
Convergent Transcription Induces Dynamic DNA Methylation at Loci
Cytosine methylation of DNA is an important epigenetic gene silencing mechanism in plants, fungi, and animals. In the filamentous fungus Neurospora crassa, nearly all known DNA methylations occur in transposon relics and repetitive sequences, and DNA methylation does not depend on the canonical RNAi pathway. disiRNAs are Dicer-independent small non-coding RNAs that arise from gene-rich part of the Neurospora genome. Here we describe a new type of DNA methylation that is associated with the disiRNA loci. Unlike the known DNA methylation in Neurospora, disiRNA loci DNA methylation (DLDM) is highly dynamic and is regulated by an on/off mechanism. Some disiRNA production appears to rely on pol II directed transcription. Importantly, DLDM is triggered by convergent transcription and enriched in promoter regions. Together, our results establish a new mechanism that triggers DNA methylation.
Published in the journal:
. PLoS Genet 9(9): e32767. doi:10.1371/journal.pgen.1003761
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003761
Summary
Cytosine methylation of DNA is an important epigenetic gene silencing mechanism in plants, fungi, and animals. In the filamentous fungus Neurospora crassa, nearly all known DNA methylations occur in transposon relics and repetitive sequences, and DNA methylation does not depend on the canonical RNAi pathway. disiRNAs are Dicer-independent small non-coding RNAs that arise from gene-rich part of the Neurospora genome. Here we describe a new type of DNA methylation that is associated with the disiRNA loci. Unlike the known DNA methylation in Neurospora, disiRNA loci DNA methylation (DLDM) is highly dynamic and is regulated by an on/off mechanism. Some disiRNA production appears to rely on pol II directed transcription. Importantly, DLDM is triggered by convergent transcription and enriched in promoter regions. Together, our results establish a new mechanism that triggers DNA methylation.
Introduction
DNA methylation at the 5th position of cytosine to form 5-methylcytosine (5mC) is an important epigenetic gene silencing mechanism conserved from plants, fungi to animals [1], [2]. Even though most of the DNA methylation is relatively stable, dynamic DNA methylation has been observed during specific stages of animal development [3]. DNA methylation occurs in three different nucleotide sequence contexts: CG, CHG, and CHH (where H is C, A, or T). Small non-coding RNAs have been shown to be involved in the establishment and maintenance of heterochromatin formation in different organisms. In the fission yeast Schizosaccharomyces pombe, small RNAs and the RNAi pathway mediate histone H3 lysine-9 methylation at the centromeric regions [4]–[6]. In plants, the asymmetrical CHH methylation is maintained by de novo DNA methylation mediated by 24-nt small interfering RNAs (siRNAs) [review in 7], [8]. In mammalian germ cells, the Dicer-independent Piwi-interacting RNAs (piRNAs) are thought to be involved in DNA methylation [9], [10].
In the filamentous fungus Neurospora crassa, about 2% of cytosines in the genome are methylated [11]. Nearly all of the known methylation sites are within transposon relics of repeat-induced point mutation (RIP) and the repetitive ribosomal DNA locus [11]–[13]. RIP is a genome defense mechanism that results in mutation and methylation of duplicated DNA sequences during the sexual cycle [11], [14]. All previously known Neurospora DNA methylation is dependent on the histone methyltransferase, DIM-5, which meditates trimethylation of histone H3 at the lysine9 (H3K9me3) [15], [16]. Heterochromatin protein 1 (HP1) recognizes H3K9me3 and recruits DIM-2, the only confirmed DNA methyltransferase in Neurospora, to methylate DNA [17]–[19]. For the natural RIP'd sequences, DNA methylation is more or less stable and is generally not required for the maintenance of H3K9 methylation [12]. In addition, the known DNA methylation events are not dependent on the canonical RNAi pathway [20].
Neurospora produces many types of small RNAs, including microRNAs, siRNAs, QDE-2 interacting RNA (qiRNAs), and dicer-independent siRNAs (disiRNAs), through diverse small RNA biogenesis pathways [reviewed in 21]. disiRNAs are a distinct class of small RNAs as they symmetrically are mapped to both strands of DNA and their production is independent of the known canonical RNAi components, including Dicer [22]. The disiRNA loci range from a few hundred base pairs to more than 10 kilobases in size and are located in gene-rich regions of the genome. The function of disiRNA is unknown.
In this study we identified a new mechanism of DNA methylation that is associated with disiRNA loci. Our results showed that this type of DNA methylation, which we call disiRNA loci DNA methylation (DLDM), is very different from the previously known DNA methylation in Neurospora. DLDM is highly dynamic, depends on transcription at disiRNA loci, and is triggered by convergent transcription in some loci.
Results
DNA methylation at the disiRNA loci
DNA methylation is an important regulatory mechanism of transcription silencing. Therefore, we examined whether in Neurospora the disiRNA loci are associated with DNA methylation using the methylation-sensitive restriction enzyme-based PCR (MSP) assays [23]. The Neurospora genomic DNA from a wild-type strain was digested with the isoschizomers DpnII or BfuCI. Both enzymes digest unmethylated GATC sites, but only DpnII can cut at sites when C is methylated. Primer sets were then used for semi-quantitative and quantitative PCR (qPCR). As shown in Figure 1A, a non-disiRNA locus (NCU06312) was not methylated, as indicated by the lack of PCR amplification product after digestion by BfuCI, whereas a PCR product was readily detected for the ζ-η region, a relic of RIP that forms constitutive heterochromatin and was previously shown to carry DNA methylation [14], [24]. For the 9 selected disiRNA loci with high level of disiRNA and with size larger than 5 kb, PCR products were detected in all samples after BfuCI digestion, indicating that these disiRNA loci are methylated. From quantitative PCR (qPCR) analyses, we estimated that percentages of methylation at the DpnII sites in these loci ranged from ∼3.5% to 28.8%, levels much lower than that of the ζ-η region (58.4%).

We then applied qPCR to MSP to further determine the DNA methylation profile in disi-47, disi-6, and disi-29 loci, which are three large loci with high levels of disiRNAs (Figure S1). The DNA methylation profiles at these loci are correlated with disiRNA profiles: DNA methylation peaked at regions of peaked disiRNA expression, and there was little or no DNA methylation outside of the disiRNA loci (Figure 1B and Figure S2).
Since the MSP results only reflect the methylation status of cytosines within the cleaved GATC sites, the detected methylation levels might be biased. To exclude this possibility, we performed methylated DNA immunoprecipitation (MeDIP) to detect DNA methylation based on 5mC density in disiRNA loci. For 13 loci (disi-6, 8, 22, 23, 29, 34, 35, 39, 42, 47–50) with relatively high disiRNA level and with size larger than 5 kb, all of them harbor DNA methylation (data not shown). We further analyzed disi-6, disi-29 and disi-47 loci with more primer sets for better resolution. As expected, the levels of DNA methylation were high in regions of high disiRNA expression and were low or absent outside of the disiRNA loci (Figure 1C and Figure S3). In constrast, no DNA methylation was detected at two negative control loci al-1 and NCU06312.
Since the previously known DNA methylation in Neurospora is located in regions derived from RIP, we calculated the RIP indices of the methylated disiRNA loci [25]. We observed no significant difference in RIP indices between that of the whole genome (1.11±0.33) and all of the 50 identified disiRNA loci (1.13±0.32). Moreover, for each disi-6, disi-29 and disi-47 loci, the lowest RIP indices are well above the threshold (0.7) for RIP-induced DNA methylation [25] (Figure S4). These data suggest that the DNA methylation in these disiRNA loci is not a result of RIP. We therefore named this type of DNA methylation DLDM (disiRNA loci DNA methylation) to distinguish it from the RIP-induced DNA methylation.
The on/off methylation pattern of DLDM
To determine the nature of DLDM, we performed bisulfite sequencing of selected genomic regions. As controls, we first sequenced the ζ-η region, a relic of RIP process and the am locus, which was previously shown to be unmethylated [26]. As expected, every clone of the DNA at the ζ-η region was methylated to various degrees at both symmetric and non-symmetric cytosine sites with an average methylation frequency of 36% (Figure 2A), similar to the estimated frequency (42%) determined in the MSP assay. In contrast, the genomic DNA was unmethylated at the am locus. In our experiment, 99.5% of all cytosines were converted to uracils. This percentage was similar to the conversion rate (99.8%) of an unmethylated PCR fragment after the bisulfite treatment, indicating that our bisulfite conversion was complete (data not shown). To our surprise, bisulfite sequencing of the disiRNA regions with peak DNA methylation revealed that vast majority of the clones were not methylated, as indicated by the nearly 100% conversion of all cytidines into uracils. For a few of the disiRNA loci clones, however, almost all cytosines were maintained after the bisulfite treatment (data not shown).
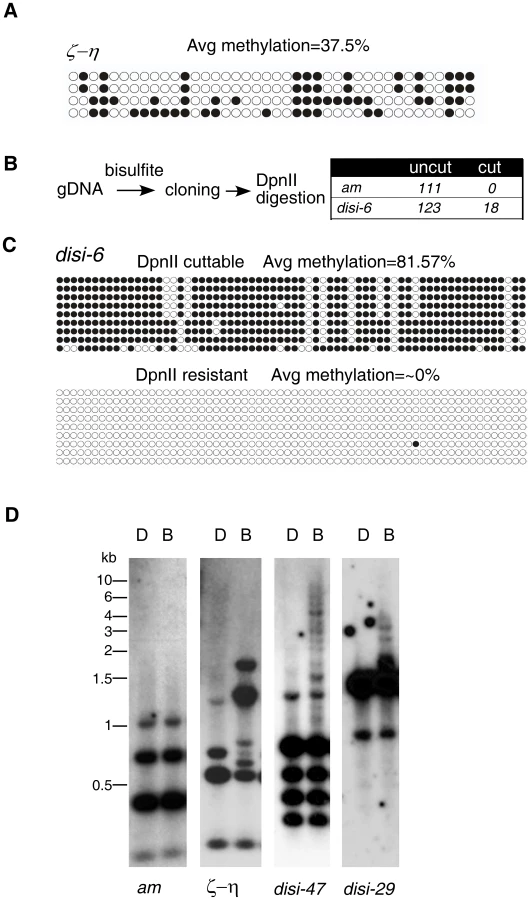
To rule out the possibility that these highly methylated clones were due to incomplete bisulfite conversion, we first treated the genomic DNA with bisulfite, then performed PCR and subcloned the fragments of either the disi-6 or the am locus into plasmids. PCR was then performed by using the plasmids as templates, and the resulting PCR products were subjected to DpnII digestion, which can only digest the PCR products if GATC site remains intact after bisulfite conversion due to the protection of 5mC (Figure 2B). As expected, all 111 clones of DNA from the am locus examined were resistant to the DpnII digestion, indicating that the bisulfite treatment of the genomic DNA was complete. For the disi-6 locus, however, 18 out of 123 DNA clones were cleaved by DpnII. Sequencing of the DpnII digestible (“cut”) and resistant (“noncut”) clones of disi-6 locus showed that in all “noncut” clones nearly 100% of cytosines were converted to uracils, indicating that the initial genomic DNA carried no 5mC (Figure 2C). In contrast, most of the cytosines in the DpnII digestible clones were methylated. Similar results were obtained from the disi-47 and disi-29 loci (Figure S5). Since Neurospora is haploid, these results indicate that the DNA methylation at disiRNA loci is actively regulated and is either on or off within each nucleus: disiRNA loci are either not methylated or are heavily methylated once DNA methylation process is switched on.
To confirm this finding, we used Southern blot analysis to visualize DNA methylation at several disiRNA loci. As shown in Figure 2D, both DpnII and BfuCI resulted in the same digestion pattern of the am locus, consistent with the lack of DNA methylation. For the ζ-η region, the BfuCI digestion resulted in the disappearance of one DNA fragment and the appearance of several additional higher molecular weight DNA fragments, consistent with all DNA molecules from this locus being methylated to some degree. The largest DNA fragment from BfuCI digestion was less than 2 kilobases (kb), indicating that methylation in the ζ-η region is limited to a small region. For the disi-47 and disi-29 loci, however, BfuCI digestion did not significantly change the levels or the relative ratios of the DpnII-digested bands, suggesting that most of the DNA lack methylation. We did observe, however, a ladder of high molecular weight (up to 8–10 kb) DNA fragments in the BfuCI-digested DNA, indicating that when methylated, the DNA at the disiRNA loci are heavily methylated across a large DNA region. Taken together, these results suggest that DLDM is highly dynamic and is regulated differently from the known DNA methylation events within relics of RIP in Neurospora.
Methylation of H3K9 at the disiRNA loci is dependent on DIM-2
The histone modification of H3K9me3 is mediated by the H3K9 methyltransferase DIM-5. This enzyme is essential for DNA methylation at relics of RIP, but H3K9 methylation at these loci is generally maintained in the absence of DNA methylation [12], [27]. To determine whether DLDM is mediated by H3K9me3 modification, we performed H3K9me3 chromatin immunoprecipitaton (ChIP) assays. Our results showed that the disiRNA loci are enriched by histones containing the H3K9me3 mark (Figure 3 and S6). However, in contrast to the ζ-η locus, in which the levels of H3K9me3 were not affected by the deletion of dim-2KO, the levels of H3K9me3 at the disi-47, -6 and -29 loci decreased dramatically in the dim-2KO mutant. These results indicate that DLDM requires DIM-2 for the maintenance of H3K9me3 at the disiRNA loci.

DLDM is linked with convergent transcription and disiRNA
As disiRNA loci are gene rich, we examined whether transcription was important for disiRNA and DLDM. Our previous EST analyses showed that at least some disiRNA loci harbor fully or partially overlapped antisense transcripts, suggesting that convergent transcription might be a trigger of disiRNA production and/or DLDM [22]. We chose disi-47 locus to test this hypothesis since it harbors the well-studied circadian clock gene frequency (frq, NCU02265), which is known to produce the sense frq transcript and an overlapping antisense transcript qrf [28], [29]. The frq gene encodes a core component of the Neurospora circadian clock. MeDIP and MSP assays showed that the promoter region of the frq gene was methylated; however, only low levels of DNA methylation were detected in the frq coding region (Figure S3).
The transcription of both frq and qrf is activated by light in a process that mediated by the WHITE COLLAR (WC) complex, which consists of two PAS domain-containing transcription factors WC-1 and WC-2 [29]. Previous studies showed that the expression of both frq and qrf are high in constant light (LL) and are low in constant darkness (DD) [28]. Our qRT-PCR assays also confirmed that the level of frq mRNA was significant higher in LL than in DD (Figures 4A). Importantly, our mRNA deep sequencing [30] and RT-qPCR assays also demonstrated the existence of light-dependent expression of transcripts from the promoter region of frq (Figure 4B and S7), where disiRNA level is high. In a wc-2KO mutant, frq mRNA and the transcripts originating from the promoter region were both abolished, indicating that, like frq mRNA, the promoter-specific transcription requires WC-2. MeDIP assays showed that the level of DNA methylation in the promoter of frq was significantly higher in LL than that in DD in the wild-type strain and was completely abolished in the wc-2KO mutant (Figure 4C). Together, these results suggest that DNA methylation at the frq promoter is dependent on WC-2-mediated transcription and that transcription at promoter region may be necessary for the DNA methylation process and disiRNA production. Similarly, divergent promoter transcripts are also clearly seen in other disiRNA loci such as disi-29 (Figure S7), suggesting that this architecture of transcription might be the reason of DLDM.

To determine whether disiRNA production is dependent on the WC-dependent transcription, we performed small RNA deep sequencing analyses in wc-2KO strain. As shown in Figure 4D, the disiRNA abundance in the mutant is completely abolished at the disi-47 locus. This result is consistent with the loss of DLDM in the wc-2KO strain (Figure 4C) and suggests that disiRNA and DLDM are tightly linked and both triggered by pol II-dependent transcription.
Convergent transcription triggers DNA methylation in the promoter region
To further test the possibility that convergent transcription is a trigger of DLDM, we created an artificial sense-antisense transcription construct Pqa-2:cul:1-gccP, in which a firefly luciferase (luc) gene is under the control of ccg-1 promoter and the quinic acid (QA) inducible promoter is used to drive the antisense transcription of the luc gene (Figure 5A). The construct was introduced into a dicer double mutant so that the convergent transcripts cannot be processed into siRNA from dsRNA [31]. As a control, a strain that contains a construct (Pqa-2:cul) lacking the ccg-1 promoter was analyzed (Figure 5B). MeDIP analyses showed that the addition of QA to growth medium resulted in a robust induction of DNA methylation in the promoter region of qa-2 in the Pqa-2:cul:1-gccP strain (Figure 5A). In contrast, no methylation of the qa-2 promoter was detected in the Pqa-2:cul strain with or without QA treatment (Figure 5B). In addition, the promoter of the endogenous qa-2 was also not methylated in a wild-type strain after QA treatment (Figure 5C). These results indicate that convergent transcription induces DNA methylation that peaks in the promoter region.

To determine the size of the methylated region in the transgene region in the Pqa-2:cul:1-gccP strain, we performed MeDIP analyses using primer sets spanning the transgene locus. As shown in Figure 5D and S8A, only low levels of DNA methylation were detected in the absence of QA; these levels are likely due to the leakage of the qa-2 promoter. The presence of QA resulted in a dramatic induction of DNA methylation. The peak of DNA methylation was upstream of the qa-2 promoter and about 3 kb upstream of the predicted qa-2 transcriptional start site (TSS). Only low levels of DNA methylation were detected in the luc gene and in the ccg-1 promoter. Interestingly, the peak of the DNA methylation in the disi-47 locus was also about 3 kb upstream of the known frq transcription start site (Figure S3). These results indicate that the transcription-induced DLDM can spread several kb upstream of the gene promoter.
To determine whether the DNA methylation induced in the artificial convergent transcription construct also showed an on/off switch pattern as observed in the disiRNA loci, we performed bisulfite sequencing. Similar to disiRNA loci, there was almost no methylated cytosine in most of the randomly selected PCR clones from genomic DNA, indicating that most of the DNA molecules were not methylated. In contrast, when the genomic DNA was first treated with BfuCI to enrich for the methylated alleles, we observed that most of cytosines upstream of the qa-2 promoter were methylated (Figure 5E). These results suggest that convergent transcription triggers dynamic DNA methylation in the disiRNA loci. In addition, in sRNA deep sequencing analyses, we observed sRNA accumulation in the upstream region of qa-2 promoter at the his-3 locus that contains Pqa-2:cul:1-gccP construct but not in that of the endogenous qa-2 locus upon QA induction (Figure S8B and S8C), indicating a link between DLDM and disiRNA production. The dependence of DLDM on transcription also provides an explanation for the on/off pattern of DLDM because DLDM may require a certain threshold of transcription from the disiRNA loci.
Discussion
Neurospora is a well-established model system for DNA methylation. All previously known DNA methylation in Neurospora occurs in relics of RIP [12], [32]. RIP is a process that silences repetitive DNA sequences during sexual stage (prior to meiosis) by converting cytosine to thymine in target sequences and occurs mostly at CpA dinucleotide context [33]. The resulted A/T rich region then serves as a signal that induces methylation of the nearby region to silence gene expression. In this study, we showed that DLDM is established and maintained very differently from the RIP-induced DNA methylation.
First, DLDM occurs in the gene-rich disiRNA loci that contain no relics of RIP or other repetitive elements. Second, in contrast to RIP'd regions in which DNA methylation is more or less constitutive and occurs in all alleles, most of the alleles are not methylated at disiRNA loci. In disiRNA loci, only a small percentage of alleles are extensively methylated with most of cytosines modified over a region that extends several kilobases. The dense cytosine methylation is similar to recently demonstrated dense methylation/hydroxylmethylation of cytosines in mouse embryonic stem cells [34]. The on-off pattern of DLDM indicates that DLDM is highly dynamic and that there is an inducible mechanism that mediates the establishment of DLDM. On the other hand, a de-methylation process may also exist to convert methylated alleles back into unmethylated alleles. Third, unlike the DNA methylation in the RIP'd regions, which is generally not required for maintenance of H3K9 methylation, DLDM is required for the maintenance of H3K9 methylation at the disiRNA loci. It suggests that an unknown mechanism should exist to recognize DNA methylation and in turn trigger H3K9me3.
Finally, DLDM is dependent on transcription. We demonstrated that DLDM is induced by convergent transcription from artificial constructs expressed in Neurospora. This conclusion is in agreement with the fact that most of disiRNA loci are known to produce sense and antisense RNAs [22]. Interestingly, the peaks of DNA methylation occurred at the promoter regions, where disiRNA expression also peaked. In addition, promoter-specific RNA transcripts were detected, and levels of these transcripts correlated with the levels of DNA methylation, suggesting that these non-coding RNA transcripts are involved in DLDM and are the precursors for disiRNAs. The induction of DLDM by transcription may explain its on/off pattern and suggests that a certain threshold level of transcription may be required for the establishment of DNA methylation. These results indicate that DLDM differs substantially from the typical DNA methylation in RIP'd DNA regions. It should be noted that transient DNA methylation was recently reported at the frq locus and was shown to be involved in setting the proper phase of circadian clock during the preparation of this paper [35]. In addition, the distribution of DNA methylation induced by convergent transcription is mainly accumulated upstream and peaks at about 3 kb from the TSS, suggesting that DLDM might also be involved in suppressing the promiscuous transcription at promoter region during transcriptional initiation. Indeed, recent studies suggest that pol II-directed gene transcription may adopt a gene loop structure by tethering promoter and terminator sequence, which enhances the transcriptional directionality toward the gene body [36]–[38]. Therefore, it is possible that the DNA methylation is a result of complex interaction between both ends of the gene for transcription initiation and termination, which strengthens the directionality of both sense and antisense transcription.
Materials and Methods
Strains and culture conditions
In this study, FGSC 4200 (a) was used as wild-type (WT) strain. Mutant strains wc-2KO, qde-1KO, qde-2RIP, qde-2RIP;sms-2RIP double mutant, and dcl-1RIP;dcl-2KO double mutant (dclDKO) were generated in previous studies [31], [39], [40]. The dim-2KO and dim-5KO strains were generously provided by Dr. Qun He [23]. Liquid cultures were grown in minimal medium (1× Vogel's, 2% glucose) at 30°C overnight and then at room temperature with shaking at 130 r.p.m. for 24 h [41]. For liquid cultures containing QA, 0.01 M QA, pH 5.8, was added to the liquid culture medium containing 1× Vogel's, 0.1% glucose and 0.17% arginine.
To make the his-3 targeting Pqa-2:cul:1-gccP constructs, a PCR fragment containing the promoter of ccg-1 was inserted into the plasmid pDE3dBH-Pqa-2 [42] to generate Pqa-2::1-ccgP. Then a PCR fragment of luciferase gene (luc) was inserted between the two promoters, with the luc sense transcripts and antisense transcripts driven by ccg-1 and qa-2 promoter, respectively. The control construct, Pqa-2:cul, was created by inserting the luc gene into pDE3dBH-Pqa-2, with qa-2 promoter driven antisense transcription of luc. The resulting constructs were introduced into the his-3 locus of dclDKO, his-3 strain [39], a his-3 strain and an eri-1lKO, his-3 recipient strain.
Southern blot analyses
Approximately 10 µg genomic DNA was digested with BfuCI or DpnII, fractioned in 1.0% agarose gels, and transferred to nylon membrane. Hybridization probes were prepared from PCR products of interest (primer sequences in Table S1) with Rediprime II DNA Labeling System (GE Healthcare).
Methylated DNA immunoprecipitation (MeDIP)
Approximately 10 µg genomic DNA was sonicated into small fragments (size ∼300–1000 bp). In each reaction, 1 µg of the 5-methylcytosine monoclonal antibody (Epigentek) was used to perform the MeDIP assay as previously described [12], [43]. MeDIP samples were analyzed with qPCR with corresponding primers listed in Table S1. In order to compare methylation in different regions, relative enrichment of DNA was calculated as the ratio of MeDIP sample over its input (set as 1), and the qPCR result of a primer pair of the am locus was used for normalization to correct for possible primer efficiency bias [44]. To compare MeDIP results of different samples or treatments, we performed the MeDIP at the same time with same batch of anti-5mC antibodies, due to the variation of MeDIP efficiency for different batches of antibodies.
Chromatin immunoprecipitation assay
The ChIP assay was performed as previously described [45]. The immunoprecipitation was performed with an H3K9me3 antibody (Abcam ab8898). The relative enrichment was calculated as the MeDIP assay and the qPCR result of a primer pair of the am locus was used for normalization.
Methylation specific PCR and qPCR
Methylation specific PCR (MSP) was performed as previously described [23]. The methylation rate, determined by quantitative PCR, was calculated as the ratio of BfuCI-digested DNA signal to its input. A primer pair for (113–114), whose PCR product carries no BfuCI/DpnII recognition site (GATC), was used to normalize for loading and primer efficiency.
Bisulfite PCR methylation analysis
The bisulfite PCR methylation analysis was carried out in three steps: 1) The bisulfite treatment of genomic DNA was carried out as described in the manual of EpiTech Bisulfite Kit (Qiagen) except that we used a modified thermal cycler condition: 99°C for 5 min followed by 60°C for 25 min; 99°C for 5 min followed by 60°C for 85 min repeated 3 times; and 99°C for 5 min followed by 60°C for 90 min. 2) Two rounds of nested PCR were performed; the PCR product of first round was diluted 10–100 fold and 1 µL was used for second round of PCR. The second round PCR products of the expected size were cloned into TOPO clone kit (Invitrogen) and individual clones were sequenced.
Two strategies were used to examine DNA methylation in disiRNA loci. Strategy 1, shown in Figure 2, used plasmids as templates to amplify the cloned fragment. The genomic counterpart of the cloned fragment carries a GATC site. If the site was methylated, the fragment would be resistant to bisulfite treatment, whereas the unmethylated sites were converted into uridine and no longer recognized by DpnII or BfuCI. By identifying whether the PCR product was resistant to DpnII or not, we could distinguish whether the cloned fragment was methylated in the GATC sequence. Strategy 2, used in experiments in Figure S5, is similar to the first strategy except that one aliquot of genomic DNA was treated and one was not treated with BfuCI before bisulfite treatment, PCR, cloning, and sequencing. The average methylation rate was calculated by dividing total number of 5-methylcytosines by the total number of cytosines in the amplified sequence. The primers used for bisulfite sequencing are shown in Table S2 and S3.
RT-qPCR
Total RNA was extracted with TRIzol (Invitrogen), digested with Turbo DNase (Ambion) and reverse transcribed into cDNA with SuperScript II (Invitrogen). β-tubulin transcripts (primer pair tub) were used as loading control for quantitative PCR.
Small RNA sequencing analyses
Total RNA of wc-2KO strain, wild-type strain and dicerDKO Pqa-2:cul:1-gccP strain were extracted with the TRIzol reagent (Invitrogen) and small RNAs were enriched with 5% polyethylene glycol (MW8000) and 500 mM NaCl as previously described [31]. Library construction and small RNA sequencing was performed by the Beijing Genomic Institute (Shenzhen, China) with Illumina standard protocol. All small RNA analyses were performed as described previously [22] except that an alignment tool Bowtie (ver 0.12.7) was used to map the small RNAs onto the N. crassa genome. In order to compare the density of small RNAs between samples, a standard normalization method was applied by scaling total reads of different samples to those of the same library size [46]–[48]. To correct bias induced by ribosomal RNA degradation products, we filtered out the reads matching rDNA regions from the total reads and used the remaining reads for scaling. The density of small RNA is presented as the relative number of small RNAs in a 100 nt non-overlapping sliding window along the Watson or Crick strand of each chromosome. The sRNA sequencing data was visualized with Generic genome browser (version 1.70) [49].
Accession number
The NCBI accession number of the sRNA deep sequencing data reported in this study is GSE47666.
Supporting Information
Zdroje
1. FreitagM, SelkerEU (2005) Controlling DNA methylation: many roads to one modification. Curr Opin Genet Dev 15 : 191–199.
2. SuzukiMM, BirdA (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9 : 465–476.
3. WuSC, ZhangY (2010) Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 11 : 607–620.
4. VolpeTA, KidnerC, HallIM, TengG, GrewalSI, et al. (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297 : 1833–1837.
5. BuhlerM, MoazedD (2007) Transcription and RNAi in heterochromatic gene silencing. Nat Struct Mol Biol 14 : 1041–1048.
6. VerdelA, JiaS, GerberS, SugiyamaT, GygiS, et al. (2004) RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303 : 672–676.
7. HeXJ, ChenT, ZhuJK (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21 : 442–465.
8. HaagJR, PikaardCS (2011) Multisubunit RNA polymerases IV and V: purveyors of non-coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 12 : 483–492.
9. AravinAA, SachidanandamR, Bourc'hisD, SchaeferC, PezicD, et al. (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31 : 785–799.
10. Kuramochi-MiyagawaS, WatanabeT, GotohK, TotokiY, ToyodaA, et al. (2008) DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22 : 908–917.
11. SelkerEU, FritzDY, SingerMJ (1993) Dense nonsymmetrical DNA methylation resulting from repeat-induced point mutation in Neurospora. Science 262 : 1724–1728.
12. LewisZA, HondaS, KhlafallahTK, JeffressJK, FreitagM, et al. (2009) Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res 19 : 427–437.
13. RountreeMR, SelkerEU (2010) DNA methylation and the formation of heterochromatin in Neurospora crassa. Heredity (Edinb) 105 : 38–44.
14. CambareriEB, JensenBC, SchabtachE, SelkerEU (1989) Repeat-induced G-C to A-T mutations in Neurospora. Science 244 : 1571–1575.
15. TamaruH, ZhangX, McMillenD, SinghPB, NakayamaJ, et al. (2003) Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet 34 : 75–79.
16. TamaruH, SelkerEU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414 : 277–283.
17. KouzminovaE, SelkerEU (2001) dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J 20 : 4309–4323.
18. FreitagM, HickeyPC, KhlafallahTK, ReadND, SelkerEU (2004) HP1 is essential for DNA methylation in neurospora. Mol Cell 13 : 427–434.
19. HondaS, SelkerEU (2008) Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Molecular and cellular biology 28 : 6044–6055.
20. FreitagM, LeeDW, KotheGO, PrattRJ, AramayoR, et al. (2004) DNA methylation is independent of RNA interference in Neurospora. Science 304 : 1939.
21. DangY, YangQ, XueZ, LiuY (2011) RNA interference in fungi: pathways, functions, and applications. Eukaryot Cell 10 : 1148–1155.
22. LeeHC, LiL, GuW, XueZ, CrosthwaiteSK, et al. (2010) Diverse Pathways Generate MicroRNA-like RNAs and Dicer-Independent Small Interfering RNAs in Fungi. Mol Cell 38 : 803–814.
23. ZhaoY, ShenY, YangS, WangJ, HuQ, et al. (2010) Ubiquitin ligase components Cullin4 and DDB1 are essential for DNA methylation in Neurospora crassa. J Biol Chem 285 : 4355–4365.
24. SelkerEU, StevensJN (1985) DNA methylation at asymmetric sites is associated with numerous transition mutations. Proc Natl Acad Sci U S A 82 : 8114–8118.
25. MargolinBS, Garrett-EngelePW, StevensJN, FritzDY, Garrett-EngeleC, et al. (1998) A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149 : 1787–1797.
26. SingerMJ, MarcotteBA, SelkerEU (1995) DNA methylation associated with repeat-induced point mutation in Neurospora crassa. Mol Cell Biol 15 : 5586–5597.
27. HondaS, SelkerEU (2008) Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Mol Cell Biol 28 : 6044–6055.
28. KramerC, LorosJJ, DunlapJC, CrosthwaiteSK (2003) Role for antisense RNA in regulating circadian clock function in Neurospora crassa. Nature 421 : 948–952.
29. HeintzenC, LiuY (2007) The Neurospora crassa circadian clock. Adv Genet 58 : 25–66.
30. YangQ, LiL, XueZ, YeQ, ZhangL, et al. (2013) Transcription of the major neurospora crassa microRNA-like small RNAs relies on RNA polymerase III. PLoS Genet 9: e1003227.
31. MaitiM, LeeHC, LiuY (2007) QIP, a putative exonuclease, interacts with the Neurospora Argonaute protein and facilitates conversion of duplex siRNA into single strands. Genes Dev 21 : 590–600.
32. SelkerEU, TountasNA, CrossSH, MargolinBS, MurphyJG, et al. (2003) The methylated component of the Neurospora crassa genome. Nature 422 : 893–897.
33. SelkerEU (1990) Premeiotic instability of repeated sequences in Neurospora crassa. Annual review of genetics 24 : 579–613.
34. FiczG, BrancoMR, SeisenbergerS, SantosF, KruegerF, et al. (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473 : 398–402.
35. BeldenWJ, LewisZA, SelkerEU, LorosJJ, DunlapJC (2011) CHD1 remodels chromatin and influences transient DNA methylation at the clock gene frequency. PLoS Genet 7: e1002166.
36. O'SullivanJM, Tan-WongSM, MorillonA, LeeB, ColesJ, et al. (2004) Gene loops juxtapose promoters and terminators in yeast. Nature genetics 36 : 1014–1018.
37. AnsariA, HampseyM (2005) A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes & development 19 : 2969–2978.
38. Tan-WongSM, ZauggJB, CamblongJ, XuZ, ZhangDW, et al. (2012) Gene loops enhance transcriptional directionality. Science 338 : 671–675.
39. ChoudharyS, LeeHC, MaitiM, HeQ, ChengP, et al. (2007) A double-stranded-RNA response program important for RNA interference efficiency. Mol Cell Biol 27 : 3995–4005.
40. HeQ, ChengP, YangY, WangL, GardnerKH, et al. (2002) White collar-1, a DNA binding transcription factor and a light sensor. Science 297 : 840–843.
41. DavisRL, deSerresD (1970) Genetic and microbial research techniques for Neurospora crassa. Methods of Enzymology 27A: 79–143.
42. ChengP, YangY, LiuY (2001) Interlocked feedback loops contribute to the robustness of the Neurospora circadian clock. Proc Natl Acad Sci U S A 98 : 7408–7413.
43. PomraningKR, SmithKM, FreitagM (2009) Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods 47 : 142–150.
44. WeberM, DaviesJJ, WittigD, OakeleyEJ, HaaseM, et al. (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37 : 853–862.
45. GuoJ, ChengP, YuanH, LiuY (2009) The exosome regulates circadian gene expression in a posttranscriptional negative feedback loop. Cell 138 : 1236–1246.
46. MarquesJT, KimK, WuPH, AlleyneTM, JafariN, et al. (2010) Loqs and R2D2 act sequentially in the siRNA pathway in Drosophila. Nat Struct Mol Biol 17 : 24–30.
47. GitA, DvingeH, Salmon-DivonM, OsborneM, KutterC, et al. (2010) Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 16 : 991–1006.
48. ZhangL, ChiaJM, KumariS, SteinJC, LiuZ, et al. (2009) A genome-wide characterization of microRNA genes in maize. PLoS Genet 5: e1000716.
49. SteinLD, MungallC, ShuS, CaudyM, MangoneM, et al. (2002) The generic genome browser: a building block for a model organism system database. Genome Res 12 : 1599–1610.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Genome-Wide Systematic Analysis Reveals Different and Predictive Proliferation Expression Signatures of Cancerous vs. Non-Cancerous Cells
- Recent Acquisition of by Baka Pygmies
- The Condition-Dependent Transcriptional Landscape of
- Histone Chaperone NAP1 Mediates Sister Chromatid Resolution by Counteracting Protein Phosphatase 2A
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy