
Npc1 Acting in Neurons and Glia Is Essential for the Formation and Maintenance of CNS Myelin
Cholesterol availability is rate-limiting for myelination, and prior studies have established the importance of cholesterol synthesis by oligodendrocytes for normal CNS myelination. However, the contribution of cholesterol uptake through the endocytic pathway has not been fully explored. To address this question, we used mice with a conditional null allele of the Npc1 gene, which encodes a transmembrane protein critical for mobilizing cholesterol from the endolysosomal system. Loss of function mutations in the human NPC1 gene cause Niemann-Pick type C disease, a childhood-onset neurodegenerative disorder in which intracellular lipid accumulation, abnormally swollen axons, and neuron loss underlie the occurrence of early death. Both NPC patients and Npc1 null mice exhibit myelin defects indicative of dysmyelination, although the mechanisms underlying this defect are incompletely understood. Here we use temporal and cell-type-specific gene deletion in order to define effects on CNS myelination. Our results unexpectedly show that deletion of Npc1 in neurons alone leads to an arrest of oligodendrocyte maturation and to subsequent failure of myelin formation. This defect is associated with decreased activation of Fyn kinase, an integrator of axon-glial signals that normally promotes myelination. Furthermore, we show that deletion of Npc1 in oligodendrocytes results in delayed myelination at early postnatal days. Aged, oligodendocyte-specific null mutants also exhibit late stage loss of myelin proteins, followed by secondary Purkinje neuron degeneration. These data demonstrate that lipid uptake and intracellular transport by neurons and oligodendrocytes through an Npc1-dependent pathway is required for both the formation and maintenance of CNS myelin.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003462
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003462
Summary
Cholesterol availability is rate-limiting for myelination, and prior studies have established the importance of cholesterol synthesis by oligodendrocytes for normal CNS myelination. However, the contribution of cholesterol uptake through the endocytic pathway has not been fully explored. To address this question, we used mice with a conditional null allele of the Npc1 gene, which encodes a transmembrane protein critical for mobilizing cholesterol from the endolysosomal system. Loss of function mutations in the human NPC1 gene cause Niemann-Pick type C disease, a childhood-onset neurodegenerative disorder in which intracellular lipid accumulation, abnormally swollen axons, and neuron loss underlie the occurrence of early death. Both NPC patients and Npc1 null mice exhibit myelin defects indicative of dysmyelination, although the mechanisms underlying this defect are incompletely understood. Here we use temporal and cell-type-specific gene deletion in order to define effects on CNS myelination. Our results unexpectedly show that deletion of Npc1 in neurons alone leads to an arrest of oligodendrocyte maturation and to subsequent failure of myelin formation. This defect is associated with decreased activation of Fyn kinase, an integrator of axon-glial signals that normally promotes myelination. Furthermore, we show that deletion of Npc1 in oligodendrocytes results in delayed myelination at early postnatal days. Aged, oligodendocyte-specific null mutants also exhibit late stage loss of myelin proteins, followed by secondary Purkinje neuron degeneration. These data demonstrate that lipid uptake and intracellular transport by neurons and oligodendrocytes through an Npc1-dependent pathway is required for both the formation and maintenance of CNS myelin.
Introduction
Ensheathment of axons by myelin is an evolutionary feature of the vertebrate nervous system that is accomplished by the extended and specialized plasma membranes of oligodendrocytes in the CNS and Schwann cells in the PNS. Myelin contains at least 70% lipids by dry weight [1], and this high ratio of lipid to protein ensures the insulating properties of myelin to maximize the efficiency of nerve conduction. Among all the lipid species found in the myelin sheath, unesterified cholesterol is a major component [1]. In the mouse CNS, cholesterol in compact myelin represents ∼78% of the total lipid pool [2], and the availability of cholesterol is the rate-limiting step for myelination [3]. Since the CNS is shielded by the blood brain barrier, cholesterol required for myelination comes entirely from local synthesis [2]. Both neurons and glia obtain the cholesterol they need either through endogenous synthesis or by uptake of lipoprotein particles produced and released within the CNS. That endogenously synthesized cholesterol is critical for CNS myelination in mice is demonstrated by deletion in oligodendrocytes of squalene synthase, the first dedicated enzyme of sterol synthesis [3]. These mutant mice exhibit delayed myelination, suggesting that exogenously supplied cholesterol also contributes to CNS myelin formation. However, whether cholesterol from exogenous sources is required for myelin synthesis, or just a compensatory source when endogenous synthesis is lacking in myelinating glia, has not been explored.
An essential component of the pathway through which cholesterol in lipoprotein particles is mobilized from the endolysosomal system is the Npc1 protein [4], [5]. This multipass transmembrane protein resides in late endosomes and lysosomes [6]–[9], and functions cooperatively with the Npc2 protein to facilitate cholesterol efflux [10], [11]. Loss of functional Npc1 disrupts intracellular lipid trafficking, and leads to the sequestration of unesterified cholesterol and glycosphingolipids in late endosomes and lysosomes [12]. Mutations in the human NPC1 gene cause Niemann-Pick type C disease (NPC), a fatal childhood-onset neurodegenerative disorder [13]. Mice with a null mutation in the Npc1 gene (Npc1−/−) recapitulate the human disease, and exhibit progressive CNS neuropathology in which intracellular lipid accumulation, abnormally swollen axons, neuron loss and gliosis underlie the occurrence of ataxia and early death [5], [14]. Notably, both NPC patients and Npc1−/− mice exhibit myelin defects indicative of dysmyelination, particularly in the forebrain [15]–[19]. However, the complex pathology resulting from Npc1 deficiency in both neurons and oligodendrocytes has limited the utility of these global null mutants to provide a detailed understanding of the contribution of exogenous cholesterol to CNS myelination.
Here we use mice with a conditional null allele of the Npc1 gene to achieve temporal and cell type specific deletion in order to define effects on CNS myelin. We show that deletion of Npc1 restricted to neurons unexpectedly recapitulates the dysmyelination phenotype of global null mutants. This effect is mediated by a block in maturation of oligodendrocyte lineage cells that is associated with decreased activation of Fyn kinase, an integrator of axon-glial signals that normally promote myelination. Furthermore, we show that deletion of Npc1 in oligodendrocytes triggers a similar, though less severe impairment of CNS myelination, as well as myelin protein loss and secondary neurodegeneration. Our analyses suggest that exogenous cholesterol entering cells and trafficking through an Npc1-dependent pathway is necessary for both the formation and maintenance of CNS myelin.
Results
Global Npc1 Deficiency Leads to CNS Dysmyelination, Followed by Late Stage Loss of Myelin Proteins
To confirm the requirement of Npc1 for proper myelination in mice during early postnatal stages, we utilized mice with a floxed Npc1 allele (Npc1flox) [20]. Cre-mediated deletion yields a null allele that is functionally indistinguishable from the spontaneous null mutation found in Npc1nih mice (Npc1−/−) [5], [20]. To generate mice with Npc1 deletion in the germline, Npc1flox/flox mice were bred with transgenic mice expressing Cre recombinase under the control of the EIIa promoter [21]. Mice mosaic for the conditionally deleted allele were bred with mice carrying the Npc1− allele to generate compound heterozygotes of the conditionally deleted and null Npc1 alleles (Npc1Δ/−). We also generated mice with Npc1 deletion in adults by using a tamoxifen-regulated Cre recombinase under the control of the cytomegalovirus (CMV) promoter (Cre-ERTM+) [22]. Cre-mediated deletion of Npc1 in adults was induced by tamoxifen injections at 6 weeks, an age at which myelination is complete. Mice with adult deletion (Npc1flox/−, Cre-ERTM+) have been shown to recapitulate most features of NPC neuropathology, and reach end-stage by ∼22 weeks [23]. To determine the effect of the timing of Npc1 deletion upon myelination, we compared 7-week-old mice with germline deletion (Npc1Δ/−), 22-week-old mice with adult deletion (Npc1flox/−, Cre-ERTM+) and age matched controls. Myelin basic protein (MBP, a standard marker for mature myelin [1]) and FluoroMyelin (a lipophilic stain for compact myelin) staining of sagittal midline brain sections revealed a dramatic reduction of myelin proteins and lipids in Npc1Δ/− mice, particularly in the forebrain (Figure 1A, 1B). This striking pattern of regionally selective myelin defects is similar to that previously reported in Npc1−/− mice [14], [15], [17]. In contrast, Npc1flox/−, Cre-ERTM+ mice exhibited a staining pattern morphologically similar to that in controls (Figure 1A, 1B). The difference in MBP staining patterns between Npc1Δ/− mice and Npc1flox/−, Cre-ERTM+ mice suggests that Npc1 is required in early postnatal stages for proper myelin formation. Further analysis of myelin-specific proteins demonstrated a decrease in MBP and CNP protein levels in Npc1flox/−, Cre-ERTM+ mice compared to littermate controls, particularly in the cortex (Figure 1C, 1D). We conclude that myelin was properly formed in Npc1flox/−, Cre-ERTM+ mice during postnatal development, but that these mice exhibit loss of myelin proteins at later stages, particularly in the cerebral cortex, after Npc1 deletion at 6 weeks. Axonal loss could contribute to the late stage pathology in Npc1flox/−, Cre-ERTM+ mice, as evidenced by decreased neurofilament levels in these aged mutants (Figure 1C). Taken together, our analysis suggests that lack of myelin in NPC mice is caused by dysmyelination at early postnatal days, followed by loss of myelin proteins at end stage.

Neuronal Deletion of Npc1 Leads to Blockade of Oligodendrocyte Maturation and Dysmyelination
We next sought to dissect the contribution of different CNS cell types to NPC dysmyelination. We started by deleting Npc1 specifically in neurons, using transgenic mice expressing Cre recombinase under the control of the Synapsin1 promoter (Syn1-Cre) [24]. We confirmed gene deletion by staining brain sections with filipin, a fluorescent dye that specifically marks accumulation of unesterified cholesterol [25]. NeuN and filipin co-staining verified that Npc1flox/−, Syn1-Cre+ mice, but not Npc1flox/+, Syn1-Cre+ controls [23], developed filipin-positive neurons throughout the brain, including brainstem and cortex (Figure S1A). A subset of neurons remained filipin negative, possibly reflecting mosaic gene deletion. To further verify neuron-specific gene deletion, Syn1-Cre+ mice were crossed to a Rosa reporter line that has been widely used to demonstrate gene deletion in both neurons and oligodendrocytes [26]. LacZ staining revealed widespread positive cells in many brain regions including the cortex, with minimal staining in the corpus callosum, where neuronal cell bodies are lacking (Figure S1B). Co-staining with NeuN or Olig2 showed that these LacZ positive cells were neurons, and not oligodendrocyte lineage cells (Figure S1C), further supporting the notion that we achieved neuron-specific deletion by using Syn1-Cre+ mice.
The effect of Npc1 deficiency in neurons upon myelination was first evaluated by MBP immunofluorescence at 3 different ages. At postnatal day 16 (P16), myelination was actively occurring in the forebrain of Npc1flox/+, Syn1-Cre+ controls, with abundant MBP-positive myelinating oligodendrocytes populating the cortex (Figure 2B). In contrast, Npc1flox/−, Syn1-Cre+ mutants exhibited a severe paucity of myelin in the same region, with most of the MBP positive cells exhibiting the morphology of pre-myelinating oligodendrocytes (Figure 2B). At 7 weeks, myelination was completed in Npc1flox/+, Syn1-Cre+ controls, but was greatly attenuated in the cortex of Npc1flox/−, Syn1-Cre+ mutants. No recovery of myelination was observed in mutants aged to 16 weeks (Figure 2B), which is end stage for these mice [23]. Similarly, FluoroMyelin staining revealed a paucity of compact myelin in the corpus callosum of Npc1flox/−, Syn1-Cre+ mutants at 16 weeks (Figure 2B, bottom panel). Although MBP staining was markedly decreased in the cortex of Npc1flox/−, Syn1-Cre+ mutants, other brain regions exhibited a normal staining pattern, reminiscent of the selective defects in myelination observed after global germline deletion (Figure 1A). Regional-specific dysmyelination was further supported by western blots showing decreased levels of myelin-specific proteins including CNP, MBP and MAG in cortex, but not brainstem of Npc1flox/−, Syn1-Cre+ mutants (Figure 2C). Electron microscopy confirmed that the density of myelinated nerve fibers in the corpus callosum was greatly reduced in Npc1flox/−, Syn1-Cre+ mutants at 3 weeks (Figure 2E). Notably, neurofilament protein levels in the cortex were similar between Npc1flox/+, Syn1-Cre+ controls and Npc1flox/−, Syn1-Cre+ mutants at P16 (Figure 2C), and neurofilament immunofluorescence staining showed no significant axonal pathology (Figure 2D). These data indicate that dysmyelination in the forebrain of Npc1flox/−, Syn1-Cre+ mutants was not secondary to axonal loss.

To characterize the mechanism underlying dysmyelination in Npc1flox/−, Syn1-Cre+ mutants, we assessed oligodendrocyte lineage cells at different stages of differentiation. At P16, Npc1flox/−, Syn1-Cre+ mutants showed a significantly reduced number of CC1-positive mature oligodendrocytes in the forebrain (Figure 3A, 3C) but a normal density of NG2-positive oligodendrocyte precursor cells (OPCs) (Figure 3A, 3B). As previously reported for global null Npc1 mutants [17], this deficit of mature oligodendrocytes was not associated with evidence of increased apoptosis (data not shown). The paucity of mature oligodendrocytes was associated with a reduced number of cells in the corpus callosum expressing Sip1, a signaling protein implicated oligodendrocyte differentiation (Figure 3C) [27]. These data indicated that Npc1 deficiency in neurons triggered a block of oligodendrocyte maturation, and prompted us to determine whether signals known to regulate oligodendrocyte maturation and myelination were perturbed in Npc1flox/−, Syn1-Cre+ mutants. We first examined proteins that mediate signaling between axons and oligodendrocyte lineage cells including PSA-NCAM [28], Lingo1 [29] and Jagged1 [30], and found no differences between Npc1flox/−, Syn1-Cre+ mutants and controls at P16 (Figure S2A). Similarly, we found no evidence of astrocyte activation in the corpus callosum of Npc1flox/−, Syn1-Cre+ mutants at P16 (Figure S2B, S2C), consistent with prior studies showing that astrogliosis is limited to the thalamus of Npc1−/− mice at two weeks [31]. In contrast, activity of the non-receptor tyrosine kinase Fyn [32] was reduced in the cortex of Npc1flox/−, Syn1-Cre+ mutants, as evidenced by decreased levels of the active form (phosphorylated at tyrosine 420) and concurrently increased levels of the inactive form (phosphorylated at tyrosine 531) (Figure 3E). As oligodendroglial Fyn is an integrator of axonal signals that promote myelination [33], the decreased activity of Fyn in Npc1flox/−, Syn1-Cre+ mutants suggests that Npc1 deficiency in axons leads to a disruption of axon-glial signaling that is crucial for oligodendrocyte differentiation and myelination.

Oligodendrocyte Deletion of Npc1 Results in a Similar, but Milder Dysmyelination Phenotype during Postnatal Development
Next, we tested if Npc1 deficiency in oligodendrocyte lineage cells contributes to the pathogenesis of dysmyelination in NPC mice. To accomplish this, we used transgenic mice expressing Cre recombinase under the control of the CNP promoter (CNP Cre/+) [34]. In these mice, Cre is abundantly and specifically expressed in postmitotic oligodendrocytes. Co-staining for Cre and Olig2, a marker of both OPCs and postmitotic oligodendrocytes, verified that Cre was specifically expressed in a subset of Olig2+ oligodendrocyte lineage cells in various brain regions including brainstem and cortex (Figure S3B). Filipin staining revealed minimal accumulation of unesterified cholesterol in Npc1flox/−, CNPCre/+ mutants (Figure S3A), a finding both consistent with a previous report showing no detectable cholesterol accumulation in oligodendrocytes of Npc1−/− mice [35] and indicative of the cell lineage specificity of this Cre line.
Deletion of Npc1 in oligodendrocytes resulted in a dysmyelination phenotype that was initially similar to that caused by Npc1 deletion in neurons. At P16, Npc1flox/−, CNPCre/+ mutants expressed markedly reduced levels of myelin-specific proteins including MBP, CNP and MAG in the cortex (Figure 4A, 4B). Similarly, compact myelin levels by FluoroMyelin staining were decreased in Npc1flox/−, CNPCre/+ mutants (Figure 4A). This dysmyelination phenotype partially recovered by 7 weeks (Figure 4A), a finding that indicates oligodendrocyte deletion delayed myelination and contrasts with the block produced by neuronal deletion. Myelination in the brainstem of Npc1flox/−, CNPCre/+ mutants was minimally affected (Figure 4B) despite robust Cre expression in this region (Figure S3B, S3C). Electron microscopy confirmed diminished density of myelinated nerve fibers in the corpus callosum of Npc1flox/−, CNPCre/+ mutants at 3 weeks (Figure 4D). Similar to neuron-specific mutants, dysmyelination in Npc1flox/−, CNPCre/+ mutants occurred without significant axonal pathology (Figure 4B, 4C). The requirement of Npc1 in oligodendrocytes for proper myelination was further confirmed by using an independent line in which Cre was highly expressed in OPCs (Olig2Cre/+ mice, Figure S4) [36]. Similar to Npc1flox/−, Syn1-Cre+ mutants, Npc1flox/−, CNPCre/+ mutants at P16 showed reduced density of mature oligodendrocytes (Figure 5A, 5C), with normal numbers of OPCs in the forebrain (Figure 5A, 5B), indicating arrest of oligodendrocyte maturation.
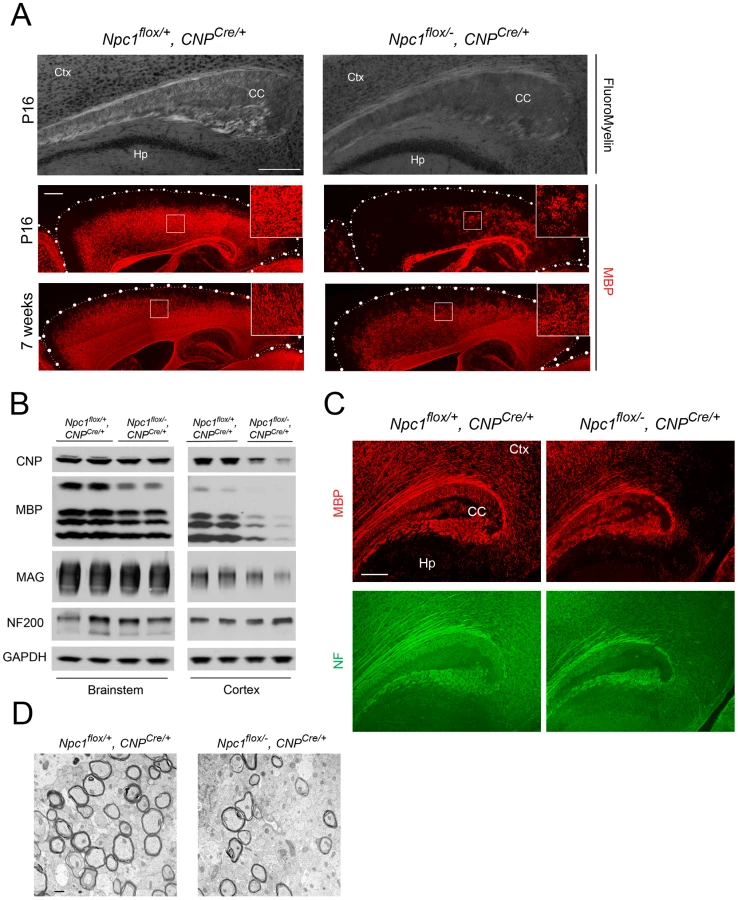

Mice with Npc1 Deletion in Oligodendrocytes Exhibit Myelin Protein Loss in Late Stages
As the Npc1flox/−, CNPCre/+ mutants aged, they developed progressive motor deficits (Figure 6C), although weight was not affected (Figure 6A, 6B). This led us to examine myelin protein levels in 23-week-old Npc1flox/−, CNPCre/+ mutants. We found decreased levels of myelin proteins not only in cortex, but also in brainstem and cerebellum (Figure 7A), where myelination in early postnatal days was nearly normal (Figure 4B). This suggested that myelin loss was taking place in several brain regions of the aged Npc1flox/−, CNPCre/+ mutants. We found this was associated with only mild changes in the pattern of MBP staining (Figure 7B). Interestingly, the total number of Olig2+ oligodendrocyte lineage cells in the cerebellar white matter was unchanged in aged mutants (Figure 7C, 7D), suggesting that loss of Npc1 did not affect the survival of oligodendrocytes in adult mice. This loss of myelin proteins was associated with secondary neuron loss in the cerebellum. We detected Purkinje cell loss in anterior lobules of 23-week-old but not 7-week-old Npc1flox/−, CNPCre/+ mutants, as demonstrated by calbindin staining of sagittal midline sections (Figure 7E, 7G) and by loss of calbindin staining on western blot (Figure 7F). Importantly, no filipin-positive Purkinje neurons were identified in these mice (not shown), supporting the conclusion that Purkinje cell loss was a consequence of non-cell autonomous toxicity. We conclude that Npc1 acts in oligodendrocytes both to promote normal myelination and to ensure the maintenance of myelin in the adult CNS.


Discussion
Here we used Npc1 conditional null mice to establish the critical role of Npc1 in both neurons and oligodendrocytes for proper CNS myelination. Our findings demonstrate that deletion of Npc1 in neurons alone is sufficient to recapitulate the dysmyelination phenotype that occurs following global germline deletion. These mice display a severe phenotype, particularly in the forebrain, characterized by a lack of mature oligodendrocytes but a normal density of OPCs, indicating that Npc1 deficiency in neurons triggers an arrest of oligodendrocyte maturation. Our data also demonstrate that deletion of Npc1 in oligodendrocytes leads to similar but milder forebrain dysmyelination that largely recovers by 7 weeks, consistent with a delay rather than a block in myelination. Furthermore, we demonstrate that these oligodendrocyte-specific mutants develop ataxia as they age, and that this is associated with decreased myelin proteins and Purkinje cell loss in anterior cerebellar lobules, establishing the occurrence of secondary neurodegeneration. Our results highlight the importance of Npc1 in both neurons and oligodendrocytes for the formation and maintenance of CNS myelin.
Significant effort has been devoted to defining the contribution of specific cell types to NPC neuropathology. Studies in chimeric mice, a conditional knock-out model, and several neuron-specific transgenic rescue experiments all demonstrate that neuronal loss can be a consequence of cell autonomous neurotoxicity [20], [23], [37]–[39]. Furthermore, these analyses indicate that brain inflammation is a consequence rather than a driver of neuron loss [20], [23], [38], [40]. The role of astroglial cells in NPC neuropathology has been more controversial. While in vitro data suggest that Npc1 deficient astrocytes fail to fully support cultured neurons [41], both conditional knockout and transgenic rescue experiments failed to establish a significant role for astrocytes in pathogenesis [23], [38]. A transgenic line that highly over-expresses Npc1 from the GFAP promoter does show some rescue [42], but the extent of cell type restricted expression in these mice remains incompletely defined. The effects of Npc1 deficiency restricted to oligodendrocytes had not been previously explored. As for effects on CNS myelin, prior transgene rescue experiments using the NSE promoter to drive Npc1 expression demonstrated partial rescue of myelination [39]. These findings are consistent with our observation that neuronal expression of Npc1 plays an important role in oligodendrocyte maturation and myelination. Finally, we note that aged, oligodendrocyte-specific null mutants show evidence of neuron loss. While prior studies firmly establish that neuronal deficiency of Npc1 is sufficient to mediate neurotoxicity [20], [23], the data reported here indicate that non-cell autonomous pathways arising from oligodendrocytes also contribute to neuropathology.
Oligodendrocyte differentiation and myelination are highly dynamic processes controlled by both intrinsic factors and extrinsic mechanisms [43]. Recent studies of axon-glial communication have identified a series of axonal signals important for regulating myelination. Oligodendroglial Fyn, a Src family kinase, has been suggested to play a central role in integrating diverse axonal signals to initiate myelination [33]. Downstream signaling from activated Fyn kinase promotes oligodendrocyte survival, alters cytoskeleton polarity and increases the expression of myelin genes. Our analysis of neuron-specific Npc1 mutants reveals decreased Fyn activity and a regionally-restricted dysmyelination phenotype similar to that of Fyn knockout mice [44]. We suggest that Npc1 deficiency in neurons disrupts an axon-glial signal vital for promoting myelination. The axonal ligand responsible for oligodendroglial Fyn activation remains elusive. The requirement of Npc1 for Fyn activation raises the possibility that a lipid species, such as cholesterol or a sphingolipid, may contribute to this signal. Additionally, recent neuron-glial co-culture studies demonstrate the role of action potentials in stimulating myelination through Fyn-dependent mechanisms [45]. It is therefore also possible that defective Fyn activation results from decreased electrical activity of axons in Npc1flox/−, Syn1-Cre+ mutants. Recently, a similar role in myelination has been demonstrated for neuron-restriction expression of the PI(3,5)P(2) phosphatase Fig4 [46], suggesting that defects in axon-glial signaling may underlie dsymyelination in several disorders.
Animal studies of cholesterol metabolism in myelinating glia have highlighted the importance of cell-autonomous production of cholesterol for myelin formation. Mice lacking oligodendroglial squalene synthase, an enzyme required for cholesterol synthesis, exhibit perturbed CNS myelination in early postnatal days [3]. Similarly, deletion of SCAP (SREBP-cleavage-activating protein) in Schwann cells, a protein that complexes with SREBP to regulate the expression of genes promoting cholesterol synthesis and lipoprotein uptake, leads to PNS hypomyelination [47]. It is notable that both mouse models partially recover at later stages, suggesting that myelinating glia have the capacity to overcome the lack of endogenous cholesterol production, probably through increased uptake. Here we present in vivo evidence indicating an important contribution of exogenous cholesterol to myelin synthesis. Our findings show that deletion of Npc1 in oligodendrocytes, which eliminates their utilization of cholesterol from the endocytosis of LDL or similar lipoprotein particles, leads to perturbed myelin formation in the CNS. Npc1 deficiency also impairs intracellular trafficking of sphingolipids [48] and endogenously synthesized cholesterol [49]. Nonetheless, the blockade of exogenous cholesterol utilization and the essential role that cholesterol plays in myelination leads us to favor the conclusion that the effects observed here are due to a disruption in the availability of exogenous cholesterol. As shown for other cell types [12], we speculate that the synthesis of endogenous cholesterol may be up-regulated in Npc1 deficient oligodendrocytes yet insufficient to overcome the lack of exogenous cholesterol, especially during the peak phase of myelination. This suggests that extracellularly-derived cholesterol is indispensible for normal CNS myelination.
Although Npc1flox/−, CNPCre/+ mutants form myelin in the brainstem and cerebellum during postnatal development, these regions exhibit loss of myelin proteins in adults. Biochemical studies have shown that in the adult CNS, myelin production and cholesterol turnover decrease to very low levels [2]. It is therefore unlikely that the loss of myelin proteins in these adult mutants results from impaired access to exogenous cholesterol as a consequence of Npc1 deficiency. Rather, we speculate that late-stage pathology stems from the unstable nature of the myelin sheath produced by mutant oligodendrocytes. Studies of cellular models of NPC have shown that cholesterol content is decreased in the plasma membrane of mutant cells [50], [51]. This change may impact myelin by disrupting membrane fluidity, altering lipid rafts or modulating the function of membrane proteins, and thereby increase the vulnerability of aged mutants. Further analysis of the biochemical composition of the myelin sheath generated by Npc1-deficient oligodendrocytes will help define the mechanism mediating late-onset loss of myelin proteins. Axonal degeneration and neuron loss in these mutants highlights the important role of oligodendrocytes in supporting neuron function and survival. Similar observations have been made in mice over-expressing alpha-synuclein in oligodendrocytes [52]. While this effect may be mediated in part through loss of myelin, other studies have shown that oligodendroglia support axons through metabolic pathways independent of myelination [53]. It is currently unclear which of these mechanisms accounts for Purkinje neuron loss in Npc1flox/−, CNPCre/+ mutants.
In summary, the data reported here extend our understanding of the role of cholesterol metabolism in myelination, and demonstrate that exogenous cholesterol is needed by both neurons and oligodendrocytes for the formation and maintenance of CNS myelin. A characteristic feature of Npc1 deficient mice, both global nulls and cell-specific knockouts, is the regionally severe dysmyelination that occurs during early postnatal stages. Fate-mapping studies have established that OPCs originate from heterogeneous regions of the subventricular zone, under the influence of different signaling pathways [54]. We speculate that these regional differences in oligodendrocyte lineage cells lead to distinct responses to axonal signals or to the need for exogenously-derived cholesterol for proper myelination, contributing to severe dysmyelination particularly in the forebrain of Npc1 mutants. While the precise mechanism underlying this regional selectivity remains to be defined, our data establish a critical role for Npc1 in both myelin formation and maintenance. Our findings have important implications for understanding the pathogenesis of NPC disease and may also inform our knowledge of other dysmyelinating/demyelinating disorders.
Materials and Methods
Ethics Statement
Animal use and procedures were approved by the University of Michigan Committee on the Use and Care of Animals.
Mice
Npc1flox/flox and Npc1Δ/− mice were generated as previously described [20]. Other mice used include tamoxifen-inducible CMV-Cre (Cre-ERTM+) (#004682, Jackson Laboratories), Sny1-Cre (#003966, Jackson Laboratories), CNPCre/+ mice [34], Olig2Cre/+ mice [36] and Rosa reporter mice (#003474, Jackson Laboratories). All mouse strains were maintained on the C57BL6/J background, except Olig2Cre/+ mice which were maintained on the 129/C3H mixed background.
Tamoxifen Induction
Tamoxifen (Sigma) was dissolved in corn oil (Sigma) at 20 mg/ml and stored at −20C in the dark. The stock solution was warmed to 37C before injection. 6-week-old mice were injected intraperitoneally with 3 mg tamoxifen per 40 g body weight for 5 consecutive days.
Phenotype Analysis
Motor function was measured using the balance beam test as described previously [20].
Western Blot
Brain lysates were homogenized in RIPA buffer (Thermo Scientific) containing Complete protease inhibitor cocktail (Roche) and phosphatase inhibitors (Thermo scientific) using a motor homogenizer (TH115, OMNI International). Samples were resolved by 4–20% Tris-glycine gradient gel and transferred to nitrocellulose membranes (BioRad) on a semidry transfer apparatus. Immunoreactivity was detected by Immobilon chemilluminescent substrate (Thermo Scientific). Antibodies used were rat anti-MBP (1∶2000, Abcam), mouse anti-CNP (1∶2000, Millipore), mouse anti-MAG (1∶5000, Millipore), mouse anti-Neurofilament 200 (1∶5000, Millipore), rabbit anti-NG2 (1∶1000, Millipore), rabbit anti-GAPDH (1∶5000, Santa Cruz), mouse anti-Cre (1∶1000, Millipore), rabbit anti-GFAP (1∶5000, Dako), mouse anti-PSA-NCAM (1∶1000, Millipore), goat anti-Jagged1(1∶1000, Santa Cruz) and rabbit anti-Lingo1 (1∶1000, Abcam).
Immunoprecipitation
200 µg brain lysates were immunoprecipitated with 10 µg anti-Fyn antibody (FYN3, Santa Cruz) overnight at 4C, followed by incubation with 20 µl Protein A beads (Santa Cruz) for 1 h at 4C. The immunoprecipitates were then washed 4 times with protein lysis buffer before being boiled with 2× sample buffer at 100C for 5 min. For the subsequent western blot analysis, anti-Fyn (FYN3, Santa Cruz), Src pY418 and pY529 antibodies (Life technologies) were used to detect total Fyn and phosphorylation of Fyn at Y420 and Y531, respectively.
Histology
Mice were perfused with 0.9% normal saline followed by 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight. Brains were bisected, with the right hemisphere processed for paraffin embedding and the left hemisphere processed for frozen sections. Prior to freezing, brain tissue was cryoprotected in 30% sucrose for 48 hr at 4C. Brains were frozen in isopentane chilled by dry ice and embedded in OCT (Tissue-Tek). Frozen sections were prepared at 14 µm in a cryostat and used for LacZ staining and subsequent eosin counter staining or immunohistochemical staining for Olig2 (1∶500, Millipore) and NeuN (1∶500, Millipore). For filipin staining, frozen sections were first used for immunofluorescence staining for NeuN or Olig2, followed by incubation for 90 min in PBS with 10% fetal bovine serum plus 25 µg/ml filipin (Sigma). For FluoroMyelin staining, frozen sections were rehyrated in PBS for 20 min, incubated with FluoroMyelin solution (1∶300, Life Technologies) at room temperature for 2 hours, and then cleared with four 30-minute washes with PBS. Paraffin-embedded sections were prepared at 5 µm and used for staining with H&E staining or MBP (1∶100, Abcam), SMI-31P (1∶200, Covance), NG2 (1∶100, Millipore), CC1 (1∶200, Calbiochem), Calbindin (1∶1000, Sigma), Sip1 (1∶100, Santa Cruz) and GFAP (1∶1000, Dako) immunofluorescence. For visualization of staining, secondary antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 488 (Molecular Probes) were used and images were captured on a Zeiss Axioplan 2 imaging system. For NG2 and CC1 co-staining and Olig2 staining, images were captured on an Olympus FluoView 500 Confocal Microscope system. Quantification of CC1+ or Olig2+ cells was performed using NIH ImageJ software. Quantification of Purkinje cell loss was performed on H&E stained sections. Counts were normalized to the length of the Purkinje layer, as measured by NIH ImageJ software, and reported as Purkinje cell density.
Electron Microscopy
Mice were perfused with 0.9% normal saline followed by 3% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M Sorensen's buffer. The corpus callosum was removed and post-fixed in perfusion solution overnight, followed by fixation in 1% osmium tetroxide solution for 1 h at room temperature. After dehydration, tissues were embedded in epoxy resin. For transmission electron microscopy, ultrathin sections were cut, and images were captured on a Philips CM-100 imaging system at 10,500× magnification.
Statistics
Statistical significance was assessed by unpaired Student's t test. Statistics were performed using the software package Prism 5 (GraphPad Software). P values less than 0.05 were considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BaumannN, Pham-DinhD (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81 : 871–927.
2. DietschyJM, TurleySD (2004) Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45 : 1375–1397.
3. SaherG, BruggerB, Lappe-SiefkeC, MobiusW, TozawaR, et al. (2005) High cholesterol level is essential for myelin membrane growth. Nat Neurosci 8 : 468–475.
4. CarsteaED, MorrisJA, ColemanKG, LoftusSK, ZhangD, et al. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277 : 228–231.
5. LoftusSK, MorrisJA, CarsteaED, GuJZ, CummingsC, et al. (1997) Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science 277 : 232–235.
6. NeufeldEB, WastneyM, PatelS, SureshS, CooneyAM, et al. (1999) The Niemann-Pick C1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J Biol Chem 274 : 9627–9635.
7. HigginsME, DaviesJP, ChenFW, IoannouYA (1999) Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab 68 : 1–13.
8. DaviesJP, IoannouYA (2000) Topological analysis of Niemann-Pick C1 protein reveals that the membrane orientation of the putative sterol-sensing domain is identical to those of 3-hydroxy-3-methylglutaryl-CoA reductase and sterol regulatory element binding protein cleavage-activating protein. J Biol Chem 275 : 24367–24374.
9. GarverWS, HeidenreichRA, EricksonRP, ThomasMA, WilsonJM (2000) Localization of the murine Niemann-Pick C1 protein to two distinct intracellular compartments. J Lipid Res 41 : 673–687.
10. KwonHJ, Abi-MoslehL, WangML, DeisenhoferJ, GoldsteinJL, et al. (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137 : 1213–1224.
11. DeffieuMS, PfefferSR (2011) Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc Natl Acad Sci U S A 108 : 18932–18936.
12. KartenB, PeakeKB, VanceJE (2009) Mechanisms and consequences of impaired lipid trafficking in Niemann-Pick type C1-deficient mammalian cells. Biochim Biophys Acta 1791 : 659–670.
13. VanierMT (2010) Niemann-Pick disease type C. Orphanet J Rare Dis 5 : 16.
14. GermanDC, LiangCL, SongT, YazdaniU, XieC, et al. (2002) Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience 109 : 437–450.
15. WeintraubH, AbramoviciA, SandbankU, PentchevPG, BradyRO, et al. (1985) Neurological mutation characterized by dysmyelination in NCTR-Balb/C mouse with lysosomal lipid storage disease. J Neurochem 45 : 665–672.
16. WeintraubH, AbramoviciA, SandbankU, BoothAD, PentchevPG, et al. (1987) Dysmyelination in NCTR-Balb/C mouse mutant with a lysosomal storage disorder. Morphological survey. Acta Neuropathol 74 : 374–381.
17. TakikitaS, FukudaT, MohriI, YagiT, SuzukiK (2004) Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol 63 : 660–673.
18. TrouardTP, HeidenreichRA, SeegerJF, EricksonRP (2005) Diffusion tensor imaging in Niemann-Pick Type C disease. Pediatr Neurol 33 : 325–330.
19. WalterfangM, FaheyM, DesmondP, WoodA, SealML, et al. (2010) White and gray matter alterations in adults with Niemann-Pick disease type C: a cross-sectional study. Neurology 75 : 49–56.
20. ElrickMJ, PachecoCD, YuT, DadgarN, ShakkottaiVG, et al. (2010) Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet 19 : 837–847.
21. LaksoM, PichelJG, GormanJR, SauerB, OkamotoY, et al. (1996) Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A 93 : 5860–5865.
22. HayashiS, McMahonAP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244 : 305–318.
23. YuT, ShakkottaiVG, ChungC, LiebermanAP (2011) Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum Mol Genet 20 : 4440–4451.
24. ZhuY, RomeroMI, GhoshP, YeZ, CharnayP, et al. (2001) Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15 : 859–876.
25. BornigH, GeyerG (1974) Staining of cholesterol with the fluorescent antibiotic “filipin”. Acta Histochem 50 : 110–115.
26. SorianoP (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21 : 70–71.
27. WengQ, ChenY, WangH, XuX, YangB, et al. (2012) Dual-mode modulation of Smad signaling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 73 : 713–728.
28. CharlesP, HernandezMP, StankoffB, AigrotMS, ColinC, et al. (2000) Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A 97 : 7585–7590.
29. LeeX, YangZ, ShaoZ, RosenbergSS, LevesqueM, et al. (2007) NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J Neurosci 27 : 220–225.
30. WangS, SdrullaAD, diSibioG, BushG, NofzigerD, et al. (1998) Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21 : 63–75.
31. BaudryM, YaoY, SimmonsD, LiuJ, BiX (2003) Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol 184 : 887–903.
32. UmemoriH, SatoS, YagiT, AizawaS, YamamotoT (1994) Initial events of myelination involve Fyn tyrosine kinase signalling. Nature 367 : 572–576.
33. Kramer-AlbersEM, WhiteR (2011) From axon-glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell Mol Life Sci 68 : 2003–2012.
34. Lappe-SiefkeC, GoebbelsS, GravelM, NickschE, LeeJ, et al. (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33 : 366–374.
35. LiaoG, CheungS, GaleanoJ, JiAX, QinQ, et al. (2009) Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1−/ − mouse brain. Brain Res 1270 : 140–151.
36. SchullerU, HeineVM, MaoJ, KhoAT, DillonAK, et al. (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14 : 123–134.
37. KoDC, MilenkovicL, BeierSM, ManuelH, BuchananJ, et al. (2005) Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet 1: e7 doi:10.1371/journal.pgen.0010007.
38. LopezME, KleinAD, DimbilUJ, ScottMP (2011) Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci 31 : 4367–4378.
39. BorbonI, TotenhagenJ, FiorenzaMT, CanteriniS, KeW, et al. (2012) Niemann-Pick C1 mice, a model of “juvenile Alzheimer's disease”, with normal gene expression in neurons and fibrillary astrocytes show long term survival and delayed neurodegeneration. J Alzheimers Dis 30 : 875–887.
40. LopezME, KleinAD, HongJ, DimbilUJ, ScottMP (2012) Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum Mol Genet 21 : 2946–2960.
41. ChenG, LiHM, ChenYR, GuXS, DuanS (2007) Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. Glia 55 : 1509–1518.
42. ZhangM, StrnatkaD, DonohueC, HallowsJL, VincentI, et al. (2008) Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1−/ − mice. J Neurosci Res 86 : 2848–2856.
43. EmeryB (2010) Regulation of oligodendrocyte differentiation and myelination. Science 330 : 779–782.
44. SperberBR, Boyle-WalshEA, EnglekaMJ, GadueP, PetersonAC, et al. (2001) A unique role for Fyn in CNS myelination. J Neurosci 21 : 2039–2047.
45. WakeH, LeePR, FieldsRD (2011) Control of local protein synthesis and initial events in myelination by action potentials. Science 333 : 1647–1651.
46. WintersJJ, FergusonCJ, LenkGM, Giger-MateevaVI, ShragerP, et al. (2011) Congenital CNS hypomyelination in the Fig4 null mouse is rescued by neuronal expression of the PI(3,5)P(2) phosphatase Fig4. J Neurosci 31 : 17736–17751.
47. VerheijenMH, CamargoN, VerdierV, NadraK, de Preux CharlesAS, et al. (2009) SCAP is required for timely and proper myelin membrane synthesis. Proc Natl Acad Sci U S A 106 : 21383–21388.
48. SunX, MarksDL, ParkWD, WheatleyCL, PuriV, et al. (2001) Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am J Hum Genet 68 : 1361–1372.
49. KartenB, VanceDE, CampenotRB, VanceJE (2002) Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem 83 : 1154–1163.
50. WojtanikKM, LiscumL (2003) The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J Biol Chem 278 : 14850–14856.
51. HawesCM, WiemerH, KruegerSR, KartenB (2010) Pre-synaptic defects of NPC1-deficient hippocampal neurons are not directly related to plasma membrane cholesterol. J Neurochem 114 : 311–322.
52. YazawaI, GiassonBI, SasakiR, ZhangB, JoyceS, et al. (2005) Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45 : 847–859.
53. LeeY, MorrisonBM, LiY, LengacherS, FarahMH, et al. (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487 : 443–448.
54. RichardsonWD, KessarisN, PringleN (2006) Oligodendrocyte wars. Nat Rev Neurosci 7 : 11–18.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy