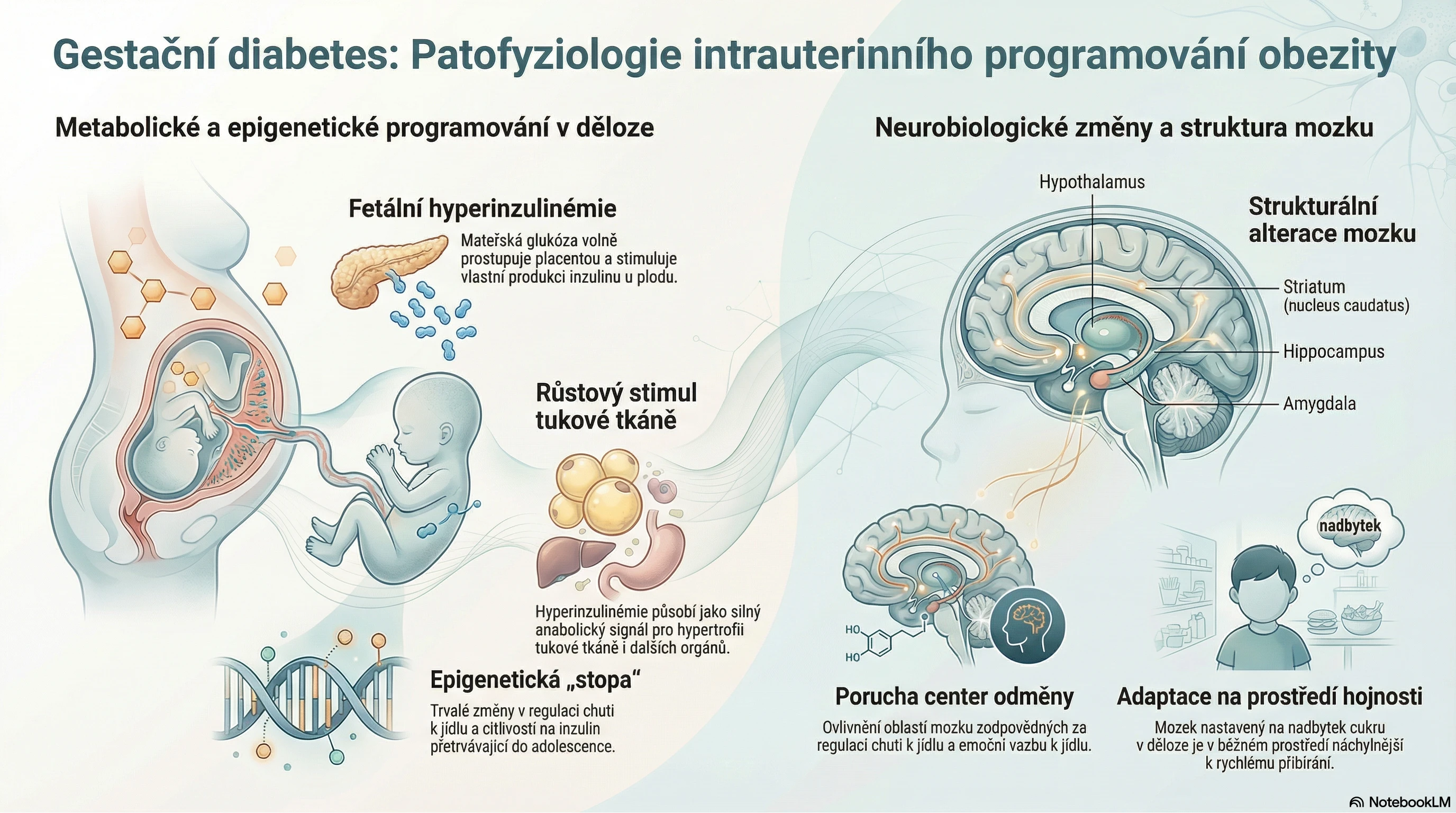
Amyloidózy charakterizuje rozmanitost klinických projevů. Co je v pozadí této heterogenity?
Klinická manifestace amyloidóz je velmi různorodá, protože nejde o jedno onemocnění. Amyloidózy se liší prekurzorovým proteinem, typem orgánového postižení i jeho závažností. Nejčastějšími typy amyloidóz jsou primární (AL), sekundární (AA) a transthyretinová (ATTR):
Které symptomy ATTR se především považují za varovné?
Mezi hlavní red flags ATTR patří progredující polyneuropatie a časné známky autonomní dysfunkce – například ortostatická hypotenze, poruchy trávení, sfinkterové či sexuální potíže (zejména erektilní dysfunkce). Významný je také oboustranný syndrom karpálního tunelu, často s recidivami i po operaci, který může předcházet jiným symptomům o několik let.
Dalším varovným znakem je restriktivní kardiomyopatie s hypertrofií levé komory u pacientů bez klasických rizikových faktorů, časté jsou také arytmie (AV blok, sick-sinus syndrom, komorové arytmie). Na ATTR mohou upozornit spontánní ruptury šlach (biceps, Achillova šlacha), opacita sklivce, proteinurie nebo nevysvětlitelný úbytek hmotnosti. Rizikovým faktorem je i familiární výskyt neuropatie, kardiomyopatie či náhlých úmrtí ve středním věku. Podezření by měla vzbudit především kombinace více těchto příznaků.
Patrně nejčastějším projevem hereditárních transthyretinových amyloidóz (hATTR) jsou neuropatie. Jsou u těchto pacientů něčím specifické?
Nejvýraznějším rysem polyneuropatie u hATTR je její rychlá progrese. Jde o nejzávažnější dědičnou polyneuropatii s nástupem v dospělosti – v neendemických oblastech pacienti často do 3 let potřebují oporu při chůzi a do 6 let bývají upoutáni na vozík. Průměrné přežití bez léčby se uvádí mezi 7 a 12 lety od výskytu prvních symptomů.
Časté jsou časné autonomní příznaky a syndrom karpálního tunelu. Polyneuropatie bývá výrazně bolestivá, což ale není specifické jen pro hATTR. Typicky jde o length-dependent senzomotorickou axonální neuropatii, začínající v akrálních částech dolních končetin a s postupným šířením. Postižena bývají nejprve tenká vlákna (bolest, teplotní čití, autonomní symptomy), později i silná vlákna (ztráta reflexů, propriocepce, ataxie, parézy). Vzácně může hATTR imitovat zánětlivou demyelinizační neuropatii, která však nereaguje na běžnou imunosupresivní terapii.
Na co si obvykle nejvíce stěžují sami pacienti? Které symptomy je dovedou k lékaři?
Nemocní s hATTR přicházejí k lékaři s různorodými obtížemi. V neendemických oblastech, jako je Česko, bývají v téměř 50 % případů prvním příznakem neuropatická bolest a další senzitivní symptomy. U 30 % dominují autonomní potíže (například ortostatická intolerance, zažívací či sexuální dysfunkce) a u 10–15 % se onemocnění zpočátku projeví kardiálně. V endemických oblastech (například Portugalsko, Španělsko /Mallorca/, Brazílie) převládají jako první právě autonomní symptomy.
Jak často bývají pacienti s ATTR v rámci neurologie nesprávně diagnostikováni? Za co bývá amyloidóza zaměňována?
Až 20–40 % pacientů s hATTR je vedeno pod jinou diagnózou. Nemocní tak často navštíví několik lékařů, než je stanovena správná příčina obtíží. Diagnostiku komplikuje i to, že většina (až 77 %) pacientů v neendemických oblastech nemá pozitivní rodinnou anamnézu, což snižuje podezření na dědičné onemocnění. Nejčastějšími chybnými diagnózami jsou chronická zánětlivá demyelinizační polyneuropatie (CIDP), diabetická či alkoholická neuropatie, případně vertebrogenní potíže, například lumbální stenóza.
Jak této chybě předcházet? Jsou nějaké projevy, které napomáhají časné diagnostice?
Prevencí chybných diagnóz je především dostatečné povědomí lékařů o hATTR a jejích projevech a aktivní časné testování pacientů na toto onemocnění. Pokud je onemocnění zahrnuto v diferenciální diagnostice a aktivně se sledují red flags, významně se snižuje riziko záměny a zkracuje se doba do stanovení správné diagnózy.
Kdy poslat pacienta na genetické vyšetření a jaké informace toto vyšetření poskytuje?
Genetické vyšetření je vhodné u pacientů s progredující axonální polyneuropatií, neuropatií tenkých vláken nebo atypickou CIDP nereagující na standardní léčbu, pokud se vyskytují red flags. Testování lze provést přes neuromuskulární centra nebo genetické laboratoře.
Výsledek odhalí přítomnost a závažnost mutace v genu pro TTR. Při absenci nebo benigní variantě je hATTR nepravděpodobná. Pokud se prokáže patogenní mutace, je nutné potvrdit přítomnost amyloidu – buď biopsií (například tuk, slinná žláza, n. suralis, srdce), nebo pomocí radionuklidových metod, jako je kostní scintigrafie (DPD sken). Tyto metody slouží jako alternativa zejména u kardiálního postižení.
Jaké terapeutické možnosti jsou aktuálně k dispozici?
Léčba ATTR se dělí na kauzální, která ovlivňuje průběh onemocnění, a symptomatickou. Historicky byla jedinou možností transplantace jater, protože játra produkují většinu transthyretinu. Tento přístup však limituje dostupnost dárců a nutnost imunosuprese.
Zásadní průlom přinesly nové účinné látky – silencery TTR (vutrisiran, patisiran, inotersen, eplontersen), které tlumí produkci patologického transthyretinu, a stabilizátory TTR (tafamidis, diflunisal), jež brání jeho rozpadu. Účinnost léčby je tím vyšší, čím dříve je zahájena. Zmíněné léky jsou určeny pro neuropatickou i kardiální formu hATTR, ne všechny jsou však schváleny pro obě tyto formy současně.
Nezbytnou součástí terapie je ovšem i symptomatická léčba (zaměřená například na bolest či autonomní dysfunkci). Péče o pacienty by měla být zajištěna prostřednictvím multidisciplinárního týmu zahrnujícího vedle specialistů jednotlivých oborů (kardiolog, neurolog, nefrolog, oftalmolog) také genetika, psychologa a fyzioterapeuta.
Jak účinná je moderní terapie hATTR? A kdy volit transplantaci jater?
Moderní léky prokázaly vysokou účinnost v klinických studiích. Stabilizátory TTR, zejména tafamidis, zpomalují progresi neuropatie a významně snižují mortalitu i nutnost hospitalizace u pacientů s kardiomyopatií – jak při hereditární, tak wild-type ATTR. Silencery TTR nejen zastavují, ale často dokonce zmírňují neurologické postižení a zvyšují kvalitu života. Vutrisiran je nově schválen rovněž pro léčbu kardiomyopatie u obou forem ATTR a také v této indikaci prokázal v provedených studiích velmi dobrou účinnost.
Význam transplantace jater výrazně poklesl a zůstává metodou volby pouze tehdy, když nejsou moderní léky dostupné. Její účinnost závisí na typu mutace a je nevhodná v případě kardiálního postižení.
Jaká je s dnešními možnostmi perspektiva pacienta z hlediska kvality a délky života?
Díky moderní léčbě se pohled na hATTR zásadně změnil – místo fatální diagnózy jej dnes lze považovat za chronické onemocnění, jehož průběh lze výrazně ovlivnit, pokud je zachyceno včas. Nové léky mají potenciál stabilizovat nebo i zlepšit stav pacientů, čímž zvyšují kvalitu jejich života a prodlužují přežití. Ve studiích přetrvávala stabilizace nebo regrese polyneuropatie až 7 let u 65 % pacientů, v publikacích dat z reálné praxe (s mírně kratší dobou sledování) dokonce až u 80 %.
Takový průběh by byl v případě přirozeného vývoje onemocnění zcela nemyslitelný a pro nemocné s ATTR jde skutečně o revoluční změnu jejich perspektiv. Odhadovaná střední délka života se dnes u časně léčených pacientů může pohybovat i více než 15 let od stanovení diagnózy, zatímco před nástupem moderních přístupů to bylo v neendemických oblastech kolem 7 let. V současnosti je dokonce pravděpodobné, že přežití pacientů s takto závažným onemocněním může být srovnatelné s běžnou populací, zvláště při časném nasazení účinné léčby.
MUDr. Andrea Skálová
redakce proLékaře.cz