
Identification of , a Retrotransposon-Derived Imprinted Gene, as a Novel Driver of Hepatocarcinogenesis
We previously utilized a Sleeping Beauty (SB) transposon mutagenesis screen to discover novel drivers of HCC. This approach identified recurrent mutations within the Dlk1-Dio3 imprinted domain, indicating that alteration of one or more elements within the domain provides a selective advantage to cells during the process of hepatocarcinogenesis. For the current study, we performed transcriptome and small RNA sequencing to profile gene expression in SB–induced HCCs in an attempt to clarify the genetic element(s) contributing to tumorigenesis. We identified strong induction of Retrotransposon-like 1 (Rtl1) expression as the only consistent alteration detected in all SB–induced tumors with Dlk1-Dio3 integrations, suggesting that Rtl1 activation serves as a driver of HCC. While previous studies have identified correlations between disrupted expression of multiple Dlk1-Dio3 domain members and HCC, we show here that direct modulation of a single domain member, Rtl1, can promote hepatocarcinogenesis in vivo. Overexpression of Rtl1 in the livers of adult mice using a hydrodynamic gene delivery technique resulted in highly penetrant (86%) tumor formation. Additionally, we detected overexpression of RTL1 in 30% of analyzed human HCC samples, indicating the potential relevance of this locus as a therapeutic target for patients. The Rtl1 locus is evolutionarily derived from the domestication of a retrotransposon. In addition to identifying Rtl1 as a novel driver of HCC, our study represents one of the first direct in vivo demonstrations of a role for such a co-opted genetic element in promoting carcinogenesis.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003441
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003441
Summary
We previously utilized a Sleeping Beauty (SB) transposon mutagenesis screen to discover novel drivers of HCC. This approach identified recurrent mutations within the Dlk1-Dio3 imprinted domain, indicating that alteration of one or more elements within the domain provides a selective advantage to cells during the process of hepatocarcinogenesis. For the current study, we performed transcriptome and small RNA sequencing to profile gene expression in SB–induced HCCs in an attempt to clarify the genetic element(s) contributing to tumorigenesis. We identified strong induction of Retrotransposon-like 1 (Rtl1) expression as the only consistent alteration detected in all SB–induced tumors with Dlk1-Dio3 integrations, suggesting that Rtl1 activation serves as a driver of HCC. While previous studies have identified correlations between disrupted expression of multiple Dlk1-Dio3 domain members and HCC, we show here that direct modulation of a single domain member, Rtl1, can promote hepatocarcinogenesis in vivo. Overexpression of Rtl1 in the livers of adult mice using a hydrodynamic gene delivery technique resulted in highly penetrant (86%) tumor formation. Additionally, we detected overexpression of RTL1 in 30% of analyzed human HCC samples, indicating the potential relevance of this locus as a therapeutic target for patients. The Rtl1 locus is evolutionarily derived from the domestication of a retrotransposon. In addition to identifying Rtl1 as a novel driver of HCC, our study represents one of the first direct in vivo demonstrations of a role for such a co-opted genetic element in promoting carcinogenesis.
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths worldwide [1]. In contrast to the downward trends in incidence observed for most cancer types, that of HCC continues to rise, particularly in the United States [2]. This is due in part to increases in obesity and hepatitis C viral infection, both of which have been implicated in HCC pathogenesis. Treatment options for patients are limited, particularly for those with advanced disease, and the five-year survival rate remains low at ∼10%.
A major goal of HCC research is to develop therapies targeted at the molecular mechanisms underlying tumor development and progression. This type of approach is expected to be much more efficacious, increasing survival rates for HCC patients. Consistent with this idea, treatment with sorafenib, a multi-kinase inhibitor, has shown survival benefits for late-stage patients [3] – a rare achievement in HCC treatment. Nevertheless, sorafenib treatment is only able to extend median survival by three months, underlying the need for improved targeted therapies. Unfortunately, the molecular drivers of HCC remain poorly characterized, precluding the development of such therapeutics. Large-scale sequencing efforts currently being undertaken by The Cancer Genome Atlas (TCGA) project will likely characterize the recurrent genetic alterations present in human liver tumors and may identify novel therapeutic targets. However, it is becoming increasingly clear that human tumors are incredibly complex, and identifying molecular drivers of carcinogenesis among the larger number of background events has proven difficult. Comparative analysis of the information gained from human tumor profiling with data from animal models provides an improved ability to distinguish driver events contributing to human disease.
The Sleeping Beauty (SB) transposon mutagenesis system has proven useful for identifying drivers of tumorigenesis in a wide variety of tissue types [4]. We have previously used SB mutagenesis to generate mice that developed HCC [5]. Subsequent genetic analysis of SB-induced liver tumors identified the Dlk1-Dio3 imprinted domain as a common target of transposon-induced mutations. This highly complex domain contains genes encoding protein-coding transcripts, long non-coding RNAs (lncRNAs), microRNAs (miRNAs), and small nucleolar RNAs (snoRNAs). Expression of domain members is regulated in an allele-specific manner and depends on epigenetic modifications established in the germline [6]. Regulation of this expression pattern is maintained, at least in part, by multiple differentially methylated regions (DMRs) throughout the domain that are methylated on the paternally inherited allele. Maintenance of imprinting is critical for normal function, as evidenced by the fact that uniparental disomy (UPD) for either parental allele leads to severe and widespread developmental defects in both mouse models [7] and human patients [8].
A link between the Dlk1-Dio3 domain and HCC has previously been identified. Interestingly, it has been reported that adeno-associated viral (AAV) vector integration within the same region of the domain as the SB transposon integrations in our model is associated with HCC development in mice [9], [10]. AAV integrations were found to alter expression of several domain members, preventing elucidation of a clear molecular mechanism of tumorigenesis. Other studies have also identified correlation between disrupted expression from the Dlk1-Dio3 domain and HCC [11]–[15], often with several domain members showing aberrant expression. The majority of these studies are correlative in nature, and no attempt is made to validate tumorigenic function of domain members through direct modulation of gene expression.
Here we describe a series of experiments that initially utilized deep-sequencing analyses to obtain detailed gene expression profiles of the SB-induced HCCs. This approach revealed that transposon integration within the Dlk1-Dio3 domain has variable effects on expression of several elements throughout the imprinted domain, but uniformly drives dramatic overexpression of Retrotransposon-like 1 (Rtl1). Validation experiments demonstrate that hepatic overexpression of Rtl1 promotes tumorigenesis in vivo. Additionally, we find that RTL1 is aberrantly expressed in ∼30% of human HCC samples, suggesting that it may be a relevant therapeutic target.
Rtl1 is a poorly characterized gene that encodes a predicted transmembrane protein with aspartic protease activity. Interestingly, this locus is derived from domestication of a sushi-ichi-related retrotransposon [16] and is unique to placental mammals [17]. This study identifies Rtl1 as a novel oncogene involved in hepatocarcinogenesis and suggests that its expression may be used as a prognostic indicator and/or targeted therapeutically to improve outcome for patients with HCC. It also represents one of the first direct in vivo demonstrations of a role for a co-opted genetic element in driving carcinogenesis.
Results/Discussion
Determining the effect of transposon integration on Dlk1-Dio3 domain members
We previously reported the identification of a 33 kilobase region of the imprinted Dlk1-Dio3 domain as a common target of transposon insertion in an SB-induced model of HCC [5] (Figure 1A). Given the domain's complexity and previous studies demonstrating altered expression of multiple domain members in response to insertion of exogenous DNA [9], [10], [18], we used both transcriptome and miRNA sequencing approaches to obtain expression profiles of eight SB-induced HCCs with Dlk1-Dio3 integrations and six normal livers for comparison (Figure 1B–1C, Figures S1 and S2, Tables S1 and S2). Expression of Dlk1-Dio3 domain miRNAs was low to undetectable in normal liver. Similar results were detected for three of eight tumors, while the remaining five tumors displayed activated expression of several imprinted miRNAs. Thus, transposon insertion in the Dlk1-Dio3 domain does not consistently alter miRNA expression. Interestingly, tumor samples with elevated expression of imprinted miRNAs also showed enhanced expression of Meg3 and Rian, suggesting a possible transposon-mediated loss of imprinting effect. Dramatic activation of expression from the locus encoding Rtl1 and Rtl1 antisense (Rtl1as) was observed in all eight SB-induced HCCs, while no significant expression was detected in normal liver. Notably, elevated expression from this locus is the only event that was consistently observed in all SB-induced HCCs with Dlk1-Dio3 integrations (Figure 1B–1C). Because transcription can occur on either strand at this locus [19], strand-specific RT-PCR was performed to determine whether the observed increase resulted from expression of Rtl1, Rtl1as, or a combination of both transcripts. As shown in Figure 2A, reads from the locus encoding Rtl1 and Rtl1as detected in HCCs were derived primarily from transcription of the protein-coding sense strand (i.e. Rtl1). The lack of detectable Rtl1 in normal liver suggests that transposon integration results in activation of a normally transcriptionally silent allele.


Integrated transposons directly drive Rtl1 expression
As we previously reported, SB transposon integration sites in HCC samples clustered near the 5′ end of Rian within the Dlk1-Dio3 domain [5]. Our initial characterization of transposon integrations was performed using ligation-mediated (LM)-PCR followed by pyrosequencing. It has been shown that this approach yields suboptimal sequencing depth for confident identification of clonal insertion sites [20]. To ensure adequate sequence coverage, the SB-induced HCCs were re-sequenced for the current study using the Illumina platform. Surprisingly, while integrations near the 5′ end of Rian were still found to be the most common event, a transposon orientation bias was revealed that had not previously been evident. For many of the tumors, multiple transposon integrations were identified in this region, and for each of the tumors at least one of these integrations was in the same orientation as Rtl1 (Figure 1A).
To validate the significance of transposon integrations upstream of Rtl1 in SB-induced HCCs, insertion sites from a larger set of tumors, as well as some normal livers (Rogers et al., in press), were sequenced using the Illumina platform. A quantitative analysis of all transposon integrations in the Dlk1-Dio3 domain for these samples is provided in Figure S3. Consistent with recent studies demonstrating minimal insertion bias for SB transposon integration [21], [22], background insertion sites identified in normal liver and subclonal insertions in HCC samples did not show any evidence for preferential integration within the Dlk1-Dio3 domain. In contrast, clonal sites identified in tumors were highly enriched upstream of Rtl1, suggesting positive selection for insertions in this region during the process of tumorigenesis. This analysis further confirmed that transposon integrations in the same transcriptional orientation as Rtl1 are preferentially detected specifically in HCCs. Based on these results, we hypothesized that the high levels of Rtl1 observed in tumors were driven directly by transposons integrated upstream. Amplification of transposon/Rtl1 fusion products from cDNA confirmed transposon-driven Rtl1 overexpression for each of the tumors harboring integrations in this region (Figure 2B). Two different sizes of fusion products were detected, representing direct splicing of the T2/Onc3 transposon into Rtl1 (smaller product) or inclusion of a cryptic upstream exon (larger product). Importantly, both fusion products encode the full Rtl1 open reading frame and are thus predicted to drive overexpression of functional Rtl1 protein.
Two additional Sleeping Beauty screens have been reported in which liver tumors were generated and characterized [23], [24]. Neither of these studies identified the Dlk1-Dio3 domain as a common site of integration. Both screens utilized T2/Onc mice as the source of mutagenic transposons. This transposon is similar in structure to that of the T2/Onc3 strain used in our study, but a distinct promoter is included within the transposon. T2/Onc transposons contain the murine stem cell virus (MSCV) 5′ long-terminal repeat (LTR) promoter, while T2/Onc3 transposons contain the cytomegalovirus (CMV) enhancer/chicken β-actin (CAG) promoter. Differences in promoter activities likely affect the profile of mutations that are selected for in tumors resulting from SB mutagenesis. We suspect that the MSCV promoter may be too weak to overcome the influence of imprinting within the Dlk1-Dio3 domain to drive sufficient hepatic Rtl1 expression to provide cells with a selective advantage and promote tumorigenesis. The CAG promoter, which has a much higher activity in epithelial cells like hepatocytes, may be better able to drive Rtl1 overexpression when integrated upstream, resulting in frequent selection of cells with such mutations in tumors. Consistent with this idea, insertional mutations upstream of Rtl1 have been linked to liver tumor development in two independent studies that utilized viral vectors containing promoters with high activity in hepatocytes [9], [15].
Rtl1 expression in cultured hepatocytes promotes growth in ECM
Our RNA profiling analyses and fusion transcript detection led us to conclude that the primary tumor-driving event under positive selection in SB-induced HCCs is activation of Rtl1. While we cannot exclude the possibility that other domain members play a role independently and/or cooperatively with Rtl1, in our model it seems to be the dominant driver of hepatocarcinogenesis. It should be noted that other models of HCC have been described in which altered expression of maternal Dlk1-Dio3 domain members is observed in the absence of Rtl1 activation [25], suggesting that distinct roles may exist for both paternal and maternal components of the domain in different subtypes of HCC.
To study the effects of Rtl1 overexpression on hepatocyte growth and morphology in vitro, we stably overexpressed it in the murine hepatocyte cell line TIB-73. Importantly, this cell line is non-tumorigenic and lacks endogenous expression of Rtl1. Based on the predicted protein structure of Rtl1, which contains an extracellular protease domain, we hypothesized that its effects may be mediated via cleavage of a substrate within the extracellular matrix (ECM). To test this hypothesis, TIB-73 cells expressing either Rtl1 or an empty vector were embedded in a matrix of Matrigel, plated in 24-well plates, and cultured in serum-free medium. Two weeks after plating, cells expressing Rtl1 had grown to form dozens of cyst-like colonies composed of several cells (Figure 3B, 3D). In contrast, cells lacking Rtl1 expression formed less than one colony per well on average, and colonies that did form were much denser and smaller (Figure 3A, 3C). These results demonstrate that Rtl1 expression promotes growth of hepatocytes in the presence of ECM in the context of physiologically relevant levels of growth factors, and they are consistent with our hypothesis that Rtl1 acts by cleaving an ECM component. ECM is an important aspect of the tumor microenvironment, particularly in the liver. The process of liver fibrosis, which involves ECM remodeling and expansion, is strongly linked to HCC, with nearly 90% of cases developing in this context [26]. One mechanism by which fibrosis may contribute to the development of HCC is through sequestration of growth factors in the newly remodeled ECM [27]. According to this model, subsequent release of growth factors through protease-mediated cleavage of ECM components promotes proliferation of adjacent hepatocytes. Our results suggest that Rtl1 may contribute to hepatocarcinogenesis via this mechanism.

In vivo hepatic Rtl1 expression drives tumorigenesis
We next sought to determine if Rtl1 overexpression is sufficient to promote hepatocarcinogenesis in vivo. Mice with stable hepatic expression of Rtl1 were generated by hydrodynamic tail vein injection of transposon-based expression constructs [28] into Fah-deficient male mice expressing SB transposase [24]. Selective repopulation of the liver was achieved through inclusion of a separate Fah expression vector that allowed stably transfected cells to survive withdrawal of NTBC [29], an event that triggers the death of Fah-null hepatocytes. Mice were euthanized nine months post-injection to assess liver tumorigenesis. Of fourteen mice injected with Rtl1 overexpression constructs, twelve (86%) developed liver tumors, with an average of 2.9 tumors per mouse (Table 1, Figure 4). In another experimental condition, a third construct encoding a short hairpin directed against Trp53 was additionally included. Loss of p53 function is one of the most commonly observed molecular abnormalities in human HCC, occurring in ∼30% of cases and making this a relevant context in which to validate putative oncogenes. Of twelve mice injected with all three transposon constructs, ten (83%) developed liver tumors, with an average of 4.3 tumors per mouse. Six of the mice from this cohort were sacrificed at time points earlier than nine months. When considering only those mice that were aged for nine months to allow direct comparison between the two experimental groups, five of six (83%) mice with p53 knockdown in addition to Rtl1 overexpression developed liver tumors, with an average of 6.7 tumors per mouse. This is significantly higher (p = 0.027) than the number of tumors per mouse developed with Rtl1 overexpression alone. Knockdown of p53 in tumors was assessed by western blot (Figure S4A). Although efficiency was somewhat variable, the majority of tumors showed significant knockdown.


It has been shown that following liver repopulation, the Fah mouse model is predisposed to tumor formation in the absence of any additional transgene [30], [31]. The tumors that develop in this context uniformly lack expression of Fah. We assessed expression of both Rtl1 and Fah by RT-PCR in fourteen tumors developed following hydrodynamic injection (Figure S4B). Of these fourteen tumors, eleven were found to express both genes. This result suggests that while a small subset of our tumors are likely background events developed independently of Rtl1 expression due to the model's predisposition, the majority of tumors were induced directly by overexpression of Rtl1. Further evidence for the tumorigenic activity of Rtl1 in vivo comes from a recently published study showing that liver tumors develop in mice following hepatic lentiviral delivery [15].
RTL1 activation in human HCC
In order to determine the prevalence of RTL1 activation in human disease, RT-PCR was performed on a collection of thirty-three human HCC RNA samples, along with matched benign adjacent liver tissue (Figure 5A, Figure S5). A lack of significant expression was observed for all but one of the benign liver samples. In contrast, significant activation of RTL1 was detected in 30% (10/33) of analyzed tumors. To assess RTL1 expression in another set of human HCCs, we utilized RNASeq data available through The Cancer Genome Atlas (TCGA) consortium. Consistent with our initial analysis, RTL1 expression was found to be significantly activated in 30% (10/33) of analyzed tumors (Figure 5B). Low-level expression was detected in two of the adjacent benign tissue samples for which sequence data was available. It should be noted that four of the tumor samples included in the TCGA dataset overlap with the initial set of 33 samples analyzed by RT-PCR. No expression of RTL1 was detected in these four samples by either analysis. A notable gender disparity is observed in human HCC, wherein men are around three times more likely to develop the disease than women [1]. We analyzed our human expression data to determine if RTL1 overexpression was associated with tumors from one gender or the other, but failed to detect evidence of any bias. Based on the combined set of human samples that we analyzed, RTL1 was found to be overexpressed in samples from 12/38 males (32%) and 8/24 females (33%).
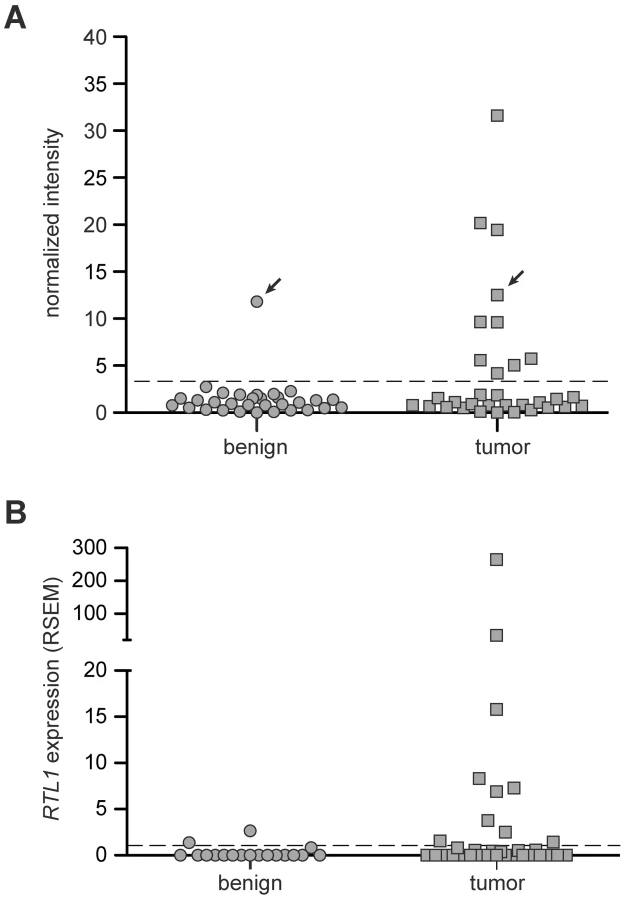
Unfortunately, there is very little existing data on the expression of RTL1 in disease states, including cancer. Most expression analyses utilize commercially available microarray platforms, the vast majority of which lack probes for RTL1. While multiple studies have identified correlative links between disrupted expression of other Dlk1-Dio3 domain members and HCC [9]–[14], expression of RTL1 has not typically been assessed. This may be due in part to the fact that RTL1 is a single exon gene, preventing straightforward design of primers that specifically amplify from cDNA and not genomic DNA. Notably, we have utilized a method for RTL1 expression analysis that adds a unique sequence tag during reverse-transcription [32], thus allowing specific amplification from cDNA and eliminating background amplification from genomic DNA.
In the setting of spontaneous hepatocarcinogenesis in humans, RTL1 activation may occur as a result of loss of imprinting (LOI) within the Dlk1-Dio3 domain. Epigenetic abnormalities are known to play a large role in driving tumor development and progression, in part through induction of LOI [33]. A direct causal role for LOI in cancer was demonstrated by Holm et al., who showed that chimeric mice created using embryonic stem cells lacking imprinting-specific DNA methylation develop multiple tumor types with nearly complete penetrance [34]. The most common tumor type observed was HCC, suggesting that LOI in the liver confers a strong predisposition to cancer. While expression from the Dlk1-Dio3 domain was not examined in the study, the results we present here suggest that hepatic activation of Rtl1 may be a driving factor in the HCCs that were developed. Interestingly, Wang et al. reported loss of methylation within the Rtl1 locus in mouse HCCs resulting from AAV integration [10], although effects on Rtl1 expression were not determined. To assess whether or not Rtl1 overexpression is associated specifically with altered expression of other imprinted genes in our SB-induced HCCs, analysis of variance (ANOVA) was conducted on the whole transcriptome to identify genes with differential expression between Rtl1-overexpressing tumors and normal liver. Following Bonferroni correction, 3 of 125 imprinted genes and 474 of 20,707 non-imprinted genes were identified as having significantly different expression between the two sample sets. By Fisher's exact test, these proportions are not significantly different (p = 0.760). This analysis shows that activation of Rtl1 does not correlate specifically with altered expression of other imprinted genes in our tumors.
Rtl1-expressing mouse HCCs resemble human S1 subclass
Next we sought to determine if Rtl1-induced HCCs in mice resemble a specific subtype of human HCC. An integrative meta-analysis of human HCC gene expression profiles has identified three major expression subtypes called S1, S2, and S3 [35]. Transcriptome sequencing data from the mouse HCCs overexpressing Rtl1 was used to determine the extent to which these SB-induced tumors resemble human HCC. Expression levels of genes defining the S1, S2, and S3 subclasses of human HCC were assessed for each of the SB-induced tumors and normal liver samples. Unsupervised clustering of samples based on expression of constituent genes was performed individually for each subclass. The results show that the SB-induced tumors resemble human HCCs within the S1 subclass (Figure 6). This was further supported by Gene Set Enrichment Analysis (GSEA) [36], [37] that showed a statistically significant association (p = 0.039) between Rtl1-induced HCCs and the S1 expression class. Immunohistochemistry was performed to validate protein expression of two S1 subclass genes in SB-induced HCC (Figure S6). This subclass of human HCC is associated with poor to moderate cellular differentiation, activation of the WNT signaling pathway, and early tumor recurrence.

Potential of RTL1 as a therapeutic target and/or biomarker
Rtl1 is a poorly characterized gene that encodes a predicted transmembrane protein with aspartic protease activity. Knockout studies in mice have demonstrated a role in the placental feto-maternal interface [38], but functional studies in other tissues are lacking. Experiments to determine the necessity of Rtl1's protease domain for its ability to promote tumorigenesis and to identify targets of its activity will help to clarify the oncogenic mechanism. If required, RTL1's protease activity represents a promising target for therapeutic intervention in HCC patients. Pepstatin is a naturally occurring bacterial peptide that demonstrates broad potential to inhibit aspartic proteases [39]. Additionally, more specific inhibitors have successfully been developed that target the activity of other aspartic proteases, including renin [40] and HIV-1 protease [41]. It is also possible that RTL1 expression could be a useful biomarker for HCC. Based on the human samples that we analyzed, its expression appears to be highly tumor-specific. Although low-level expression was detected in three non-tumor liver samples, all of the benign samples came from HCC patients and are therefore unlikely to be representative of truly normal liver.
Conclusion
In this study we identify Rtl1, a co-opted imprinted gene, as a novel driver of hepatocarcinogenesis. Mutations resulting in its overexpression were highly selected for in liver tumors developed using a forward genetic screen. While several correlative results linking the Dlk1-Dio3 domain to HCC development have been reported, our study provides direct evidence that modulation of a domain member in vitro and in vivo promotes a tumorigenic phenotype. We show here that overexpression of Rtl1 in cultured hepatocytes results in an increased growth ability in extracellular matrix. We also show that overexpression via hydrodynamic gene delivery results in highly penetrant liver tumor formation in mice. Additionally, a subset of human HCCs displays overexpression of RTL1, suggesting it may be a relevant therapeutic target for patients.
Materials and Methods
Mice
SB-induced mouse HCCs used in this study were generated as previously described [5]. All tumors used in this study came from male mice and were collected using procedures approved and monitored by the Institutional Animal Care and Use Committees at the National Cancer Institute-Frederick and the University of Minnesota.
Human tissue samples
Paired tumor and benign liver tissues were obtained from 33 patients undergoing resections for HCC at Mayo Clinic between 1987 and 2003, snap-frozen in liquid nitrogen, and stored at −80°C. The Mayo Clinic Institutional Review Board approved the study.
RNA sequencing and data analysis
Transcriptome sequencing
Total RNA was collected from SB-induced HCC and normal liver samples using the miRNeasy kit (Qiagen). Library preparation and sequencing were performed using Illumina's mRNA-Seq workflow. For data normalization, the raw number of reads for each transcript was converted to reads per kilobase per million mapped reads (RPKM) [42]. This was followed by log2 transformation of the RPKM value +1. Unsupervised clustering was performed on samples based on normalized expression of genes with variation in Euclidean distance among samples of at least 2.5 standard deviations using Cluster 3 software [43]. Heat maps were generated using Java TreeView software [44].
miRNA sequencing
Total RNA was collected from SB-induced HCC and normal liver samples using the miRNeasy kit (Qiagen). The flashPAGE Fractionator system (Life Technologies) was used to isolate RNAs shorter than 40 nt. Library preparation and sequencing were performed using the SOLiD small RNA expression workflow (Life Technologies). For data normalization, the raw number of reads for each miRNA was converted to reads per 100,000 mapped reads. This was followed by log2 transformation of the normalized value +1. Unsupervised clustering was performed on samples based on normalized expression of genes with variation in Euclidean distance among samples of at least 1.5 standard deviations using Cluster 3 software [43]. Heat maps were generated using Java TreeView software [44].
RT–PCR
Strand-specific RT–PCR to detect expression of Rtl1 and Rtl1as
One nanogram total RNA was used as template for cDNA synthesis with AMV reverse transcriptase (New England Biolabs). The cDNA synthesis reaction was primed with oligonucleotides complementary to Gapdh (Gapdh_R: 5′ – TGTAGGCCATGAGGTCCACCAC – 3′) and either Rtl1 (Rtl1_R: 5′ – GGAGCCACTTCATGCCTAAGACGA – 3′) or Rtl1as (Rtl1as_R: 5′ – GTGGAGAACTTCGCTGTCATCGC – 3′). PCR was performed with primers to detect transcripts for Gapdh (Gapdh_F: 5′ – TTGTCTCCTGCGACTTCAA – 3′ and Gapdh_R (amplicon 150 bp)), Rtl1 (Rtl1_F: 5′ – TACTGCTCTTGGTGAGAGTGGACCC – 3′ and Rtl1_R (amplicon 297 bp)), or Rtl1as (Rtl1as_F: 5′ – TCTCCACTCGAGGGTACTCCACCT – 3′ and Rtl1as_R (amplicon 298 bp)).
Transposon/Rtl1 fusion transcript detection
One microgram total RNA was used as template for oligodT-primed cDNA synthesis with Superscript III reverse transcriptase (Life Technologies). Control reactions lacking the RT enzyme were also performed. PCR was performed with a forward primer within the transposon splice donor (SD_F: 5′ – AAGCTTGCTACTAGCACCAGAACGCC – 3′) and reverse primer within Rtl1 (Rtl1_R2 : 5′ – TTCCTGGGCTGGGCCACTATC – 3′) (amplicon 394 bp or 459 bp, depending on splicing pattern).
Detection of RTL1 in human HCCs
Five hundred nanograms total RNA was used as template for cDNA synthesis with AMV reverse transcriptase (New England Biolabs). The cDNA synthesis reaction was primed with oligonucleotides complementary to TBP (TBP_R: 5′ – GCCATAAGGCATCATTGGAC – 3′) and RTL1 (RTL1_RT_tag: 5′ – GTAATACGACTCACTATAGGGCCTCGATAGGGGAGATGTTGC – 3′). The RTL1_RT_tag primer adds a unique 22 base sequence (underlined) to the 5′ end of the newly synthesized cDNA. Control reactions lacking the RT enzyme were also performed. PCR was performed with primers to detect transcripts for TBP (TBP_F: 5′ – GCTGAGAAGAGTGTGCTGGA – 3′ and TBP_R (amplicon 204 bp)) and RTL1 (RTL1_F: 5′ – TTCTACTGGGGAGTCGAGGA – 3′ and RT_tag_R: 5′ – GTAATACGACTCACTATAGGGC – 3′ (amplicon 238 bp)). RT_tag_R binds to the unique sequence tag added during cDNA synthesis, minimizing the potential for amplification from contaminating genomic DNA. Formamide was included in the PCR mix at a final concentration of 3% or 5% for amplification of TBP or RTL1, respectively. Gel images were processed using VisionWorks®LS image analysis software (UVP) to obtain intensity values for each lane. For each sample, values were normalized by subtracting the - RT lane from the +RT lane, then dividing each corrected value by the average corrected intensity value in benign samples lacking detectable expression.
Detection of Rtl1 and Fah in tumors induced by hydrodynamic injection
One microgram total RNA was used as template for oligodT-primed cDNA synthesis with Superscript III reverse transcriptase (Life Technologies). Control reactions lacking the RT enzyme were also performed. PCR was performed with primers to detect Rtl1 (Rtl1_F2 : 5′ – GTGGAGAACTTCGCTGTCATCGC – 3′ and Rtl1_R2 : 5′ – TCTCCACTCGAGGGTACTCCACCT – 3′ (amplicon 298 bp)), Fah (Fah_F: 5′ – CTTCTGCGACAATGCACCT – 3′ and Fah_R: 5′ – ACCACAATGGAGGAAGCTCG – 3′ (amplicon 172 bp)), or Tbp (Tbp_F: 5′ – CTATCACTCCTGCCACACCA – 3′ and Tbp_R: 5′ – CAGTTGTCCGTGGCTCTCTT – 3′ (amplicon 189 bp)).
Illumina sequencing of transposon insertions
DNA from SB-induced tumors was prepared for sequencing of transposon integration sites as previously described [20].
Matrigel growth assay
Stable cell lines were generated by delivery of piggyBac transposon constructs encoding either Rtl1 or an empty vector into TIB-73 (ATCC: BNL CL.2) cultured mouse hepatocytes. 24-well plates were coated with a thin layer of Matrigel basement membrane mix (BD Biosciences) and allowed to set up for 30 minutes at 37°C. For each stable cell line, cells were trypsinized and washed with PBS before resuspension of 5,000 cells in additional Matrigel. The resuspended cells were plated on top of the thin layer of basement membrane mix and allowed to set up, followed by addition of serum-free, low-glucose DMEM (Life Technologies). Images were taken two weeks after plating.
Hydrodynamic gene delivery
Hydrodynamic tail vein injection into Fah-deficient male mice expressing SB11 transposase was performed as previously described [24]. A plasmid expressing Rtl1 from the human PGK promoter and flanked by SB transposon inverted repeat/direct repeats (IR/DRs) was generated by amplifying the open reading frame of Rtl1 from C57Bl/6J mouse genomic DNA and subcloning it into pT2/PGK-pA. This plasmid was co-injected with PT2/PGK-FAHIL, a plasmid containing an SB IR/DR-flanked expression cassette for Fah and firefly luciferase. Some mice were additionally injected with pT2/shp53, a plasmid containing an SB IR/DR-flanked expression cassette for a short-hairpin RNA directed against Trp53 [29], [45].
Western blotting
Total protein was collected from liver tumor samples by homogenization in RIPA lysis buffer. Samples were boiled for five minutes in a reducing buffer and SDS-PAGE was performed. Proteins were transferred to nitrocellulose membranes for blotting. Primary antibodies used were anti-p53 (Cell Signaling Technology #2524), anti-GFP (Clontech #632380), and anti-β-tubulin (Sigma-Aldrich #T4026).
Gene Set Enrichment Analysis (GSEA)
GSEA [36], [37] was performed using default parameters. Analyzed gene sets were comprised of all the genes defining human HCC subclasses S1, S2, and S3 [35] for which mouse orthologs have been annotated.
Immunohistochemistry
Formalin-fixed, paraffin-embedded liver samples were sectioned to a thickness of 4 µm and baked onto glass slides. Samples were de-paraffinized, rehydrated, and treated with citrate antigen unmasking solution (Vector Laboratories). Endogenous peroxidase activity was blocked by treatment with a 3% solution of hydrogen peroxide for fifteen minutes. The anti-rabbit ImmPRESS reagent kit (Vector Laboratories) was used for immunolabeling with primary antibodies anti-Fyb (Abgent #AJ1306a) and anti-Ier3 (Abgent #AP11790a). Both primary antibodies were diluted 1∶100 and incubated with samples for one hour at room temperature. The ImmPACT DAB kit (Vector Laboratories) was used for detection. Sections were counterstained with hematoxylin QS (Vector Laboratories) and mounted in Permount (Fisher Scientific) for light microscopy.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FerlayJ, ShinHR, BrayF, FormanD, MathersC, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127 : 2893–2917.
2. AltekruseSF, McGlynnKA, ReichmanME (2009) Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 27 : 1485–1491.
3. LlovetJM, RicciS, MazzaferroV, HilgardP, GaneE, et al. (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359 : 378–390.
4. HowellVM (2012) Sleeping beauty–a mouse model for all cancers? Cancer Lett 317 : 1–8.
5. DupuyAJ, RogersLM, KimJ, NannapaneniK, StarrTK, et al. (2009) A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res 69 : 8150–8156.
6. da RochaST, EdwardsCA, ItoM, OgataT, Ferguson-SmithAC (2008) Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24 : 306–316.
7. GeorgiadesP, WatkinsM, SuraniMA, Ferguson-SmithAC (2000) Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 127 : 4719–4728.
8. OgataT, KagamiM, Ferguson-SmithAC (2008) Molecular mechanisms regulating phenotypic outcome in paternal and maternal uniparental disomy for chromosome 14. Epigenetics 3 : 181–187.
9. DonsanteA, MillerDG, LiY, VoglerC, BruntEM, et al. (2007) AAV vector integration sites in mouse hepatocellular carcinoma. Science 317 : 477.
10. WangPR, XuM, ToffaninS, LiY, LlovetJM, et al. (2012) Induction of hepatocellular carcinoma by in vivo gene targeting. Proc Natl Acad Sci U S A 109 : 11264–11269.
11. BraconiC, KogureT, ValeriN, HuangN, NuovoG, et al. (2011) microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene 30 : 4750–4756.
12. HuangJ, ZhangX, ZhangM, ZhuJD, ZhangYL, et al. (2007) Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis 28 : 1094–1103.
13. LukJM, BurchardJ, ZhangC, LiuAM, WongKF, et al. (2011) DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem 286 : 30706–30713.
14. YuF, HaoX, ZhaoH, GeC, YaoM, et al. (2010) Delta-like 1 contributes to cell growth by increasing the interferon-inducible protein 16 expression in hepatocellular carcinoma. Liver Int 30 : 703–714.
15. RanzaniM, CesanaD, BartholomaeCC, SanvitoF, PalaM, et al. (2013) Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods 10 : 155–161.
16. YoungsonNA, KocialkowskiS, PeelN, Ferguson-SmithAC (2005) A small family of sushi-class retrotransposon-derived genes in mammals and their relation to genomic imprinting. J Mol Evol 61 : 481–490.
17. EdwardsCA, MungallAJ, MatthewsL, RyderE, GrayDJ, et al. (2008) The evolution of the DLK1-DIO3 imprinted domain in mammals. PLoS Biol 6: e135 doi:10.1371/journal.pbio.0060135.
18. SteshinaEY, CarrMS, GlickEA, YevtodiyenkoA, AppelbeOK, et al. (2006) Loss of imprinting at the Dlk1-Gtl2 locus caused by insertional mutagenesis in the Gtl2 5′ region. BMC Genet 7 : 44.
19. SeitzH, YoungsonN, LinSP, DalbertS, PaulsenM, et al. (2003) Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet 34 : 261–262.
20. BrettBT, Berquam-VriezeKE, NannapaneniK, HuangJ, ScheetzTE, et al. (2011) Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors. PLoS ONE 6: e24668 doi:10.1371/journal.pone.0024668.
21. LiX, EwisH, HiceRH, MalaniN, ParkerN, et al. (2013) A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc Natl Acad Sci U S A 110: E478–487.
22. WoodardLE, LiX, MalaniN, KajaA, HiceRH, et al. (2012) Comparative analysis of the recently discovered hAT transposon TcBuster in human cells. PLoS ONE 7: e42666 doi:10.1371/journal.pone.0042666.
23. O'DonnellKA, KengVW, YorkB, ReinekeEL, SeoD, et al. (2012) A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A 109: E1377–1386.
24. KengVW, VillanuevaA, ChiangDY, DupuyAJ, RyanBJ, et al. (2009) A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol 27 : 264–274.
25. HarriL, PhilippeC, FedericoB, ArneM, ValerieD, et al. (2012) Identification of Dlk1-Dio3 imprinted gene cluster non-coding RNAs as novel candidate biomarkers for liver tumor promotion. Toxicol Sci 131 : 375–86.
26. SeitzHK, StickelF (2006) Risk factors and mechanisms of hepatocarcinogenesis with special emphasis on alcohol and oxidative stress. Biol Chem 387 : 349–360.
27. ZhangDY, FriedmanSL (2012) Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 56 : 769–775.
28. BellJB, Podetz-PedersenKM, AronovichEL, BelurLR, McIvorRS, et al. (2007) Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc 2 : 3153–3165.
29. WangensteenKJ, WilberA, KengVW, HeZ, MatiseI, et al. (2008) A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology 47 : 1714–1724.
30. GrompeM, OverturfK, al-DhalimyM, FinegoldM (1998) Therapeutic trials in the murine model of hereditary tyrosinaemia type I: a progress report. J Inherit Metab Dis 21 : 518–531.
31. KengVW, TschidaBR, BellJB, LargaespadaDA (2011) Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology 53 : 781–790.
32. ShuldinerAR, NirulaA, RothJ (1990) RNA template-specific polymerase chain reaction (RS-PCR): a novel strategy to reduce dramatically false positives. Gene 91 : 139–142.
33. FeinbergAP, TyckoB (2004) The history of cancer epigenetics. Nat Rev Cancer 4 : 143–153.
34. HolmTM, Jackson-GrusbyL, BrambrinkT, YamadaY, RideoutWM3rd, et al. (2005) Global loss of imprinting leads to widespread tumorigenesis in adult mice. Cancer Cell 8 : 275–285.
35. HoshidaY, NijmanSM, KobayashiM, ChanJA, BrunetJP, et al. (2009) Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69 : 7385–7392.
36. MoothaVK, LindgrenCM, ErikssonKF, SubramanianA, SihagS, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34 : 267–273.
37. SubramanianA, TamayoP, MoothaVK, MukherjeeS, EbertBL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102 : 15545–15550.
38. SekitaY, WagatsumaH, NakamuraK, OnoR, KagamiM, et al. (2008) Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet 40 : 243–248.
39. MarciniszynJJr, HartsuckJA, TangJ (1976) Mode of inhibition of acid proteases by pepstatin. J Biol Chem 251 : 7088–7094.
40. WoodJM, MaibaumJ, RahuelJ, GrutterMG, CohenNC, et al. (2003) Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem Biophys Res Commun 308 : 698–705.
41. RobinsonBS, RiccardiKA, GongYF, GuoQ, StockDA, et al. (2000) BMS-232632, a highly potent human immunodeficiency virus protease inhibitor that can be used in combination with other available antiretroviral agents. Antimicrob Agents Chemother 44 : 2093–2099.
42. MortazaviA, WilliamsBA, McCueK, SchaefferL, WoldB (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 : 621–628.
43. de HoonMJ, ImotoS, NolanJ, MiyanoS (2004) Open source clustering software. Bioinformatics 20 : 1453–1454.
44. SaldanhaAJ (2004) Java Treeview–extensible visualization of microarray data. Bioinformatics 20 : 3246–3248.
45. DickinsRA, HemannMT, ZilfouJT, SimpsonDR, IbarraI, et al. (2005) Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37 : 1289–1295.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy