
Rtt109 Prevents Hyper-Amplification of Ribosomal RNA Genes through Histone Modification in Budding Yeast
The genes encoding ribosomal RNA are the most abundant in the eukaryotic genome. They reside in tandem repetitive clusters, in some cases totaling hundreds of copies. Due to their repetitive structure, ribosomal RNA genes (rDNA) are easily lost by recombination events within the repeated cluster. We previously identified a unique gene amplification system driven by unequal sister-chromatid recombination during DNA replication. The system compensates for such copy number losses, thus maintaining proper copy number. Here, through a genome-wide screen for genes regulating rDNA copy number, we found that the rtt109 mutant exhibited a hyper-amplification phenotype (∼3 times greater than the wild-type level). RTT109 encodes an acetyl transferase that acetylates lysine 56 of histone H3 and which functions in replication-coupled nucleosome assembly. Relative to unequal sister-chromatid recombination-based amplification (∼1 copy/cell division), the rate of the hyper-amplification in the rtt109 mutant was extremely high (>100 copies/cell division). Cohesin dissociation that promotes unequal sister-chromatid recombination was not observed in this mutant. During hyper-amplification, production level of extra-chromosomal rDNA circles (ERC) by intra-chromosomal recombination in the rDNA was reduced. Interestingly, during amplification, a plasmid containing an rDNA unit integrated into the rDNA as a tandem array. These results support the idea that tandem DNA arrays are produced and incorporated through rolling-circle-type replication. We propose that, in the rtt109 mutant, rDNA hyper-amplification is caused by uncontrolled rolling-circle-type replication.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003410
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003410
Summary
The genes encoding ribosomal RNA are the most abundant in the eukaryotic genome. They reside in tandem repetitive clusters, in some cases totaling hundreds of copies. Due to their repetitive structure, ribosomal RNA genes (rDNA) are easily lost by recombination events within the repeated cluster. We previously identified a unique gene amplification system driven by unequal sister-chromatid recombination during DNA replication. The system compensates for such copy number losses, thus maintaining proper copy number. Here, through a genome-wide screen for genes regulating rDNA copy number, we found that the rtt109 mutant exhibited a hyper-amplification phenotype (∼3 times greater than the wild-type level). RTT109 encodes an acetyl transferase that acetylates lysine 56 of histone H3 and which functions in replication-coupled nucleosome assembly. Relative to unequal sister-chromatid recombination-based amplification (∼1 copy/cell division), the rate of the hyper-amplification in the rtt109 mutant was extremely high (>100 copies/cell division). Cohesin dissociation that promotes unequal sister-chromatid recombination was not observed in this mutant. During hyper-amplification, production level of extra-chromosomal rDNA circles (ERC) by intra-chromosomal recombination in the rDNA was reduced. Interestingly, during amplification, a plasmid containing an rDNA unit integrated into the rDNA as a tandem array. These results support the idea that tandem DNA arrays are produced and incorporated through rolling-circle-type replication. We propose that, in the rtt109 mutant, rDNA hyper-amplification is caused by uncontrolled rolling-circle-type replication.
Introduction
The ribosome is an abundant macromolecular protein-RNA complex that translates mRNA into protein. In Saccharomyces cerevisiae, ribosomal proteins (RP) account for approximately 50% of total protein and ribosomal RNA (RNA) represents approximately 80% of total RNA [1]. While the amount of RP increases during translation from mRNA, the amount of rRNA is dependent on transcription level. To meet this huge biosynthetic demand for rRNA in eukaryotic cells, the rRNA genes are present in hundreds of copies and are transcribed by a highly efficient RNA polymerase, RNA pol I [1]. We recently reported that about half the rDNA genes are transcriptionally silent, but serve as a “foothold” for repair enzymes to maintain the integrity of the rDNA region [2]. Nonetheless, the highly repetitive structure of the rDNA makes it fragile and vulnerable to loss of copies following homologous recombination events within the repeat.
As an apparent adaptation for a requirement for many copies of the rRNA genes and their simultaneous genetic instability, an rDNA-specific amplification system has evolved in eukaryotic cells. In Saccharomyces cerevisiae, the approximately 150 tandemly-repeated rDNA genes are located on chomosome XII (Figure 1A) [3]. One unit of rDNA is 9.1 kb and is composed of a region encoding a pre-35S rRNA, a small 5S rRNA and two intergenic spacer (IGS). The IGS region encodes the key elements involved in the amplification system that maintains rDNA gene copy number [4]. During S phase, progress of the DNA replication fork is blocked at the replication fork barrier sequence (RFB) through the action of the fork-blocking protein, Fob1 [5]. This inhibition induces DNA double-strand breaks (DSBs) and subsequent repair by unequal sister-chromatid recombination which increases rDNA copy number through re-replication of neighbor copies [6]. Moreover, this amplification is promoted by RNA pol II dependent transcription from E-pro, a non-coding bi-directional promoter near the RFB sequence [7]. Transcription from E-pro is proposed to clear cohesin proteins from nearby, which in turn may promote unequal sister-chromatid recombination, because cohesin proteins suppress unequal sister chromatid recombination [7]. In a wild type strain with ∼150 rDNA copies, E-pro transcription is repressed by a histone deacetylase, Sir2. Once rDNA copy number is reduced, Sir2 repression is relieved and amplification by unequal sisiter-chromatid recombination is enhanced. This amplification gradually increases copy number at a rate of ∼ one per cell division until a maximum of about 150 copies is reached [3].

To elucidate the mechanism by which rDNA copy number is regulated, we sought and found mutants in which copy number increased abnormally by screening a genome-wide deletion library consisting of ∼4,800 mutants (OpenBiosystems). Among these mutants, the rtt109Δ strain exhibited a prominent phenotype in which rDNA copy number is ∼400 in contrast to ∼150 in the wild type. RTT109 encodes an acetyltransferase that is stimulated by the histone chaperone Asf1 [8]–[11] to acetylate histone H3 on residue K56. The acetylated histones are incorporated into newly-synthesized DNA to promote replication-coupled nucleosome assembly by chromatin assembly factor 1 (CAF-1) and histone chaperone Rtt106 [12]. It is known that the defect in acetylation affects DNA damage sensitivity, stability of the replication fork, and recombination between sister-chromatids [8], [13]–[16]. Recently, Houseley and Tollervey also independently found this phenotype in the asf1 and rtt109 mutants [17].
We analyzed the relationship between histone acetylation and rDNA expansion in depth. We found that mutation of K56 of histone H3 also induced “hyper-amplification” of rDNA. Moreover, the histone deacetylase double mutant, hst3 hst4, had an increased rDNA copy number as well. Interestingly, the rate of the hyper-amplification in the rtt109 mutant was much faster (∼100 copies per cell division), than the ∼1 copy per cell division in wild-type. During hyper-amplification, extra chromosomal rDNA circles (ERC) generated from the rDNA by Fob1-dependent recombination did not accumulate. Instead, integration of plasmids with rDNA sequences was detected. They replicated sequentially and repeatedly during the integration step. These observations suggest that intra-chromosomal recombinational repair of Fob1-driven DSB that produces ERC does not occur. Instead, the data suggest that break-induced replication (BIR) takes place between the DSB end and ERC, resulting in rolling circle replication and hyper-amplification. We conclude that Rtt109 prevents such unusual replication and facilitates completion of recombinational repair with a sister-chromatid after a DNA double-stranded break occurs at the blocking site.
Results
Identification of mutants with high rDNA copy number
To elucidate the mechanism which regulates rDNA copy number, we measured the size of rDNA in the ∼4,800 mutants that comprise the yeast deletion library (S288c genetic background) by pulsed-field gel electrophoresis (CHEF, Figure S1). We identified eight mutants with higher-than-usual copy numbers (∼150), and presumed that they were defective in copy number regulation (Table 1). The rtt109 mutant was among those with the highest rDNA copy number. Chromosome XII of the rtt109 mutant is bigger than the biggest size maker (3.13 Mb) and sharper than other chromosome XIIs (lane 6 in Figure S1). This indicates that the chromosome XII is too big to be analyzed under these electrophoretic conditions and failed to migrate.

To confirm the “hyper-copy” rDNA phenotype in the rtt109 mutant, we deleted RTT109 in a different genetic background (W303) and tested the size of chromosome XII in this mutant by CHEF. As shown in Figure 2A, chromosome XII in the rtt109 strain was much longer than in the wild type strain. Moreover, we measured rDNA copy number in the rtt109 mutant based on signal intensities of Southern blots after BglII digestion (Figure 2B). The copy number was ∼2.5 times greater (∼400 copies) than in the wild type strain (∼150 copies, Figure 2B).

To determine whether hyper-amplification of the rDNA was a direct result of the deletion of RTT109, we transformed the rtt109 mutant with plasmid-encoded RTT109 and monitored the size of chromosome XII by CHEF. Chromosome XII remained longer up to ∼24 generations following the transformation (Figure 2C, lane 6–10), but decreased in size to that observed in the wild-type strain after ∼90 generations (Figure 2D, lane 5–6 in the W303 background, lane 13–16 in the S288c background), indicating that the hyper-copy rDNA phenotype was due to deletion of RTT109.
Acetylation and deacetylation of histone H3K56 inhibit hyper-amplification
Rtt109 acetylates the K56 residue of newly-synthesized histone H3 and plays an important role in chromatin remodeling during DNA replication, repair, and recombination [8], [12], [14], [15]. Acetylation of K56 no longer occurs in the rtt109 mutant [8]–[10]. To determine whether the acetylation activity is related to the hyper-amplification of rDNA, we measured rDNA copy number in the histone H3 mutants, H3K56R (non-acetylated) and H3K56Q (acetylated mimic). As expected, the H3K56R mutant had a longer chromosome XII similar to the rtt109 mutant (Figure 3A, lane 2), indicating that the hyper-amplification of rDNA occurred by inhibition of acetylation of the histone H3K56 residue. However, unexpectedly, the H3K56Q mutant also had a longer chromosome XII (Figure 3A, lane 3), although the rDNA copy number did not reach the level observed in the histone deacetylase mutant. Moreover, in the W303 genetic background, the double H3K56 histone deacetylase (HDAC) mutant, hst3 hst4, in which almost all H3 K56 residues were acetylated, exhibited the hyper-amplification phenotype as well (Figure 3B) [18]. These results indicate that deacetylation of K56 on histone H3 on the sister chromatin also prevents hyper-amplification and maintains proper copy number.

Hyper-amplification is accelerated by Fob1
We have previously shown that rDNA amplification from low copy number to wild type levels is dependent on the replication fork blocking activity of Fob1 and subsequent recombination [3]. To determine whether hyper-amplification starting from wild type copy number levels also requires this blocking activity, we deleted RTT109 in the fob1 mutant. As shown in Figure 4A (right, lanes 9–10), hyper-amplification in the double mutant was not observed. However, after more than 50 generations, some rtt109Δ fob1 clones had a longer chromosome XII than the single fob1 mutant, although they were still shorter than chromosome XII in the rtt109 single mutant (Figure S2, lanes 15–16). To confirm the contribution of Fob1 to the hyper-amplification phenotype, we monitored amplification rate in the rtt109 fob1 GAL-FOB1 double mutant over a time course after induction of FOB1 transcription. Amplification was observed 6 hrs post-induction (Figure 4B, asterisks). Because the doubling time of the mutant we measured was 3.4 hrs, the hyper-amplification phenotype was detectable within ∼2 generations. On the other hand, in cells grown on glucose where FOB1 was not induced, chromosome XII appeared slightly smeared in the gel, but the longer chromosome XII variant was not detected even after 32 hrs (Figure 4B). Taken together, these observations indicate that Fob1 plays an essential role in the hyper-amplification phenotype. We also tested whether hyper-amplification occurred in mutants belonging to the RTT109 epistasis group. A histone chaperone, Asf1, functions with Rtt109 to acetylate histone H3K56 [19], [20]. In an asf1 mutant, the rDNA was also hyper-amplified, although the length of chromosome XII was much shorter than in the rtt109 mutant (Figure 4A, lane 4–5). Deletion of the other histone chaperone Vps75/Rtt109 complex that acetylates H3 N-terminal residues did not promote a hyper-amplification phenotype (Figure 4A, lane 6–7) [21]–[23]. A similar result was reported by Houseley & Tollervey (2011) [17]. In the double fob1 asf1 mutant, amplification was substantially repressed, while in the vps75 mutant, no hyper-amplification was observed (Figure 4A, lane 11–14). This is consistent with a previous report that showed that an Rtt109-Asf1, but not an Rtt109-Vps75 complex, acetylated the histone H3/H4 dimer, which is a key event required for genome stability [9].

The rtt109 mutation does not affect replication fork stability, non-coding transcription, or cohesin association in the intergenic region
Because histone H3K56 acetylation is important for replisome integrity at a stalled replication fork under replication stress [8], [10], we monitored stability of a replication fork arrested at the RFB site in the rtt109 mutant, in which FOB1 is induced by galactose as used in Figure 4B, by two-dimensional gel electrophoresis during hyper-amplification. In both the wild type and rtt109 mutant, the RFB spot on the Y-arc appeared only when FOB1 was induced (cells grown on galactose). Because the signal intensities were similar (Figure 5A), the fork stability at the RFB appears not to be affected by RTT109 function. In addition, the amount of recombination intermediates was not increased in the rtt109 mutant (Figure 5A).

Non-coding transcription from the IGS region (E-pro) is up-regulated during amplification from low copy number rDNA [7]. The transcription reduces association of the cohesin complex, resulting in unequal sister-chromatid recombination [7]. Therefore, we tested whether transcription increased in the rtt109 mutant. By northern analysis, the RNA increased in the rtt109 mutant (Figure 5B). However, high level E-pro expression was not observed in the rtt109 fob1 double mutant (Figure 5B) in which rDNA copy number is maintained at wild type levels (Figure 4A, lanes 9–10). This suggests that the increased rDNA copy number causes the high level of E-pro transcription rather than deletion of RTT109. To address this possibility, we measured the level of E-pro in the wild type strain with a higher rDNA copy number constructed by introduction of a plasmid-encoded RTT109 into the rtt109 mutant shown in Figure 2C. As the result (Figure S3), the strain with the longer rDNA had a high level of E-pro expression as well as the rtt109 mutant. Therefore, we conclude that Rtt109 is not involved in repression of E-pro transcription, but that the abnormally increased rDNA copy number leads to a failure to repress transcription. Moreover, we examined the effect of deletion of RTT109 on the association of cohesin with the IGS region of rDNA by chromatin immunoprecipitation (ChIP) assay. To exclude the indirect effect of increaed rDNA copy number, we compared cohesin (Mcd1) association level between the rtt109 mutant and the wild type in a FOB1 defective background. As shown in Figure 5D, we confirmed cohesin association in the IGS in the mutant. Because the association represses unequal sister-chromatid recombination [7], the hyper-amplification in the rtt109 mutant is most likely not induced by the recombination.
Rolling circle amplification occurs during hyper-amplification
It appears to be difficult for break-induced unequal sister-chromatid amplification at the standard level to achieve the high rate of amplification observed in the rtt109 mutant (>100 copies/cell division). As mentioned above, we detected cohesin association that repressed the frequency of unequal sister-chromatid recombination. Rather, rolling circle type amplification appears to occur in such a manner that a large increase in copy number occurs in just a few cell divisions. In fact, the rolling circle structure was observed in the fraction remaining in the agrose gel plug subjected to pulsed-field gel electrophoresis (Figure 6). To initiate rolling circle replication, the broken end at the RFB must recombine with the intra-sister-chromatid (Figure 7B) or with popped-out circular molecules (ERC, extra chromosomal rDNA circle, Figure 7C). In the former case, the recombination usually produces an ERC [4]. Therefore, we monitored ERC formation during hyper-amplification by 2D gel electrophoresis in which the non-linear DNA molecules are separated from genomic DNA. In the wild type control, ERCs were detected in a Fob1-dependent manner as previously described [24], [25]. In contrast, in the rtt109 mutant, more ERCs were detected than in the wild type strain before FOB1 induction, while after induction, little increase in ERC level was observed (Figure 8A and 8B). This suggests that Fob1-induced DSB does not change the ERC level, but promotes an alternative pathway, perhaps rolling circle replication.



To test the possibility of a rolling circle amplification for which the ERC serves as a template (Figure 7C), we transformed a URA3-plasmid containing an intact rDNA unit (ERC-like plasmid) into the rtt109 fob1 double mutant and induced FOB1. As shown in Figure 8C by Southern hybridization with a URA3 probe, a signal was detected at the position of the hyper-amplified chromosome XII (ChrXII hyp.) in the rtt109 mutant. This indicates that the ERC (in this case ERC-like plasmid) was integrated into the rDNA during hyper-amplification in the mutant and the integration may explain little increase of ERC level after Fob1 induction (Figure 8A and 8B). To monitor the status of integration, we performed 2D gel analysis as shown in Figure 8A. For this purpose, the rtt109 mutant was transformed with another URA3-plasmid (IGS-plasmid) containing only the IGS region, after which the DNA was analyzed by digestion with BglII. The emzyme cuts only chromosomal rDNA units because the IGS region lacks a BglII recognition site. If rolling circle integration were to occur with the plasmid, tandem arrays of the plasmid sequence would be expected to form, generating long fragments following BglII digestion (Figure 8D, upper). In contrast, if the integration did not accompany rolling circle replication, a few integrated plasmid sequences would be expected. As shown in Figure 8D lower, signals for the plasmid-specific probe (amp) were observed both at positions corresponding to circular (IGS plasmid) and linear DNAs. Apparently, the copy number for the linear position in the rtt109 mutant is much higher than in the wild type strain (Figure 8D). This indicates that in the rtt109 mutant, rolling circle replication occurs more efficiently than in wild type and contributes to hyper-amplification.
The rtt109 mutation changes ratio of active to silent copies of rDNA but does not affect transcription rate
It has been reported that some mutants missing transcription factors for rRNA genes (Rrn9, Rrn5, Rrn6, Uaf30), acquired more copies of rDNA to compensate for less efficient transcription [26], [27]. Therefore, we examined whether deletion of RTT109 decreased transcription of rRNA genes, resulting in compensatory hyper-amplification of rDNA. To exclude indirect effects leading to copy amplification, we compared nascent rRNA in the rtt109 mutant and in wild type in a FOB1-defective background, where hyper-amplification is repressed temporarily. As shown in Figure 9A and 9B, transcript abundance did not decrease in the rtt109 mutant. Therefore, the amplification is not due to transcriptional compensation.
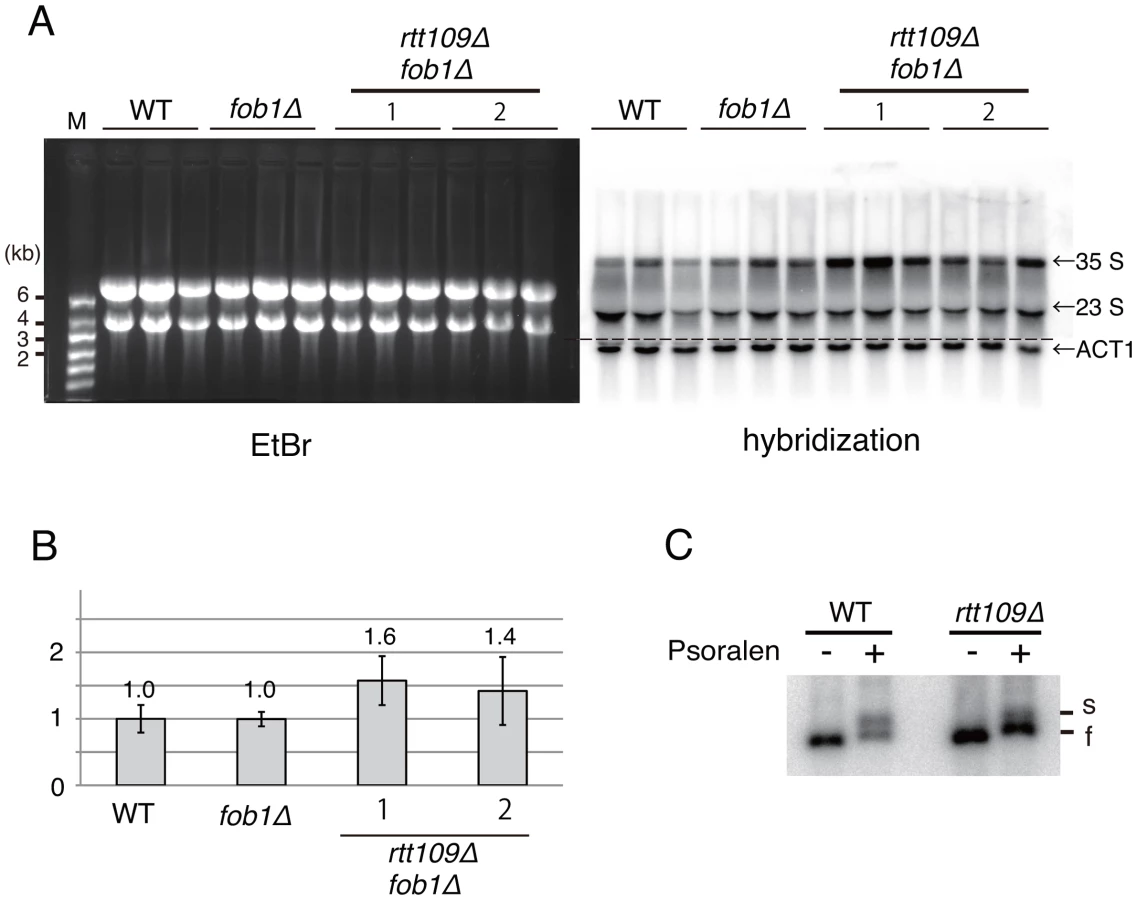
Further, we examined the transcription status of the rRNA genes in the rtt109 mutant by psoralen crosslinking [28]. In this assay, because the transcribed (active) rDNA copies have fewer nucleosomes, more psoralen can intercalate into the DNA and retard electrophoretic mobility. As shown in Figure 9C, the intensity of the lower band (silent copies) increased ∼3-fold in the rtt109 mutant, while that of the upper band (active copies) did not change significantly. This indicates that the hyper-amplification does not increase the number of copies with an open chromatin conformation.
Discussion
The ribosomal RNA gene is the most abundant and unstable gene in cells. Abundance is maintained in spite of the instability through several mechanisms. Gene amplification after deleterious recombination is one. We found that modification of a histone represses rolling circle replication and maintains wild type rDNA copy number.
Study of the relationship between histone modification, as a phenotype of the sir2 mutant, and rDNA stability was initiated by Gottlieb and Esposito (1989) [29]. They found that rDNA recombination was greatly enhanced in a sir2 mutant. Fritze et al. (1997) found that the mutation affected chromatin structure and speculated that it prevented recombination (instability) in the repeat [30]. Later, Sir2 was identified as a histone deacetylase [31]. Subsequently, we found that such alterations of chromatin structure repressed the noncoding promoter (E-pro) whose transcription reduces cohesion association with rDNA, and induces recombination between unequal sister-chromatids, because cohesin proteins hold sister-chromatids together and suppress unequal sister-chromatid recombination [7]. Therefore, one putative mechanism for rDNA maintenance by histone modification is through E-pro regulation. As reported here, however, deletion of RTT109 alone did not activate E-pro expression when copy number was normal (Figure 5B). Instead, as the result of the hyper-amplification, E-pro transcription was elevated in the rtt109 mutant. This may be because the copy number is too high to be silenced transcriptionally. It is known that the shorter rDNA also induces a high level of E-pro and induces rDNA amplification [7]. In this case, the activation can be explained by self-regulation of SIR2 as follows. When rDNA copy number is reduced, a large amount of Sir2 is released from the rDNA and may silence other parts of the genome. In fact, Michell et al. reported that telomere silencing is enhanced in a low rDNA copy strain (2005) [32]. They also showed that SIR2 itself is a target for repression. Therefore, we propose that autoregulated less SIR2 expression induces more E-pro transcription to amplify rDNA in a low rDNA copy strain. As copy number increases, more Sir2 is consumed by interaction with rDNA and repression of SIR2 is reduced. When copy number reaches wild type levels, SIR2 expression is maximal which represses E-pro resulting in a halt to further amplification. Therefore, in the case of hyper-amplified rDNA, Sir2 levels may be too low to interact with all rDNA copies resulting in a net partial loss of E-pro repression.
Hyper-amplification was observed in the mms22 mutant which has been shown to interact genetically with RTT109 (Table 1) [33]. In both rtt109 and mms22 mutants, cells become more sensitive to DNA damage during S phase than in wild type. Both Rtt109 and Mms22 are also involved in regulation of homologous recombination induced by replication inhibition [14]–[16], [34]. Our results show that the hyper amplification is accelerated by replication fork-blocking activity in the RFB. Therefore, one explanation is that the blocked fork induces DNA double stranded breaks and that recombinational repair of the broken sister-chromatid can be modulated by these proteins through histone modification. However, as shown in our 2D gels, no significant abnormality was observed in amount of recombination intermediates in the rtt109 mutant (Figure 5A), e.g., the early steps of recombination (DSB, homologous annealing, strand invasion and formation of Holliday structure). This suggests that Rtt109 and Mms22 function in later steps of the recombination reaction. For example, after formation of the Holliday structure, the donor strand may not separate from the template strand, or, alternatively, replication, such as break-induced replication (BIR) may occur [35]. In case of repetitive sequences such as rDNA, this BIR initiates rolling circle replication which generates an enormous increase in copy number during the cell cycle (Figure 7B).
The pattern of integration of the IGS plasmid into the chromosomal region indicates that rolling circle replication occurs in the rtt109 mutant (Figure 8D). However, integration of the ERC-like plasmid does not seem to be the sole or main contribution to rDNA amplification. The total signal intensities of both an integrated ERC-like plasmid into chromosome XII [Figure 8C, Chr XII(hyp)] and intermediates of on-going rolling circle replication in the well were ∼15 times greater than that of single copy URA3 on Chromosome V (Figure 8C; lanes 7–8 in the right pannel, Chr. V). Of course, because both the ERC-like plasmid and native ERC become templates for rolling circle replication, the actual extent of replication is expected to be greater. Intra-chromosomal recombination may also contribute to promote an additional pathway of rolling circle replication. As shown in Figure 7B, the broken end selects the intra-sister chromatid as a recombination template, instead of ERC, and converts into a Break-Induced Replication (BIR) event. Thus, we think that such template switch from sister-chromatid to ERC (results in ERC integration) and BIR conversion (results in less ERC production) reduce Fob1-induced ERC level during hyper-amlification in the rtt109 mutant (Figure 8A and 8B).
Our rolling circle replication model is supported by other reports concerning Rtt109 function. Wurtele et al. (2012) showed that the histone H3K56R mutation reduced marker loss rate in rDNA [36]. As we showed in Figure 2A, the mutation induced rDNA hyper-amplification which should have reduced loss of rDNA copies. Another study reported that mutated RTT109 reduced the frequency of unequal recombination between sister chromatids in non-rDNA regions [16]. This is consistent with our observation that cohesin associates with the IGS region of rDNA. This suggests that rDNA amplification by unequal sister-chromatid recombination (Figure 7A) does not seem elevated in the mutant. Houseley and Tollervey (2011) recently reported that hyper-amplification occurred in an asf1 mutant and proposed a model based on unequal sister chromatid interaction [17]. Because the rate of amplification is much lower in the asf1 mutant than in the rtt109 mutant, their model appears plausible. But in the case of the rtt109 mutant, models other than one based on rolling circle replication would not seem compatible with such a high amplification rate. In addition to the rtt109 mutant and the histone H3K56R mutant, we found that the double deletion mutant of histone deacetylase that acts on the K56 residue of histone H3, Hst3 Hst4, and the H3K56Q mutant all exhibited a hyper-amplification phenotype. These mutations also increased sensitivity to DNA damage relative to wild type cells [18]. Either an excess or an absence of acetylation on H3K56 of sister chromatids during recombination at the replication fork blocking site leads to rolling-circle amplification of rDNA. While we currently lack a satisfying explanation, the downstream event of the unusual H3K56 modification is important for the completion of recombination at the RFB in order to inhibit BIR-based rolling circle replication.
Rolling circle replication is known to play important roles in cell physiology. The most well known example may be rDNA amplification during amphibian oogenesis [37]. In this stage, extra-chromosomal rolling circle amplification increases copy number up to 2–3 orders of magnitude resulting in a huge increase in rRNAs (and ribosomes) needed for early development [38], [39]. This mechanism of replication is not limited to rDNA but occurs elsewhere in the genome and in some cases, helps cells adapt to various stresses (for review, see [40]). In cancer cells, gene amplification is frequently observed and may be mediated in part by rolling circle replication [41]. Abnormal histone modification has also been reported in malignant tissue [42] consistent with the possibility that gene amplification plays a role in acceleratating tumorigenesis.
Materials and Methods
Yeast strains, plasmids, growth conditions for hyper amplification
Yeasts strains used in this study were derived from NOY408-1b in the W303 genetic background and from strain S288c (OpenBiosystems) and are listed in Table S1. The strains were grown at 30°C in YPD medium except for the hst3 hst4 double mutant that was cultured at 23°C. To monitor rDNA hyper-amplification, the rtt109 mutant with the plasmid containing galactose-inducible FOB1 (YCplac22-Gal-FOB1) was precultured in synthetic complete medium minus tryptophan with raffinose (SC-trp+raf). The strain was then transferred to SC-trp with galactose instead of raffinose (at time zero) and harvested at the indicated times (0, 6, 12, 18, 24, 36 hr) to prepare genomic DNA in agrose plugs [43]. As a control, the RTT109 strain was monitored in parallel. Raffinose (1/10 volume of a 10X raffinose stock) and galactose were added just before use. Yeast strains with amino acid substitutions at histone K56 were generated by plasmid shuffling [44].
Genome-wide screening for yeast mutants with high rDNA copy number
Genomic DNA was purified from ∼4,800 yeast mutants from the genome-wide deletion library (OpenBiosystems) as described [43]. They were then subjected to pulsed-field gel electrophoresis as described below. Gels were stained with ethidium bromide (EtBr). The mutant strains with a longer chromosome XII than the 3.13 Mb size marker (H. wingei) were classified as hyper-amplified rDNA mutants (Table 1). We used a wide gel with a long comb (45 teeth) to perform the electrophoresis. Three thin gels were stacked on one another in the electrophoresis tank permitting simultaneous running of ∼130 samples.
Pulsed-field gel electrophoresis and Southern hybridization
Pulsed-field gel electrophoresis (CHEF) using CHEF Mapper XA (BioRad) and Southern hybridization were performed as described [25], [43]. The conditions were a 300–900 sec pulse time and 100 V for 68 hrs at 14°C in a 0.8% agarose gel.
2D gel electrophoresis
To detect replication and recombination intermediates by 2D gel electrophoresis during hyper-amplification, DNA was prepared from cells (YSI204, YSI205) growing in SC-trp+Gal (9 hrs) and SC-trp+Raf, and was digested with NheI in agarose plugs [43]. rDNA was detected with an rDNA-specific probe. DNA was isolated and digested in agarose plugs.
To detect ERC during hyper-amplification by 2D gel electrophoresis, DNA was isolated from the cells before and 9 hours after release into SC–trp +Gal. To separate large DNAs between 5 and 50 kb by 1D gel electrophoresis, a 0.3% SeaKem Gold Agarose gel (Lonza) was used in TAE at 1 V/cm for 20 hours. After excising each lane above 5 kb from the 1D gel, 2D gel electrophoresis was performed in 1.0% SeaKem LE Agarose gel (Lonza) with EtBr at 6 V/cm for 5 hours at 4°C. The gel was blotted and part of the membrane containing DNAs longer than 7 kb was hybridized with an rDNA-specific probe. The other part of the membrane containing DNAs shorter than 7 kb was probed with URA3. The ERC signal was normalized to the URA3 signal in the each blot. The relative amounts of ERC were determined from three independent experiments as shown in Figure 8B. To detect rolling circle amplification (Figure 8D) by 2D gel electrophoresis, cells with the IGS plasmid, YEplac195 containing the IGS region of rDNA were used. DNA was isolated from cells 48 hrs after release into SC–trp +Gal. Conditions used for 2D gel electrophoresis and induction of hyper-amplification were similar to those described above, except that DNA was digested with BglII before electrophoresis. BglII does not cut the plasmid but cuts the chromosomal rDNA, such that only plasmids integrated on the chromosome form linear structures following BglII digestion. The plasmid sequence was detected using a specific probe (amp).
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as described [6].
Atomic force microscopy (AFM)
DNA fraction remaining in the agrose gel plug after pulsed-field gel electrophoresis was extracted by crushing. AFM observation was performed as described [45]. During the sample preparation, DNA was sheared into smaller fragments (<∼50 kb). Among these fragments (∼1,500), we looked for the circle with the proper size (8–11 kb). Imaging was carried out with a Didital Instrument MultiMode AFM in the tapping mode. Image J (NIH) was used for measurement of DNA length.
Supporting Information
Zdroje
1. WarnerJR (1999) The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 24 : 437–40.
2. IdeS, MiyazakiT, MakiH, KobayashiT (2010) Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327 : 693–6.
3. KobayashiT, HeckDJ, NomuraM, HoriuchiT (1998) Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev 12 : 3821–30.
4. KobayashiT (2006) Strategies to maintain the stability of the ribosomal RNA gene repeats–collaboration of recombination, cohesion, and condensation. Genes Genet Syst 81 : 155–61.
5. KobayashiT (2003) The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol 23 : 9178–88.
6. KobayashiT, HoriuchiT, TongaonkarP, VuL, NomuraM (2004) SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117 : 441–53.
7. KobayashiT, GanleyAR (2005) Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309 : 1581–4.
8. DriscollR, HudsonA, JacksonSP (2007) Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315 : 649–52.
9. TsubotaT, BerndsenCE, ErkmannJA, SmithCL, YangL, FreitasMA, DenuJM, KaufmanPD (2007) Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell 25 : 703–12.
10. HanJ, ZhouH, HorazdovskyB, ZhangK, XuRM, ZhangZ (2007) Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315 : 653–5.
11. HanJ, ZhouH, LiZ, XuRM, ZhangZ (2007) Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J Biol Chem 282 : 28587–96.
12. LiQ, ZhouH, WurteleH, DaviesB, HorazdovskyB, VerreaultA, ZhangZ (2008) Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 134 : 244–55.
13. MasumotoH, HawkeD, KobayashiR, VerreaultA (2005) A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436 : 294–8.
14. PradoF, Cortes-LedesmaF, AguileraA (2004) The absence of the yeast chromatin assembly factor Asf1 increases genomic instability and sister chromatid exchange. EMBO Rep 5 : 497–502.
15. DuroE, VaisicaJA, BrownGW, RouseJ (2008) Budding yeast Mms22 and Mms1 regulate homologous recombination induced by replisome blockage. DNA Repair (Amst) 7 : 811–8.
16. EndoH, KawashimaS, SatoL, LaiMS, EnomotoT, SekiM, HorikoshiM (2010) Chromatin dynamics mediated by histone modifiers and histone chaperones in postreplicative recombination. Genes Cells 15 : 945–58.
17. HouseleyJ, TollerveyD (2011) Repeat expansion in the budding yeast ribosomal DNA can occur independently of the canonical homologous recombination machinery. Nucleic Acids Res 39 : 8778–91.
18. CelicI, VerreaultA, BoekeJD (2008) Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics 179 : 1769–84.
19. RechtJ, TsubotaT, TannyJC, DiazRL, BergerJM, ZhangX, GarciaBA, ShabanowitzJ, BurlingameAL, HuntDF, KaufmanPD, AllisCD (2006) Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Nat Acad Sci USA 103 : 6988–93.
20. SchneiderJ, BajwaP, JohnsonFC, BhaumikSR, ShilatifardA (2006) Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem 281 : 37270–4.
21. BerndsenCE, TsubotaT, LindnerSE, LeeS, HoltonJM, KaufmanPD, KeckJL, DenuJM (2008) Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat Struct Mol Biol 15 : 948–56.
22. ParkYJ, SudhoffKB, AndrewsAJ, StargellLA, LugerK (2008) Histone chaperone specificity in Rtt109 activation. Nat Struct Mol Biol 15 : 957–64.
23. FillinghamJ, RechtJ, SilvaAC, SuterB, EmiliA, StagljarI, KroganNJ, AllisCD, KeoghMC, GreenblattJF (2008) Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 28 : 4342–53.
24. DefossezPA, PrustyR, KaeberleinM, LinSJ, FerrignoP, SilverPA, KeilRL, GuarenteL (1999) Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol Cell 3 : 447–55.
25. TakeuchiY, HoriuchiT, KobayashiT (2003) Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev 17 : 1497–506.
26. OakesM, SiddiqiI, VuL, ArisJ, NomuraM (1999) Transcription factor UAF, expansion and contraction of ribosomal DNA (rDNA) repeats, and RNA polymerase switch in transcription of yeast rDNA. Mol Cell Biol 19 : 8559–69.
27. BernsteinKA, ReidRJ, SunjevaricI, DemuthK, BurgessRC, RothsteinR (2011) The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase. Mol Biol Cell 22 : 1599–607.
28. DammannR, LucchiniR, KollerT, SogoJM (1993) Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res 21 : 2331–38.
29. GottliebS, EspositoRE (1989) A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell 56 : 771–6.
30. FritzeCE, VerschuerenK, StrichR, Easton EspositoR (1997) Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J 16 : 6495–509.
31. ImaiS, JohnsonFB, MarciniakRA, McVeyM, ParkPU, GuarenteL (2000) Sir2: an NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol 65 : 297–302.
32. MichelAH, KornmannB, DubranaK, ShoreD (2005) Spontaneous rDNA copy number variation modulates Sir2 levels and epigenetic gene silencing. Genes Dev 19 : 1199–210.
33. ZaidiIW, RabutG, PovedaA, ScheelH, MalmstromJ, UlrichH, HofmannK, PaseroP, PeterM, LukeB (2008) Rtt101 and Mms1 in budding yeast form a CUL4(DDB1)-like ubiquitin ligase that promotes replication through damaged DNA. EMBO Rep 9 : 1034–40.
34. DuroE, LundinC, AskK, Sanchez-PulidoL, MacArtneyTJ, TothR, PontingCP, GrothA, HelledayT, RouseJ (2010) Identification of the MMS22L-TONSL complex that promotes homologous recombination. Mol Cell 40 : 632–44.
35. KrausE, LeungW-Y, HaberJE (2001) Break-induced replication: a review and an example in budding yeast. Pro Natl Acad Sci 98 : 8255–62.
36. WurteleH, et al. (2012) Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol 32 : 154–72.
37. HourcadeD, DresslerD, WolfsonJ (1973) The amplification of ribosomal RNA genes involves a rolling circle intermediate. Proc Natl Acad Sci U S A 70 : 2926–30.
38. BrownDD, DawidI (1968) Specific gene amplification in oocytes. Oocyte nuclei contain extrachromosomal replicas of the genes for ribosomal RNA. Science 160 : 272–80.
39. GallJG (1968) Differential synthesis of the genes for ribosomal RNA during amphibian oögenesis. Proc Nat Acad Sci USA 60 : 553–60.
40. AnderssonDI, HughesD (2009) Gene Amplification and Adaptive Evolution in Bacteria. Ann Rev Genet 43 : 167–95.
41. AlbertsonDG (2006) Gene amplifiction and cancer. Trend Genet 22 : 447–55.
42. ArcherSY, HodinRA (1999) Histon acetylation and cancer. Cur Opin Genet & Dev 9 : 171–74.
43. IdeS, KobayashiT (2010) Analysis of DNA replication in Saccharomyces cerevisiae by two-dimensional and pulsed-field gel electrophoresis. Curr Protoc Cell Biol Chapter 22: Unit 22 14.
44. MillerA, YangB, FosterT, KirchmaierAL (2008) Proliferating cell nuclear antigen and ASF1 modulate silent chromatin in Saccharomyces cerevisiae via lysine 56 on histone H3. Genetics 179 : 793–809.
45. NettikadanS, TokumasuF, TakeyasuK (1996) Quantitative analysis of the transcription factor AP2 binding to DNA by atomic force microscopy. Biochem Biophys Res Commun 1226 : 645–9.
46. CherryJM, HongEL, AmundsenC, BalakrishnanR, BinkleyG, et al. (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40(Database issue): D700–5.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy