
Regulates Synaptic Development and Endocytosis by Suppressing Filamentous Actin Assembly
The formation of synapses and the proper construction of neural circuits depend on signaling pathways that regulate cytoskeletal structure and dynamics. After the mutual recognition of a growing axon and its target, multiple signaling pathways are activated that regulate cytoskeletal dynamics to determine the morphology and strength of the connection. By analyzing Drosophila mutations in the cytoplasmic FMRP interacting protein Cyfip, we demonstrate that this component of the WAVE complex inhibits the assembly of filamentous actin (F-actin) and thereby regulates key aspects of synaptogenesis. Cyfip regulates the distribution of F-actin filaments in presynaptic neuromuscular junction (NMJ) terminals. At cyfip mutant NMJs, F-actin assembly was accelerated, resulting in shorter NMJs, more numerous satellite boutons, and reduced quantal content. Increased synaptic vesicle size and failure to maintain excitatory junctional potential amplitudes under high-frequency stimulation in cyfip mutants indicated an endocytic defect. cyfip mutants exhibited upregulated bone morphogenetic protein (BMP) signaling, a major growth-promoting pathway known to be attenuated by endocytosis at the Drosophila NMJ. We propose that Cyfip regulates synapse development and endocytosis by inhibiting actin assembly.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003450
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003450
Summary
The formation of synapses and the proper construction of neural circuits depend on signaling pathways that regulate cytoskeletal structure and dynamics. After the mutual recognition of a growing axon and its target, multiple signaling pathways are activated that regulate cytoskeletal dynamics to determine the morphology and strength of the connection. By analyzing Drosophila mutations in the cytoplasmic FMRP interacting protein Cyfip, we demonstrate that this component of the WAVE complex inhibits the assembly of filamentous actin (F-actin) and thereby regulates key aspects of synaptogenesis. Cyfip regulates the distribution of F-actin filaments in presynaptic neuromuscular junction (NMJ) terminals. At cyfip mutant NMJs, F-actin assembly was accelerated, resulting in shorter NMJs, more numerous satellite boutons, and reduced quantal content. Increased synaptic vesicle size and failure to maintain excitatory junctional potential amplitudes under high-frequency stimulation in cyfip mutants indicated an endocytic defect. cyfip mutants exhibited upregulated bone morphogenetic protein (BMP) signaling, a major growth-promoting pathway known to be attenuated by endocytosis at the Drosophila NMJ. We propose that Cyfip regulates synapse development and endocytosis by inhibiting actin assembly.
Introduction
To establish functional neural circuits, synapses must form at specific locations and grow to an appropriate size and strength. A multitude of signaling pathways are required to achieve and maintain these precise patterns of synaptic connectivity [1]–[3]. Many of these signals regulate local actin cytoskeletal networks, which are crucial for both synapse formation and plasticity [4]–[6]. Precisely how the actin cytoskeleton integrates various signaling pathways to regulate synaptic formation and function remains to be elucidated.
At Drosophila neuromuscular junctions (NMJs), dysregulation of actin dynamics results in morphological defects, including the formation of excess satellite boutons. For example, mutants of the actin regulator nervous wreck (Nwk), an N-WASP (neuronal Wiskott–Aldrich syndrome protein) interacting protein, show excess satellite boutons at NMJs [7]. Nervous wreck activates WASP-Arp2/3-mediated actin polymerization and coordinates with Cdc42 to regulate actin assembly [8]. Additional actin regulatory proteins implicated in synapse formation include WASP, spectrin, and adducin [6], [9], [10]. Moreover, these proteins and their interactors are conserved across species, indicating a seminal role for the actin cytoskeleton in synaptic development.
In addition to regulating synaptic development, multiple lines of evidence show that actin and its regulators function in synaptic endocytosis. First, filamentous actin (F-actin) is observed around synaptic vesicle clusters where it facilitates vesicle endocytosis or mobility [11], [12]. Second, many actin regulators bind endocytic proteins directly or indirectly. For example, Cdc42, WASP, and Nwk all interact directly with the endocytic protein intersectin-1/Dap160, an important binding partner of dynamin [7], [8], [13]. Third, disruption of the actin cytoskeleton impairs vesicle recycling at both vertebrate and invertebrate synapses [12], [14]. Fourth, actin regulator mutants such as twinfilin, dap160/intersectin, and nwk show defects in synaptic endocytosis [7], [15]–[17]. In addition to endocytosis of synaptic vesicle membrane, receptors must be retrieved from the presynaptic membrane to downregulate specific signaling pathways. At the Drosophila NMJ for example, actin-mediated endocytosis downregulates the bone morphogenetic protein (BMP) signaling pathway that normally promotes synaptic growth [1], [7], [8], suggesting that actin cytoskeleton may contribute to synaptic development by regulating endocytosis.
The heteropentameric WAVE complex, composed of WAVE (WASP/verprolin homologous protein), Cyfip/Sra-1/Pir121, Kette/Nap1, Abi, and HSPC300 [18]–[21], relays signals from the Rac GTPase to the actin nucleator Arp2/3 complex to control de novo F-actin assembly. The organization of the WAVE complex is well established in vitro. Specifically, Abi and Nap1 form the core sub-complex and Cyfip binds Nap1, while both WAVE and HSPC300 bind the N-terminus of Abi [20], [21]. In the resting state, the verprolin-homology, central, and acidic (VCA) domain of the WAVE protein is sequestered by binding to Cyfip and/or Nap1 [18], [21]. Upon Rac1 binding to the N-terminus of Cyfip, together with other coincident signals, the VCA domain is released from the WAVE complex to activate the actin nucleator Arp2/3 [18], [21]–[23]. However, this transduction mechanism has only been demonstrated in vitro, while the exact role of each component in regulating the activity of the WAVE complex in vivo is poorly understood.
We provide evidence that loss of Cyfip leads to enhanced F-actin assembly in Drosophila, resulting in altered NMJ morphology. We also report that Cyfip loss disrupts synaptic endocytosis, likely by regulating presynaptic F-actin networks. The bone morphogenetic protein (BMP) signaling attenuated by endocytosis is upregulated in cyfip mutants, consistent with an endocytic defect. Reducing the level of SCAR, the Drosophila homolog of WAVE, partially rescues the morphological anomalies, endocytic defects, and enhanced actin dynamics at cyfip mutant NMJs. Thus, our findings demonstrate that Cyfip regulates synapse formation and endocytosis by inhibiting actin dynamics.
Results
Cyfip regulates NMJ development
Drosophila cyfip85.1 null mutants were early pupal lethal, consistent with a previous report [24]. Tissue-specific expression of Cyfip in muscles driven by C57-Gal4 or in neurons driven by elav-Gal4 allowed survival of cyfip85.1 nulls to late pupal stages, but no adults emerged (Table 1). In contrast, 85.63% of homozygous cyfip85.1 nulls ubiquitously expressing Cyfip under the control of act-Gal4 survived to adulthood. Thus, the lethality of cyfip nulls was specifically caused by loss of Cyfip.

The muscle 4 NMJ of wild-type (w1118) wandering third instar larvae shows a stereotyped “beads-on-a-chain” pattern of boutons (Figure 1A). In contrast to the wild-type controls, NMJ boutons in cyfip85.1 null homozygotes appeared clustered together with little or no spacing between them (Figure 1B). The total NMJ length was reduced by 50% in cyfip85.1 mutants relative to wild type (p<0.001; Figure 1A, 1B, and 1F), in agreement with a previous report [24]. cyfip hemizygous mutants in which cyfip85.1 was on one chromosome, while Df(3R)Exel6174, which uncovers cyfip, was on the other also showed reduced NMJ length (p<0.001; Figure 1F). Muscle expression of Cyfip partially rescued, while neuronal or ubiquitous expression of Cyfip fully rescued the short NMJ phenotype of cyfip85.1 mutants (Figure 1F).

Small ectopic boutons that bud from synaptic branches or primary boutons are normally rare and referred to as satellite boutons [7], [25], [26]. At cyfip85.1 homozygous mutant NMJ4, there was a 15-fold increase in the number of these satellite boutons (p<0.001; Figure 1A, 1B, and 1G), described previously as “supernumerary buds” [24]. cyfip hemizygotes (cyfip85.1/Df(3R)Exel6174) showed a similar increase in the number of satellite boutons (p<0.001; Figure 1G). Neuronal or muscular expression of Cyfip partially rescued the phenotype, as there was still a 3 - or 4-fold increase in the number of satellite boutons compared to wild-type controls (Figure 1C, 1D, and 1G), whereas ubiquitous expression of cyfip fully rescued the excess satellite bouton of cyfip85.1 nulls (Figure 1E and 1G). The satellite boutons form primarily in the late larval stages of cyfip mutants (Figure S1). These phenotypes were highly penetrant, as other NMJ terminals such as NMJ6/7 were also shorter and exhibited excess satellite boutons.
We then quantified the number of superboutons, defined as parental primary boutons from which at least three satellite boutons formed. The average number of superboutons was significantly higher in cyfip85.1 homozygous mutants (2.9±0.27 per NMJ4) compared to wild type (0.35±0.13) (Figure 1H; p<0.001). Additionally, nearly 30% of superboutons in mutants possessed five or more satellite boutons (Figure 1H). Again, ubiquitous expression of Cyfip fully rescued, while neuronal or muscular expression partially rescued the superbouton phenotype of cyfip85.1 mutants (Figure 1C–1E and 1H). These results demonstrate that cyfip regulates synapse formation from both pre - and postsynaptic sides.
cyfip mutants display enlarged synaptic vesicles at active zones and more presynaptic cisternae
To understand the synaptic defects in cyfip mutants at the ultrastructural level, we examined NMJ synapses in wandering third instar larvae by electron microscopy. Many features, such as the number and structure of mitochondria, the number and morphology of active zones, and vesicle density in the whole bouton appeared largely normal in cyfip85.1 mutants compared to wild-type controls (Figure 2A and 2B). The mean number of synaptic vesicles (SVs) within a 200 nm radius of the active zone was also normal (n≥20; p>0.05), but the mean SV diameter was 19.8% larger in cyfip85.1 mutants (p<0.001; Figure 2C, 2D, and 2G). Ubiquitous expression of Cyfip by act-Gal4 partially but significantly rescued the increased SV diameter in cyfip85.1 mutants (Figure 2E and 2G). Histograms and cumulative probability plots of the SV size distribution showed that 89% of wild-type SVs and 80% of SVs in rescued animals were <40 nm, whereas only 65% of cyfip mutant SVs were <40 nm in diameter (Figure 2H and 2I). In addition, the number of cisternae (presumably endosomes) >60 nm in diameter per bouton cross-section was dramatically higher in mutants (arrow in Figure 2B). Enlarged SVs and more cisternae have been reported in mutants with deficient endocytosis, including AP180/lap, dap160, eps15, tweek, and flower mutants [15], [27]–[30], suggesting that cyfip may regulate endocytosis and/or vesicle recycling.

Increased quantal size and decreased quantal content at cyfip mutant NMJs
To determine if synaptic transmission was impaired in cyfip mutants, we stimulated the motor neurons and recorded excitatory junctional potentials (EJPs) from NMJ6/7 in abdominal segments A2 or A3 of wandering third instar larvae. We first recorded evoked EJPs at 0.3 Hz in a modified HL3 saline containing 0.23 mM Ca2+. At this Ca2+ concentration, the EJP amplitudes of wild-type controls and cyfip85.1 mutants were similar (Figure 3A–3C).

We then analyzed quantal size by recording the amplitudes of miniature EJPs (mEJPs). Mean mEJP amplitudes were 40% larger in both homozygous and hemizygous cyfip85.1 mutants compared to wild type (p<0.001; Figure 3A, 3B, and 3E), whereas the mEJP frequency in cyfip85.1 mutants was not altered (Figure 3A, 3B, and 3D). The increased quantal size in cyfip85.1 mutants was fully rescued by presynaptic expression of cyfip driven by elav-Gal4 (Figure 3E). Histogram and cumulative probability plot of mEJP amplitudes revealed that large-amplitude mEJPs occurred more often in cyfip85.1 mutants than in wild type. Specifically, mEJP amplitudes larger than 4 mV occurred in cyfip85.1 mutants but were never seen in wild type (Figure 3G–3I). Quantal size is closely correlated with vesicle size, a likely determinant of transmitter content per vesicle [15], [30]. As there were no changes in the expression of postsynaptic glutamate receptors detected by immunostaining (data not shown), the increased quantal size suggests that the enlarged SVs in cyfip mutants release more neurotransmitter.
The normal EJP amplitude and increased quantal size in cyfip mutants indicate that the number of vesicles released per stimulus (quantal content) was reduced by 35% compared to wild type (p<0.05; Figure 3F). The reduced quantal content in cyfip85.1 mutants was rescued to the wild-type level by presynaptic expression of Cyfip controlled by elav-Gal4 (Figure 3F). The reduced quantal content may arise from a homeostatic control mechanism that compensates for the abnormally enlarged vesicles to maintain a constant synaptic output.
Cyfip is required for SV recycling
The increased number of satellite boutons, the enlarged SVs, and the increased mEJP amplitudes (Figure 1, Figure 2, Figure 3) in cyfip85.1 mutants all indicate a possible role for Cyfip in endocytosis. If so, then cyfip85.1 mutant synapses may be more prone to EJP rundown during high-frequency stimulation. During a 10 min train of 10 Hz stimulation in modified HL3 saline containing high Ca2+ (10 mM), wild-type synapses exhibited a slow EJP decline to 65.4±6.6% of the initial amplitude (Figure 4A). In homozygous cyfip85.1 nulls, however, EJPs evoked by the same 10 Hz stimulus train exhibited a much faster decline within the first 120 s and decreased to 24.8±2.4% of the initial amplitude by the end of stimulation (Figure 4A). Hemizygous cyfip85.1/Df(3R)Exel6174 mutants also displayed an enhanced rundown in EJP amplitude similar to that of homozygous mutants. Furthermore, neuronal expression of cyfip driven by elav-Gal4 largely reversed the faster decline in EJP amplitude observed in homozygous cyfip85.1 null mutants, indicating that this phenotype is specifically caused by cyfip loss-of-function. The enhanced rundown of EJPs shows that cyfip participates in the replenishment of SVs.

If endocytosis and concomitant membrane recycling were compromised, then the NMJ vesicle pool would not recover as quickly as wild type to the prestimulus level after depletion [31]. We stimulated the nerve at 0.3 Hz for 10 min following the 10 min tetanic stimulation to measure recovery from synaptic depletion. Indeed, EJP amplitudes in wild-type larvae recovered within 60 s to reach 94.2±7.7% of the prestimulus EJP amplitude during the low-frequency (0.3 Hz) stimulus train (Figure 4B and 4C), while EJP amplitude at cyfip85.1 NMJs reached only 65.9±4.8% of the prestimulus level after 10 min of 0.3 Hz stimulation (Figure 4B and 4C). The enhanced rundown and slower recovery of EJP amplitudes in cyfip85.1 mutant NMJs strongly suggest that Cyfip is required for normal vesicle recycling.
Excess satellite boutons in cyfip mutants result from upregulated BMP signaling
The excess satellite boutons in cyfip mutants likely result from dysregulation of signaling pathways that modulate NMJ growth. Accumulating evidence demonstrates that retrograde BMP signaling is the primary growth-promoting pathway at developing Drosophila NMJs [1], [7]. Given that the supernumerary satellite bouton phenotype of cyfip85.1 mutants resembled that observed in animals with upregulated BMP signaling [7], we examined if BMP signaling was over-activated in cyfip mutants.
Activation of BMP receptors upon ligand binding leads to the phosphorylation of the receptor-regulated R-Smad mothers against Dpp (Mad), which in turn activates the transcription of downstream genes. Therefore, the level of phosphorylated Mad (pMad) serves as a readout of BMP signaling at synaptic terminals. To test if BMP signaling was increased in cyfip mutants, we first quantified the pMad level at NMJ synapses by fluorescence immunohistochemistry and found that it was indeed significantly higher in cyfip85.1 homozygous and hemizygous mutants than in wild type (p<0.001; Figure 5A–5C). We then examined the genetic interaction between cyfip and the components of the BMP signaling pathway. Mutating one copy of mad or tkv, which encodes the type I BMP receptor thickveins, significantly suppressed satellite bouton formation in cyfip85.1 mutants (p<0.05 for both madk00237/+; cyfip85.1 and tkv7/+; cyfip85.1 lines compared to cyfip85.1 mutants; Figure 5D–5F and 5H). On the other hand, trans-heterozygotes of cyfip85.1 and dadJ1E4 showed significantly more satellite boutons compared to wild type, while cyfip85.1 or dadJ1E4 heterozygotes showed normal NMJ morphology (Figure 5G and 5H). dad encodes an inhibitory Smad that negatively regulates BMP signaling at the Drosophila NMJ synapses [32]. Together, these results indicate that the excess satellite bouton formation in cyfip mutants is caused by upregulated BMP signaling.

Disruption of the presynaptic F-actin cytoskeleton in cyfip mutants
Cyfip is a component of the heteropentameric WAVE complex that relays Rac1 signaling to the Arp2/3 nucleating complex to promote de novo actin polymerization. We therefore asked whether the synaptic phenotypes described above might result from a defect in the actin cytoskeleton. As the endogenous presynaptic actin cytoskeleton cannot be visualized by immunostaining, we expressed the GFP-moe reporter in motoneurons using elav-Gal4. A transgenic line expressing GFP fused to the C-terminal actin-binding domain of moesin (GFP-moe) has been used widely as an actin reporter [6], [14], [33], [34]. As shown in Figure 6A, when overexpressed in presynaptic neurons, GFP-moe concentrated into discrete puncta that distributed throughout wild-type NMJ terminals. These patches consist of F-actin, as they are stained by phalloidin and diffused after treatment with the F-actin disrupting drug latrunculin A [9], [14].
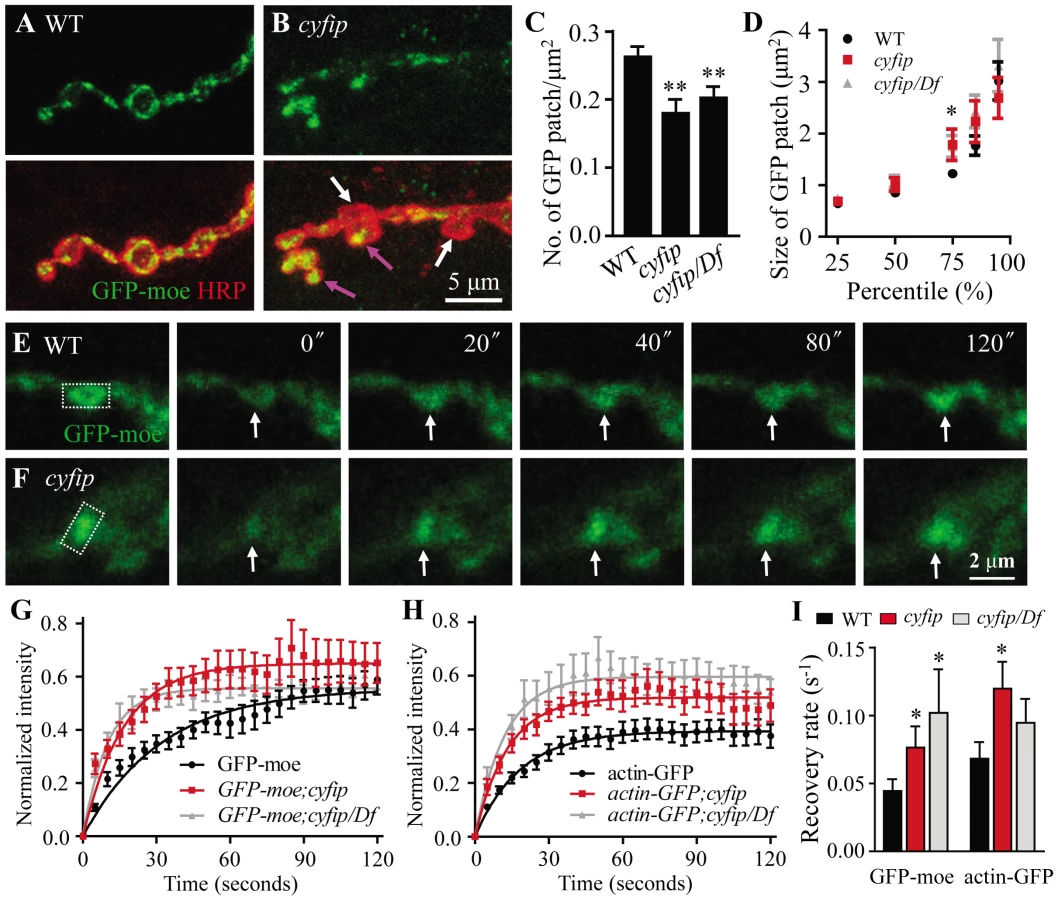
In cyfip85.1 mutants, however, the presynaptic distribution of GFP-moe differed markedly between boutons; in some boutons, GFP-moe patches were large and spread throughout the whole bouton (pink arrow in Figure 6B), whereas in other boutons, no GFP-moe patches were observed (white arrow in Figure 6B). Quantitative analysis showed that the number of GFP-moe patches normalized to the synaptic area was significantly reduced in cyfip mutants (p<0.01; Figure 6C). The size distribution of F-actin patches was comparable between cyfip mutants and the control except that the patch size at the 75th percentile was significantly increased from 1.22±0.08 µm2 in wild type to 1.78±0.30 µm2 and 1.74±0.21 µm2 in cyfip85.1 homozygous and hemizygous mutants, respectively (p<0.05; Figure 6D). The uneven distribution of GFP-moe patches in different boutons of cyfip85.1 mutants was also observed when an actin-GFP transgene was used as an actin marker (data not shown), confirming the abnormal presynaptic F-actin distribution in cyfip mutant NMJ synapses.
Increased F-actin formation at the NMJ terminals of cyfip mutants
To evaluate F-actin dynamics, we performed fluorescent recovery after photobleaching (FRAP) assay in living synapses of GFP-moe labeled F-actin. GFP-moe was ectopically expressed in motoneurons driven by elav-Gal4. The region of interest (ROI, rectangles in Figure 6E and 6F) labeled with strong GFP signals at NMJ4 was used for photobleaching. A significant difference in the recovery of GFP intensity was observed within the first 20 s after photobleaching. The recovery from photobleaching was accelerated in mutants; fluorescence intensity within the ROI at 20 s returned to only 35.1±3.8% of the pre-bleached level in wild type (Figure 6E and 6G) but to 43.1±4.6% (cyfip) and 46.6±6.5% (cyfip/Df) in mutants (p<0.05; Figure 6F and 6G). The GFP intensity recovery rate was increased from 0.044±0.0087 s−1 in the wild-type controls to 0.076±0.015 s−1 in cyfip85.1 homozygotes and 0.10±0.048 s−1 in hemizygotes, respectively (p<0.05; Figure 6I). To confirm that this fast recovery in cyfip mutants is caused by the dynamics of endogenous actin, we repeated the analysis using actin-GFP, an independent actin reporter. Again, a faster fluorescence recovery was observed in cyfip85.1 homozygous mutants (p<0.05; Figure 6I). Collectively, the increased GFP fluorescence recovery indicates accelerated F-actin dynamics in cyfip mutants.
New F-actin assembly is enhanced in cyfip mutants
To verify the accelerated actin dynamics in cyfip mutants, we applied a method developed to measure new F-actin assembly [35]. Endogenous F-actin at the presynaptic NMJ terminals is undetectable, but postsynaptic F-actin can be clearly visualized by phalloidin staining. The membrane-permeable drug jasplakinolide binds to and stabilizes pre-existing F-actin and prevents F-actin binding to phalloidin [36]. Thus, following jasplakinolide treatment, phalloidin will bind to only newly assembled F-actin. The distribution pattern and intensity of postsynaptic F-actin were similar in wild type and cyfip85.1 mutants when treated with the vehicle DMSO (Figure 7A and 7D). After incubation with jasplakinolide for 15 min, postsynaptic F-actin was almost undetectable by phalloidin staining in both genotypes (Figure 7B and 7E). After 1 h recovery in jasplakinolide-free medium, phalloidin staining (selective for newly formed F-actin) was significantly higher in cyfip85.1 mutants than in wild type (Figure 7C and 7F). We quantified newly formed F-actin by the fluorescence recovery index, defined as the ratio of phalloidin staining intensity at 1 h after jasplakinolide washout over the phalloidin staining intensity after vehicle-treatment. The recovery index indicated a significant increase in F-actin assembly in cyfip85.1 homozygous and hemizygous mutants (p<0.01), which was rescued by ubiquitous expression of Cyfip driven by act-Gal4 (Figure 7G). After 90 min recovery from jasplakinolide treatment, however, the F-actin intensity in homozygous cyfip85.1 mutant NMJ terminals was comparable to the wild-type control (n≥18; p>0.05), suggesting that the increased F-actin formation in cyfip mutants is transient.

To further evaluate the role for Cyfip in new F-actin formation, we analyzed cyfip mosaic eye discs after drug treatment as described in a recent paper [37]. Development of Drosophila eye begins in the morphogenetic furrow (MF), where new ommatidia are formed as cells aggregate in periodic groupings. The new rows of ommatidial clusters are added anterior to older ones; consequently, the MF sweeps across the eye disc in a posterior to anterior direction [38]. In the region posterior to the MF, F-actin concentrates in the center of ommatidial clusters which will develop into rhabdomeres (Figure 7H). When the eye discs were treated with the vehicle DMSO, we observed similar intensity and distribution pattern of F-actin in cyfip mutant clones compared with the adjacent wild-type tissue (Figure 7H), consistent with the results observed at the postsynapse (Figure 7A and 7D). After incubation of jasplakinolide for 10 min, phalloidin staining detected no signal in the whole disc as the existing F-actin was totally masked by jasplakinolide binding (Figure 7I). Following 90 min recovery in jasplakinolide-free medium, F-actin started to form in the region posterior to the MF (Figure 7K). In cyfip85.1 mutant clones, we found enhanced and advanced F-actin recovery in a subpopulation of cyfip mutant cells, presumably undergoing active morphogenesis, compared to the nearby wild-type tissue (arrows in Figure 7J). As the recovery continues, additional phalloidin labeling could be detected in the more posterior ommatidial clusters. These results together show elevated F-actin formation within the examined time window in cyfip mutants.
Cyfip functionally antagonizes SCAR
Cyfip is a component of the WAVE complex. In vitro biochemical studies show that in the resting state, the WAVE complex is inactive. Upon binding of Rac1 to the N-terminus of Cyfip, the VCA domain of WAVE protein is released from the complex to activate F-actin assembly through the actin nucleator Arp2/3 [18], [21]–[23]. However, it is unknown if Cyfip regulates WAVE activity and subsequent F-actin assembly in vivo through the same mechanism.
Enhanced F-actin assembly (Figure 6 and Figure 7) suggests an increased activity of SCAR, the Drosophila homolog of WAVE, in cyfip mutants. We demonstrated that this was indeed the case by examining NMJ morphology and synaptic transmission in cyfip85.1 null mutants with a copy of SCAR mutated. Heterozygous SCARΔ37 (a null allele) or SCARk13811 (a hypomorphic allele) mutations reversed the increased number of satellite boutons in cyfip85.1 mutants (p<0.001 for SCARΔ37/+; cyfip85.1 and p<0.01 for SCARk138117/+; cyfip85.1 compared to cyfip85.1 mutants; Figure 8A–8D). Similarly, the enhanced rundown of EJP amplitude under tetanic stimulation was also partially restored from 24.8±2.4% to 40.3±2.9% of the initial EJP amplitude when a copy of SCAR was mutated in the cyfip null background (SCARΔ37/+; cyfip85.1) (Figure 8E). Reducing the dose of SCAR also rescued the accelerated F-actin assembly at the postsynaptic site of cyfip85.1 mutants. The fluorescence recovery index was 112.52±3.49% in SCARΔ37/+; cyfip85.1 mutants, significantly lower than 128.72±7.93% in cyfip85.1 mutants (p<0.05; Figure 8F). As a control, heterozygous SCARΔ37 or SCARk13811 mutants showed no obvious defects in synaptic morphology, endocytosis or F-actin assembly (Figure 8D–8F). Together, these data indicate that the synaptic defects and accelerated F-actin assembly in cyfip85.1 mutants result at least partially from increased SCAR activity.

Discussion
Supernumerary buds, i.e., excess satellite boutons at NMJs are observed in both cyfip null mutants and animals with cyfip knocked down specifically in presynaptic neurons by RNA interference [24], [39]. However, little is known of how Cyfip regulates synapse development. Here, we provide experimental evidence for a model in which Cyfip regulates the development and endocytosis of NMJ synapses by inhibiting F-actin assembly.
Cyfip suppresses F-actin assembly by antagonizing WAVE
The Arp2/3 complex, which is activated by the WASP family proteins including WASP and WAVE, mediates nucleation of de novo F-actin assembly. WASP is auto-inhibited, whereas the activity of the WAVE protein is inhibited in the heteropentameric WAVE complex. Two models have been proposed to explain WAVE complex activation in vitro. One model proposes that Rac1 binding to Cyfip causes dissociation of the WAVE complex, releasing the active WAVE-containing subcomplex and resulting in actin nucleation by Arp2/3 [19]. The other model hypothesizes that upon binding of Rac1 to Cyfip, the WAVE complex is activated through an allosteric change rather than by dissociation of the complex to expose the VCA domain [21], [23]. However, the mechanisms regulating the activity of the WAVE complex in vivo remain unclear.
Like its mammalian counterpart, Drosophila Cyfip is a component of the heteropentameric WAVE complex as evidenced by co-immunoprecipitation assays and the strong mutual dependence on the protein levels of the individual components [40], [41], which we confirmed (Figure S2). If the integrity of the WAVE complex is necessary for its activity, then loss of Cyfip would disrupt the complex, leading to reduced Arp2/3 activity and slower F-actin assembly. However, we showed that loss of Cyfip accelerated F-actin formation in vivo. First, FRAP analysis at NMJ synapses revealed a faster recovery of GFP-moe fluorescence in cyfip mutants than in wild type (Figure 6). Second, new F-actin assembly was accelerated at NMJ terminals and eye discs of cyfip mutants (Figure 7). Third, genetic analysis showed that mutating a copy of SCAR reversed the cyfip mutant phenotypes including excessive satellite bouton formation and accelerated F-actin assembly (Figure 8). The antagonistic interaction between cyfip and SCAR resembles that between SCAR and kette, the Drosophila homolog of Nap1. Drosophila kette mutants show fused commissures in the embryonic nervous system that are rescued by reducing the dose of SCAR [42]. Together, in vivo studies from independent assays reveal an upregulation of SCAR activity when the WAVE/SCAR complex is disrupted in cyfip mutants. We envision that in the absence of Cyfip, the WAVE complex is dissociated, leaving the VCA domain exposed to activate Arp2/3 and promote F-actin assembly.
Klambt and colleagues reported a role for Cyfip in F-actin formation [39]. When cyfip is knocked down by RNAi in S2 cells, there is an accumulation of cytosolic F-actin; when the expression of Cyfip is up - or downregulated, F-actin formation during bristle development is altered; they concluded that Cyfip affects F-actin formation though the specific mechanism was unclear [39]. In agreement with their findings, our results from phalloidin staining of steady-state samples did not show consistent alterations in the level of F-actin in cyfip mutants (i.e. normal F-actin at the postsynapse, abnormal ring canal and radial F-actin fibers in nurse cells, but absence of F-actin in the cortex of nurse cells; Figure 7 and Figure S3), suggesting that Cyfip-regulated F-actin formation is dependent on cellular contexts. However, our analyses of live imaging and pharmacological treatment independently showed increased new F-actin assembly, though the steady-state F-actin level was normal at NMJ terminals and eye discs of cyfip mutants (Figure 6 and Figure 7), suggesting that the increased F-actin assembly in cyfip mutants could be temporary. We suppose that a complex regulatory network is at work to maintain the dynamics of F-actin. Lack of cyfip impairs the “brake” and results in inappropriate or ectopic F-actin polymerization. However, enhanced formation of F-actin in cyfip mutants could be quickly mitigated by proteasomal degradation of SCAR ([40] and Figure S2), activation of depolymerizing factors, or both.
In addition to being a component of the WAVE complex that regulates actin dynamics, Cyfip also interacts with the translational regulator FMRP and the translation initiation factor eIF4E [24], [43]. It is possible that Cyfip interacts with different partners to mediate different cellular processes. We note that while a synaptic role of eIF4E has not been demonstrated, cyfip and dfmr1 mutants show distinct NMJ phenotypes [24], [44]. How Cyfip coordinates with these different partners to regulate synapse formation and function remains to be elucidated.
Cyfip plays a regulatory role in synaptic endocytosis
Multiple lines of evidence suggest a role for Cyfip in endocytosis at Drosophila NMJ synapses. First, cyfip mutant NMJ synapses exhibited prominent satellite boutons, a phenotype well documented in several endocytic mutants [15], [16], [26], [27], [29]. Second, genetic interaction analysis showed a synergistic effect between cyfip and endocytic genes such as dynamin, dap160, and endophilin in the control of satellite bouton formation (Figure S4). Third, EM analysis revealed enlarged SVs at active zones and a significant increase in the number of cisternae in cyfip mutant NMJ boutons (Figure 2). Consistent with enlarged SVs, there was a higher frequency of large mEJPs in cyfip mutants (Figure 3). Last, cyfip mutant synapses could not sustain neurotransmission during high-frequency stimulation and displayed a slower recovery of neurotransmission following tetanic stimulation (Figure 4). We also performed FM1–43 dye loading assay and found normal dye uptake at cyfip mutant NMJs (data not shown). FM dye loading is commonly used for examining endocytosis. However, there are reports documenting that endocytic mutants showed normal FM dye uptake. For example, clathrin is a critical component of endocytic machinery, but acute inactivation of clathrin showed normal FM loading [45]. These results together support a role for Cyfip in synaptic endocytosis.
The actin cytoskeleton has been implicated in multiple steps of the endocytic pathway, from membrane invagination to vesicle fission and subsequent trafficking [46]–[48]. At which step of endocytosis might Cyfip act? Synaptic vesicles are generated either directly from plasma membrane through clathrin-mediated endocytosis or from endosomal compartments derived from bulk endocytosis during intense stimulation. Electron microscopic analysis revealed normal SV density in cyfip mutants, indicating that membrane retrieval capacity is not altered by loss of Cyfip (Figure 2). Consistently, the FM1–43 dye uptake assay showed largely normal endocytosis and vesicle pool in cyfip mutants (data not shown). However, cyfip mutant NMJ boutons show increased vesicle size, which is tightly controlled by clathrin and its adaptor proteins during endocytosis [30], [45]. It is well documented that endocytic mutants of Dap160/intersectin and Eps15 exhibit enlarged vesicle size and these endocytic proteins interact with actin regulators Nwk and WASP [7], [15], [16], [27]. Thus, Cyfip-regulated actin cytoskeleton may affect SV size through endocytosis directly or indirectly.
Cyfip inhibits satellite bouton formation
We report here that Cyfip normally suppresses F-actin assembly to restrain satellite bouton formation. We showed that F-actin distributed unevenly among different boutons of cyfip mutants (Figure 6). The F-actin cytoskeleton was also disrupted in egg chambers (Figure S3). Furthermore, we found upregulated F-actin assembly in cyfip mutants, which might cause the mislocalization of F-actin filaments in neuronal and non-neuronal cells (Figure 6, Figure 7, and Figure S3). Both the enhanced actin dynamics and aberrant NMJ morphology in cyfip mutants were rescued by reducing the dose of SCAR. Based on these findings, we propose that Cyfip normally restrains satellite bouton formation by suppressing F-actin assembly through the SCAR-Arp2/3 pathway.
It is well established that an actin-dependent endocytic mechanism contributes to the formation of satellite boutons. Loss of Nwk, an SH3 adaptor protein that interacts with Cdc42 and WASP, also impairs endocytic attenuation of BMP signaling and results in excess satellite bouton formation [7], [8], but SV endocytosis was normal in nwk mutants as evidenced by FM1–43 dye uptake and EJP recordings under high-frequency stimulation [25]. Here, we report that, like nwk mutants, the excess satellite boutons in cyfip mutants are also due to upregulated BMP signaling (Figure 5). In contrast to nwk null mutants, however, cyfip mutants showed defective endocytosis as revealed by EM and electrophysiological analysis (Figure 2, Figure 3, Figure 4). Thus, nwk and cyfip may regulate synaptic endocytosis through distinct actin-mediated pathways; nwk primarily affects endocytic regulation of BMP signaling, while cyfip regulates endocytosis of both SVs and BMP receptors. In any case, mutations in both genes lead to formation of excess satellite boutons as a result of dysregulated actin dynamics.
Materials and Methods
Drosophila strains and genetics
Fly cultures were raised on conventional cornmeal medium and maintained at 25°C unless specified. w1118 flies were used as the wild-type controls in all experiments. cyfip85.1 and UAS-cyfip were gifts of Dr. A. Giangrande [24]. Df(3R)Exel6174 (88F1–88F7) covering the cyfip locus (88F1) was obtained from the Bloomington Stock Center. The BMP pathway mutants madk00237, tkv7, and dadJ1E4, and the SCAR mutants SCARΔ37 (from the Bloomington Stock Center) and SCARK13811 (from the Kyoto Drosophila Genetic Resource Center) were used for genetic interaction analysis. Transgenic flies carrying UAS-GFP-moesin were from S. Hayashi [33]. A line carrying UAS-actin-GFP was from the Kyoto Drosophila Genetic Resource Center. For overexpression or rescue experiments, the pan-neuronal elav-Gal4, the muscle-specific C57-Gal4, and the ubiquitously expressed act-Gal4 drivers were used. hs-FLP; FRT82B cyfip85.1 females were crossed to FRT82B ubi-GFP males to generate mosaic clones in eye discs [37].
Immuno-histochemical analysis
For immunostaining of NMJ synapses, wandering third-instar larvae were dissected in Ca2+-free standard saline, and then fixed in fresh 4% paraformaldehyde for 30 min. The monoclonal mouse antibody anti-DLG (4F3; 1∶1000) was from the Developmental Studies Hybridoma Bank at the University of Iowa. FITC-conjugated goat anti-HRP and Alexa 568-conjugated goat anti-HRP (1∶100 for both) were from Jackson ImmunoResearch. Texas-red phalloidin (1∶6) was from Molecular Probes. The secondary antibodies used were Alexa 488-, or 568-conjugated anti-mouse or anti-rabbit from Invitrogen (1∶1000). F-actin in eye discs was labeled with Texas-red phalloidin at 1∶400. All images were collected using a Leica SP5 laser scanning confocal microscope and processed with Adobe Photoshop 8.0.
For statistical analysis, type Ib terminals of NMJ4 in abdominal segment A2 or A3 were quantified. A donut-shaped anti-DLG staining pattern indicated a single bouton. Satellite boutons were defined as small boutons budding from major synaptic terminals or primary boutons. Superboutons were defined as the parental boutons around which three or more satellite boutons formed. The superboutons were categorized into three groups bearing 3, 4, or ≥5 satellite boutons. The length of the NMJ was measured by Image J based on HRP-stained terminals. For presynaptic GFP-moe patch analysis, we quantified the number and area of GFP patches over 0.5 µm2 by ImageJ. The number of GFP patches was normalized to the synaptic areas delineated by anti-HRP staining.
Electron microscopy
Larval neuromusculature was prepared for EM analysis according to procedures described previously [49]. Dissected larvae were fixed for 2 h with 2.5% glutaraldehyde (Sigma-Aldrich) in cacodylate buffer (pH 7.4) at room temperature followed by several rinses with cacodylate buffer. Right and left hemi-segments from abdominal segments A3 and A4 were separated from the larval fillets and post-fixed with 1% OsO4 in cacodylate buffer for 2 h. The preparations were stained en bloc for 1 h with saturated uranyl acetate in 50% ethanol before dehydration in an ethanol series. The samples were embedded in Spurr resin (Sigma). Longitudinal ultra-thin sections were made on an LKB ultra-microtome or Leica UC6 ultra-microtome using a diamond knife. Grids were post-stained with saturated uranyl acetate in 50% ethanol and 1% lead citrate (pH 12) and examined under a JEOL 1010 electron microscope. Electron micrographs were acquired using a Ganton792 digital CCD. For quantification, we analyzed cross-sections through the midline of more than 20 individual boutons from 5 animals of each genotype. The number and diameter of synaptic vesicles within a 200 nm radius of the active zone were measured by ImageJ. Vesicle structures with diameters >60 nm were defined as cisternae.
Electrophysiological assays
Intracellular recordings were performed at 20°C following published procedures [17], [49] with minor modifications. Briefly, to assess basal neuromuscular transmission, wandering third instar larvae were dissected in modified HL3 saline (NaCl 70 mM, KCl 5 mM, MgCl2 10 mM, NaHCO3 10 mM, sucrose 115 mM, D-trehalose 5 mM, and HEPES 5 mM, pH 7.2) and evoked responses recorded in modified HL3 saline containing 0.23 mM Ca2+. Intracellular microelectrodes with a resistance of 8–20 MΩ when filled with 3 M KCl were used for recording. Recordings were performed using an Axoclamp 2B amplifier (Axon Instruments) in Bridge mode. Data were filtered at 1 kHz and digitized using Digitizer 1322A (Axon Instruments). Data acquisition was controlled by Clampex 9.1 software. Excitatory junctional potentials (EJPs) were evoked at 0.3 Hz by a suction electrode using a depolarizing pulse delivered by a Grass S48 stimulator (Astro-Grass Inc.). Both EJPs and miniature EJPs (mEJPs) were recorded from muscle 6 of abdominal segment A2 or A3, and processed with Clampfit 10.2 software. Quantal content was calculated by dividing the EJP amplitude (after correction for nonlinear summation) by the mEJP amplitude according to a classic protocol [50]. The EJP correction for nonlinear summation assumed a reversal potential of 10 mV. To examine synaptic transmission under high frequency stimulation, synapses were stimulated at 10 Hz for 10 min and evoked EJPs recorded in modified HL3 saline with 10 mM Ca2+. Immediately following the high-frequency train, EJPs in response to 0.3 Hz stimulation were recorded for 10 min. The amplitudes of EJPs within successive 30 s intervals were averaged and normalized to the initial amplitude to yield the time course of recovery. At least 10 NMJ preparations were recorded for each genotype.
FRAP analysis
For fluorescence recovery after photobleaching (FRAP) analysis, actin-GFP or GFP-moe was expressed presynaptically by elav-GAL4. Third instar larvae were dissected in modified HL3 saline (NaCl 70 mM, KCl 5 mM, MgCl2 10 mM, NaHCO3 10 mM, sucrose 115 mM, D-trehalose 5 mM, and HEPES 5 mM, pH 7.2) and imaged using a 40× water immersion lens on a Leica SP5 confocal microscope. The GFP fluorescence at NMJ synapses was bleached by scanning the region of interest (ROI, 1×2 µm) using full power of the 488 nm Argon laser line for 8 s with a pinhole setting of 2.0 airy units. The images were captured at zoom 8 with a resolution of 512×512 pixels. Recovery images were collected with low-intensity 488 nm excitation light to avoid additional bleaching. The fluorescence intensity within the ROI was calculated using ImageJ software. The relative fluorescence emission intensity F(t) was calculated as follows: F(t) = (Ft–Fb)/(Fi–Fb)×100%, where Ft is the fluorescence intensity within the ROI at some time (t) after bleaching, Fb is the background intensity, and Fi is the initial fluorescence intensity before bleaching [51]. The recovery data points were fitted to a one-phase exponential equation. The recovery rate k was calculated from k = 0.693/t1/2 [52], in which t1/2 represents half time, the time for the recovered GFP to reach 50% of the fluorescence intensity plateau. At least 17 serial recordings from 4 animals were used for statistical analysis.
Examination of new F-actin assembly
New F-actin assembly was examined as previously described [35] with minor modifications. Briefly, jasplakinolide (Invitrogen) was dissolved in DMSO to make a 1 mM stock solution. To visualize newly polymerized F-actin, dissected samples were pretreated with jasplakinolide at a concentration of 10 µM for 15 min for NMJ synapses or 10 min for eye discs. After recovery in the drug-free medium (60 min for NMJ synapses, 90 min for eye discs), preparations were fixed and stained with Alexa-546-conjugated phalloidin (1∶400, Invitrogen) to detect newly-polymerized F-actin. Controls were treated with the vehicle DMSO only. For quantitative analysis of fluorescence intensity, samples were processed simultaneously and imaged using identical acquisition parameters. Serial images of eye discs and 1 µm single section of NMJ4 boutons in abdominal segments A2 or A3 were collected using a Leica SP5 laser scanning confocal microscope. The intensity of phalloidin staining in individual boutons was analyzed using ImageJ software (NIH). The recovery index RI was calculated as follows: RI = (Ire–Ijasp)/Ictl×100%, where Ire is the phalloidin intensity after 1 h recovery, Ijasp is the phalloidin intensity immediately after jasplakinolide treatment, and Ictl is the phalloidin intensity with vehicle treatment. Statistical analysis was performed using ANOVA for comparison of multiple group means.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CollinsCA, DiAntonioA (2007) Synaptic development: insights from Drosophila. Curr Opin Neurobiol 17 : 35–42.
2. GiagtzoglouN, LyCV, BellenHJ (2009) Cell adhesion, the backbone of the synapse: “vertebrate” and “invertebrate” perspectives. Cold Spring Harb Perspect Biol 1: a003079.
3. PackardM, MathewD, BudnikV (2003) Wnts and TGF beta in synaptogenesis: old friends signalling at new places. Nat Rev Neurosci 4 : 113–120.
4. BallRW, Warren-PaquinM, TsurudomeK, LiaoEH, ElazzouziF, et al. (2010) Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron 66 : 536–549.
5. DillonC, GodaY (2005) The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28 : 25–55.
6. PielageJ, BulatV, ZucheroJB, FetterRD, DavisGW (2011) Hts/Adducin controls synaptic elaboration and elimination. Neuron 69 : 1114–1131.
7. O'Connor-GilesKM, HoLL, GanetzkyB (2008) Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron 58 : 507–518.
8. RodalAA, Motola-BarnesRN, LittletonJT (2008) Nervous wreck and Cdc42 cooperate to regulate endocytic actin assembly during synaptic growth. J Neurosci 28 : 8316–8325.
9. KhuongTM, HabetsRL, SlabbaertJR, VerstrekenP (2010) WASP is activated by phosphatidylinositol-4,5-bisphosphate to restrict synapse growth in a pathway parallel to bone morphogenetic protein signaling. Proc Natl Acad Sci U S A 107 : 17379–17384.
10. PielageJ, FetterRD, DavisGW (2005) Presynaptic spectrin is essential for synapse stabilization. Curr Biol 15 : 918–928.
11. RichardsDA, RizzoliSO, BetzWJ (2004) Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J Physiol 557 : 77–91.
12. ShupliakovO, BloomO, GustafssonJS, KjaerulffO, LowP, et al. (2002) Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci U S A 99 : 14476–14481.
13. HussainNK, JennaS, GlogauerM, QuinnCC, WasiakS, et al. (2001) Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat Cell Biol 3 : 927–932.
14. NunesP, HainesN, KuppuswamyV, FleetDJ, StewartBA (2006) Synaptic vesicle mobility and presynaptic F-actin are disrupted in a N-ethylmaleimide-sensitive factor allele of Drosophila. Mol Biol Cell 17 : 4709–4719.
15. KohTW, VerstrekenP, BellenHJ (2004) Dap160/intersectin acts as a stabilizing scaffold required for synaptic development and vesicle endocytosis. Neuron 43 : 193–205.
16. MarieB, SweeneyST, PoskanzerKE, RoosJ, KellyRB, et al. (2004) Dap160/intersectin scaffolds the periactive zone to achieve high-fidelity endocytosis and normal synaptic growth. Neuron 43 : 207–219.
17. WangD, ZhangL, ZhaoG, WahlstromG, HeinoTI, et al. (2010) Drosophila twinfilin is required for cell migration and synaptic endocytosis. J Cell Sci 123 : 1546–1556.
18. ChenZ, BorekD, PadrickSB, GomezTS, MetlagelZ, et al. (2010) Structure and control of the actin regulatory WAVE complex. Nature 468 : 533–538.
19. EdenS, RohatgiR, PodtelejnikovAV, MannM, KirschnerMW (2002) Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418 : 790–793.
20. GautreauA, HoHY, LiJ, SteenH, GygiSP, et al. (2004) Purification and architecture of the ubiquitous Wave complex. Proc Natl Acad Sci U S A 101 : 4379–4383.
21. IsmailAM, PadrickSB, ChenB, UmetaniJ, RosenMK (2009) The WAVE regulatory complex is inhibited. Nat Struct Mol Biol 16 : 561–563.
22. KobayashiK, KurodaS, FukataM, NakamuraT, NagaseT, et al. (1998) p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J Biol Chem 273 : 291–295.
23. LebensohnAM, KirschnerMW (2009) Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell 36 : 512–524.
24. SchenckA, BardoniB, LangmannC, HardenN, MandelJL, et al. (2003) CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38 : 887–898.
25. CoyleIP, KohYH, LeeWC, SlindJ, FergestadT, et al. (2004) Nervous wreck, an SH3 adaptor protein that interacts with Wsp, regulates synaptic growth in Drosophila. Neuron 41 : 521–534.
26. DickmanDK, LuZ, MeinertzhagenIA, SchwarzTL (2006) Altered synaptic development and active zone spacing in endocytosis mutants. Curr Biol 16 : 591–598.
27. KohTW, KorolchukVI, WairkarYP, JiaoW, EvergrenE, et al. (2007) Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol 178 : 309–322.
28. VerstrekenP, OhyamaT, HaueterC, HabetsRL, LinYQ, et al. (2009) Tweek, an evolutionarily conserved protein, is required for synaptic vesicle recycling. Neuron 63 : 203–215.
29. YaoCK, LinYQ, LyCV, OhyamaT, HaueterCM, et al. (2009) A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell 138 : 947–960.
30. ZhangB, KohYH, BecksteadRB, BudnikV, GanetzkyB, et al. (1998) Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21 : 1465–1475.
31. DickmanDK, HorneJA, MeinertzhagenIA, SchwarzTL (2005) A slowed classical pathway rather than kiss-and-run mediates endocytosis at synapses lacking synaptojanin and endophilin. Cell 123 : 521–533.
32. SweeneyST, DavisGW (2002) Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron 36 : 403–416.
33. ChiharaT, KatoK, TaniguchiM, NgJ, HayashiS (2003) Rac promotes epithelial cell rearrangement during tracheal tubulogenesis in Drosophila. Development 130 : 1419–1428.
34. EdwardsKA, DemskyM, MontagueRA, WeymouthN, KiehartDP (1997) GFP-moesin illuminates actin cytoskeleton dynamics in living tissue and demonstrates cell shape changes during morphogenesis in Drosophila. Dev Biol 191 : 103–117.
35. LeeCW, HanJ, BamburgJR, HanL, LynnR, et al. (2009) Regulation of acetylcholine receptor clustering by ADF/cofilin-directed vesicular trafficking. Nat Neurosci 12 : 848–856.
36. BubbMR, SenderowiczAM, SausvilleEA, DuncanKL, KornED (1994) Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem 269 : 14869–14871.
37. ChuD, PanH, WanP, WuJ, LuoJ, et al. (2012) AIP1 acts with cofilin to control actin dynamics during epithelial morphogenesis. Development 139 : 3561–3571.
38. WolffT, ReadyDF (1991) The beginning of pattern formation in the Drosophila compound eye: the morphogenetic furrow and the second mitotic wave. Development 113 : 841–850.
39. BogdanS, GreweO, StrunkM, MertensA, KlambtC (2004) Sra-1 interacts with Kette and Wasp and is required for neuronal and bristle development in Drosophila. Development 131 : 3981–3989.
40. KundaP, CraigG, DominguezV, BaumB (2003) Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol 13 : 1867–1875.
41. SchenckA, QurashiA, CarreraP, BardoniB, DieboldC, et al. (2004) WAVE/SCAR, a multifunctional complex coordinating different aspects of neuronal connectivity. Dev Biol 274 : 260–270.
42. BogdanS, KlambtC (2003) Kette regulates actin dynamics and genetically interacts with Wave and Wasp. Development 130 : 4427–4437.
43. NapoliI, MercaldoV, BoylPP, EleuteriB, ZalfaF, et al. (2008) The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134 : 1042–1054.
44. ZhangYQ, BaileyAM, MatthiesHJ, RendenRB, SmithMA, et al. (2001) Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107 : 591–603.
45. HeerssenH, FetterRD, DavisGW (2008) Clathrin dependence of synaptic-vesicle formation at the Drosophila neuromuscular junction. Curr Biol 18 : 401–409.
46. CingolaniLA, GodaY (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9 : 344–356.
47. FergusonSM, RaimondiA, ParadiseS, ShenH, MesakiK, et al. (2009) Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell 17 : 811–822.
48. KaksonenM, ToretCP, DrubinDG (2006) Harnessing actin dynamics for clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 7 : 404–414.
49. LiuZ, HuangY, ZhangY, ChenD, ZhangYQ (2011) Drosophila Acyl-CoA synthetase long-chain family member 4 regulates axonal transport of synaptic vesicles and is required for synaptic development and transmission. J Neurosci 31 : 2052–2063.
50. MartinAR (1955) A further study of the statistical composition on the end-plate potential. J Physiol 130 : 114–122.
51. AxelrodD, KoppelDE, SchlessingerJ, ElsonE, WebbWW (1976) Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16 : 1055–1069.
52. HotulainenP, LlanoO, SmirnovS, TanhuanpaaK, FaixJ, et al. (2009) Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185 : 323–339.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy