
System-Wide Analysis Reveals a Complex Network of Tumor-Fibroblast Interactions Involved in Tumorigenicity
Many fibroblast-secreted proteins promote tumorigenicity, and several factors secreted by cancer cells have in turn been proposed to induce these proteins. It is not clear whether there are single dominant pathways underlying these interactions or whether they involve multiple pathways acting in parallel. Here, we identified 42 fibroblast-secreted factors induced by breast cancer cells using comparative genomic analysis. To determine what fraction was active in promoting tumorigenicity, we chose five representative fibroblast-secreted factors for in vivo analysis. We found that the majority (three out of five) played equally major roles in promoting tumorigenicity, and intriguingly, each one had distinct effects on the tumor microenvironment. Specifically, fibroblast-secreted amphiregulin promoted breast cancer cell survival, whereas the chemokine CCL7 stimulated tumor cell proliferation while CCL2 promoted innate immune cell infiltration and angiogenesis. The other two factors tested had minor (CCL8) or minimally (STC1) significant effects on the ability of fibroblasts to promote tumor growth. The importance of parallel interactions between fibroblasts and cancer cells was tested by simultaneously targeting fibroblast-secreted amphiregulin and the CCL7 receptor on cancer cells, and this was significantly more efficacious than blocking either pathway alone. We further explored the concept of parallel interactions by testing the extent to which induction of critical fibroblast-secreted proteins could be achieved by single, previously identified, factors produced by breast cancer cells. We found that although single factors could induce a subset of genes, even combinations of factors failed to induce the full repertoire of functionally important fibroblast-secreted proteins. Together, these results delineate a complex network of tumor-fibroblast interactions that act in parallel to promote tumorigenicity and suggest that effective anti-stromal therapeutic strategies will need to be multi-targeted.
Published in the journal:
. PLoS Genet 9(9): e32767. doi:10.1371/journal.pgen.1003789
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003789
Summary
Many fibroblast-secreted proteins promote tumorigenicity, and several factors secreted by cancer cells have in turn been proposed to induce these proteins. It is not clear whether there are single dominant pathways underlying these interactions or whether they involve multiple pathways acting in parallel. Here, we identified 42 fibroblast-secreted factors induced by breast cancer cells using comparative genomic analysis. To determine what fraction was active in promoting tumorigenicity, we chose five representative fibroblast-secreted factors for in vivo analysis. We found that the majority (three out of five) played equally major roles in promoting tumorigenicity, and intriguingly, each one had distinct effects on the tumor microenvironment. Specifically, fibroblast-secreted amphiregulin promoted breast cancer cell survival, whereas the chemokine CCL7 stimulated tumor cell proliferation while CCL2 promoted innate immune cell infiltration and angiogenesis. The other two factors tested had minor (CCL8) or minimally (STC1) significant effects on the ability of fibroblasts to promote tumor growth. The importance of parallel interactions between fibroblasts and cancer cells was tested by simultaneously targeting fibroblast-secreted amphiregulin and the CCL7 receptor on cancer cells, and this was significantly more efficacious than blocking either pathway alone. We further explored the concept of parallel interactions by testing the extent to which induction of critical fibroblast-secreted proteins could be achieved by single, previously identified, factors produced by breast cancer cells. We found that although single factors could induce a subset of genes, even combinations of factors failed to induce the full repertoire of functionally important fibroblast-secreted proteins. Together, these results delineate a complex network of tumor-fibroblast interactions that act in parallel to promote tumorigenicity and suggest that effective anti-stromal therapeutic strategies will need to be multi-targeted.
Introduction
Solid tumors are aberrant tissues where stromal cell types co-develop with and influence cancer cells [1]. Significant epigenetic alterations and gene expression changes occur in stromal cells as tumors progress, and the stromal changes are as strikingly different as those observed in the cancer epithelial compartments [2]–[5]. Many of these stromal cell changes are elicited by factors secreted by cancer cells, such as vascular endothelial growth factor (VEGF; MIM: 192240), which helps recruit and induce proliferation of endothelial cells [6]. Cancer cells also secrete factors that alter surrounding fibroblasts, such as transforming growth factor (TGF)-β (MIM: 190180) which induces fibroblasts to differentiate into myofibroblasts and secrete collagen, thereby contributing to the abundant extracellular matrix often observed in epithelial tumors [7]. In addition to TGF-β factors secreted by cancer cells that influence stromal fibroblasts include platelet derived growth factor (PDGF; MIM: 173430), interleukin (IL)-6 (MIM:147620), IL1-α (MIM: 147760), and WNT1 inducible signaling pathway protein (WISP)-1 and -2 (MIM: 603398 and 603399) [8], [9].
Tumor associated fibroblasts have been shown to promote cancer cell proliferation, angiogenesis, extracellular matrix (ECM) remodeling, inflammation, invasion and metastasis [10], [11]. Several fibroblast-secreted or membrane-bound factors that mediate these effects have been identified, including CXCL12 (MIM: 600835), hepatocyte growth factor (HGF; MIM:142409), matrix metalloproteinase MMP14 (MIM: 600754), osteopontin (MIM: 166490), TGF-β, and CCL2 (MIM: 158105) [12]–[17].
Some basic underlying processes involved in regulating the interactions between the epithelial cancer cells and the stromal fibroblasts have been identified. For example, several fibroblast-secreted factors are inflammatory cytokines whose expression is driven by NF-kappaB-dependent transcription in a process similar to the senescent secretory phenotype observed in aging fibroblasts [18], [19]. Additionally, a study of how fibroblasts co-evolve with tumor cells determined that fibroblasts gradually implement two signaling loops, involving TGF-β and CXCL12, which act together through both autocrine and cross-signaling mechanisms [20]. What remains unclear is whether the different factors involved in cancer cell-fibroblast interactions reflects a requirement of a multitude of fibroblast factors acting in parallel to promote tumorigenicity, or whether it reflects the diversity of the approaches and systems used to identity important interactions. Here, we designed a study to explore how the entire repertoire of fibroblast-secreted factors that are induced by human cancer cells function as a whole and compared the factors. This systems-level study was designed to be complimentary to approaches that focus on single genes or single processes. Our study indicates that the majority of induced fibroblast-secreted factors play a role in promoting tumorigenicity and that they do so through diverse effects on the tumor tissue.
Results
Comparative genomic analysis of pro-tumorigenic fibroblast factors induced by breast cancer cells
We adapted previously used systems of human cancer cells and fibroblasts [21]–[23], by using fibroblast lines that were amenable for the use of stable RNA interference (shRNA) so the relevance of candidate mediators of tumor-stromal interactions could be tested. We determined using co-injection assays that two human fibroblast lines previously shown to promote tumorigenicity in other systems (HFFF2 and HFF1) were able to promote tumorigenicity of two basal breast cancer subtype cell lines, MDA-MB-231 and Cal51, whereas two other human fibroblast lines (Wi-38 and CCD1112Sk) were not (Figure S1). We used the two tumor-supportive fibroblast lines (HFFF2 and HFF1) as models for patient-derived breast carcinoma associated fibroblasts and the two non-supportive fibroblasts as models for normal breast tissue derived fibroblasts. Using fibroblasts cell lines allowed shRNA transfection, selection and subsequent validation of gene silencing, whereas patient-derived breast fibroblasts cannot be passaged in culture long enough for such manipulations (data not shown; Ahmet Acar, personal communication). To determine at which point the host murine fibroblasts replaced the co-injected human fibroblasts, we tagged the human fibroblasts with green fluorescent protein (GFP) so that we could visualize how long they survived ensconced within developing tumors. We found that after three weeks, the number of co-injected GFP-tagged human fibroblasts comprised roughly 20% of the tumor stroma as judged by co-localization of GFP and the fibroblast stromal marker alpha-smooth muscle actin (α-SMA) (Figure S2), but the relative contribution steadily declined from week 3 to week 8, although the GFP-fibroblasts never completely disappear. The level of α-SMA positive cells within tumor stroma persisted during this same time period (Figure S2).
We performed transcriptional profiling and pathway analysis to determine if exposure of our model tumor-supportive fibroblasts to breast cancer cells in co-culture resembled expression changes seen in patient-derived breast carcinoma fibroblasts. We compared expression profiles of HFF1 and HFFF2 tumor-supportive fibroblasts co-cultured with either Cal51 or MDA-MB-231 (four combinations total) to those of Wi-38 and CCD1112Sk non-supportive fibroblasts co-cultured with either Cal51 or MDA-MB-231 (another four combinations). We chose to work with basal subtype breast cancer cell lines due to the greater need to develop new treatments for this poor prognosis subtype. We then compared the pathways selectively enriched in tumor-supportive fibroblasts co-cultured with Cal51 and MDA-MB-231 to pathways enriched by comparing cultures of patient-derived breast carcinoma fibroblasts to their normal counterparts. Strikingly, the top four pathways identified by gene-set enrichment analysis (GSEA) that are activated by exposure of tumor-promoting human fibroblasts to breast cancer cells are also amongst the top ten pathways activated in cultures of patient-derived breast carcinoma fibroblasts relative to normal fibroblasts (Figure 1). These pathways are ECM-receptor interaction, focal adhesion, integrin signaling and integrin cell-surface interactions; interrelated pathways that have been shown to be involved in the activation of cancer associated fibroblasts [21], [24], [25]. Additionally, most of the other top ten activated pathways in both systems were activated in the other system at lower ranking but still significant levels. These pathways included cytokine cytokine-receptor interactions, PDGF signaling, and Rho GTPase signaling; which likewise have shown to be involved in activation of cancer associated fibroblasts [7], [13], [26] (Figure 1). On the whole, the overlap in all significantly activated pathways is 44% (Table S1). This affirmed that this system of tumor-promoting human fibroblasts resembled in vitro cultures of patient-derived breast cancer fibroblasts.

We next wanted to test whether this system also reflected changes observed in vivo in human primary breast cancer stroma. To address this, we used gene-set enrichment analysis to determine the pathways that were activated in microdissected human breast stroma relative to normal breast stroma [3]. Despite the fact that human breast stroma contains many cell types in addition to fibroblasts, four of the eight significantly activated pathways in human breast stroma were also significantly activated by exposure of tumor-promoting fibroblasts to breast cancer cells, including cytokine/cytokine-receptor interactions and JAK-STAT signaling (Figure 1). Additionally, we developed a gene signature based on stimulation of tumor-promoting fibroblasts by breast cancer cells. Based on both clustering and principal component analysis, this gene signature was able to correctly predict whether microdissected human breast stroma was derived from cancerous or normal breast tissue for 98 of 99 samples (Figure 1). These comparative genomic analyses establish the close correspondence of our system of interaction of breast cancer cells with tumor-promoting fibroblasts to stroma and stromal fibroblasts isolated from primary human breast cancer.
Determining the repertoire of fibroblast-secreted factors that are induced by breast cancer cells
We used genome-wide analysis to determine the full repertoire of secreted factors induced by breast cancer cells in our tumor-promoting fibroblasts. As the first step, we compared genes induced in tumor-promoting fibroblast lines to genes induced in fibroblast lines incapable of promoting tumorigenicity. We found 320 genes that were more than 2-fold greater induced in tumor-promoting fibroblasts (Table S2) and within this group were 62 genes encoding secreted proteins. Of these, 42 were also significantly upregulated (p<0.05) in stromal cells isolated from primary breast cancer relative to normal breast stroma cells [3], [4], [27]. This group contained cytokines (15), extracellular matrix proteins (7), proteases (6), and growth factors and hormones (6) (Table 1). Cytokines as a class were significantly enriched in our screen relative to their representation in the set of all secreted proteins (36% vs. 12%, p = 5.6e-6). This enrichment is consistent with the prior reports that cytokines play key roles in the tumor-supportive function of tumor-associated fibroblasts [19]. In contrast, the other functional classes of secreted proteins were not significantly enriched. The cytokines upregulated in tumor-supportive fibroblasts included CC - and CXC-chemokines (CCL-2, -5, -7, -8, -20, and CXCL-5, -10; MIM: 158105, 187011, 158106, 602283, 600324 and 147310 respectively), pro-inflammatory interleukins (IL-1α, -1β, -8, -11, -24; MIM: 147760, 147720, 146930, 147681 and 604136 respectively), colony stimulating factor (G-CSF; MIM: 138970), interleukin receptor antagonist (IL1RN; MIM: 147679), and TNF superfamily member TNSF15 (MIM: 604052) (Table 1). Systematic literature searches revealed that 16 of the 42 secreted proteins had one or more publication(s) implicating them in the tumor-supportive function of fibroblasts while 26 did not (Table 1). The genes not previously implicated as mediators of the tumor-supportive function of fibroblasts included the majority (8/15) of cytokines, one-half (3/6) of the proteases, and the majority (4/6) of the growth factors and hormones. Examples include CCL8, encoding a chemokine not previously linked to cancer that is involved in homing of memory T lymphocytes to inflamed skin [28]; pregnancy-associated plasma protein-a (PPAPA; MIM: 176385), a protease that degrades IGF-binding proteins and acts as a positive modulator of local IGF signaling in skin repair [29]; and EGFL6 (MIM: 300239), encoding an epidermal growth factor (EGF) repeat protein expressed in osteoblastic-like cells and capable of inducing migration of endothelial cells [30].Some of the fibroblast-secreted candidate tumor-supportive factors fall outside of the known classes of proteins involved in tumor-stromal interactions, including ISG15 (MIM: 147571), an interferon-inducible, ubiquitin-like protein whose secretion plays a critical role in mediating an effective immune response to mycobacteria [31]; and complement component C3 (MIM: 120700), which in addition to its role in the complement cascade helps mobilize hematopoietic stem/progenitor cells to wounds [32].

Development of functional assays to test the effects of selected candidate mediators of fibroblasts on tumor growth and tumor microenvironment
In order to characterize the effects of fibroblasts on tumor progression, we co-injected tumor-promoting fibroblasts and examined their effects on both tumor cells and associated non-tumor cells within the tumor microenvironment. Consistent with a faster tumor growth rate, cancer cells in the co-injected tumors exhibited a two-fold higher proliferative rate based on Ki-67 labeling (Figure 2). To visualize and quantify the presence of three of the most important features of the tumor microenvironment - inflammation, vascularization and fibroblast activation - we performed immunohistochemical analysis at week six after co-injection. We used antibodies to the 7/4-antigen, which is highly expressed in neutrophils and inflammatory monocytes [33], the CD31 endothelial antigen, which visualizes blood vessels [34], and α-SMA, which is expressed by activated fibroblasts [35]. Strikingly, the presence of inflammatory cells, degree of vascularization, and number of activated fibroblasts were all 3 to 4 fold higher in the co-injected tumors (Figure 2), indicating a profound influence of tumor-supportive fibroblasts on the composition of the tumor microenvironment.

We decided to analyze a mix of previously implicated and novel candidate pro-tumorigenic fibroblast-secreted factors. Additionally, we wanted to test whether seemingly redundant factors were indeed functionally redundant. We focused on cytokines, growth factors, and hormones, and chose CCL2 and CCL7 (both previously validated as functionally important fibroblast secreted factors [17], [36]); the related factor CCL8 which, like CCL7, binds CCR1 (MIM: 601159); amphiregulin (AREG; MIM: 104640), which has been implicated in tumor stromal-interactions but as a ligand produced by cancer cells acting on fibroblasts [37]; and stanniocalcin1 (STC1; MIM: 601185), which has been shown to act as a cancer cell autonomous factor, but not as a stromally produced factor [38]. For each of these five genes, we confirmed by quantitative RT-PCR that there was a significant induction in the co-cultured tumor supportive fibroblasts relative to the co-cultured neutral fibroblasts (Figure S3).
CCL2, CCL7 and CCL8 chemokines mediate tumor supportive abilities of fibroblasts with distinct effects on the tumor microenvironment
CCL2, CCL7, and CCL8 are structurally related chemokines that share a common function of recruitment of monocytes to areas of injury and inflammation [39]. Based on their structural similarity and overlapping functions in inflammation, we wanted to determine if they had redundant roles in the tumor supportive function of co-injected fibroblasts. Remarkably, shRNAs directed against each of these three cytokines suppressed tumorigenicity in the co-injection assay, with the strongest effects observed when silencing CCL2 (53%) or CCL7 (66%) compared to weaker effects exerted by silencing of CCL8 (25%) (two validated shRNAs per gene, Figure 3). Interestingly, despite their related structure, silencing of each of the three cytokines had a distinct impact on the tumors: silencing of CCL2 suppressed recruitment of innate immune cells and angiogenesis (Figure 4) almost to the same levels as in tumors without co-injected fibroblasts. In contrast, silencing of CCL8 suppressed only the recruitment of innate immune cells, while silencing of CCL7 reduced tumor cell proliferation almost to the levels observed in tumors growing in the absence of co-injected fibroblasts (Figure 4 and Figure S4). Our data therefore showed that these related chemokines had non-redundant roles in mediating fibroblast-supportive functions.
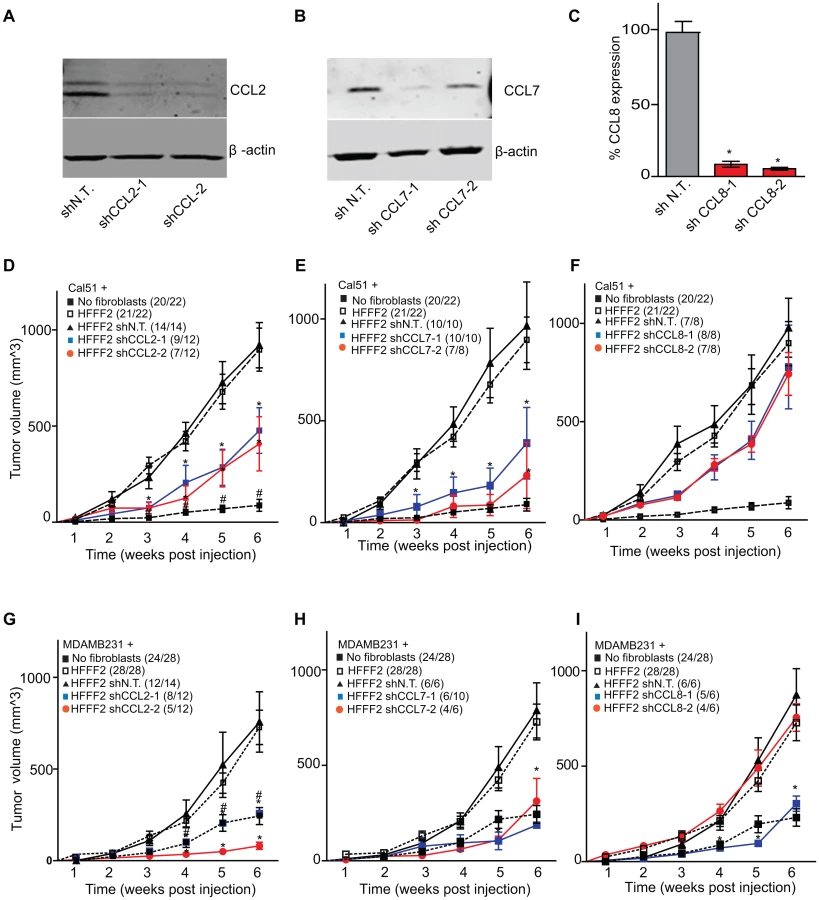

Suppressing AREG reduces the ability of fibroblasts to support tumorigenicity
We next tested whether silencing of AREG in fibroblasts affected tumor supportive function. Silencing of AREG had a pronounced effect, with an average reduction in tumor size at six weeks of 55% to 65% (Figure 5). In contrast to the chemokines, silencing AREG in fibroblasts had no effect on the number of blood vessels or innate immune cells (Figure 5). However, immunohistochemical analysis of the percentage of cells within the tumor that expressed α-SMA, a marker of mesenchymal-derived cells such as activated fibroblasts, revealed a 78% reduction. This level was comparable to the low levels of α-SMA positive cells in tumors formed in the absence of co-injected fibroblasts (Figure 5). Although α-SMA is expressed by activated pericytes, a cell type that surrounds endothelial cells of blood vessels, we did not observe a difference in number of α-SMA cells associated with blood vessels (Figure 5). This suggested that secretion of amphiregulin by the co-injected human fibroblasts plays a major role in establishing a tumor microenvironment that is enriched for activated fibroblasts, and since the co-injected human fibroblasts are almost entirely replaced by mouse fibroblasts at the point of excision (6 weeks), this effect must be propagated through recruited mouse fibroblasts. Consistent with this, we found that amphiregulin increased proliferation of mouse fibroblasts, as well as human fibroblasts (Figure 5). We also found that amphiregulin was a chemoattractant for mouse fibroblasts in migration and invasion assays, suggesting a mechanism of recruitment into the tumor, and that amphiregulin directly activated fibroblasts as judged by induction of α-SMA expression (Figure 6).


In addition to the pronounced reduction of activated fibroblasts when AREG was silenced, we also noted changes in activation of the amphiregulin receptor EGFR (MIM: 131550) on tumor cells. Compared to tumor cells co-injected with control tumor-supportive fibroblasts, tumor cells when co-injected with AREG-silenced fibroblasts showed a <2-fold reduction of activated, phospho-EGFR as measured by immunohistochemistry (Figures 6). This was accompanied by a minor, barely significant (p = 0.06), negative effect on tumor cell proliferative rate in vivo, with no direct effect of amphiregulin on tumor cell proliferation in vitro (Figure 5 and Figure S5). In contrast, we found a significant increase in necrosis in tumors after silencing of fibroblast-secreted amphiregulin (Figure 6). Notably, amphiregulin significantly protected the breast cancer cells from cell death induced by detachment from their normal extracellular matrix (anoikis, Figure 6). Together, these results suggest that fibroblast-secreted amphiregulin has potent effects on tumor progression, with autocrine effects leading to activation of fibroblasts and paracrine effects protecting cancer cells from cell death.
Strong tumor suppressive responses are achieved by simultaneously targeting different mediators of tumor-fibroblast interactions on both cell types
Co-culturing also lead to changes in gene expression in the breast cancer cells, which upregulated a shared receptor for CCL2 and CCL7, the chemokine receptor CCR1 upon co-culturing with tumor-supportive fibroblasts (Figure 7). We therefore tested whether CCR1 expression by cancer cells was critical for some of the tumor supportive functions of fibroblasts by stably expressing shRNAs targeting CCR1 in Cal51 breast cancer cell line (knockdown efficiency was quantified by both qRT-PCR and immunoblotting, Figure S6). Silencing of CCR1 had no effect on tumor growth when cancer cells were injected alone. However, in the context of co-injection with fibroblasts, silencing of cancer cell CCR1 resulted in a 3-fold reduction in tumor size, almost eliminating the effect of the fibroblasts (Figure 7 and Figure S6). We further observed a 40% reduction in the proliferative index of cancer cells and a 2-fold reduction in recruitment of neutrophils and inflammatory monocytes to the tumor, but only minor effects on the number of blood vessels and mesenchymal cells (Figure 7 and Figure S6). Thus, expression of CCR1 by cancer cells plays a critical role in enabling fibroblasts to exert tumor supportive function, through increased tumor cell proliferation and potentially indirectly through recruitment of leukocytes.

We next asked whether blocking two interactions between fibroblasts and cancer cells was more effective than blocking either one alone. We therefore asked whether simultaneously silencing fibroblast-secreted amphiregulin and cancer cell expressed CCR1 was more efficacious than blocking either alone. We found that tumor growth was significantly more reduced when both pathways were targeted (Figure 7).
Cancer cell induction of the fibroblast-secreted proteins requires parallel activation
A number of studies have implicated specific cancer-cell secreted factors in the activation of neighboring fibroblasts, including TGF-β [40] and IL-1β [19]. We wanted to determine whether the repertoire of tumor-promoting fibroblast secreted factors could be induced by single specific inducers, or whether multiple pathways were acting in parallel. To test this, we used quantitative RT-PCR to measure gene expression changes in tumor-promoting fibroblasts of the five factors (CCL-2, -7, -8, AREG, and STC1) that we had determined all had functional relevance, along with two others factors (NRG1; MIM: 142445 and WISP1) that also were induced in the system. Surprisingly, TGF-β, did not induce expression of any of the seven factors (Table 2), despite its ability to induce activation of fibroblasts to myofibroblasts, which resembles many aspects of carcinoma-associated fibroblasts [10]. AREG also did not induce expression of any of the seven factors, despite its key role in stimulating mammary fibroblasts during normal mammary development [37]. IL-1β, a potent activator of NF-κB signaling, produced an upregulation in chemokines CCL-2, -7, -8 similar to that seen by co-culture with breast cancer cells (Table 2). However, IL-1β did not induce expression of WISP1, STC1, AREG, or NRG1. Interestingly, a combination of IL-1β and AREG lead to significant upregulation of WISP1 in addition to stimulation of CCL-2, -7, -8. In contrast, none of the fibroblast factors were induced when TGF-β and IL-1β were combined (Table 2). Based on these results, it seems likely that the ability of breast cancer cells to induce the full spectrum of pro-tumorigenic fibroblast-secreted proteins involves a multitude of interacting factors, including some not previously identified. We also found that co-culture of tumor-promoting fibroblasts with a normal, non-malignant breast epithelial cell line, MCF10A, was able to induce fibroblast expression of two out of the seven factors induced by breast cancer cells, namely AREG and WISP2 (Table 2). For these two factors, it would appear that breast epithelial cells per se, regardless of tumorigenic properties, elicit the same response in fibroblasts. This result is consistent with the key role that stromal AREG plays in normal mammary development [37].

Discussion
Numerous studies have used the co-injection assay to identify the factors produced by fibroblasts that are responsible for promoting tumorigenicity. These studies have identified single factors that when inhibited strongly suppress the ability of activated fibroblasts to promote tumorigenicity, and they include scatter factor, SDF-1, MMP14, NF-κB, osteopontin, TGF-β, and CCL2 [12]–[17]. However, these studies did not address whether the single factors were unique in their capacity of promoting fibroblast-supported tumorigenicity, nor did they compare different factors. Here, we used comparative genomics to identify 42 candidate mediators of fibroblast-promoted tumorigenicity and we tested the functional impact of a set of five of these factors. Surprisingly, we found that four of the five tested factors promoted tumorigenicity, three of them strongly (CCL7, CCL2, and AREG) and one of them weakly (CCL8) (Table 3). Although the fifth factor STC1 significantly affected tumorigenicity by the area under the tumor growth curve test (Table 3), it failed to show significant effects using t-tests at any single time points (data not shown). Since we only tested five of the 42 fibroblast-secreted-factors that were induced by breast cancer cells, it seems highly likely that an even greater number of fibroblast-secreted factors play a role in promoting tumorigenicity. Thus, our study indicates that even in a single system there are a large number of secreted factors involved in the ability of fibroblasts to promote carcinomas, rather than a single important mediator.

Intriguingly, our results also indicate widely diverse mechanisms for fibroblast-secreted factors in the promotion of tumorigenicity. The strong effects of fibroblast CCL7 appeared to be caused by a significant effect on cancer cell proliferation (Table 3), which was unique among the factors tested. We also found that reducing cancer cell expression of the CCL7 receptor, CCR1, also reduced fibroblast-induced proliferation. In contrast, the strong tumor promoting effects of fibroblast CCL2 was associated with different effects on the tumor microenvironment, as we found significant decreases in both angiogenesis and recruitment of innate immune cell upon silencing of CCL2. CCL8 had weaker effects on tumor growth than the other tested chemokines, which likely reflects it inability to affect either tumor proliferation or vascularity (Table 3). Fibroblast AREG also had very strong effects on tumor growth, and influenced both the total number of activated fibroblasts in the tumors and the survival of the cancer cells, a combination not observed with any other tested factor. Interestingly, secretion of amphiregulin by fibroblasts appeared to potentially act as a chemoattractant to recruit new fibroblasts and induce their proliferation and activation, resulting in a tumor microenvironment that is enriched in activated fibroblasts. Thus, in a single system of carcinoma cells and tumor-supportive fibroblasts, several factors play key roles.
Our study also sheds light on how cancer cells modify the stromal cells to enable them to promote tumorigenicity. We tested several cancer cell-secreted factors, previously reported to influence stromal cells, but none of them were able to induce the full panel of verified tumor-promoting fibroblast factors as well as the cancer cells themselves. Even in one simple system, it appears that cancer cells act on fibroblasts through multiple factors, resulting in the secretion of another complex set of factors that influence cancer cells and other components of the tumor microenvironment.
One of the key findings of our study was that inhibiting multiple interactions between cancer cells and fibroblasts is more efficacious than blocking individual pathways. This finding is not completely unexpected in light of the complexity of the tumor microenvironment and in fact previous reports have suggested this as a possibility [20], [41]. Nevertheless, this result highlight that the different interactions are non-redundant and act in parallel, but it also suggests that effective anti-stromal fibroblast therapeutic strategies can be achieved by taking a multi-targeted approach. Future research towards this end will need to employ models that closely resemble the type of tumors to be targeted in human patients. This presents many challenges, including the ability to selectively target endogenous fibroblasts in tumor tissues, along with the ability to monitor the in vivo response of tumor-associated fibroblasts. Despite these challenges, our study shows that there are several potential combinatorial targets for future fibroblast-targeted therapeutic approaches.
Materials and Methods
Data files
All genomic data for this study, including expression analysis of both fibroblasts and breast cancer cells, have been deposited in the Gene Expression Omnibus (GEO) repository (GSE41678). http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=bnepxkssyoamgdu&acc=GSE41678
Cell lines, co-culture, flow cytometry and gene expression analysis
Breast cancer cell lines and human fibroblast strains were obtained from ATCC (Manassas, VA), DSMZ (Braunschweig, Germany) or Sigma (St.Louis, MO) and grown under standard tissue culture conditions in growth medium recommended by the supplier. For dual-color co-culture experiments, breast cancer cells stably expressing Discosoma sp. red fluorescent protein (DsRed) and human fibroblasts stably expressing Zoanthus sp. green fluorescent protein (ZsGreen) were generated using pRetroX-IRES-DsRedExpress vector and pRetroX-IRES-ZsGreen1 vector (Clontech, CA) respectively via retroviral transduction (detailed description in Supplemental Experimental Procedures). None of the experiments utilized multiple retroviral transfections. For the co-culture experiments, we transfected the original fibroblasts with a fluorescent marker in order to facilitate cell separation before transcriptome analysis (the breast cancer cells were transfected with a different fluorescent marker). For the shRNA experiments, we transfected the original fibroblasts with validated shRNA constructs from the Broad library. Co-culture of fluorescently-tagged breast cancer cells and fibroblasts was initiated by plating 1.5 million fibroblasts into 10 cm dishes, and after 18 hours 1 million breast cancer cells were added and then incubated for six days. Monocultures were performed in parallel for the same duration. Following co-culture or mono-culture, cells were trypsinized and resuspended in FACS sorting buffer (PBS+1% FBS) for separation into DsRed+ and ZsGreen+ populations using an ARIA II flow cytometer and analyzed using FACS DiVA software (Becton Dickenson, CA). Total RNA was isolated using RNeasy kit (Qiagen, Netherlands) and hybridized to Gene 1.0 ST arrays (Affymetrix, CA). Data was extracted, background corrected, normalized, and converted from probe values to gene values using the AROMA R package (www.aroma-project.org).
Tumorigenicity assays
All studies utilizing human xenograft experiments were approved by and in accordance with Cold Spring Harbor Laboratory's Institutional Animal Care and Use Committee. Five to six week old female nude mice (NCR nu/nu; Charles River Inc., Wilmington, MA) were irradiated at 400 cGy 24–36 hours prior to injections. One million breast cancer cells were trypsinized, resuspended with or without 1.5 million fibroblasts in 100 µl DMEM and injected subcutaneously into both flanks of irradiated, nude mice. Growth was followed over time by taking caliper measurements at indicated time points. Tumor volume was measured as 0.52×length×width2. Tumors were excised six-eight weeks post injections or when one of the measurements reached 2 cm.
Immunostaining and quantification
Immunostaining procedure is described in detail in Supplementary Experimental Procedures. The primary antibodies used for immunostaining are as follows: α-SMA (1∶2000; # 1A4; Sigma-Aldrich), CD31 (1∶100; ab28364; Abcam), antigen 7/4 (1∶400, CL8993AP, Cedarlane), Ki-67 (1∶2000; MIB5; Dako), pEGFR (1∶100; 1138-1; Epitomics) and GFP (1∶1000; ab290; Abcam). Immunostained slides were quantified by counting (for 7/4, Ki-67 and CD31), by percentage of stained area (for α –SMA and pEGFR) using Image J software (NIH, Bethesda, MD).
Additional methods are found in the file Text S1.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. EgebladM, NakasoneES, WerbZ (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18 : 884–901.
2. CaseyT, BondJ, TigheS, HunterT, LintaultL, et al. (2009) Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat 114 : 47–62.
3. FinakG, BertosN, PepinF, SadekovaS, SouleimanovaM, et al. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14 : 518–527.
4. MaXJ, DahiyaS, RichardsonE, ErlanderM, SgroiDC (2009) Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 11: R7.
5. AllinenM, BeroukhimR, CaiL, BrennanC, Lahti-DomeniciJ, et al. (2004) Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6 : 17–32.
6. FerraraN, GerberHP, LeCouterJ (2003) The biology of VEGF and its receptors. Nat Med 9 : 669–676.
7. ElenbaasB, WeinbergRA (2001) Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res 264 : 169–184.
8. GiannoniE, BianchiniF, MasieriL, SerniS, TorreE, et al. (2010) Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 70 : 6945–6956.
9. KumarS, KishimotoH, ChuaHL, BadveS, MillerKD, et al. (2003) Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am J Pathol 163 : 2531–2541.
10. EgebladM, LittlepageLE, WerbZ (2005) The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol 70 : 383–388.
11. PolyakK, KalluriR (2010) The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol 2: a003244.
12. StueltenCH, BuschJI, TangB, FlandersKC, OshimaA, et al. (2010) Transient tumor-fibroblast interactions increase tumor cell malignancy by a TGF-Beta mediated mechanism in a mouse xenograft model of breast cancer. PLoS One 5: e9832.
13. OrimoA, GuptaPB, SgroiDC, Arenzana-SeisdedosF, DelaunayT, et al. (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121 : 335–348.
14. ChengN, ChytilA, ShyrY, JolyA, MosesHL (2007) Enhanced hepatocyte growth factor signaling by type II transforming growth factor-beta receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res 67 : 4869–4877.
15. ZhangW, MatrisianLM, HolmbeckK, VickCC, RosenthalEL (2006) Fibroblast-derived MT1-MMP promotes tumor progression in vitro and in vivo. BMC Cancer 6 : 52.
16. PazolliE, LuoX, BrehmS, CarberyK, ChungJJ, et al. (2009) Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res 69 : 1230–1239.
17. TsuyadaA, ChowA, WuJ, SomloG, ChuP, et al. (2012) CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res 72 : 2768–2779.
18. CoppeJP, DesprezPY, KrtolicaA, CampisiJ (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5 : 99–118.
19. ErezN, TruittM, OlsonP, ArronST, HanahanD (2010) Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 17 : 135–147.
20. KojimaY, AcarA, EatonEN, MellodyKT, ScheelC, et al. (2010) Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 107 : 20009–20014.
21. KalluriR, ZeisbergM (2006) Fibroblasts in cancer. Nat Rev Cancer 6 : 392–401.
22. MartinTA, ParrC, DaviesG, WatkinsG, LaneJ, et al. (2003) Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis 24 : 1317–1323.
23. HollidayDL, HughesS, ShawJA, WalkerRA, JonesJL (2007) Intrinsic genetic characteristics determine tumor-modifying capacity of fibroblasts: matrix metalloproteinase-3 5A/5A genotype enhances breast cancer cell invasion. Breast Cancer Res 9: R67.
24. KimBG, GaoMQ, ChoiYP, KangS, ParkHR, et al. (2012) Invasive breast cancer induces laminin-332 upregulation and integrin beta4 neoexpression in myofibroblasts to confer an anoikis-resistant phenotype during tissue remodeling. Breast Cancer Res 14: R88.
25. StokesJB, AdairSJ, Slack-DavisJK, WaltersDM, TilghmanRW, et al. (2011) Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 10 : 2135–2145.
26. Sanz-MorenoV, GaggioliC, YeoM, AlbrenguesJ, WallbergF, et al. (2011) ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20 : 229–245.
27. BauerM, SuG, CasperC, HeR, RehrauerW, et al. (2010) Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene 29 : 1732–1740.
28. IslamSA, ChangDS, ColvinRA, ByrneMH, McCullyML, et al. (2011) Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+ T(H)2 cells. Nat Immunol 12 : 167–177.
29. BoldtHB, ConoverCA (2011) Overexpression of pregnancy-associated plasma protein-A in ovarian cancer cells promotes tumor growth in vivo. Endocrinology 152 : 1470–1478.
30. ChimSM, QinA, TicknerJ, PavlosN, DaveyT, et al. (2011) EGFL6 promotes endothelial cell migration and angiogenesis through the activation of extracellular signal-regulated kinase. J Biol Chem 286 : 22035–22046.
31. BogunovicD, ByunM, DurfeeLA, AbhyankarA, SanalO, et al. (2012) Mycobacterial Disease and Impaired IFN-gamma Immunity in Humans with Inherited ISG15 Deficiency. Science
32. RatajczakMZ, RecaR, WysoczynskiM, YanJ, RatajczakJ (2006) Modulation of the SDF-1-CXCR4 axis by the third complement component (C3)–implications for trafficking of CXCR4+ stem cells. Exp Hematol 34 : 986–995.
33. RosasM, ThomasB, StaceyM, GordonS, TaylorPR (2010) The myeloid 7/4-antigen defines recently generated inflammatory macrophages and is synonymous with Ly-6B. J Leukoc Biol 88 : 169–180.
34. HorakER, LeekR, KlenkN, LeJeuneS, SmithK, et al. (1992) Angiogenesis, assessed by platelet/endothelial cell adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet 340 : 1120–1124.
35. SugimotoH, MundelTM, KieranMW, KalluriR (2006) Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 5 : 1640–1646.
36. JungDW, CheZM, KimJ, KimK, KimKY, et al. (2010) Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int J Cancer 127 : 332–344.
37. SternlichtMD, SunnarborgSW (2008) The ADAM17-amphiregulin-EGFR axis in mammary development and cancer. J Mammary Gland Biol Neoplasia 13 : 181–194.
38. LiuG, YangG, ChangB, Mercado-UribeI, HuangM, et al. (2010) Stanniocalcin 1 and ovarian tumorigenesis. J Natl Cancer Inst 102 : 812–827.
39. FernandezEJ, LolisE (2002) Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol 42 : 469–499.
40. Ronnov-JessenL, PetersenOW (1993) Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest 68 : 696–707.
41. ChouJ, LinJH, BrenotA, KimJW, ProvotS, et al. (2013) GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol 15 : 201–213.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Genome-Wide Systematic Analysis Reveals Different and Predictive Proliferation Expression Signatures of Cancerous vs. Non-Cancerous Cells
- Recent Acquisition of by Baka Pygmies
- The Condition-Dependent Transcriptional Landscape of
- Histone Chaperone NAP1 Mediates Sister Chromatid Resolution by Counteracting Protein Phosphatase 2A
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy