
Hsp104 Suppresses Polyglutamine-Induced Degeneration Post Onset in a Drosophila MJD/SCA3 Model
There are no effective therapeutics that antagonize or reverse the protein-misfolding events underpinning polyglutamine (PolyQ) disorders, including Spinocerebellar Ataxia Type-3 (SCA3). Here, we augment the proteostasis network of Drosophila SCA3 models with Hsp104, a powerful protein disaggregase from yeast, which is bafflingly absent from metazoa. Hsp104 suppressed eye degeneration caused by a C-terminal ataxin-3 (MJD) fragment containing the pathogenic expanded PolyQ tract, but unexpectedly enhanced aggregation and toxicity of full-length pathogenic MJD. Hsp104 suppressed toxicity of MJD variants lacking a portion of the N-terminal deubiquitylase domain and full-length MJD variants unable to engage polyubiquitin, indicating that MJD-ubiquitin interactions hinder protective Hsp104 modalities. Importantly, in staging experiments, Hsp104 suppressed toxicity of a C-terminal MJD fragment when expressed after the onset of PolyQ-induced degeneration, whereas Hsp70 was ineffective. Thus, we establish the first disaggregase or chaperone treatment administered after the onset of pathogenic protein-induced degeneration that mitigates disease progression.
Published in the journal:
. PLoS Genet 9(9): e32767. doi:10.1371/journal.pgen.1003781
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003781
Summary
There are no effective therapeutics that antagonize or reverse the protein-misfolding events underpinning polyglutamine (PolyQ) disorders, including Spinocerebellar Ataxia Type-3 (SCA3). Here, we augment the proteostasis network of Drosophila SCA3 models with Hsp104, a powerful protein disaggregase from yeast, which is bafflingly absent from metazoa. Hsp104 suppressed eye degeneration caused by a C-terminal ataxin-3 (MJD) fragment containing the pathogenic expanded PolyQ tract, but unexpectedly enhanced aggregation and toxicity of full-length pathogenic MJD. Hsp104 suppressed toxicity of MJD variants lacking a portion of the N-terminal deubiquitylase domain and full-length MJD variants unable to engage polyubiquitin, indicating that MJD-ubiquitin interactions hinder protective Hsp104 modalities. Importantly, in staging experiments, Hsp104 suppressed toxicity of a C-terminal MJD fragment when expressed after the onset of PolyQ-induced degeneration, whereas Hsp70 was ineffective. Thus, we establish the first disaggregase or chaperone treatment administered after the onset of pathogenic protein-induced degeneration that mitigates disease progression.
Introduction
Many neurodegenerative diseases, such as Alzheimer's Disease, Parkinson's Disease (PD), prion disease, and the collection of polyglutamine (PolyQ) disorders, including Huntington's Disease (HD) and the Spinal Cerebellar Ataxias (SCAs), are characterized by the formation of protein inclusions in the nervous system [1]–[3]. Moreover, despite vastly different primary sequences, many of the proteins implicated in these diseases adopt the stereotypical amyloid conformation in the aggregated state [1]. Amyloid is defined by a highly stable cross-β conformation, in which proteins polymerize via intermolecular contacts of β-strands that align orthogonal to the fiber axis. Amyloid is typically a stable structure that is resistant to denaturation by heat, detergents (up to 2% sodium dodecyl sulfate (SDS)), and proteases [2], [4].
Despite the extraordinary structural stability of amyloid, a protein disaggregase from yeast, Hsp104, can rapidly solubilize amyloid. Hsp104 is a hexameric AAA+ (ATPases Associated with diverse cellular Activities) protein that couples ATP hydrolysis to translocation of substrate through a central pore, thus prying individual monomers from the amyloid fiber [5]–[8]. In yeast, Hsp104 is a heat shock protein (HSP), promoting survival following stresses by resolubilizing denatured protein aggregates and restoring proteins to native form and function [9], [10]. Hsp104 also maintains beneficial prion states by controlling the disassembly and dissemination of amyloid aggregates [11]–[13].
Curiously, Hsp104 has no homologue in metazoa. Indeed, until recently it was unclear whether the metazoan proteostasis network possessed any coupled protein disaggregase and reactivation machinery. It is now clear that Hsp110, Hsp70, and Hsp40 collaborate to promote the dissolution and reactivation of disordered aggregates [14], [15], and can even slowly depolymerize amyloid fibrils from their ends [16]. However, these disaggregase activities are slow and ineffective compared to Hsp104 [15], [16]. In particular, amyloid depolymerization by Hsp110, Hsp70, and Hsp40 is many orders of magnitude slower (weeks versus minutes) than amyloid dissolution by Hsp104 [16]. Importantly, Hsp104 can synergize with metazoan Hsp110, Hsp70, and Hsp40 to promote dissolution of amyloid and nonamyloid aggregates [15], [16]. Thus, introduction of Hsp104 into an animal system may provide an unprecedented opportunity to directly and rapidly target the intractable protein aggregates that underlie amyloid diseases [17], [18].
Spinocerebellar Ataxia Type 3 or Machado-Joseph Disease (MJD/SCA3) is the most prevalent dominantly inherited ataxia [19], [20]. The genetic basis of MJD/SCA3 is an expansion of the polyglutamine (PolyQ) tract of ataxin-3 (also known as Machado-Joseph Disease protein; MJD). When the PolyQ tract surpasses 50 consecutive Qs it is associated with the formation of amyloid aggregates and development of disease [21]–[23]. The normal physiological function of MJD is as a deubiquitylase (DUB) that catalyzes the cleavage of polyubiquitin (poly-ub) chains to promote proteostasis. It has a chain-editing function, preferentially cleaving certain poly-ub linkages to increase the presence of poly-ub chains that signal for degradation via the ubiquitin-proteasome system (UPS) [24], [25]. MJD has DUB activity in the N-terminal Josephin domain, plus two ubiquitin-interacting motifs (UIMs) that present poly-ub chains to the Josephin domain, as well as the C-terminal PolyQ tract that is associated with disease [26]. The PolyQ domain is known to form amyloid fibers, and interestingly, MJD aggregation occurs in a two-step process in vitro, with the Josephin domain forming SDS-soluble linear polymers that then convert into SDS-insoluble PolyQ-driven amyloid fibers [27]–[29]. As such, Hsp104 may be well suited to combating MJD protein aggregation because it antagonizes non-amyloid aggregates, pre-amyloid conformers, and amyloid fibers [5], [13], [30], [31].
Hsp104 has been introduced to combat protein-aggregation disease in metazoan systems with various levels of success [31]–[35]. In C. elegans, Hsp104 prevented aggregation and toxicity of GFP-tagged PolyQ [33]. In a lentiviral rat model, co-expression of Hsp104 with a PolyQ fragment implicated in HD resulted in the accumulation of more but smaller aggregates and rescue of striatal dysfunction [32]. In mouse, animals transgenic for both an HD fragment and Hsp104 showed limited suppression of PolyQ inclusion formation and a lifespan prolonged by ∼20% [34]. While these studies suggest promise for Hsp104 as a therapeutic against disease-associated protein aggregation, none has provided mechanistic insight into how Hsp104 interacts with amyloidogenic proteins in an animal system. Further, studies to date have looked only at antagonism of aggregation by concomitant co-expression of Hsp104. There has not been an evaluation of the potential of Hsp104 to modulate disease phenotypes in vivo after aggregates have already formed and degeneration has begun; a situation likely to mimic an actual therapy. Therefore, we created novel Hsp104 Drosophila lines to exploit well-characterized models of disease in combination with powerful genetic tools to temporally control the expression of Hsp104 after disease-associated aggregation and degeneration has begun.
Our studies reveal surprisingly distinct interactions of Hsp104 with the full-length versus a truncated version of the MJD protein. Importantly, we establish that Hsp104 possesses the ability to suppress the progression of degeneration when activated subsequent to onset of expression of the disease protein. These data indicate that protein context is central in Hsp104 interactions, and that Hsp104 displays the ability to halt the progression of pre-established disease in vivo.
Results
Hsp104 mitigates toxicity of truncated MJD, but enhances toxicity of the full-length MJD
The disaggregase Hsp104 efficiently antagonizes protein aggregates in yeast, and while homologues are present in bacteria, plants, fungi, chromista, and protozoa, no functional homologue has been found in metazoa [17], [36]. We stably introduced Hsp104 into Drosophila to evaluate its ability to prevent and potentially reverse aggregation of disease-associated human proteins, readily available in various fly models of disease. To achieve strong expression of the Hsp104 protein in the fruit fly, we codon-optimized the transgene for Drosophila (see Materials and Methods), and added a fly-optimal Kozak sequence (ACAAA) before the start codon [37]. The Hsp104 transgene was then expressed in Drosophila using the GAL4/UAS system [38]. Because we achieved high expression of Hsp104, expression by the gmr-GAL4 driver in the eye had a mild disruptive effect (Fig. 1A), which has also been observed for another AAA+ protein, p97 [39]. As the gmr-GAL4 driver line has multiple copies of the glass gene element for driving GAL4 expression, we instead used a driver line with reduced expression (Fig. 1B) bearing only a single glass element, 1×gr-GAL4. Using this driver, the effect of Hsp104 was minimized (Fig. 1A and Fig. 2A). Thus, we used the 1×gr-GAL4 driver line for our experiments to evaluate the impact of Hsp104 on protein-aggregation disease in vivo.


Hsp104 dissolves PolyQ amyloid in vitro [5], [16] and has been expressed in various PolyQ animal models, with results ranging from minimal beneficial effect to strong abrogation of PolyQ aggregation [32]–[34]. However, a detailed analysis of the underlying protein interactions is lacking in vivo. We sought to dissect the ability of Hsp104 to antagonize PolyQ aggregation and toxicity using MJD as a model protein. Pathogenic MJD with expanded PolyQ has been previously established in fly models of MJD/SCA3, and induces progressive neurodegeneration with the formation of nuclear inclusions [40], [41]. We also examined a truncated C-terminal fragment of MJD that is predominantly comprised of the PolyQ tract because fragmentation of the protein may be associated with MJD/SCA3 pathogenesis [40], [42], [43].
We examined interactions of Hsp104 with the pathogenic, full-length MJD containing an expanded glutamine tract (MJDnQ78) and the truncated C-terminal region of the protein containing the expanded glutamine tract (MJDtrQ78) [40]. Hsp104 had no effect on non-pathogenic forms of MJD containing non-expanded PolyQ tracts (Fig. 2B), confirming that the interaction is PolyQ length-dependent. With expanded PolyQ domains, the pathogenic MJDtrQ78 and MJDnQ78 both caused degeneration of the external eye and disruption to internal retinal structure (Fig. 2C, D). Unexpectedly, we found that Hsp104 had opposite effects on these two forms of MJD that have an identical PolyQ expansion: Hsp104 mitigated MJDtrQ78 degeneration (Fig. 2C), yet enhanced degeneration associated with the full-length MJDnQ78 (Fig. 2D). This effect is in contrast to human Hsp70, a molecular chaperone that suppresses PolyQ disease in multiple systems [44]–[47]. Despite more severe degeneration due to stronger expression by the gmr-GAL4 driver, Hsp70 suppressed the toxicity of both MJDtrQ78 and MJDnQ78 (Fig. 2E).
The opposite effects of Hsp104 on MJDtrQ78 and MJDnQ78 correspond to distinct modulation of underlying protein accumulations
To probe the mechanism underlying the dichotomous results found for the Hsp104 interaction with MJDtrQ78 and MJDnQ78, an in-depth investigation of the protein aggregates was performed. To slow protein aggregation such that we could analyze underlying protein accumulations in detail, we expressed the transgenes in the eye with an adult-onset driver rhodopsin1(rh1)-GAL4. Analysis of the PolyQ protein accumulations showed that Hsp104 altered the kinetics of inclusion formation for both MJD protein isoforms. By cryosectioning and subsequent immunohistochemistry (IHC), MJDtrQ78 formed compact inclusions that increased in size over time (Fig. 3A, top row). Quantification of inclusion size over time (Fig. 3A, gray bars in graph) reveals distinct inclusion size populations (small <2.5 µm, medium 2.5–5 µm, large >5 µm in optical diameter), demonstrating that inclusions became larger and more numerous with time. Consistent with previous studies, co-expression of Hsp70 delayed the kinetics and significantly reduced MJDtrQ78 protein aggregation (Fig. 3A, bottom row; green bars in graph, p = 0.03). By contrast, Hsp104 initially delayed inclusion formation (p = 0.004, but then significantly enhanced the formation of small inclusions (p = 0.04), eventually reaching accumulation levels similar to that with MJDtrQ78 alone (Fig. 3A, center row; red bars in graph, n.s. p = 0.5).

To examine protein accumulation by biochemical methods, we used SDD-AGE (Semi-Denaturing Detergent–Agarose Gel Electrophoresis), a protein agarose gel technique that can resolve amyloid aggregates [48]. This technique is useful for resolving high molecular weight polymer assemblies that maintain stable contacts in 2% SDS (a feature of highly stable amyloid). SDD-AGE revealed that the truncated MJDtrQ78 protein formed SDS-resistant amyloid structures that accrue with time (Fig. 3C). Unlike Hsp70, which significantly suppressed amyloid formation (p = 0.003), Hsp104 did not change the overall kinetics of MJDtrQ78 amyloid formation or the overall level of aggregation (Fig. 3C). Confirming that insoluble amyloid material was increased, the reduction of SDS-soluble levels of protein by immunoblot matched the concomitant increase in amyloid formation observed by SDD-AGE (Fig. 3C). Thus, Hsp104 rescues MJDtrQ78 toxicity, but the relationship to MJDtrQ78 aggregation is complex. IHC revealed that Hsp104 initially delays MJDtrQ78 inclusion formation, but then significantly enhances the formation of small inclusions (Fig. 3A). However, when amyloidogenesis was tracked by SDD-AGE, Hsp104 affected neither the rate nor the extent of amyloid formation (Fig. 3C). This finding indicates that to rescue toxicity Hsp104 might reduce formation of soluble and toxic oligomeric MJDtrQ78 species that are populated during amyloidogenesis, just as it does with the yeast prion proteins Sup35 and Ure2 [12], [13].
Next, we assessed MJDnQ78 misfolding. In contrast to the truncated MJDtrQ78 isoform, the pathogenic full-length MJDnQ78 initially formed amorphous inclusions that did not become more numerous after day 1 (Fig. 3B, top row; gray bars in graph). These MJDnQ78 amorphous aggregates appeared early by IHC, and insoluble amyloid aggregates developed later as observed by SDD-AGE (Fig. 3B and D). Thus, the early MJDnQ78 aggregates are non-amyloid in nature but later convert into the insoluble amyloid structure, closely resembling the two-step aggregation kinetics observed in vitro [27]–[29]. As with MJDtrQ78, co-expression of Hsp70 delayed the kinetics of aggregation and significantly suppressed inclusion formation (Fig. 3B, bottom row; green bars in graph, p = 0.02). However, in marked contrast, co-expression of Hsp104 significantly increased the formation of large aggregates at early time points (p = 0.002), and then significantly increased the number of small inclusions over time (Fig. 3B, center row; red bars in graph, p<0.001). Consistent with IHC results, SDD-AGE analysis demonstrated that Hsp104 significantly promoted the early formation of insoluble MJDnQ78 amyloid aggregates (Fig. 3D, p<0.001), whereas Hsp70 delayed kinetics of amyloid formation (Fig. 3D, p = 0.01). Hsp70, but not Hsp104, stably colocalized with both MJDtrQ78 and MJDnQ78 inclusions (Fig. 3A and B; see Fig. 4 for channel breakdown). The striking contrast between the effects of Hsp104 and Hsp70 on inclusion formation reinforces their functional differences [30], [49].

Domains neighboring the expanded PolyQ tract hinder protective Hsp104 activities
While it is known that in select conditions, Hsp104 promotes amyloid formation of specific yeast prions [12], [13], [30], we did not anticipate that Hsp104 would have opposite actions on two constructs of the same PolyQ protein. Thus, we assessed which domains of the full-length MJD protein prevented rescue by Hsp104 by employing a series of expression-matched MJD variants with disruptions to specific motifs (Fig. 5, 6, and see summary in Fig. 7A). Because the PolyQ domains are pure CAG repeats, they are subject to instability. Given this instability, the repeat lengths have been matched as closely as possible with matching protein expression levels (see [41]). Because of the reduced expression level by the 1×gr-GAL4 driver used for these experiments, MJDnQ84 (the pathogenic protein for this set of expression matched proteins) now showed mild degeneration. However, as before, analysis of retinal integrity demonstrated that the toxicity of MJDnQ84 was enhanced upon co-expression of Hsp104 (Fig. 5A). In contrast, an MJD variant in which both UIMs were mutated and unable to engage poly-ub, MJD-Q80-UIM* (Fig. 7A), exhibited mild toxicity that was suppressed by Hsp104 (Fig. 5A). Thus, the ability of the UIM domains to engage poly-ub hinders protective Hsp104 activity.



We also examined variants lacking DUB activity through mutation of the active site in the Josephin domain, MJD-Q88-C14A (Fig. 7A), which causes more severe toxicity than MJDnQ84 due to the loss of the physiological UPS function [41]. This occurs because DUB activity of MJD can suppress its own PolyQ toxicity; the C14A mutation is innocuous when MJD has a normal length, non-expanded Q repeat (Fig. 6A) [41]. Co-expression of Hsp104 did not affect the severe MJD-Q88-C14A toxicity (Fig. 5A). However, when the active site mutation was combined with the UIM mutations, in MJD-Q80-C14A-UIM* (Fig. 7A), Hsp104 now suppressed toxicity (Fig. 5A). This result reiterates that functional UIMs hinder rescue by Hsp104. We further examined a separate splice variant lacking DUB activity through an exon deletion that includes the active site, MJD-Q79-Δexon2 (missing amino acids 9–63) (Fig. 6B, 7A) [50], [51], which, like MJD-Q88-C14A, conferred severe toxicity (Fig. 5A). Co-expression of Hsp104 with MJD-Q79-Δexon2 strongly suppressed degeneration (Fig. 5A). Thus, Hsp104 mitigated MJD toxicity when the exon containing the active site was deleted (MJD-Q79-Δexon2) but had no effect when the active site was inactivated by a single point mutation (MJD-Q88-C14A). Taken together, these data indicate that functional UIMs and an intact Josephin domain both prohibit Hsp104 from rescuing full-length MJDnQ84 toxicity.
To uncover additional mechanistic insight into the interactions with Hsp104, we examined inclusion formation and kinetics with adult-onset rh1-GAL4 expression. By IHC, the MJD variants formed accumulations in a manner roughly consistent with severity of eye degeneration (Fig. 5B, top row; gray bars in graph). Those variants with mutated UIMs, MJD-Q80-UIM* and MJD-Q80-C14A-UIM*, showed significantly reduced levels of aggregate formation with Hsp104 (Fig. 5B, bottom row; red bars in graph, p = 0.003 for both). SDD-AGE analysis revealed that Hsp104 significantly enhanced the conversion of soluble protein to SDS-resistant polymers for variants with intact UIMs: MJDnQ84 and MJD-Q88-C14A (Fig. 5C, p = 0.002 and p = 0.005, respectively). Moreover, Hsp104 significantly reduced the formation of amyloid material by MJD-Q80-C14A-UIM* (Fig. 5C, p = 0.01). The MJD-Q79-Δexon2 protein was not detectable by immunoblot (but was confirmed by genotyping, Fig. 6C, D), precluding aggregate analysis of this variant. These findings underscore the role of active ubiquitin binding in obstructing productive remodeling by Hsp104.
A summary of the effect of Hsp104 on MJD variants is presented in Fig. 7B. In the two cases in which Hsp104 enhanced aggregation (MJDnQ84, MJD-Q88-C14A), the MJD protein has both functional UIMs and an intact Josephin domain. By contrast, protein variants whose toxicity and underlying protein accumulations were suppressed by Hsp104 (MJD-Q80-UIM*, MJD-Q80-C14A-UIM*, MJD-Q70-Δexon2) each lack UIM binding or a portion of the Josephin domain. This supports a model in which an inflexible or “closed” loop is formed, possibly through associations with a poly-ub chain, between the functional UIMs and the intact Josephin domain (Fig. 7B, red circle). Our hypothesis is that Hsp104 is able to effectively remodel a more flexible or “open” conformation of select protein variants (e.g., MJD-Q80-UIM*, MJD-Q80-C14A-UIM*, MJD-Q70-Δexon2, or the truncated MJDtrQ78), but that the inflexible/closed conformation of other proteins (e.g., MJDnQ78, MJDnQ84, or MJD-Q88-C14A) obstructs protective Hsp104 activities.
Active remodeling by Hsp104 is required for modulation of protein pathogenicity
To verify the critical role of active remodeling by Hsp104, we created an ATPase-Dead and substrate-binding defective Hsp104 transgenic fly. We introduced four mutations (Y257A:E285Q:Y662A:E687Q) into Hsp104 to ensure that Hsp104 could not engage substrate or hydrolyze ATP, creating the mutant known as Double Pore Loop Double Walker B (Hsp104DPLDWB), which is structurally identical to wild-type but functionally inactive [5]. Unlike wild-type Hsp104, which caused mild retinal disruption by the gmr-GAL4 driver, similar expression levels of Hsp104DPLDWB were innocuous (Fig. 8A, B). Moreover, Hsp104DPLDWB did not modulate the toxicity of either MJDtrQ78 or MJDnQ78 (Fig. 8A), underscoring the importance of substrate translocation for Hsp104 to mitigate MJDtrQ78 or worsen MJDnQ78-associated degeneration. Thus, ATPase activity and substrate binding are required in vivo for modulatory effects of Hsp104.

Hsp104 suppresses progression of pre-existing degenerative disease in vivo
Hsp104 is unique in its capacity to reverse pre-existing amyloids in yeast and in vitro [5], [52]. However, the potential of Hsp104 to affect pre-existing protein-aggregation disease in a metazoan, i.e., a genuine in vivo treatment situation, has never been addressed. To address this deficit, we constructed fly lines containing three elements: (1) the toxic MJDtrQ78 protein driven directly by a gmr element such that the disease-associated protein was constitutively expressed in the eye; (2) a drug-inducible gmr-GAL4 driver known as “GeneSwitch” (gmr-GS) to activate GAL4 expression only in the presence of the drug RU486 (mifepristone) [53], [54]; and (3) the UAS-HSP treatment molecule (here, Hsp104 or Hsp70), such that the HSP will be expressed conditionally only when RU486 is present in the fly food (Fig. 9A). This system allows the activation of HSP expression sequential to disease-associated protein onset. In this manner, we could test the ability of exogenous HSPs to mitigate the toxicity of the pathogenic PolyQ protein after the pathogenic protein was already accumulating in aggregated forms and causing degeneration. We hypothesized that, due to its disaggregation rather than chaperone activity, Hsp104 may have the potential to markedly mitigate degeneration associated with pre-existing PolyQ protein aggregates.

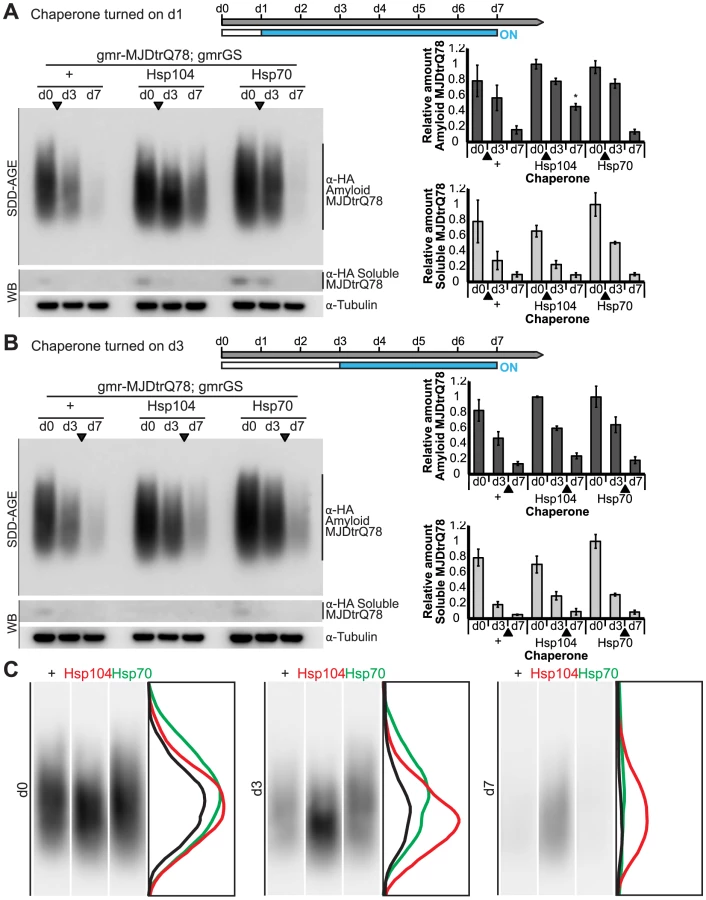
We established that retinal degeneration associated with gmr-MJDtrQ78 had begun at the time of adult fly emergence (d0) and progressed in severity to d7 (Fig. 9B, Fig. 10). We then activated Hsp104 or Hsp70 expression at an early time point (d1), or a later time point (d3), and examined the pathogenic impact of the MJDtrQ78 protein at d7 by retinal section. When activated at d1, Hsp104 was able to significantly mitigate retinal degeneration associated with MJDtrQ78 (p = 0.001), while Hsp70 did not have a significant effect (Fig. 10A, n.s. p = 0.06). These data show that Hsp104 is significantly more effective than Hsp70 at mitigating toxicity once disease progression has begun (Fig. 10A, p = 0.01). Importantly, inactive Hsp104DPLDWB had no effect (Fig. 10A). Hsp104 significantly improved tissue structure even when expression was induced at a later time point of d3 when degeneration was even more severe (Fig. 10B, p = 0.003). Moreover, while MJDtrQ78 treated with Hsp70 continued to degenerate with time, induction of Hsp104 arrested disease progression (Fig. 10A, d7 vs d1; Fig. 10B, d7 vs d3). Thus, Hsp104 mitigates pathogenesis even when administered after the onset of pathogenic protein-induced degeneration.

Next, we examined the underlying protein aggregates by SDD-AGE and Western immunoblot. We observed that gmr-MJDtrQ78 had high levels of amyloid, and this was lessened with time (potentially due to tissue loss) (Fig. 11). When turned on at d1, Hsp104 did not reverse MJDtrQ78 amyloid formation, but rather significantly increased the amyloid present by d7 (p = 0.02), while Hsp70 had no effect (Fig. 11A). Neither molecule significantly altered amyloid load when turned on at d3 (Fig. 11B). These results imply that Hsp104 is not acting as a MJDtrQ78-amyloid disaggregase, but rather is mitigating toxicity in a distinct manner, which is also consistent with our earlier results where MJDtrQ78 and Hsp104 are expressed at the same time (see Fig. 2, 3). Intriguingly, representative densitometry traces for the amyloid smears for each treatment condition turned on at d1 suggest that the peak of MJDtrQ78 amyloid species shifts downward to indicate smaller amyloid accumulations by d3 following Hsp104 activation (Fig. 11C). No shift in the densitometry trace is observed for the control or Hsp70 treatment (Fig. 11C), suggesting that Hsp104 is indeed altering the character of amyloid species although not eliminating these fibrils completely. In summary, this novel system of temporally controlled HSP expression demonstrates that although concomitant expression of Hsp70 is more successful than Hsp104 at preventing degeneration (see Fig. 2), inducible expression of Hsp104 is more effective than Hsp70 at suppressing disease progression once protein aggregation and degeneration are already established (Fig. 10).
Discussion
Here, we reveal key novel insights into the efficacy and interactions of Hsp104 with the pathogenic PolyQ protein MJD. Our studies reveal the surprising finding that Hsp104 interacts differentially with different forms of the MJD protein. Hsp104 is a potent suppressor of toxicity of the truncated protein, but an enhancer of toxicity of the full-length protein. These differences are determined by specific domains of MJD that are not directly implicated in aggregation. Our findings have also uncovered a heretofore unrecognized and important application of Hsp104 in vivo, which is its ability to mitigate the course of protein-aggregation disease even after it has already initiated. Indeed, our studies show that Hsp104 is able to mitigate disease progression once it has begun, unlike the classical metazoan chaperone Hsp70. These studies provide new insight into the in vivo effects of Hsp104 in the context of a therapeutic agent.
Hsp104 has opposite effects on two constructs of the same disease protein
Our detailed investigations of the effects of Hsp104 on the MJD protein led to the unexpected result that Hsp104 has opposite effects on the toxicity of different versions of the MJD protein (MJDtrQ78 and MJDnQ78), despite the fact that these proteins contain the same pathogenic PolyQ stretch. These disparate actions indicate that Hsp104 might be a useful probe to understand the nature of aggregates and the toxicity imposed by them. Hsp70 suppressed both MJDtrQ78 aggregation and toxicity (Fig. 2E, 3C). By contrast, Hsp104 mitigated toxicity of MJDtrQ78 without suppressing the extent of MJDtrQ78 aggregation (Fig. 2C, 3C). This result suggests that MJDtrQ78 aggregation per se need not be deleterious. Uncoupling of aggregation and toxicity has also been observed in other settings. For example, numerous genetic suppressors of FUS and TDP-43 toxicity, which are connected with amyotrophic lateral sclerosis and frontotemporal dementia, rescue toxicity without affecting FUS or TDP-43 aggregation in yeast [55]–[57]. We suggest that Hsp104 likely mitigates toxicity of MJDtrQ78 accumulations via subtle biochemical changes rather than gross changes in aggregation levels.
What might these biochemical changes be? Hsp104 can disrupt toxic soluble oligomers of various proteins, including Sup35, which may help explain why Sup35 prion formation is not intrinsically toxic to yeast [5], [12], [13], [31], [58]. Thus, Hsp104 might eliminate toxic soluble oligomers formed by MJDtrQ78 just as it does for Sup35 and alpha-synuclein [5], [12], [13], [31]. Furthermore, the amyloid-remodeling activity of Hsp104 can selectively amplify some amyloid strains (i.e. different cross-β structures formed by the same polypeptide) at the expense of others [59]. Given that PolyQ can access both toxic and benign amyloid strains [60], it is plausible that the presence of Hsp104 might amplify benign amyloid strains of MJDtrQ78 at the expense of toxic strains [59], [60]. Indeed, we observed that Hsp104 activity visibly altered the distribution of MJDtrQ78 amyloid species (Fig. 11C) suggesting that Hsp104 may be selectively eliminating certain fibril strains. Finally, to promote toxicity amyloid structures typically sequester large metastable proteins with unstructured regions, which occupy key nodes in functional networks linked to transcription, translation, chromatin organization, cell structure, and proteostasis [61], [62]. Hsp104 might disaggregate and rescue these proteins sequestered by MJDtrQ78 amyloid or promote the formation of MJDtrQ78 amyloid strains that do not deplete such an essential constellation of proteins. Further studies are needed to distinguish these non-mutually exclusive possibilities.
In other settings, it has been suggested that chaperone-initiated formation of large, insoluble amyloid aggregates can actually be protective by sequestering potentially toxic pre-fibrillar conformers [63]–[65]. Our results, however, indicate that, at least for Hsp104-driven enhancement of MJDnQ78, increased and accelerated aggregation is more toxic than MJDnQ78 aggregation that occurs in the absence of Hsp104. Our findings illustrate the complex relationship between aggregation and toxicity, which likely extends to other neurodegenerative disease models [64]. Moreover, our studies suggest that cautious interpretation is required when translating findings from cell culture experiments to neurodegeneration in animal models [34], [66]. Although Hsp104 is not found in the metazoan proteostasis network, our observations could help inform how to manipulate existing components of the metazoan proteostasis network for therapeutic purposes. Thus, components that suppress MJDnQ78 aggregation are likely beneficial, whereas MJDtrQ78 toxicity can be mitigated without having to suppress MJDtrQ78 aggregation.
Moreover, our findings demonstrate that an agent with a mitigating effect on the truncated version of the MJD protein may act in a different manner against the full-length MJD protein. Thus, what is good for one may not be beneficial for the other. In MJD/SCA3, as well as other neurodegenerative diseases, fragmentation of the disease protein may initiate aggregation and this process is critical for disease progression [42], [43], [67], [68]. Therefore, agents that effectively eliminate one specific sub-population of toxic protein accumulation but enhances another toxic sub-population may not be therapeutically viable in the complicated mixed populations that occur in disease. Our results highlight the complexity in developing therapeutic agents for neurodegenerative disorders.
Protein context is critical in evaluating disease-associated proteins
Although Hsp104 enhances MJDnQ78 amyloidogenesis and toxicity, we found that elimination of functional domains not implicated in PolyQ aggregation facilitated the ability of Hsp104 to suppress MJD-associated degeneration. Elimination of UIM functionality or removal of a component of the Josephin domain (exon 2) restored the remodeling capacity of Hsp104. This suggests that MJDnQ78 pathogenicity is not intrinsically intractable, but is capable of being suppressed by Hsp104 if other domains of the protein are inactivated (e.g., the UIMs). Alternatively, potentiated or MJDnQ78-optimized Hsp104 variants might be developed that are able to overcome these hindrances via increased unfolding power [5], [17].
Our studies underscore the importance of protein context in studying protein-misfolding diseases. Within the protein itself, neighboring domains not thought to be involved in aggregation may be impacting accumulation kinetics and the biochemical properties of inclusions [69], as well as accessibility of the aggregation domain to potential disaggregase therapeutics. That Hsp104 efficiently mitigates toxicity of MJD variants with ubiquitin-binding defects also demonstrates that, in addition to protein context, the cellular context of the protein is critical to consider; for example, the interaction between poly-ub chains and MJD may hinder protective Hsp104 modalities.
Previous studies in vitro have characterized aggregation of the full-length, pathogenic MJD protein as a two-step process in which the protein assembles first into SDS-soluble fibrillar polymers associating via the Josephin domain, and then converts to SDS-insoluble amyloid fibers driven by the PolyQ domain [27]–[29], [70]. We propose that this two-step process occurs in vivo as well. Indeed, it is consistent with our observation that full-length MJDnQ78 forms amorphous accumulations that appear visually by IHC before they can be observed as SDS-insoluble amyloid aggregates by SDD-AGE (see Fig. 3B and D). We suspect that full-length MJD initially forms SDS-soluble, Josephin-driven non-amyloid accumulations that initiate Hsp104 remodeling.
The initial formation of non-amyloid polymers is also compatible with our model of Hsp104 interacting differentially with the “open” and “closed” conformations of the protein discussed above (see Fig. 7B). We hypothesize that a poly-ub chain creates the closed loop by mutually interacting with the UIMs and the flexible helical hairpin encoded by exon 2 in the Josephin domain (Fig. 6B), as this arm is thought to be important for interacting with substrates [71]. Further experiments are required to confirm a poly-ub-mediated interaction between the two domains. According to our model, Hsp104 is able to efficiently translocate and release proteins that are more flexible (e.g., MJD-Q80-UIM*), resulting in fewer aggregates (see Fig. 5B). But due to an inflexible conformation imposed by the UIMs and the Josephin domain, Hsp104 is unable to efficiently remodel proteins containing the closed loop (e.g., MJDnQ84). This incomplete or slow translocation may expose or “prime” the PolyQ region to drive the formation SDS-insoluble amyloid inclusions (see Fig. 3D, Fig. 5C).
Our model suggests that the UIMs and the Josephin domain act together to obstruct Hsp104 remodeling, but we cannot rule out a separate function of the Josephin domain outside of poly-ub interactions. For example, removal of 55 amino acids might destabilize the Josephin domain such that it gets proteolytically cleaved and Hsp104 would then encounter a protein similar to MJDtrQ78 and rescue toxicity. Alternatively, because the MJD-Q79-Δexon2 protein could not be detected by biochemical methods, we cannot exclude the possibility that the deletion within the Josephin domain disrupts the proposed process of Josephin-domain-driven polymerization. In this case, toxicity of this variant may be dependent on highly soluble, possibly oligomeric species, which are effectively targeted by Hsp104.
These findings indicate that there is an opportunity to tailor therapies that are optimized for a specific disease scenario. In the case of full-length MJD, if inefficient translocation by Hsp104 does indeed drive the switch from less toxic SDS-soluble aggregates to highly toxic SDS-insoluble amyloid inclusions, then the development of substrate-optimized Hsp104 mutants (or Hsp104 mutants with altered ATPase rates or unfolding power) may increase efficiency of such interactions and enable Hsp104 to rescue disease phenotypes. Moreover, if UIM binding to poly-ub chains is impairing access of Hsp104 to MJD, this suggests that co-administering an agent to modulate function of a neighboring domain may affect the access of a treatment to the aggregation-prone domain. Indeed, increasing global DUB activity coupled with Hsp104 induction could overcome antagonism due to poly-ub chains.
Implications for Hsp104 as a therapeutic agent
Chaperone treatment, and examination of Hsp70 in particular, has been an exciting avenue of research in the battle to combat and contain neurodegenerative disease [72], [73]. However, all studies investigating Hsp70 as a modulator of disease have looked only at the chaperone transgenically co-expressed or activated prior to the disease insult. In a study seeking to activate existing Hsp70 rather than introducing exogenic expression, researchers sought to evaluate the chaperone in a mouse model of PD by boosting endogenous Hsp70 expression by treating animals with geldanamycin [74]. While this procedure has the potential to test true reversal of disease course, the authors found that beneficial effects were only observed if upregulation of Hsp70 was initiated prior to pharmacological induction of the PD phenotype [74]. In fact, in a cell culture PD model, geldanamycin was required at least 24 hours prior to disease protein transfection to provide any protection against inclusion formation [75]. In addition, pharmacologic activation of Hsp70 has been shown to suppress both PD and PolyQ disease in Drosophila [76], [77], but again, these manipulations were performed prior to disease onset. Despite the existing pharmacological paradigms, and other genetic tools available, such as the tetracycline-inducible system in mouse, no group has evaluated specific chaperone or disaggregase expression induced subsequent to expression of a disease-associated protein.
An inducible system is particularly well suited for Hsp104 because of its unique ability to rapidly dismantle pre-existing amyloid aggregates. Since metazoan chaperones can only very slowly depolymerize amyloid [16], Hsp104 may be more effective in an environment with pre-existing aggregation than a chaperone such as Hsp70, which is more adapted to prevent the initial aggregation. Here, we address the value of temporally controlled induction of disaggregase function after the initiation of PolyQ protein aggregation and the beginning of disease progression. To our knowledge, previous research has been performed with concomitant expression of a therapeutic gene, and thus does not distinguish prevention of disease from halting the progression of the disease state. Neurodegenerative diseases are not detected until later in life, and symptoms may not be apparent until pathological damage has accumulated beyond a tolerable point [78]–[80]. Thus, an agent that can rapidly impact the existing trajectory would be more valuable than one that can only prevent the development of the disease.
Our experimental paradigm offers the exciting possibility to address the efficacy of Hsp104 (or other molecules) in a more genuine therapeutic setting. Indeed, we found that turning on Hsp104 was able to significantly suppress disease-associated degeneration. Interestingly, however, Hsp104 did not disaggregate MJDtrQ78 amyloid in these experiments (Fig. 11A, B). Thus, Hsp104 might mitigate disease progression in this setting by: (a) eradicating toxic soluble MJDtrQ78 oligomers, (b) amplifying benign amyloid forms of MJDtrQ78 at the expense of toxic MJDtrQ78 amyloid, (c) by disaggregating and rescuing essential metastable proteins sequestered by MJDtrQ78 aggregates. Our observation that activation of Hsp104 shifted the MJDtrQ78 amyloid smears resolved by SDD-AGE toward smaller species without eliminating the total amyloid population (Fig. 11C) suggests that Hsp104 may indeed have strain selectivity. Further studies are required to define precisely how Hsp104 mitigates disease progression. Our data show that Hsp70 induction after MJD-associated degeneration has already initiated was unable to significantly mitigate disease progression. Optimization of Hsp70 expression, or administration of the suite of chaperones (for example, Hsp70 with Hsp110 and Hsp40), may improve the outcome, but our findings are consistent with other reports that Hsp70 must be administered before disease initiation to have a positive effect [74], [75]. Our observation that even a later-onset induced expression of Hsp104 is able to significantly suppress progressive PolyQ degeneration suggests that it is possible to mitigate disease phenotypes even after aggregates have begun accumulating and marked pathological degeneration is underway.
Naturally, several barriers must be surmounted to translate Hsp104 into a therapeutic agent for human neurodegenerative disease [17], [18]. Not least is the issue that gene therapy might be required to introduce Hsp104 (or any other genetic modifier) as a therapeutic agent. Gene therapy has yielded encouraging preclinical results for several disorders including congenital blindness [81]–[83]. However, technical and safety issues restrict facile translation to the clinic. Indeed, gene therapy for neurodegenerative diseases remains in early developmental stages and considerable caution is essential at this time. However, initial studies have generated cautious optimism that gene therapy in the adult brain might be safe for various neurodegenerative disorders, including Parkinson's disease [84]–[88]. Thus, even though we await several key advances before any Hsp104 gene therapy (or any other gene therapy) becomes truly viable it is, nonetheless, important to develop solutions to protein misfolding and to test these solutions both in vitro and in the most appropriate animal models. Moreover, the fact that Hsp104 is well tolerated by mammalian systems is encouraging [31], [32], [34]–[36], [66]. Ultimately, we envision that only transient expression of Hsp104 (or a substrate-optimized variant) would be required to provide therapeutic benefit. In this way, long-term expression of an exogenous agent and potential off-target side effects would be minimized. Alternatively, methods could be developed to deliver pure Hsp104 (or a substrate-optimized variant) to targeted areas in a single or multiple doses, and thereby avoid issues connected with long-term expression. These various issues and others highlight the complexities of designing therapeutics to treat human neurodegenerative disease.
Finally, the concept of using a yeast protein as the basis for a therapeutic agent might at first glance seem implausible. However, it must also have seemed equally implausible to use a lethal protein toxin from the bacterium, Clostridium botulinum, as a therapeutic agent. Despite being a deadly toxin, botulinum toxin variants have found key clinical applications due to their highly potent and selective ability to cleave SNARE proteins and prevent secretion [89]. Importantly, they are used to treat a variety of neuromuscular disorders including: blepharospasm, strabismus, muscle spasms, upper motor neuron syndrome, cervical dystonia and chronic migraine [90]–[95]. Indeed, the massive clinical success of botulinum toxin variants suggests it is critical to identify potentially therapeutic biological activities that originate in the microbial world and utilize and develop them to treat human disease.
Materials and Methods
Drosophila transgenic lines and crosses
Transgenic flies expressing UAS-Hsp104 and UAS-Hsp104DPLDWB were generated by standard techniques using the pUAST vector [38]. In order to boost expression of the transgene, pUAST-Hsp104 was codon-optimized (via gene synthesis; GenScript) for expression in Drosophila and a Kozak sequence (ACAAA) was added prior to the start codon [37].
The full sequence of codon-optimized Hsp104 is:
ATGAACGATCAGACCCAGTTCACCGAGCGCGCCCTGACCATCCTGACCCTGGCCCAGAAGCTGGCCAGCGATCACCAGCACCCCCAGCTGCAGCCCATCCACATCCTGGCCGCCTTCATCGAGACCCCCGAGGATGGCAGCGTGCCCTACCTGCAGAACCTGATCGAGAAGGGCCGCTACGATTACGATCTGTTCAAGAAGGTGGTGAACCGCAACCTGGTGCGCATCCCCCAGCAGCAGCCAGCCCCAGCCGAGATCACCCCAAGCTACGCCCTGGGCAAGGTGCTGCAGGATGCCGCCAAGATCCAGAAGCAGCAGAAGGATAGCTTCATCGCCCAGGATCACATCCTGTTCGCCCTGTTCAACGATAGCAGCATCCAGCAAATCTTCAAGGAGGCCCAGGTGGATATCGAGGCCATCAAGCAGCAGGCCCTGGAGCTGCGCGGAAACACCCGCATCGATAGCCGCGGAGCCGATACCAACACCCCCCTGGAGTACCTGAGCAAGTACGCCATCGATATGACCGAGCAGGCCCGCCAGGGAAAGCTGGACCCAGTGATCGGACGCGAGGAGGAGATCCGCAGCACCATCCGCGTGCTGGCCCGCCGCATCAAGAGCAACCCATGCCTGATCGGAGAGCCAGGAATCGGCAAGACCGCCATCATCGAGGGAGTGGCCCAGCGCATCATCGATGATGATGTGCCAACCATCCTGCAGGGAGCCAAGCTGTTCAGCCTGGATCTGGCCGCCCTGACCGCCGGCGCCAAGTACAAGGGCGATTTCGAGGAGCGCTTCAAGGGCGTGCTGAAGGAGATCGAGGAGAGCAAGACCCTGATCGTGCTGTTCATCGATGAGATCCACATGCTGATGGGCAACGGCAAGGATGATGCCGCCAACATCCTGAAGCCAGCCCTGAGCCGCGGACAGCTGAAGGTCATCGGAGCCACCACCAACAACGAGTACCGCAGCATCGTGGAGAAGGATGGAGCCTTCGAGCGCCGCTTCCAGAAGATCGAGGTGGCCGAGCCAAGCGTGCGCCAGACCGTGGCCATCCTGCGCGGACTGCAGCCCAAGTACGAGATCCACCACGGCGTGCGCATCCTGGATAGCGCCCTGGTGACCGCCGCCCAGCTGGCCAAGCGCTACCTGCCATACCGCCGCCTGCCAGATAGCGCCCTGGATCTGGTGGATATCAGCTGCGCCGGAGTGGCCGTGGCCCGCGATAGCAAGCCAGAGGAGCTGGATAGCAAGGAGCGCCAGCTGCAGCTGATCCAGGTGGAGATCAAGGCCCTGGAGCGCGATGAGGATGCCGATAGCACCACCAAGGATCGCCTGAAGCTGGCCCGCCAGAAGGAGGCCAGCCTGCAGGAGGAGCTGGAGCCACTGCGCCAGCGCTACAACGAGGAGAAGCACGGCCACGAGGAGCTGACCCAGGCTAAGAAAAAGCTGGATGAGCTGGAGAACAAGGCCCTGGATGCCGAGCGCCGCTACGATACCGCCACCGCCGCCGATCTGCGCTACTTCGCCATCCCCGATATCAAGAAGCAGATCGAGAAGCTGGAGGATCAGGTGGCCGAGGAGGAGCGCCGCGCCGGCGCCAACAGCATGATCCAGAACGTGGTGGATAGCGATACCATCAGCGAGACCGCCGCCCGCCTGACCGGCATCCCCGTGAAGAAGCTGAGCGAGAGCGAGAACGAGAAGCTGATCCACATGGAGCGCGATCTGAGCAGCGAGGTGGTGGGCCAGATGGATGCCATCAAGGCCGTGAGCAACGCCGTGCGCCTGAGCCGCAGCGGACTGGCCAACCCACGCCAGCCAGCCAGCTTCCTGTTCCTGGGCCTGAGCGGCAGCGGCAAGACCGAGCTGGCCAAGAAGGTGGCCGGCTTCCTGTTCAACGATGAGGATATGATGATCCGCGTGGATTGCAGCGAGCTGAGCGAGAAGTACGCCGTGAGCAAGCTGCTGGGCACCACCGCCGGCTACGTGGGCTACGATGAGGGCGGCTTCCTGACCAACCAGCTGCAGTACAAGCCCTACAGCGTGCTGCTGTTCGATGAGGTGGAGAAGGCCCACCCCGATGTGCTGACCGTGATGCTGCAGATGCTGGATGATGGCCGCATCACCAGCGGCCAGGGCAAGACCATCGATTGCAGCAACTGCATCGTGATCATGACCAGCAACCTGGGCGCCGAGTTCATCAACAGCCAGCAGGGCAGCAAGATCCAGGAGAGCACCAAGAACCTGGTCATGGGCGCCGTGCGCCAGCACTTCCGCCCCGAGTTCCTGAACCGCATCAGCAGCATCGTGATCTTCAACAAGCTGAGCCGCAAGGCCATCCACAAGATCGTGGATATCCGCCTGAAGGAGATTGAGGAGCGCTTCGAGCAGAACGATAAGCACTACAAGCTGAACCTGACCCAGGAGGCCAAGGATTTCCTGGCCAAGTACGGCTACAGCGATGATATGGGCGCCCGCCCCCTGAACCGCCTGATCCAGAACGAGATCCTGAACAAGCTGGCCCTGCGCATCCTGAAGAACGAGATCAAGGATAAGGAGACCGTGAACGTGGTGCTGAAGAAGGGCAAGAGCCGCGATGAGAACGTGCCAGAGGAGGCCGAGGAGTGCCTGGAGGTGCTGCCAAACCACGAGGCCACCATCGGAGCCGATACCCTGGGCGATGATGATAACGAGGATAGCATGGAGATCGATGATGATCTGGATTAA
Multiple insertion lines were characterized for each transgene. To create the 1×gr-GAL4 driver line, the pGMR (glass multimer reporter) vector [96] was digested by XhoI/Acc651 to remove the insert containing five glass-binding sites. Complementary oligonucleotides (5′-TCGAACCCAGTGGAAACCCTTGAAATGCCTTTAACTCGAGACGG-3′ and 5′-GTACCCGTCTCGAGTTAAAGGCATTTCAAGGGTTTCCACTGGGT-3′), with a single copy of the 31 bp glass-binding site from the Rh1 proximal enhancer, were duplexed and ligated into the vector, producing p1×GR (1 copy of glass reporter). This plasmid was then modified to introduce the GAL4 coding sequence, excised from pGaTN [38] with HindIII, to create p1×gr-GAL4. MJD lines are from [41]. Experiments were performed at 25°C except for a select few conducted at 29°C as indicated. All results were confirmed with multiple UAS-Hsp104 insertion lines.
Evaluation of eye degeneration
Eye images were obtained on day 7 of adulthood using a Leica Z-16 apo zoom microscope. To view internal retinal structure, heads were embedded in paraffin according to standard protocols, sectioned at 8 µm, and autofluorescence was viewed with a Leica fluorescence microscope. To quantify tissue loss, a standard area (3×15 rectangle; 7,000 µm2) (see also Fig. 9) was selected within the paraffin retinal section and the percentage of area covered by tissue was measured in ImageJ. Statistical analysis was performed using one-way ANOVA and unpaired t-test.
Cryosections and immunohistochemistry
Heads were frozen in Tissue Freezing Medium (Electron Microscopy Sciences) and sectioned at 12 µm by cryotome, and the tissue sections were then fixed with 4% paraformaldehyde. Immunohistochemistry was performed according to standard procedures using primary antibodies anti-HA 5B1D10 (1∶100, Invitrogen 32-6700) or anti-myc 9E10 (1∶100, Santa Cruz sc-40) (both mouse) alongside either anti-Hsp104 (1∶100, Enzo Life Sciences ADI-SPA-1040) or anti-Hsp70 (1∶100, Enzo Life Sciences ADI-SPA-812) (both rabbit). Hsp70 staining was confirmed with human-specific anti-Hsp70 (1∶100, Santa Cruz sc-24) (mouse) alongside anti-HA Y11 (1∶100, Santa Cruz sc-805) or anti-myc A14 (1∶100, Santa Cruz sc-789) (both rabbit). Rabbit primary antibodies were preadsorbed at 1∶25 with fixed, dissected wild-type larvae. Secondary antibodies were Alexa Fluor 594 Goat-anti-Mouse IgG (1∶100, Life Technologies A-11032), Alexa Fluor 488 Goat-anti-Rabbit IgG (1∶100, Life Technologies A-11008), Alexa Fluor 594 Goat-anti-Rabbit IgG (1∶100, Life Technologies A-11037), and Alexa Fluor 488 Goat-anti-Mouse IgG (1∶100, Life Technologies A-11029). Sections were co-stained with Hoechst nuclear dye (1∶1000, Molecular Probes 33342) and viewed with a Leica fluorescence microscope. A 75 µm×75 µm square (5625 µm2) area was selected; particle analysis was performed with ImageJ and statistics performed with one-way ANOVA and unpaired t-test.
Immunoblots and SDD-AGE
For Hsp104 expression level characterization, heads were ground with a pestle in NuPage LDS Sample Buffer, boiled for 3 min, run on NuPage 4–12% Bis-Tris gel, and semi-dry transferred onto nitrocellulose membrane. Antibodies used were anti-Hsp104 (1∶2000, Enzo Life Sciences ADI-SPA-1040) and anti-actin (1∶2000, Abcam ab8227) with secondary antibody Goat-anti-Rabbit-HRP (1∶5000, Chemicon AP307P). For MJD aggregation analysis through SDD-AGE (Semi-Denaturing Detergent Agarose Gel Electrophoresis) and accompanying Western immunoblots, heads were ground in lysis buffer (100 mM Tris pH 7.5, 50 mM NaCl, 10 mM β-Mercaptoethanol, and Roche complete mini EDTA-free protease inhibitor cocktail tablets) [48] and an aliquot was taken for evaluation of soluble material by Western immunoblot, as above. To the remaining sample, 4× Sample Buffer (2× TAE, 20% glycerol, 8% SDS, bromophenol blue) was added to final concentration 1×. The samples were run on a 1.5% agarose gel with 0.1% SDS in a running buffer of 1× TAE (40 mM Tris, 20 mM Acetic acid, 1 mM EDTA, pH 8.3) containing 0.1% SDS, and then transferred overnight onto nitrocellulose membrane using downward capillary transfer [48]. Antibodies used were anti-HA-conj-HRP 3F10 (1∶500, Roche 12013819001), anti-myc 9E10 (1∶500, Santa Cruz sc-40) followed by Goat-anti-Mouse-HRP (1∶2000, Jackson ImmunoResearch 115-035-146), and anti-tubulin-conj-HRP (1∶1000, Cell Signaling 11H10). All immunoblots were imaged using a FujiFilm LAS-3000 imaging system and quantification was performed in ImageJ and statistically analyzed by one-way ANOVA and unpaired t-test.
Gene switch protocol
A 4.0 mg/ml stock solution of RU486 (Sigma M8046) was prepared in 100% ethanol, and then 50 µl (200 µg) was added to pre-prepared food vials containing ∼12 ml of food and gently shaken overnight [97]. For control conditions, 50 µl of 100% ethanol was added to vials. Adult flies were aged in food treated with either RU486 or ethanol for the time periods indicated.
Zdroje
1. FormanMS, TrojanowskiJQ, LeeVM (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 10 : 1055–1063.
2. ChitiF, DobsonCM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75 : 333–366.
3. CushmanM, JohnsonBS, KingOD, GitlerAD, ShorterJ (2010) Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 123 : 1191–1201.
4. WestermarkP, BensonMD, BuxbaumJN, CohenAS, FrangioneB, et al. (2005) Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 12 : 1–4.
5. DeSantisME, LeungEH, SweenyEA, JackrelME, Cushman-NickM, et al. (2012) Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151 : 778–793.
6. TessarzP, MogkA, BukauB (2008) Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol Microbiol 68 : 87–97.
7. WendlerP, ShorterJ, PlissonC, CashikarAG, LindquistS, et al. (2007) Atypical AAA+ subunit packing creates an expanded cavity for disaggregation by the protein-remodeling factor Hsp104. Cell 131 : 1366–1377.
8. WendlerP, ShorterJ, SneadD, PlissonC, ClareDK, et al. (2009) Motor mechanism for protein threading through Hsp104. Mol Cell 34 : 81–92.
9. SanchezY, LindquistSL (1990) HSP104 required for induced thermotolerance. Science 248 : 1112–1115.
10. ParsellDA, KowalAS, SingerMA, LindquistS (1994) Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372 : 475–478.
11. AlbertiS, HalfmannR, KingO, KapilaA, LindquistS (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137 : 146–158.
12. ShorterJ, LindquistS (2004) Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304 : 1793–1797.
13. ShorterJ, LindquistS (2006) Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell 23 : 425–438.
14. RampeltH, Kirstein-MilesJ, NillegodaNB, ChiK, ScholzSR, et al. (2012) Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 31 : 4221–4235.
15. ShorterJ (2011) The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 6: e26319.
16. DuennwaldML, EcheverriaA, ShorterJ (2012) Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol 10: e1001346.
17. VashistS, CushmanM, ShorterJ (2010) Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol 88 : 1–13.
18. ShorterJ (2008) Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals 16 : 63–74.
19. BettencourtC, LimaM (2011) Machado-Joseph Disease: from first descriptions to new perspectives. Orphanet J Rare Dis 6 : 35.
20. PaulsonH (2012) Machado-Joseph disease/spinocerebellar ataxia type 3. Handb Clin Neurol 103 : 437–449.
21. PaulsonHL, DasSS, CrinoPB, PerezMK, PatelSC, et al. (1997) Machado-Joseph disease gene product is a cytoplasmic protein widely expressed in brain. Ann Neurol 41 : 453–462.
22. PaulsonHL, PerezMK, TrottierY, TrojanowskiJQ, SubramonySH, et al. (1997) Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19 : 333–344.
23. TakiyamaY, NishizawaM, TanakaH, KawashimaS, SakamotoH, et al. (1993) The gene for Machado-Joseph disease maps to human chromosome 14q. Nat Genet 4 : 300–304.
24. WinbornBJ, TravisSM, TodiSV, ScaglioneKM, XuP, et al. (2008) The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J Biol Chem 283 : 26436–26443.
25. KuhlbrodtK, JanieschPC, KeveiE, SegrefA, BarikbinR, et al. (2011) The Machado-Joseph disease deubiquitylase ATX-3 couples longevity and proteostasis. Nat Cell Biol 13 : 273–281.
26. MasinoL, MusiV, MenonRP, FusiP, KellyG, et al. (2003) Domain architecture of the polyglutamine protein ataxin-3: a globular domain followed by a flexible tail. FEBS Lett 549 : 21–25.
27. EllisdonAM, PearceMC, BottomleySP (2007) Mechanisms of ataxin-3 misfolding and fibril formation: kinetic analysis of a disease-associated polyglutamine protein. J Mol Biol 368 : 595–605.
28. EllisdonAM, ThomasB, BottomleySP (2006) The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem 281 : 16888–16896.
29. MasinoL, NicastroG, MenonRP, Dal PiazF, CalderL, et al. (2004) Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J Mol Biol 344 : 1021–1035.
30. ShorterJ, LindquistS (2008) Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J 27 : 2712–2724.
31. Lo BiancoC, ShorterJ, RegulierE, LashuelH, IwatsuboT, et al. (2008) Hsp104 antagonizes alpha-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 118 : 3087–3097.
32. PerrinV, RegulierE, Abbas-TerkiT, HassigR, BrouilletE, et al. (2007) Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol Ther 15 : 903–911.
33. SatyalSH, SchmidtE, KitagawaK, SondheimerN, LindquistS, et al. (2000) Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A 97 : 5750–5755.
34. VacherC, Garcia-OrozL, RubinszteinDC (2005) Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington's disease. Hum Mol Genet 14 : 3425–3433.
35. Dandoy-DronF, BogdanovaA, BeringueV, BaillyY, ToveyMG, et al. (2006) Infection by ME7 prion is not modified in transgenic mice expressing the yeast chaperone Hsp104 in neurons. Neurosci Lett 405 : 181–185.
36. MosserDD, HoS, GloverJR (2004) Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress-induced proapoptotic signaling. Biochemistry 43 : 8107–8115.
37. TrinhK, MooreK, WesPD, MuchowskiPJ, DeyJ, et al. (2008) Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson's disease. J Neurosci 28 : 465–472.
38. BrandAH, PerrimonN (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 : 401–415.
39. RitsonGP, CusterSK, FreibaumBD, GuintoJB, GeffelD, et al. (2010) TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci 30 : 7729–7739.
40. WarrickJM, PaulsonHL, Gray-BoardGL, BuiQT, FischbeckKH, et al. (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93 : 939–949.
41. WarrickJM, MorabitoLM, BilenJ, Gordesky-GoldB, FaustLZ, et al. (2005) Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell 18 : 37–48.
42. HaackeA, HartlFU, BreuerP (2007) Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J Biol Chem 282 : 18851–18856.
43. JungJ, XuK, LessingD, BoniniNM (2009) Preventing Ataxin-3 protein cleavage mitigates degeneration in a Drosophila model of SCA3. Hum Mol Genet 18 : 4843–4852.
44. CummingsCJ, ManciniMA, AntalffyB, DeFrancoDB, OrrHT, et al. (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19 : 148–154.
45. WarrickJM, ChanHY, Gray-BoardGL, ChaiY, PaulsonHL, et al. (1999) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23 : 425–428.
46. ChanHY, WarrickJM, Gray-BoardGL, PaulsonHL, BoniniNM (2000) Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 9 : 2811–2820.
47. MuchowskiPJ, SchaffarG, SittlerA, WankerEE, Hayer-HartlMK, et al. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A 97 : 7841–7846.
48. HalfmannR, LindquistS (2008) Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J Vis Exp 17: e838 doi:810.3791/3838
49. SanchezY, ParsellDA, TaulienJ, VogelJL, CraigEA, et al. (1993) Genetic evidence for a functional relationship between Hsp104 and Hsp70. J Bacteriol 175 : 6484–6491.
50. BettencourtC, SantosC, MontielR, Costa MdoC, Cruz-MoralesP, et al. (2010) Increased transcript diversity: novel splicing variants of Machado-Joseph disease gene (ATXN3). Neurogenetics 11 : 193–202.
51. HarrisGM, DodelzonK, GongL, Gonzalez-AlegreP, PaulsonHL (2010) Splice isoforms of the polyglutamine disease protein ataxin-3 exhibit similar enzymatic yet different aggregation properties. PLoS One 5: e13695.
52. DiSalvoS, DerdowskiA, PezzaJA, SerioTR (2011) Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat Struct Mol Biol 18 : 486–492.
53. RomanG, DavisRL (2002) Conditional expression of UAS-transgenes in the adult eye with a new gene-switch vector system. Genesis 34 : 127–131.
54. OsterwalderT, YoonKS, WhiteBH, KeshishianH (2001) A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A 98 : 12596–12601.
55. JuS, TardiffDF, HanH, DivyaK, ZhongQ, et al. (2011) A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol 9: e1001052.
56. ArmakolaM, HigginsMJ, FigleyMD, BarmadaSJ, ScarboroughEA, et al. (2012) Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44 : 1302–1309.
57. SunZ, DiazZ, FangX, HartMP, ChesiA, et al. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 9: e1000614.
58. ShorterJ, LindquistS (2005) Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 6 : 435–450.
59. DeSantisME, ShorterJ (2012) Hsp104 drives “protein-only” positive selection of Sup35 prion strains encoding strong [PSI(+)]. Chem Biol 19 : 1400–1410.
60. Nekooki-MachidaY, KurosawaM, NukinaN, ItoK, OdaT, et al. (2009) Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A 106 : 9679–9684.
61. OlzschaH, SchermannSM, WoernerAC, PinkertS, HechtMH, et al. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144 : 67–78.
62. VabulasRM, HartlFU (2011) Aberrant protein interactions in amyloid disease. Cell Cycle 10 : 1512–1513.
63. DouglasPM, TreuschS, RenHY, HalfmannR, DuennwaldML, et al. (2008) Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci U S A 105 : 7206–7211.
64. WolfeKJ, CyrDM (2011) Amyloid in neurodegenerative diseases: friend or foe? Semin Cell Dev Biol 22 : 476–481.
65. DobsonCM (2003) Protein folding and misfolding. Nature 426 : 884–890.
66. CarmichaelJ, ChatellierJ, WoolfsonA, MilsteinC, FershtAR, et al. (2000) Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc Natl Acad Sci U S A 97 : 9701–9705.
67. WellingtonCL, EllerbyLM, HackamAS, MargolisRL, TrifiroMA, et al. (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem 273 : 9158–9167.
68. GrahamRK, DengY, SlowEJ, HaighB, BissadaN, et al. (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125 : 1179–1191.
69. DuennwaldML, JagadishS, MuchowskiPJ, LindquistS (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc Natl Acad Sci U S A 103 : 11045–11050.
70. MasinoL, NicastroG, De SimoneA, CalderL, MolloyJ, et al. (2011) The Josephin domain determines the morphological and mechanical properties of ataxin-3 fibrils. Biophys J 100 : 2033–2042.
71. NicastroG, MenonRP, MasinoL, KnowlesPP, McDonaldNQ, et al. (2005) The solution structure of the Josephin domain of ataxin-3: structural determinants for molecular recognition. Proc Natl Acad Sci U S A 102 : 10493–10498.
72. MuchowskiPJ, WackerJL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6 : 11–22.
73. BroadleySA, HartlFU (2009) The role of molecular chaperones in human misfolding diseases. FEBS Lett 583 : 2647–2653.
74. ShenHY, HeJC, WangY, HuangQY, ChenJF (2005) Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem 280 : 39962–39969.
75. McLeanPJ, KluckenJ, ShinY, HymanBT (2004) Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun 321 : 665–669.
76. AuluckPK, BoniniNM (2002) Pharmacological prevention of Parkinson disease in Drosophila. Nat Med 8 : 1185–1186.
77. WangAM, MiyataY, KlinedinstS, PengHM, ChuaJP, et al. (2012) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9 : 112–8.
78. BraakH, Del TrediciK, RubU, de VosRA, Jansen SteurEN, et al. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24 : 197–211.
79. NaslundJ, HaroutunianV, MohsR, DavisKL, DaviesP, et al. (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283 : 1571–1577.
80. DaviesSW, TurmaineM, CozensBA, DiFigliaM, SharpAH, et al. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90 : 537–548.
81. BainbridgeJW, SmithAJ, BarkerSS, RobbieS, HendersonR, et al. (2008) Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med 358 : 2231–2239.
82. Hacein-Bey-AbinaS, Le DeistF, CarlierF, BouneaudC, HueC, et al. (2002) Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med 346 : 1185–1193.
83. MaguireAM, SimonelliF, PierceEA, PughENJr, MingozziF, et al. (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 358 : 2240–2248.
84. FeiginA, KaplittMG, TangC, LinT, MattisP, et al. (2007) Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson's disease. Proc Natl Acad Sci U S A 104 : 19559–19564.
85. KaplittMG, FeiginA, TangC, FitzsimonsHL, MattisP, et al. (2007) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet 369 : 2097–2105.
86. StoesslAJ (2007) Gene therapy for Parkinson's disease: early data. Lancet 369 : 2056–2058.
87. SimonatoM, BennettJ, BoulisNM, CastroMG, FinkDJ, et al. (2013) Progress in gene therapy for neurological disorders. Nat Rev Neurol 9 : 298.
88. San SebastianW, SamaranchL, KellsAP, ForsayethJ, BankiewiczKS (2013) Gene Therapy for Misfolding Protein Diseases of the Central Nervous System. Neurotherapeutics 10 : 498–510.
89. SchiavoG, BenfenatiF, PoulainB, RossettoO, Polverino de LauretoP, et al. (1992) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359 : 832–835.
90. YehFL, ZhuY, TeppWH, JohnsonEA, BerticsPJ, et al. (2011) Retargeted clostridial neurotoxins as novel agents for treating chronic diseases. Biochemistry 50 : 10419–10421.
91. HallettM (1999) One man's poison–clinical applications of botulinum toxin. N Engl J Med 341 : 118–120.
92. EsquenaziA, MayerNH, EliaAE, AlbaneseA (2009) Botulinum toxin for the management of adult patients with upper motor neuron syndrome. Toxicon 54 : 634–638.
93. EliaAE, FilippiniG, CalandrellaD, AlbaneseA (2009) Botulinum neurotoxins for post-stroke spasticity in adults: a systematic review. Mov Disord 24 : 801–812.
94. BentivoglioAR, AlbaneseA (1999) Botulinum toxin in motor disorders. Curr Opin Neurol 12 : 447–456.
95. OsborneSL, LathamCF, WenPJ, CavaignacS, FanningJ, et al. (2007) The Janus faces of botulinum neurotoxin: sensational medicine and deadly biological weapon. J Neurosci Res 85 : 1149–1158.
96. HayBA, WolffT, RubinGM (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development 120 : 2121–2129.
97. ShenJ, CurtisC, TavareS, TowerJ (2009) A screen of apoptosis and senescence regulatory genes for life span effects when over-expressed in Drosophila. Aging (Albany NY) 1 : 191–211.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Genome-Wide Systematic Analysis Reveals Different and Predictive Proliferation Expression Signatures of Cancerous vs. Non-Cancerous Cells
- Recent Acquisition of by Baka Pygmies
- The Condition-Dependent Transcriptional Landscape of
- Histone Chaperone NAP1 Mediates Sister Chromatid Resolution by Counteracting Protein Phosphatase 2A
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy