Jakými kardiovaskulárními projevy se manifestuje transthyretinová amyloidóza (ATTR)? Kdy by měl kardiolog či internista pojmout podezření na toto onemocnění?
Postižení srdce transthyretinovou amyloidózou se nejčastěji projevuje známkami srdečního selhání, fibrilací síní a/nebo atrioventrikulárními blokádami. Podezření na srdeční amyloidózu vzniká při echokardiografickém vyšetření, pokud je přítomné ztluštění stěn levé komory srdeční (LKS) ≥ 12 mm, zvláště při absenci EKG známek hypertrofie LKS. Vysvětlením pro chybějící nárůst voltáže v EKG je infiltrace intersticia myokardu amyloidními fibrilami, jež neprodukují elektrický potenciál. Na přítomnost srdeční amyloidózy může ukázat také ztluštění volné stěny pravé komory, septa síní nebo vzácněji chlopenního aparátu, protože amyloidóza vede k difuznímu postižení stěn většiny srdečních oddílů. Relativně specifickou známkou srdeční amyloidózy je porucha kontrakce LKS v dlouhé ose s maximem postižení v bazálních segmentech a menším postižením hrotových segmentů (takzvaný apical sparing).
Nálezy na zobrazovacích metodách však nejsou specifické pro ATTR, v klinické praxi je třeba vždy vyloučit přítomnost AL amyloidózy při monoklonální gamapatii. Tyto dvě jednotky podmiňují většinu klinických případů srdeční amyloidózy.
Onemocnění bývá provázeno i dalšími systémovými, nekardiálními projevy. Které jsou pro něj typické?
ATTR je často doprovázena extrakardiálními příznaky, jakými jsou syndrom karpálního tunelu či ruptura distální šlachy bicepsu, někdy i hluchotou nebo stenózou páteřního kanálu a také progredující senzomotorickou a vegetativní polyneuropatií u některých hereditárních forem.
Co je třeba zvažovat v rámci diferenciální diagnostiky? Za která onemocnění bývá ATTR nejčastěji zaměňována?
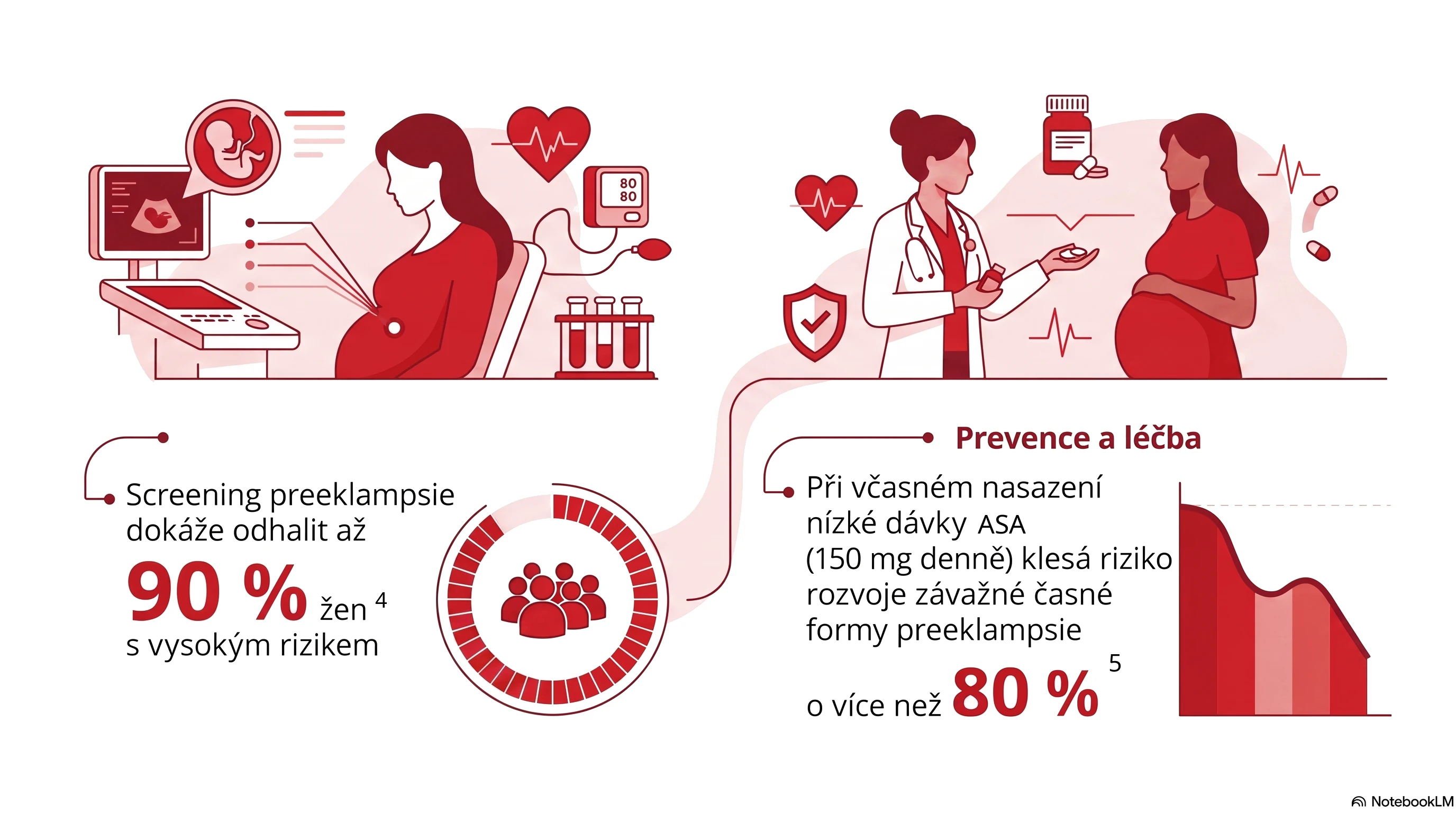
Při podezření na srdeční amyloidózu je třeba na prvním místě rozhodnout, zda se jedná u ATTR, nebo AL amyloidózu. U nemocných proto testujeme přítomnost monoklonální gamapatie pomocí imunofixace séra a moči, dále provádíme vyšetření na volné řetězce. Současně indikujeme scintigrafii s kostními izotopy (například 99mTc-DPD sken), která je vysoce senzitivním testem na přítomnost ATTR kardiomyopatie. Pokud vyloučíme monoklonální gamapatii, můžeme při jednoznačně pozitivní 99mTc-DPD scintigrafii stanovit diagnózu ATTR kardiomyopatie neinvazivně. V ostatních případech musíme typ srdečního amyloidu ověřit v biopsii, nejčastěji endomyokardiální.
Někdy jsou klinické známky a nálezy na zobrazovacích metodách nevýrazné. Nemocní se srdeční amyloidózou jsou potom sledováni pod jinými diagnózami, jakými jsou hypertrofická kardiomyopatie, srdeční selhání se zachovanou ejekční frakcí (HFpEF), méně významná aortální stenóza se symptomatologií srdečního selhání, fibrilace síní nebo pokročilá atrioventrikulární blokáda s implantovaným kardiostimulátorem. Část nemocných s ATTR můžeme najít také mezi jedinci se syndromem karpálního tunelu či polyneuropatií.
Můžete pro ilustraci uvést, jaký podíl pacientů s polyneuropatií, kardiomyopatií nebo HFpEF může mít současně ATTR?
Hereditární ATTR se vyskytuje zhruba u 2 % nemocných s progredující idiopatickou polyneuropatií. ATTR můžeme diagnostikovat u 2−3 % nemocných s hypertrofickou kardiomyopatií a přibližně u 13 % pacientů s HFpEF ve věku nad 65 let s tloušťkou septa komor ≥ 12 mm. ATTR je častým nálezem také u nemocných referovaných ke katetrizační náhradě aortální chlopně, s prevalencí 8−12 %.
Jakou úlohu má genetické testování a u koho jej indikovat? Které vyšetření definitivně potvrdí diagnózu ATTR?
Genetické vyšetření je indikované u všech nemocných s prokázanou ATTR. Je důležité pro objasnění etiologie onemocnění, indikaci screeningu v rodině, má prognostický význam a ovlivňuje rovněž volbu terapie. Definitivním průkazem ATTR je průkaz transthyretinového amyloidu v tkáni. Jak jsem již zmínil, existuje i možnost neinvazivní diagnostiky ATTR kardiomyopatie při akumulaci kostního izotopu v srdci a absenci monoklonální gamapatie.
Můžete v této souvislosti upřesnit rozdíl mezi wild-type a hereditární formou?
ATTR je podmíněna nestabilitou cirkulujících tetramerů plazmatické bílkoviny transthyretinu (TTR). Děje se tak vlivem stárnutí organismu u formy wild-type (wt; dříve senilní) nebo na základě genetické mutace u formy hereditární. V našich podmínkách má wt formu srdeční ATTR asi 97 % nemocných, ve zbývajících 3 % se jedná o formu hereditární (hATTR). Ta je autosomálně dominantně dědičná, s variabilní penetrancí a expresivitou.
Jaký je další postup u jedinců se zjištěnou mutací genu pro transthyretin?
U osob s hereditární ATTR doporučujeme genetický screening v rodině a genotypově pozitivní rodinné příslušníky dále vyšetřujeme a sledujeme. Podle typu mutace u nich aktivně pátráme po známkách srdečního postižení a/nebo polyneuropatie. Na základě nálezů upravujeme management onemocnění.
K dispozici je dnes specifická léčba dostupná ve specializovaných centrech. Za jakých podmínek ji lze nasadit a jak se liší terapeutická strategie podle typu onemocnění?
U jedinců s izolovanou hereditární ATTR polyneuropatií je podle tíže postižení indikovaná léčba stabilizátorem TTR tafamidisem nebo inhibitory syntézy TTR vutrisiranem či inotersenem. U obou forem srdeční ATTR je dosud jediným registrovaným lékem tafamidis, ale narůstá evidence také pro inhibitory syntézy TTR (ve 2. čtvrtletí 2025 bude vutrisiran schválen Evropskou lékovou agenturou /EMA/ na základě výsledků registrační studie HELIOS B pro pacienty s hATTR a ATTRwt kardiomyopatiemi ve třídě NYHA I−III). Léčba tafamidisem má u srdeční ATTR zatím úhradu jen ve funkční třídě NYHA I−II, u hereditární formy srdeční ATTR je třeba jeho úhradu schvalovat přes revizního lékaře.
Specifická léčba srdeční ATTR je poskytována ve 4 specializovaných centrech: Klinika kardiologie IKEM, 2. interní klinika − klinika kardiologie a angiologie 1. LF UK a VFN v Praze, 1. interní klinika − kardiologická LF UP a FN Olomouc, 1. interní kardioangiologická klinika LF MU a FN u sv. Anny. Léčba izolovaných neuropatických forem ATTR je zprostředkována v síti neuromuskulárních center (zatím nejvíce zkušeností s nimi mají ve FN Brno a FN Motol v Praze).
Jedná se o vzácné onemocnění, které může zůstat nediagnostikované. Jaký je osud nemocných, kteří unikají diagnóze a nejsou léčeni?
Medián přežívání u neléčené ATTRwt kardiomyopatie se pohybuje kolem 3−4 let v závislosti na vstupní pokročilosti onemocnění. Jedinci s hATTR kardiomyopatií potom mají v případě přítomnosti varianty V122I genu TTR významně horší přežívání než nemocní s ATTRwt kardiomyopatií nebo s hereditární ATTR kardiomyopatií na podkladě jiných variant TTR než V122I.
Jak může napomoci k včasnému záchytu mezioborová spolupráce a kde jsou její styčné body?
Pro dobrý management ATTR je zásadní spolupráce kardiologa, neurologa a praktického lékaře. Přítomnost syndromu karpálního tunelu u pacienta se srdečním selháním a ztluštěním stěn LKS při echokardiografii by praktického lékaře a neurologa měla vést k indikaci podrobnějšího kardiologického vyšetření. Naopak diagnóza hATTR by měla iniciovat kvalitní neurologické vyšetření a praktický lékař by měl koordinovat vyšetření a péči u dalších specialistů, jako jsou gastroenterologové, oftalmologové a nutricionisti. Genetické testování pak může potvrdit či vyloučit hereditární formu ATTR – v současné době je v Česku již plně dostupné vyšetření s výsledkem do 1 měsíce.
MUDr. Andrea Skálová
redakce proLékaře.cz