease resistance genes in lettuce. Plant Cell 16 : 2870–2894.
64. Mondragon-PalominoM, GautBS (2005) Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol Biol Evol 22 : 2444–2456.
65. BergelsonJ, KreitmanM, StahlEA, TianD (2001) Evolutionary dynamics of plant R-genes. Science 292 : 2281–2285.
66. KochMA, MatschingerM (2007) Evolution and genetic differentiation among relatives of Arabidopsis thaliana. Proc Natl Acad Sci U S A 104 : 6272–6277.
67. DassanayakeM, OhDH, HaasJS, HernandezA, HongH, et al. (2011) The genome of the extremophile crucifer Thellungiella parvula. Nat Genet 43 : 913–918.
68. NairS, NashD, SudimackD, JaideeA, BarendsM, et al. (2007) Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol Biol Evol 24 : 562–573.
69. CookDE, LeeTG, GuoX, MelitoS, WangK, et al. (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338 : 1206–1209.
70. TurnerTL, BourneEC, Von WettbergEJ, HuTT, NuzhdinSV (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet 42 : 260–263.
71. SuginoRP, InnanH (2006) Selection for more of the same product as a force to enhance concerted evolution of duplicated genes. Trends Genet 22 : 642–644.
72. BertV, MacNairMR, De LaguérieP, Saumitou-LapradeP, PetitD (2000) Zinc tolerance and accumulation in metallicolous and non metallicolous populations of Arabidopsis halleri (Brassicaceae). New Phytol 146 : 225–233.
73. The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 : 796–815.
74. MorelM, CrouzetJ, GravotA, AuroyP, LeonhardtN, et al. (2009) AtHMA3, a P1B-ATPase allowing Cd/Zn/Co/Pb vacuolar storage in Arabidopsis. Plant Physiol 149 : 894–904.
75. BradleyRD, HillisDM (1997) Recombinant DNA sequences generated by PCR amplification. Mol Biol Evol 14 : 592–593.
76. LibradoP, RozasJ (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 : 1451–1452.
77. TamuraK, PetersonD, PetersonN, StecherG, NeiM, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28 : 2731–2739.
78. ClementM, PosadaD, CrandallKA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9 : 1657–1659.
79. ExcoffierL, LischerHE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10 : 564–567.
80. RamakersC, RuijterJM, DeprezRH, MoormanAF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339 : 62–66.
81. HellemansJ, MortierG, De PaepeA, SpelemanF, VandesompeleJ (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8: R19.
82. D'HaeneB, VandesompeleJ, HellemansJ (2010) Accurate and objective copy number profiling using real-time quantitative PCR. Methods 50 : 262–270.
83. VandesompeleJ, De PreterK, PattynF, PoppeB, Van RoyN, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy