
Comparative Genomic and Functional Analysis of 100 Strains and Their Comparison with Strain GG
Lactobacillus rhamnosus is a lactic acid bacterium that is found in a large variety of ecological habitats, including artisanal and industrial dairy products, the oral cavity, intestinal tract or vagina. To gain insights into the genetic complexity and ecological versatility of the species L. rhamnosus, we examined the genomes and phenotypes of 100 L. rhamnosus strains isolated from diverse sources. The genomes of 100 L. rhamnosus strains were mapped onto the L. rhamnosus GG reference genome. These strains were phenotypically characterized for a wide range of metabolic, antagonistic, signalling and functional properties. Phylogenomic analysis showed multiple groupings of the species that could partly be associated with their ecological niches. We identified 17 highly variable regions that encode functions related to lifestyle, i.e. carbohydrate transport and metabolism, production of mucus-binding pili, bile salt resistance, prophages and CRISPR adaptive immunity. Integration of the phenotypic and genomic data revealed that some L. rhamnosus strains possibly resided in multiple niches, illustrating the dynamics of bacterial habitats. The present study showed two distinctive geno-phenotypes in the L. rhamnosus species. The geno-phenotype A suggests an adaptation to stable nutrient-rich niches, i.e. milk-derivative products, reflected by the alteration or loss of biological functions associated with antimicrobial activity spectrum, stress resistance, adaptability and fitness to a distinctive range of habitats. In contrast, the geno-phenotype B displays adequate traits to a variable environment, such as the intestinal tract, in terms of nutrient resources, bacterial population density and host effects.
Published in the journal:
. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003683
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003683
Summary
Lactobacillus rhamnosus is a lactic acid bacterium that is found in a large variety of ecological habitats, including artisanal and industrial dairy products, the oral cavity, intestinal tract or vagina. To gain insights into the genetic complexity and ecological versatility of the species L. rhamnosus, we examined the genomes and phenotypes of 100 L. rhamnosus strains isolated from diverse sources. The genomes of 100 L. rhamnosus strains were mapped onto the L. rhamnosus GG reference genome. These strains were phenotypically characterized for a wide range of metabolic, antagonistic, signalling and functional properties. Phylogenomic analysis showed multiple groupings of the species that could partly be associated with their ecological niches. We identified 17 highly variable regions that encode functions related to lifestyle, i.e. carbohydrate transport and metabolism, production of mucus-binding pili, bile salt resistance, prophages and CRISPR adaptive immunity. Integration of the phenotypic and genomic data revealed that some L. rhamnosus strains possibly resided in multiple niches, illustrating the dynamics of bacterial habitats. The present study showed two distinctive geno-phenotypes in the L. rhamnosus species. The geno-phenotype A suggests an adaptation to stable nutrient-rich niches, i.e. milk-derivative products, reflected by the alteration or loss of biological functions associated with antimicrobial activity spectrum, stress resistance, adaptability and fitness to a distinctive range of habitats. In contrast, the geno-phenotype B displays adequate traits to a variable environment, such as the intestinal tract, in terms of nutrient resources, bacterial population density and host effects.
Introduction
The current development and application of high-throughput sequencing technologies allow to intensively investigate complex microbial ecosystems, such as the human gastro-intestinal (GI) microbiota, consisting of over 3 million genes from mainly Gram-positive bacteria [1]–[4]. This and other metagenomic approaches obviate the necessity to culture bacterial isolates to comprehend the richness and the diversity of such ecosystem. However, detailed analysis at the strain level still requires isolation and growth of bacterial residents. Gram-positive lactobacilli are naturally found among ∼1000 phylotypes identified in the human intestinal tract [2], but only a fraction is represented in the present metagenomic sequences that derive from faecal samples. Lactobacilli mainly reside in the intestinal mucosa and were detected in the ileum metagenome [5], [6]. The limitations of metagenomic approach, i.e. sequencing depth, are well described and can be obviated by 16S rRNA sequencing and phylogenetic microarrays [7], [8]. Thus, Heilig et al. provided clear sequence-based evidence for the presence of L. rhamnosus related species in the human intestinal tract [9]. As a consequence of their interactions and ecological role in the human intestinal tract [10]–[13], lactobacilli are increasingly used in food production, food preservation and nutritional complement formulation [14]–[18]. One of the most used and documented lactobacilli marketed as a probiotic is Lactobacillus rhamnosus GG, which has been isolated from the human intestine and characterized extensively [19]–[21]. L. rhamnosus contains a 3.0-Mbp genome, among the largest of the lactic acid bacteria, and has the ability to persist in the human intestinal mucosa, as it produces pili that are decorated with the mucus-binding protein SpaC [22]–[26]. This significantly impacts the intestinal microbiota, via the displacement of pathogenic bacteria [27], modulation of epithelial barrier functions [28] and potential stimulation of the host immune system via bacteria-host surface molecule crosstalk [16], [29]–[31]. Since the interaction between host and bacteria has a pivotal role in the impact on the host, much research efforts are presently focused on characterizing the different interaction mechanisms, including the metabolic properties and host-signalling components of L. rhamnosus [30]. However, no studies have actually addressed the genomic diversity of the species L. rhamnosus, in spite of its extensive use in a variety of food products. While some Lactobacillus species have been found in only one dedicated niche, such as the milk-adapted L. helveticus [32], other lactobacilli such as L. rhamnosus, L. casei or L. plantarum have the capacity to colonize multiple habitats [15], [33]–[35]. More specifically, L. rhamnosus has been isolated from a large variety of ecological niches, e.g. human intestinal tract, vaginal cavity, oral cavity and cheese, exemplifying its remarkable ecological adaptability as a generalist [19], [36]–[39].
Genome sequence analysis of a number of lactobacilli revealed that their adaptation to diverse ecological niches is promoted by the acquisition of new genes by horizontal gene transfer and the decay or loss of non-essential genes [33], [35], [40], [41]. The domestication of some lactobacilli to the dairy environment is a typical example of a niche specialization, where milk-adapted strains have unusually high number of pseudogenes, reflected by the loss of metabolic pathways and transport systems that are non-essential in dairy niches rich in nutrients [40], [42]. In contrast, bacteria from the intestinal tract, a very dynamic habitat in terms of nutrient availability and bacterial population density, have broad metabolic capacities and lifestyle traits essential for survival, persistence and colonization in this niche, e.g. bile resistance [25], [43], anti-microbial activity [44], and mucus-binding pili expression [19]. In some cases, gene sets could even be specifically linked to a particular ecological niche, i.e. intestine vs. dairy environment, as reported for the related L. acidophilus and L. helveticus [40]. In L. reuteri, Frese and colleagues also demonstrated a host specialization between L. reuteri strains isolated from different vertebrates [45].
The present study of the species L. rhamnosus aimed at: (a) investigating the genomic diversity of the species and, (b) examining variable chromosomal regions associated with phenotypic and/or lifestyle traits found in L. rhamnosus isolates. Four complete L. rhamnosus genomes have been fully sequenced and assembled allowing us to have a glance at the diversity within the species [19], [46], [47]. In an effort to further comprehend the diversity and versatility of L. rhamnosus species, we sequenced and compared the genomes of 100 Lactobacillus rhamnosus strains that were isolated from different ecological niches and analyzed their phenotypes. This study represents the first large-scale genomic and functional analysis of L. rhamnosus, providing new insight in the genetics and lifestyle of this species that has a long history associated with human lifestyle and health.
Results and Discussion
General genomic features of the species L. rhamnosus
To comprehensively depict the phenotypic and genomic diversity of the L. rhamnosus species, 100 L. rhamnosus strains were isolated from a broad spectrum of ecological niches, e.g. 77 strains of various sites of the human body (oral cavity, vaginal cavity, blood and intestinal tract) and 23 strains of dairy origins, including artisanal cheeses and products marketed as probiotics (Table S1). The genomes of all strains were sequenced using the SOLiD sequencing technology and reads were mapped onto the L. rhamnosus GG chromosome [48]. This allowed detailed comparative genomic analysis and data mining as described in the Materials and Methods section. The number of shared genes between the 100 L. rhamnosus isolates and L. rhamnosus GG ranged from 2622/3016 (86.9%) to 3016/3016 (100%) genes with a median number of 2918/3016 (96.7%) genes. In terms of relative gene content, the dairy isolates significantly showed the most diversity with L. rhamnosus GG (average of 92.4%) than the human isolates (average of 96.04%, excluding clinical isolates), indicating that the dairy isolates are genetically most distant (p<0.001 between the two groups). It is noteworthy that 11 strains of human origin, 3 strains isolated from products marketed as probiotics, and only 1 strain isolated from artisanal cheese shared the complete set of 3016 genes present in L. rhamnosus GG. However, it has to be kept in mind that orthologous genes present in these isolates may carry mutations, i.e. single nucleotide polymorphisms, insertion and deletions that were not addressed in detail in this study. Therefore, the presence of a gene may not necessarily reflect its functionality, as observed within these 11 human strains, which showed significant phenotypic variations, i.e. sugar metabolism, indicating that these strains are not L. rhamnosus GG (see below). Moreover, strain-specific genes are likely to be present in these isolates, conferring additional phenotypic traits not present in L. rhamnosus GG. Based on comparative gene content, the hierarchical clustering of the L. rhamnosus species resulted in four distinct clusters (Figure 1). Remarkably, most dairy strains were found to belong to the cluster 1 and show marked differences with other clusters. In contrast, intestinal isolates, including L. rhamnosus strains marketed as probiotics shared similarities with other human isolates (Figures 1 and 2). This is in line with the hypothesis that the genomes of probiotic-marketed strains still reflect their adaptation to their original isolation source, i.e. the human intestinal tract [19]. The distribution of the clinical isolates all across the clustering rather reflects their original ecological niche than their isolation source, since infections are extremely rare events and evolutionary dead ends. The clusters 3 and 4 consist predominantly of L. rhamnosus strains closely related to L. rhamnosus GG (Figure 1). In Figure 2, comparison of hierarchical clustering and phylogenetic tree shows some degree of conservation in the grouping of the strains. The phylogenetic tree reflects slow evolution within the genome, i.e. point mutations, whereas the genomic tree (or hierarchical clustering) describes major genetic re-arrangement events, i.e. insertions or deletions. Hierarchical clustering therefore shows more recent chromosomal changes, where recombination events contribute to the diversity of the species. Similar differences have been observed in other species, such as L. casei [41].


Based on the 100 mapped genomes, we defined a set of all orthologous genes that are shared by all L. rhamnosus strains. We observed that the shared gene set (core) of the L. rhamnosus species consists of 2419 genes, which represents 80.2% of L. rhamnosus GG genome. The larger the set of strains used, the smaller the core genome becomes, a trend observed in other genomes as well, such as the core-genome of Streptococcus agalactiae and other bacterial species [49], [50]. However, the size of the core genome remained stable above ∼20 genomes (data not shown). The full comparative genomic results are shown in Tables S2 and S3. Although the characterization of L. rhamnosus pan-genome would bring further insights into the species, we did not address it in the present study, as this would require complementary sequencing techniques. Further deep and full-coverage sequence analysis of a selected subset of heterogeneous L. rhamnosus strains is now on-going to report the pan-genome of the species (data not shown). The initial read mapping to the reference genome L. rhamnosus strain GG clearly give a GG-centric view of the genome diversity within the species. However, the additional read mapping to the dairy strain LC705 of a selected set of L. rhamnosus strains revealed a similar clustering as in Figures 1 and 2 (data not shown). This suggests that the use of one strain or another as a reference does not impact on the hierarchical clustering of the isolates and also supports the validity of the experimental design approach chosen in the present study.
The distribution of Clusters of Orthologous Groups of proteins (COG) was determined for L. rhamnosus GG genome, the L. rhamnosus core-genome and the non-core gene set (Figure S1). Although no major differences in the relative COG distribution between the different subsets were found, it is noteworthy that 87 L. rhamnosus GG genes (30.2%) out of 288 genes assigned to the COG ‘Carbohydrate transport and metabolism’ are not in the estimated core genome and are predicted to encode mostly phosphotransferase system (PTS) and other sugar transport systems, possibly essential for the persistence in the intestinal tract. These genes were located in highly variable regions of the L. rhamnosus genome, reflecting the metabolic diversity of this species (Figure 3). The 17 most variable chromosomal regions include all genomic islands (GIs), typically rich in transposases and other mobile genetic elements (Figure 1 and Table 1). In L. rhamnosus GG, 5 GIs had previously been identified [19]. The presence of these genomic islands greatly varies among strains of the species L. rhamnosus, as observed previously for the strains LC705 and GG [19]. This suggests that horizontal gene transfer events have contributed significantly to the diversity of the L. rhamnosus species. The GIs identified here were associated with specific biological functions, including interaction and signalling with the host, optimal use of available nutrients and protection against autochthonous phages and mobile genetic elements. Hence they may be considered as lifestyle islands, as their predicted function may specifically contribute to the persistence and colonization in intestinal and other habitats. Other variable regions consisted mostly of transposases and conserved proteins with no clear function and were not further addressed (Figure S2).


Metabolic islands, carbohydrate transport and metabolism and niches
Comparative genomic analysis of the 100 strains revealed the loss of genes encoding various carbohydrate PTS system and metabolism-associated proteins compared to L. rhamnosus GG. To study the impact of these genomic characteristics, the metabolic capability to utilize different carbon sources was investigated. Carbohydrate utilization profiling showed that most L. rhamnosus strains use a large range of simple and complex carbohydrates (Figure 3). However, some differences may reflect their genomic diversity and also at some extent how they evolved in different ecological niches, by the acquisition or the loss of metabolic-associated genes. The ability to utilize carbohydrates mostly relies on the presence of functional transporter machinery and intact metabolic pathways. The clustering of L. rhamnosus strains (Figure 3) revealed strong associations between genome diversity, carbohydrate metabolism and their origins. Typically, strains belonging to cluster 4 utilize D-arabinose, dulcitol and L-fucose, whereas other strains lost these functions but possess the ability to ferment L-sorbose, D-maltose, D-lactose, D-turanose, methyl-α-D-glucopyranoside, L-rhamnose and D-saccharose (Figure 3). Hence, we detail the differences in carbohydrate utilization within the L. rhamnosus species below.
The genome of L. rhamnosus GG harbors a tagatose-6-phosphate pathway (lacABCD) and a lactose PTS (lacFEG) but the antiterminator lacT and the phospho-β-galactosidase encoding lacG genes are altered and non-functional, preventing GG from metabolizing D-lactose [19]. Strains belonging to the cluster 4 also show a poor ability or incapacity to use D-lactose, whereas other isolates, including most dairy ones utilize this disaccharide, which is found in milk and milk-derived products. We propose that the lacT and lacG genes have been kept intact in these strains, as lactose represents an important carbon source and provides a real benefit for L. rhamnosus strains residing in dairy niches. The maltose locus was predicted to be non-functional in L. rhamnosus GG due to the insertion of a conserved gene (LGG_00950) between genes encoding the maltose-specific malEFGK transporter and the hydrolase (LGG_0954-LGG_0951 and LGG_00949, respectively) [19]. Similarly, we found that most L. rhamnosus strains unable to use maltose also contained a maltose locus disrupted by LGG_00950. In contrast, the majority of strains belonging to other sublineage contained an intact maltose locus and were able to utilize maltose, indicating that the insertional inactivation by LGG_00950 may have played a significant role in L. rhamnosus species ecology. Comparative genome sequencing of L. rhamnosus GG also showed that the rhamnose locus is altered: the galactitol-specific gatABCD PTS and a DeoR transcriptional regulator are missing while the rhaB gene is duplicated, possibly explaining the inability to use rhamnose compared to some other L. rhamnosus strains, such as LC705 [19]. Combination of the genomic and metabolic data indicates that most strains of the cluster 4 similarly contain a defective rhamnose locus. It is noteworthy that 74% of all isolates can partially or fully utilized L-rhamnose, a carbohydrate from which the species name derives. In contrast, fucosylated compounds such as human mucin and other glycoproteins play an important role in the human gut ecology, as a carbon source for intestinal bacterial species [30]. Close inspection of the L-fucose metabolism revealed that a large number of dairy-associated strains are unable to use L-fucose due to the lack of one or multiple genes required to transport and to metabolize L-fucose: the fucU and fucI isomerases, fcsR fucose operon repressor and α-L-fucosidase (LGG_02652). Most strains closely related to L. rhamnosus GG retained the capacity to use L-fucose, whereas dairy strains lost this ability, since L-fucose is not as abundant in bovine milk. Dulcitol, a polyol also known as galactitol, is used by the cluster 4 (Figure 3). In some strains unable to use dulcitol, the function loss was associated with the lack of an intact gatABC PTS system. Other carbohydrates such as turanose and sorbose were not metabolized by strains related to GG (Figure 3). In L. rhamnosus LC705, an intact sorbose sorABCDEFGR locus is present, explaining its ability to utilize sorbose, whereas L. rhamnosus GG lacks such machinery [19]. L. rhamnosus strains with similar capabilities may therefore possess an intact sorbose locus. Remarkably, the strains from the cluster 1 present a similar metabolic profile as the industrial dairy strain L. rhamnosus LC705 [19]. This suggests that dairy-related strains characterized in the present study underwent similar niche adaptation as LC705 in terms of acquisition, decay or loss of genes.
Diversity of the Clustered Regularly Interspaced Short Palindromic Repeats-Cas system: A spacer oligotyping analysis
CRISPR (clustered regularly interspaced short palindromic repeats) loci are present in a large number of prokaryote genomes [51], playing an important role in controlling horizontal gene transfer. It has been well established that some bacteria acquired the CRISPR-Cas system as a protection/immunization system against plasmid conjugation and phage predation [52]–[55]. The CRISPR-Cas system usually consists of a leader sequence, an array of CRISPRs interspaced by spacers and a cas gene cluster encoding the Cas protein complex (Figure 4A) [56]. The role and mechanistic of the CRISPR-Cas system in bacterial species have been extensively studied and indicate that the spacer sequences can be considered as a signature of past exposure to exogenous DNA [57]. L. rhamnosus GG has a single Type II-A CRISPR-Cas locus, consisting of 4 cas genes and one CRISPR array containing 24 spacers [19]. To determine whether the CRISPR sequences could be used as an indicator of a specific niche, we determined their diversity and the presence of the cas genes. CRISPR genotyping has been previously developed for epidemiological purposes and strain differentiation for Mycobacterium tuberculosis [58], enterohemorrhagic Escherichia coli [59] and Salmonella enterica [60]. We were able to generate a CRISPR profile (based on spacer oligotyping) for each strain and this revealed a high degree of diversity among the various strains (Figure 4B, C). Remarkably, all strains from cluster 4 were sharing a comparable CRISPR spacer set, whereas the genetically more distant L. rhamnosus strains were only harbouring few of the spacers found in L. rhamnosus GG and a poor conservation of the cas genes. The overall CRISPR-Cas typing analysis showed that strains from the same sublineage mostly shared identical CRISPR-Cas loci. Interestingly, strains H1093 and H4692 did not have any of L. rhamnosus GG spacers but some of the cas genes remained present, whilst strain H1275 lacked the entire CRISPR-Cas locus. It has to be kept in mind that only sequences homologous to the CRISPR-Cas locus from strain L. rhamnosus were identified, allowing the possibility that additional spacers, cas genes or even additional CRISPR loci may be present. To determine the function of the CRISPR-Cas system in protecting L. rhamnosus from exogenous DNA, blastn searches on all 24 spacers were performed against virus and plasmid database at GenBank. Out of 24 spacers, 11 spacer sequences showed substantial sequence identity with plasmid or phage sequences (Table S4). Eight spacer sequences fully or partially matched known bacteriophages genomes: L. rhamnosus phage Lc-Nu, L. casei phage φ AT3, L. casei phage Lrm1, L. casei phage A2 and L. casei phage PL-1. The identified CRISPR spacers thus belonged to phages from L. rhamnosus strains or closely related bacterial species, i.e. L. casei, highlighting the role of the CRISPR-Cas system as an immunity system against phage predation. Some spacers (4, 12, 18, 21 and 22) have multiple phage hits, showing that the corresponding phage genomes share the same region, preventing us to predict from which bacteriophage these particular spacers were acquired. One match for plasmids was also found: the conjugative plasmid pSB102. The data also indicates that the CRISPR-Cas system may play a role in the L. rhamnosus species diversity by controlling horizontal gene transfer and providing phage resistance, thereby contributing to diversification of the species. Our data also showed that the degree of CRISPR diversity correlated with the genomic clustering of the 100 isolates and at some extent with their ecological niche (Figure 5). Most dairy isolates shared only 6–7 spacers with L. rhamnosus GG, indicating that the variety and the exposure to phages and other mobile genetic elements differ in each habitat, i.e. the intestinal tract and cheese. We anticipate that some of the dairy strains may have an entirely different set of CRISPR sequences, representative of their own habitat and possibly additional CRISPR-Cas Types, as seen across the lactic acid bacteria [61].


Bile resistance, a persistence trait
All 100 L. rhamnosus isolates were tested for resistance to bile salts, a property that is usually associated with the intestinal tract environment (Figure 5). A majority of L. rhamnosus strains were bile resistant (45% resistant and 30% moderately resistant) and different bile resistance profiles were observed in each niche (Figure 5). No clear association could be seen when combining the bile salt resistance data with the hierarchical clustering. A similar distribution was observed in strains isolated from clinical specimens and cheeses, even though a slightly higher proportion of bile salt-sensitive strains could be observed in the cheese isolate group. As expected, all strains from the human intestinal tract were resistant to bile salts, illustrating that such trait is essential for persisting in the intestinal tract. All vaginal isolates also showed bile resistance, suggesting that L. rhamnosus strains of the colonic microbiota may possibly have colonized the vaginal cavity as previously reported [62]. The low number of isolates from oral cavities (n = 3) did not allow us to draw any conclusions, but revealed a different profile in terms of bile sensitivity. One of the hyper-variable regions in GG contained genes encoding the taurine transport system tauABC, potentially involved in the bile salt conjugation. Nine out of 25 bile-sensitive strains had a defective tauABC locus, suggesting that the tauABC locus may affect the bile sensitivity of these strains although most likely additional genes are involved.
Pilosotype diversity
Pili in L. rhamnosus strains play a significant role in terms of interaction, colonization, persistence and potential signalling in the human intestinal tract [19]–[21]. The spaCBA pili gene cluster is flanked by numerous IS elements, suggesting that L. rhamnosus might have acquired the spaCBA pili gene cluster by horizontal gene transfer [41], [63], where the integration of the iso-IS30 element had constituted a promoter that allowed the expression of the pili genes in L. rhamnosus GG [63]. It also indicates that this IS element-rich chromosomal region may be subject to important genetic recombination events within the species [19], [64]. Hence, we examined the pili diversity among all 100 isolates, providing a detailed picture on the conservation of the pili genes in each strain, since as little as one mutation is potentially sufficient to prevent the pili production or to affect the mucus binding abilities (Figure 6). Moreover, to support the genomic data, we investigated the mucus adhesion abilities of all L. rhamnosus isolates and also verified the presence of pili in a number of these strains by immunoblotting analysis (n = 64), transmission electron microscopy (n = 10) and in vitro blocking mucus binding assays (n = 22) (Figures 6, S3 and S4). The mucus binding capacity ranged from 0.05% to 29.9% in all tested strains and was clearly correlated with the presence of a functional SpaCBA pili gene cluster, as shown at both genomic and phenotypic levels (Figure 6). To further demonstrate that the mucus binding capacity of these strains was mediated by SpaCBA pili, we performed in vitro blocking mucus binding assays on 22 SpaCBA-positive isolates using SpaC anti-serum as previously described (Figures 6 and S3) [19]. In all 22 strains tested, the addition of SpaC anti-serum significantly reduced mucus binding, indicating that the SpaCBA pili has a major role in the interaction between L. rhamnosus and the human intestinal mucus. Remarkably, some strains displayed significant mucus-binding capacity but lacked the canonical SpaCBA pili structures, suggesting that alternative interaction players are involved. The genes encoding the SpaCBA pili of some strains such as H1242, H1304, F1178 and H6110 are highly conserved but, however, with some subtle sequence differences. We propose that the sequence polymorphism of the pili genes in these strains might modulate mucus binding capacity or affinity. Alternatively, we cannot rule out that additional strain-specific traits might be involved in the mucus binding, especially in strain F1178 where the residual binding in the presence of SpaC anti-serum still remained high (Figure S3). In contrast, strains with poor mucus-binding abilities appeared to have some remnants of pili genes in a more or less decayed form (Figure 6). In strains H1275, H4689 and H1100, the spaCBA pili gene cluster is highly conserved (>98%), but show a very poor binding, indicating that the pili production may be impaired by critical mutation(s) or a defective promoter.
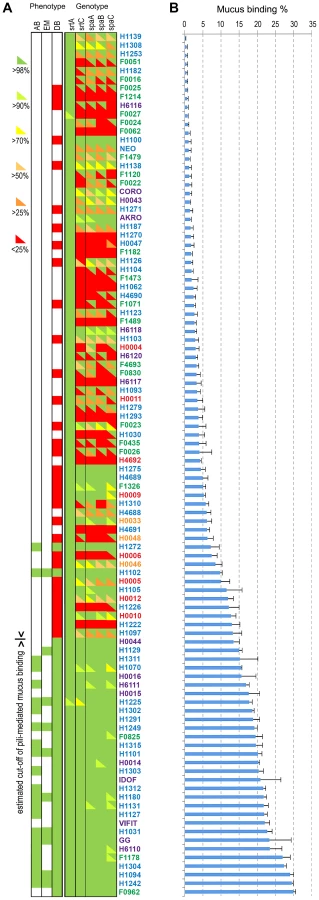
The L. rhamnosus strains were further classified according to two main criteria, i.e. their ecological niche and their pilosotype, defined as the presence of pili genes that encode functional pili (Table 2). The results indicate that the production of functional SpaCBA pili was significantly more prevalent in human isolates (40.2% or 31/77) than in dairy isolates (13% or 3/23). This suggests that the lack of the SpaCBA pili gene cluster in most dairy strains reflects a possible niche specialization to a habitat where pili structures are not essential and do not bring any benefit for persistence and colonization. Among all niche groups, the intestinal strains are the most prevalent group to produce SpaCBA pili, which would confer the ability to efficiently colonize and persist in the intestinal tract. In contrast, none of the strains originated from the oral and vaginal cavities produces functional pili, indicating that such trait may not be required in these two ecological niches. Our observations support the hypothesis that the human-mucus binding properties of pili may constitute an advantage to the lactobacilli to persist in the intestinal tract, but may be lost in strains evolving in other ecological niches, such as dairy products, through the decay or loss of the non-essential SpaCBA pili gene cluster.

Cross-talk between L. rhamnosus and intestinal cells
Due to the intimate interaction between L. rhamnosus and the intestinal mucosa [30], we studied the potential signalling pathways that could be triggered by the L. rhamnosus strains. This was realized by determining the signal transduction in intestinal epithelial cells via Toll-like Receptors (TLRs) TLR-2, TLR-4 and TLR-5. All 100 isolates were tested for signallings via TLR-4 and TLR-5 receptors, but no significant responses were observed, which is in agreement with the identified ligands for these two TLRs, i.e. lipopolysaccharides and flagellins respectively (data not shown). Clearly, L. rhamnosus-host signallings are mediated through different receptors. Signalling via the TLR-2 receptor in L. rhamnosus species was observed and greatly varied among isolates (Figure S5). More than half of the isolates mediated a TLR-2 response very similar to the level observed for strain GG after 1 h (fold-induction of ∼1.5). Six strains (H6111, H0009, H4692, H1311, H1226 and H1131) triggered a stronger TLR-2 response in this assay system. We did not determine the nature of the ligand recognized by TLR2 but assume in analogy with what has been found in L. rhamnosus GG that the signalling might be mediated by the lipoteichoic acids [65]. The levels of TLR2 signalling could not be correlated with any other traits, such as EPS production, pili production or the presence of other membrane-associated proteins. No links between the TLR2 response, hierarchical clustering and ecological niches of the various strains were either identified. This suggests that the TLR-2 response triggered by L. rhamnosus does not reflect its adaptation to one particular niche, but is rather a trait acquired, maintained, altered or exacerbated by other factors that remains yet to be identified.
L. rhamnosus vs. other bacterial populations
L. rhamnosus isolates have been isolated from various ecological habitats, showing its large ecological versatility. Niche-specialized strains have developed distinctive metabolic traits, phage resistance system, stress-resistance mechanisms and colonization traits to efficiently persist in an ecological habitat. However, the microbiota of habitats such as the human intestinal tract or the vaginal cavity are rich and complex, consisting of many phylotypes [2], [66]. L. rhamnosus strains may therefore compete with other bacterial species by producing bacteriocins that prevent growth of other bacterial populations. In contrast, the diversity and richness of the microbiota in dairy products is much lower, suggesting less competition [67]. When testing the anti-microbial activity of 92 L. rhamnosus strains, we found that most strains displayed anti-microbial activity against pathogens E. coli, Yersinia enterocolitica and Listeria monocytogenes at different pH (Figure S6). This is in line with previous studies on L. rhamnosus anti-microbial activity [44], [68], [69]. Remarkably, most dairy isolates shared comparable anti-microbial capabilities and clustered together, e.g. poor anti-microbial activity against E. coli and, to a lesser degree, against L. monocytogenes. The human strains displayed a differential spectrum and level of antimicrobial activity against the three human pathogens tested than most dairy strains. This illustrates the fitness of human isolates to compete with other bacteria potentially present in the human body cavities. In contrast, a high proportion of dairy isolates seems to have lost the ability to produce antimicrobial compounds against these three human pathogens, suggesting that such trait might not be essential in an environment with a lower and different microbiota diversity than in human body cavities. It, however, does not imply that those isolates do not produce antimicrobial compounds active against other pathogens more prevalent in their respective niche.
Concluding remarks: Strain diversity and niche adaptation
The species L. rhamnosus has been isolated in various dairy products and human body cavities, highlighting the close association and frequent interactions between L. rhamnosus and the human body. The analysis of the genomes and phenotypes of 100 strains of the species L. rhamnosus provides then a wealth of information with respect to the traits that are beneficial or essential in different ecological niches and, also allowed us to depict in details the species from an anthropocentric perspective. As expected, close inspection of the hierarchical clustering of the 100 L. rhamnosus strains showed that this can be paralleled to some extent by clustering of phenotypic data, i.e. carbohydrate metabolism, antagonistic activity, CRISPR oligotyping, bile salt resistance or pilosotype. Interestingly, the integration of both phenotypic and genomic data of each strain revealed the presence of two prevailing geno-phenotypes called A & B in the L. rhamnosus species (Figure 7). The strains belonging to the geno-phenotype A are characterized by a lack of SpaCBA pili, a different carbohydrate metabolism (D-lactose, D-maltose and L-rhamnose) and a distinct CRISPR system profile, indicative a possible adaptation to dairy-like environment. In contrast, the geno-phenotype B depicts strains with a specific set of lifestyle traits that would confer them adequate fitness to the intestinal tract, such as bile resistance, pili production and L-fucose utilization. The geno-phenotype B showed a high similarity with L. rhamnosus GG in terms of genomes and phenotypes.

The geno-phenotype A is prevalent in the cheese group, indicating their adaptation to the dairy environment. The PTS and metabolic-related genes non-essential in dairy products were lost or decayed, i.e. loss of L-fucose utilization. In parallel, we hypothesized that additional functions were acquired possibly through horizontal gene transfers, genetic mobile elements or plasmids, i.e. the ability to use lactose, a major carbon source in milk-derivative products. The loss of pili in these dairy strains is another characteristic example of a trait lost during niche-adaptation, where the absence of mucosa surfaces is reflected by the decay or complete loss of non-essential pili. In dairy niches, phage predation is ubiquitous as showed in many studies of lactic acid bacteria [70], [71] and the CRISPR system might evolve by the acquisition of spacers representative of phages or plasmids of a particular niche. This is the case as the CRISPR locus profile between both geno-phenotypes differs considerably.
Interestingly, L. rhamnosus from the vaginal cavity and urethra have a geno-phenotype A, which is in agreement with previous studies showing that the rectal microbiota is a potential reservoir of bacteria that may colonize the vaginal cavity [62]. This also suggests that the intestinal isolates (geno-phenotype A) may be more adapted to the vaginal environment, possibly due to their distinct metabolic abilities. This remains speculative, as at individual level, we do not know which L. rhamnosus strains these women possibly have in the intestinal tract. Interestingly, the oral isolates also possess a geno-phenotype A. Due to the low number of strains, it is difficult to draw any definitive conclusions for the oral group. However, the prevalence of the geno-phenotype A in these three niches highlights a close link between them, indicating that the geno-phenotype A strains may likely originate from either dairy products but also oral or vaginal cavities.
Both geno-phenotypes A and B were found among the intestinal isolates (Figure 7). We proposed that the geno-phenotype A strains were likely introduced in the intestinal tract via consumption of foods. Bile resistant and with different metabolic capabilities, they are able to survive in the intestinal tract but may not be able to compete with other autochthonous intestinal bacteria to colonize the intestinal tract, i.e. lack of mucus-binding pili. This would indicate the most of these isolates are transient in the intestinal tract and further eliminated along with the faecal material. Other L. rhamnosus dairy isolates that are bile sensitive may also be introduced in the gastro-intestinal tract through the diet but cannot survive the intestinal conditions. On the other hand, geno-phenotype B strains are likely to be autochonous, as they possess phenotypic traits, promoting resistance and persistence in the human intestinal gut. Still, we cannot exclude the hypothesis that geno-phenotype B strains may also be transient in the gut. But this would then indicate alternative functions for the SpaCBA pili, such as binding to other mucosa. This brings us to raise one question: is L. rhamnosus only specific to the human host or are there any other potential animal reservoirs? Addressing the host specificity of L. rhamnosus would potentially lead to identifying novel host-specific strains and remarkable adaptation patterns as reported in the species L. reuteri [45].
The clinical isolates constitute a very eclectic pool of strains, whose genotype and phenotype do not reflect adaptation patterns of their source of isolation, i.e. blood or pus, but rather of their original ecological niche. Thirty-three clinical strains could be assigned to one distinct geno-phenotype, i.e. 10 isolates with geno-phenotype A, 9 isolates with geno-phenotype B and 14 isolates with geno-phenotype BΔspaCBA. The geno-phenotypes B and BΔspaCBA differ by the presence or lack of the genomic island containing the spaCBA pili gene cluster, which is located in an unstable genomic region [64]. A number of other strains (n = 17) were not be assigned any geno-phenotype, as they possess transitory geno-phenotypes or may have atypical history. It is noteworthy, that some of the clinical isolates have similar gene content to L. rhamnosus GG. However, differences in phenotypes clearly show that these strains are not identical to GG. This indicates that they may have additional genes and/or nucleotide variations in their respective genomes and share close ancestor to L. rhamnosus GG. This is in line with a previous study that showed that the widespread and increasing use of probiotic strain L. rhamnosus GG was not associated with the augmentation of Lactobacillus bacteremia [37].
To conclude, this work represents the first extensive genomic and functional analysis of the species L. rhamnosus and provides further insights into the genetics and lifestyle of this species. The data and model presented here may serve as a basis to understand the ecology of novel L. rhamnosus isolates, to identify novel probiotic candidates and also to examine the functional properties of current commercial L. rhamnosus strains.
Materials and Methods
L. rhamnosus isolate collection, DNA isolation and molecular typing
All 100 Lactobacillus rhamnosus strains used in this study were obtained from various institutions, universities and hospitals (Table S1). Well-characterized, L. rhamnosus GG was used as reference strain throughout the study [15], [19], [43]. Strains VIFIT, IDOF, AKRO, CORO and NEO were isolated from probiotic-marketed products (Table S1), whereas a number of strains were made available from strain collections or institutions. All isolates were routinely propagated in anaerobic conditions at 37°C in MRS medium (Difco BD, NJ, USA). Chromosomal DNA from each isolate was extracted using Wizard Genomic DNA Purification Kit (Promega, WI, USA) following the manufacturer's instructions. Initial bacterial identification at the species level was performed by amplification of tuf gene as described by Ventura et al. [72], [73] using standard PCR amplification conditions and multiplex PCR amplification (data not shown).
Fermentative profile
Sugar metabolism and other catabolic properties of the L. rhamnosus strains were investigated using API CH 50 kit (bioMerieux, Marcy L'Etoile, France). All strains were grown until logarithmic phase and then inoculated in API galleries following the manufacturer's instructions. API galleries were further incubated at 37°C in anaerobic conditions for 48 h prior to colorimetric analysis.
Genome SOLiD sequencing and bioinformatic sequence analysis
Genomes of all L. rhamnosus isolates were sequenced on a SOLiD sequencer platform (Life Technologies) at the Institute of Biotechnology (Helsinki, Finland). Sequence alignments and consensus sequences were generated by mapping color-space reads to the L. rhamnosus GG reference genome, using the SOLiD BioScope software (Life Technologies) and the SAM tools [74]. In order to transfer annotation from a reference genome (L. rhamnosus GG) to each un-annotated mapped genome, sequences were compared with ‘nucmer’ to identify regions that share synteny [75]. Those regions were extracted as base range in the mapped genome and in the reference genome (L. rhamnosus GG). In-house custom-made scripts were then used to transfer annotation. Synteny blocks had a nucleotide sequence identity more than or equal to 40%. For each query genome, a set of shared L. rhamnosus GG orthologous genes was obtained and further analyzed. Similarly, for a number of strains, we mapped the SOLiD reads onto the LC705 genome sequence and obtained an additional set of shared L. rhamnosus LC705 orthologous genes. The L. rhamnosus GG genome was assigned to COGs using Reverse Position Specific blast and Conserved Domain Database from NCBI. Mapped genome sequences are available upon request.
Human mucus binding assay
Human intestinal mucus was kindly collected and provided by S. Vesterlund (University of Turku, Finland) and H. Huhtinen (Turku University Central Hospital, Turku, Finland) as previously described [27], [76]. L. rhamnosus strains were propagated and radiolabeled overnight in MRS broth supplemented with 10 µl.ml−1 [5′-3H] thymidine (16.7 Ci .mmol−1). MaxiSorp microtiter plates (Nunc, Denmark) were coated with 100 µL of human mucus solution prepared in PBS at a final concentration of 0.5 mg/mL and further incubated overnight at 4°C. The wells were then washed with PBS to remove unbound mucus and 100 µL of 3H-radiolabeled bacterial suspensions at optical density (OD600) 0.25±0.01 were added to the wells. The microtiter plate was further incubated at 37°C for 1 h and then wells were washed with PBS in order to remove unbound bacteria. Bacteria adhering to mucus were incubated at 60°C for 1 h in 1% SDS-0.1 M NaOH solution and the radioactivity level of lyzed bacterial suspensions was measured by liquid scintillation counting in a Wallac 1414 liquid scintillation counter (PerkinElmer). The percentage ratio between radioactivity values of lysed L. rhamnosus suspension (mucus-bound fraction) and L. rhamnosus suspension (unbound fraction) reflects the adhesion ability to human intestinal mucus. For each isolate the experiment was performed in quadruplicate.
Antiserum-mediated human mucus binding assay
Human mucus binding assay was performed for L. rhamnosus isolates in the presence of polyclonal SpaC antibody as described above. 3H radio radiolabeled bacteria were co-incubated with the immobilized mucus in the presence of a 1∶100 dilution of anti-SpaC serum.
Immunoblotting analysis of cell wall proteins
For each isolate, bacterial suspension adjusted to an optical density (OD600) of 1.0 was used to extract cell wall-associated proteins. Cell pellets were washed once with PBS and disrupted mechanically by bead-beating using sterile quartz beads (Merck KGaA, Germany). Cell wall material was resuspended in 500 µL of PBS and further pelleted by centrifugation at high speed for 30 min. Next, the samples were digested for 3 h at 37°C in a 50 µL enzymatic mixture containing 50 mM Tris-HCl, 5 mM MgCl2, 5 mM CaCl2, 10 mg/mL lysozyme and 150 U/mL mutanolysin. Samples were mixed with 12.5 µL of 4× Laemmli loading buffer (BioRad, CA, USA) and heated at 99°C for 10 min. Cell wall proteins were resolved on 10% acrylamide gel and electroblotted onto 0.2 µm nitrocellulose membrane (BioRad, CA, USA). Polyclonal rabbit SpaA antiserum (1∶10,000) and peroxidase-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, USA) (1∶10,000) were respectively used as a primary and secondary antibody in 2% (w/v) ECL Prime Blocking Reagent (GE Healthcare Life Science, UK). Membranes were blocked with 2% (w/v) ECL Prime Blocking Reagent, and washed with 0.1% Tween 20 – PBS solution in-between incubations. Membranes were analyzed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Science, UK).
Host signallings
HEK-Blue hTLR2/4/5 cell lines (Invivogen, CA, USA) were used in this assay. All cell lines were grown and subcultured up to 70–80% of confluency using as a maintenance medium Dulbecco's Modified Eagle Medium (DMEM) supplemented with 4.5 g/L D-glucose, 50 U/mL penicillin, 50 µg/mL streptomycin, 100 µg/mL Normocin, 2 mM L-glutamine, and 10% (v/v) of heat-inactivated fetal bovine serum. For each cell line, the immune response assay was carried out by splitting HEK-Blue cells in flat-bottom 96-well plates and stimulating them by addition of 20 µl bacterial suspensions adjusted to OD600 1, 1∶10, 1∶100. The 96-well plates were incubated for 20–24 h at 37°C in a 5% CO2 incubator. Receptor ligands as Pam3CSK4 (100 ng/mL for hTLR2), LPS-EB (100 ng/mL for hTLR4) and RecFLA-ST (10 ng/mL for hTLR5) were used as positive control while maintenance medium without any selective antibiotics was used as negative control. SEAP secretion was detected by measuring the OD600 at 15 min, 1 h, 2 h, and 3 h after addition of 180 µL of QUANTI-Blue (Invivogen, CA, USA) to 20 µL of induced HEK-Blue hTLR2/4/5 supernatant. All cell lines were stimulated in triplicate for each isolate.
TEM sample preparation
Selected L. rhamnosus isolates were analyzed by transmission electron microscopy (TEM) as previously described by Reunanen et al. [20]. Briefly, 20 µL of overnight bacterial cultures were added to Formvar-carbon-coated copper grids for 30 min at room temperature. Grids were then washed three times with 0.02 M glycine solution and further incubated for 15 min in a blocking solution containing 1% (w/v) of bovin serum albumin (BSA). Next, a 1∶100 dilution of SpaA antibody was prepared in 1% (w/v) BSA solution and added to the grids for 1 h, washed with 0.1% (w/v) BSA and incubated for 20 min with protein A conjugated to 10 nm gold particles. Grids were washed several times in PBS, fixed for 5 min using 1% glutaraldehyde, washed again with deionized water and stained with a solution containing 1.8% methycellulose and 0.4% uranyl acetate. Grids were visualized using JEOL JEM-1400 transmission electron microscope (JEOL Ltd., Japan).
Bile resistance assay
L. rhamnosus strains were cultured in MRS broth at 37°C in anaerobic conditions. The OD600 of the bacterial culture suspensions were equalized to 1.5 and 3 µl of cell suspensions were spotted onto MRS agar plates containing 0.5% (w/v) Ox gall (Sigma, MO, USA). Plates were incubated anaerobically at 37°C for two days and visually examined.
Antagonistic assay
L. rhamnosus strains were grown until stationary phase as described above. Next, the cell suspensions were thoroughly homogenized and the OD600 was equalized. Cell mixtures were then centrifuged for 20 min at 650×g at +5°C and the supernatants were pH-adjusted at 5.0 and 6.20 by addition of NaOH and HCl solutions, filtered (0.22 µm filter) and stored at −20°C for further analysis. Antagonistic assays were performed in microtiter well plates as previously described [77]. E. coli O157:H7 (ATCC 43894), L. monocytogenes R14-2-2 and Y. enterocolitica R5-9-1 were incubated for 15 h at 37°C in the presence of 30 µl of L. rhamnosus pH-adjusted supernatant. As positive controls, 30 µl of sterile MRS broth at pH 6.20 or pH 5.0 was added the dedicated medium (TSB or LB) inoculated with one of the pathogenic strains. As a negative control, 300 µl of medium (TSB or LB) was used. The OD600 values were measured in an automatic reader (Bioscreen C, Oy Growth Curves Ab Ltd, Finland) every 30 min. The bacterial growth was quantified using growth curves and the area under curve (AUC) values, automatically processed by the BioLink software (Oy Growth Curves Ab). Inhibition was expressed as an area reduction percentage (ARP) compared to control samples grown without the addition of supernatant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Clemente JoseC, Ursell LukeK, Parfrey LauraW, KnightR (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148 : 1258–1270.
2. Rajilić-StojanovićM, SmidtH, De VosWM (2007) Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol 9 : 2125–2136.
3. WilliamsonSJ, YoosephS (2011) From bacterial to microbial ecosystems (metagenomics) Bacterial Molecular Networks. Meth Mol Biol 35–55.
4. VaughanEE, SchutF, HeiligHG, ZoetendalEG, de VosWM, et al. (2000) A molecular view of the intestinal ecosystem. Curr Issues Intest Microbiol 1 : 1–12.
5. KleerebezemM, VaughanEE (2009) Probiotic and gut lactobacilli and bifidobacteria: molecular approaches to study diversity and activity. Annu Rev Microbiol 63 : 269–290.
6. ZoetendalEG, RaesJ, van den BogertB, ArumugamM, BooijinkCC, et al. (2012) The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 6 : 1415–1426.
7. ClaessonMJ, O'SullivanO, WangQ, NikkilaJ, MarchesiJR, et al. (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 4 (8) e6669.
8. SalonenA, SalojarviJ, LahtiL, de VosWM (2012) The adult intestinal core microbiota is determined by analysis depth and health status. Clin Microbiol Infect 4 : 16–20.
9. HeiligHG, ZoetendalEG, VaughanEE, MarteauP, AkkermansAD, et al. (2002) Molecular diversity of Lactobacillus spp. and other lactic acid bacteria in the human intestine as determined by specific amplification of 16S ribosomal DNA. Appl Environ Microbiol 68 : 114–123.
10. WalterJ (2008) Ecological Role of Lactobacilli in the gastrointestinal tract: implications for fundamental and biomedical research. Appl Environ Microbiol 74 : 4985–4996.
11. WalterJ, BrittonRA, RoosS (2010) Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc Natl Acad Sci U S A 108 Suppl 1 : 4645–4652.
12. KalliomakiM, SalminenS, ArvilommiH, KeroP, KoskinenP, et al. (2001) Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 357 : 1076–1079.
13. KalliomakiM, SalminenS, PoussaT, ArvilommiH, IsolauriE (2003) Probiotics and prevention of atopic disease: 4-year follow-up of a randomised placebo-controlled trial. Lancet 361 : 1869–1871.
14. BernardeauM, GuguenM, VernouxJP (2006) Beneficial lactobacilli in food and feed: long-term use, biodiversity and proposals for specific and realistic safety assessments. FEMS Microbiol Rev 30 : 487–513.
15. LebeerS, VanderleydenJ, De KeersmaeckerSC (2010) Adaptation factors of the probiotic Lactobacillus rhamnosus GG. Benef Microbes 1 : 335–342.
16. SaxelinM, TynkkynenS, Mattila-SandholmT, de VosWM (2005) Probiotic and other functional microbes: from markets to mechanisms. Curr Opin Biotechnol 16 : 204–211.
17. RandazzoCL, De LucaS, TodaroA, RestucciaC, LanzaCM, et al. (2007) Preliminary characterization of wild lactic acid bacteria and their abilities to produce flavour compounds in ripened model cheese system. J Appl Microbiol 103 : 427–435.
18. PitinoI, RandazzoCL, CrossKL, ParkerML, BisignanoC, et al. (2012) Survival of Lactobacillus rhamnosus strains inoculated in cheese matrix during simulated human digestion. Food Microbiol 31 : 57–63.
19. KankainenM, PaulinL, TynkkynenS, von OssowskiI, ReunanenJ, et al. (2009) Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human-mucus binding protein. Proc Natl Acad Sci U S A 106 : 17193–17198.
20. ReunanenJ, von OssowskiI, HendrickxAPA, PalvaA, de VosWM (2012) Characterization of the SpaCBA pilus fibers in the probiotic Lactobacillus rhamnosus GG. Appl Environ Microbiol 78 : 2337–2344.
21. von OssowskiI, ReunanenJ, SatokariR, VesterlundS, KankainenM, et al. (2010) Mucosal adhesion properties of the probiotic Lactobacillus rhamnosus GG SpaCBA and SpaFED pilin subunits. Appl Environ Microbiol 76 : 2049–2057.
22. MillarMR, BaconC, SmithSL, WalkerV, HallMA (1993) Enteral feeding of premature infants with Lactobacillus GG. Arch Dis Child 69 : 483–487.
23. PetschowBW, FigueroaR, HarrisCL, BeckLB, ZieglerE, et al. (2005) Effects of feeding an infant formula containing Lactobacillus GG on the colonization of the intestine: a dose-response study in healthy infants. J Clin Gastroenterol 39 : 786–790.
24. AlanderM, SatokariR, KorpelaR, SaxelinM, Vilpponen-SalmelaT, et al. (1999) Persistence of colonization of human colonic mucosa by a probiotic strain, Lactobacillus rhamnosus GG, after oral consumption. Appl Environ Microbiol 65 : 351–354.
25. LebeerS, VanderleydenJ, De KeersmaeckerSCJ (2008) Genes and molecules of lactobacilli supporting probiotic action. Microbiol Mol Biol Rev 72 : 728–764.
26. MackDR, AhrneS, HydeL, WeiS, HollingsworthMA (2003) Extracellular MUC3 mucin secretion follows adherence of Lactobacillus strains to intestinal epithelial cells in vitro. Gut 52 : 827–833.
27. VesterlundS, KarpM, SalminenS, OuwehandAC (2006) Staphylococcus aureus adheres to human intestinal mucus but can be displaced by certain lactic acid bacteria. Microbiology 152 : 1819–1826.
28. Johnson-HenryKC, DonatoKA, Shen-TuG, GordanpourM, ShermanPM (2008) Lactobacillus rhamnosus strain GG prevents enterohemorrhagic Escherichia coli O157:H7-induced changes in epithelial barrier function. Infect Immun 76 : 1340–1348.
29. YoungVB (2012) The intestinal microbiota in health and disease. Curr Opin Gastroenterol 28 : 63–69.
30. LebeerS, VanderleydenJ, De KeersmaeckerSC (2010) Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat Rev Microbiol 8 : 171–184.
31. KlaenhammerTR, KleerebezemM, KoppMV, RescignoM (2012) The impact of probiotics and prebiotics on the immune system. Nat Rev Immunol 12 : 728–734.
32. CallananM, KaletaP, O'CallaghanJ, O'SullivanO, JordanK, et al. (2008) Genome sequence of Lactobacillus helveticus, an organism distinguished by selective gene loss and insertion sequence element expansion. J Bacteriol 190 : 727–735.
33. CaiH, ThompsonR, BudinichMF, BroadbentJR, SteeleJL (2009) Genome sequence and comparative genome analysis of Lactobacillus casei: insights into their niche-associated evolution. Genome Biol Evol 1 : 239–257.
34. SiezenR, TzenevaV, CastioniA, WelsM, PhanH, et al. (2010) Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ Microbiol 12 : 758–773.
35. SiezenR, van Hylckama VliegJ (2011) Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microbial Cell Fact 10: S3.
36. SucciM, TremonteP, RealeA, SorrentinoE, GraziaL, et al. (2005) Bile salt and acid tolerance of Lactobacillus rhamnosus strains isolated from Parmigiano Reggiano cheese. FEMS Microbiol Lett 244 : 129–137.
37. SalminenMK, TynkkynenS, RautelinH, SaxelinM, VaaraM, et al. (2002) Lactobacillus bacteremia during a rapid Increase in probiotic use of Lactobacillus rhamnosus GG in Finland. Clin Infect Dis 35 : 1155–1160.
38. PascualLM, DanieleM, iacute, aB, RuizF, et al. (2008) Lactobacillus rhamnosus L60, a potential probiotic isolated from the human vagina. J Gen Appl Microbiol 54 : 141–148.
39. RichardB, GroisillierA, BadetC, DorignacG, Lonvaud-FunelA (2001) Identification of salivary Lactobacillus rhamnosus species by DNA profiling and a specific probe. Res Microbiol 152 : 157–165.
40. O'SullivanO, O'CallaghanJ, Sangrador-VegasA, McAuliffeO, SlatteryL, et al. (2009) Comparative genomics of lactic acid bacteria reveals a niche-specific gene set. BMC Microbiol 9 : 50.
41. BroadbentJ, Neeno-EckwallE, StahlB, TandeeK, CaiH, et al. (2012) Analysis of the Lactobacillus casei supragenome and its influence in species evolution and lifestyle adaptation. BMC Genomics 13 : 533.
42. DouglasGL, KlaenhammerTR (2010) Genomic evolution of domesticated microorganisms. Annu Rev Food Sci Technol 1 : 397–414.
43. KoskenniemiK, LaaksoK, KoponenJ, KankainenM, GrecoD, et al. (2011) Proteomics and transcriptomics characterization of bile stress response in probiotic Lactobacillus rhamnosus GG. Mol Cell Prot 10: M110.002741.
44. De KeersmaeckerSCJ, VerhoevenTLA, DesairJ, MarchalK, VanderleydenJ, et al. (2006) Strong antimicrobial activity of Lactobacillus rhamnosus GG against Salmonella typhimurium is due to accumulation of lactic acid. FEMS Microbiol Lett 259 : 89–96.
45. FreseSA, BensonAK, TannockGW, LoachDM, KimJ, et al. (2011) The Evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet 7: e1001314.
46. MoritaH, TohH, OshimaK, MurakamiM, TaylorTD, et al. (2009) Complete genome sequence of the probiotic Lactobacillus rhamnosus ATCC 53103. J Bacteriol 191 : 7630–7631.
47. PittetV, EwenE, BushellBR, ZiolaB (2012) Genome Sequence of Lactobacillus rhamnosus ATCC 8530. J Bacteriol 194 : 726.
48. de VosW (2011) Systems solutions by lactic acid bacteria: from paradigms to practice. Microb Cell Fact 10: S2.
49. TettelinH, MasignaniV, CieslewiczMJ, DonatiC, MediniD, et al. (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 102 : 13950–13955.
50. KantR, BlomJ, PalvaA, SiezenRJ, de VosWM (2011) Comparative genomics of Lactobacillus. Microb Biotechnol 4 : 323–332.
51. JansenR, EmbdenJDAv, GaastraW, SchoulsLM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43 : 1565–1575.
52. HorvathP, RomeroDA, Coûté-MonvoisinA-C, RichardsM, DeveauH, et al. (2008) Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190 : 1401–1412.
53. DeveauH, BarrangouR, GarneauJE, LabontéJ, FremauxC, et al. (2008) Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190 : 1390–1400.
54. BarrangouR, FremauxC, DeveauH, RichardsM, BoyavalP, et al. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315 : 1709–1712.
55. MarraffiniLA, SontheimerEJ (2008) CRISPR Interference limits horizontal gene transfer in Staphylococci by targeting DNA. Science 322 : 1843–1845.
56. MarraffiniLA, SontheimerEJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11 : 181–190.
57. WestraER, SwartsDC, StaalsRHJ, JoreMM, BrounsSJJ, et al. (2012) The CRISPRs, they are A-changin': how prokaryotes generate adaptive immunity. Annu Rev Genet 46 : 311–339.
58. ZhangJ, AbadiaE, RefregierG, TafajS, BoschiroliML, et al. (2010) Mycobacterium tuberculosis complex CRISPR genotyping: improving efficiency, throughput and discriminative power of ‘spoligotyping’ with new spacers and a microbead-based hybridization assay. J Med Microbiol 59 : 285–294.
59. DelannoyS, BeutinL, FachP (2012) Use of CRISPR sequence polymorphisms for specific detection of enterohemorrhagic E. coli (EHEC) strains of serotypes O26:H11, O45:H2, O103:H2, O111:H8, O121:H19, O145:H28 and O157:H7 by real time PCR. J Clin Microbiol 12 : 4035–4040.
60. FabreL, ZhangJ, GuigonG, Le HelloS, GuibertV, et al. (2012) CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLoS ONE 7: e36995.
61. HorvathP, Coûté-MonvoisinA-C, RomeroDA, BoyavalP, FremauxC, et al. (2009) Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol 131 : 62–70.
62. El AilaN, TencyI, ClaeysG, VerstraelenH, SaerensB, et al. (2009) Identification and genotyping of bacteria from paired vaginal and rectal samples from pregnant women indicates similarity between vaginal and rectal microflora. BMC Infect Dis 9 : 167.
63. DouillardFP, RibberaA, JärvinenHM, KantR, PietiläTE, et al. (2013) Comparative genomic and functional analysis of Lactobacillus casei and Lactobacillus rhamnosus strains marketed as probiotics. Appl Environ Microbiol 79 (6) 1923–1933.
64. SybesmaW, MolenaarD, van IJckenW, VenemaK, KortR (2013) Genome instability in Lactobacillus rhamnosus GG. Appl Environ Microbiol 79 (7) 2233–2239.
65. LebeerS, ClaesI, TytgatHL, VerhoevenTL, MarienE, et al. (2012) Functional analysis of Lactobacillus rhamnosus GG pili in relation to adhesion and immunomodulatory interactions with intestinal epithelial cells. Appl Environ Microbiol 78 (1) 185–193.
66. HickeyRJ, ZhouX, PiersonJD, RavelJ, ForneyLJ (2012) Understanding vaginal microbiome complexity from an ecological perspective. Transl Res 160 : 267–282.
67. PoznanskiE, CavazzaA, CappaF, CocconcelliPS (2004) Indigenous raw milk microbiota influences the bacterial development in traditional cheese from an alpine natural park. Int J Food Microbiol 92 : 141–151.
68. SilvaM, JacobusNV, DenekeC, GorbachSL (1987) Antimicrobial substance from a human Lactobacillus strain. Antimicrob Agents Chemother 31 : 1231–1233.
69. LehtoEM, SalminenSJ (1997) Inhibition of Salmonella typhimurium adhesion to Caco-2 cell cultures by Lactobacillus strain GG spent culture supernate: only a pH effect? FEMS Immunol Med Microbiol 18 : 125–132.
70. BrüssowH (2001) Phages of dairy bacteria. Annu Rev Microbiol 55 : 283–303.
71. PfeilerEA, KlaenhammerTR (2007) The genomics of lactic acid bacteria. Trends Microbiol 15 : 546–553.
72. SheuS-J, HwangW-Z, ChiangY-C, LinW-H, ChenH-C, et al. (2010) Use of tuf gene-based primers for the PCR detection of probiotic Bifidobacterium species and enumeration of Bifidobacteria in fermented milk by cultural and quantitative Real-Time PCR methods. J Food Sci 75: M521–M527.
73. VenturaM, CanchayaC, MeylanV, KlaenhammerTR, ZinkR (2003) Analysis, characterization, and loci of the tuf genes in Lactobacillus and Bifidobacterium species and their direct application for species identification. Appli Environ Microbiol 69 : 6908–6922.
74. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
75. KurtzS, PhillippyA, DelcherA, SmootM, ShumwayM, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12.
76. VesterlundS, PalttaJ, KarpM, OuwehandAC (2005) Measurement of bacterial adhesion — in vitro evaluation of different methods. J Microbiol Meth 60 : 225–233.
77. SkyttäE, Mattila-SandholmT (1991) A quantitative method for assessing bacteriocins and other food antimicrobials by automated turbidometry. J Microbiol Meth 14 : 77–88.
78. SturnA, QuackenbushJ, TrajanoskiZ (2002) Genesis: cluster analysis of microarray data. Bioinformatics 18 : 207–208.
79. TamuraK, DudleyJ, NeiM, KumarS (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24 : 1596–1599.
80. SalminenMK, RautelinH, TynkkynenS, PoussaT, SaxelinM, et al. (2006) Lactobacillus bacteremia, species identification, and antimicrobial susceptibility of 85 blood isolates. Clinical Infect Dis 42: e35–e44.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy