
The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
Embryonic development is tightly regulated by transcription factors and chromatin-associated proteins. H3K4me3 is associated with active transcription and H3K27me3 with gene repression, while the combination of both keeps genes required for development in a plastic state. Here we show that deletion of the H3K4me2/3 histone demethylase Jarid1b (Kdm5b/Plu1) results in major neonatal lethality due to respiratory failure. Jarid1b knockout embryos have several neural defects including disorganized cranial nerves, defects in eye development, and increased incidences of exencephaly. Moreover, in line with an overlap of Jarid1b and Polycomb target genes, Jarid1b knockout embryos display homeotic skeletal transformations typical for Polycomb mutants, supporting a functional interplay between Polycomb proteins and Jarid1b. To understand how Jarid1b regulates mouse development, we performed a genome-wide analysis of histone modifications, which demonstrated that normally inactive genes encoding developmental regulators acquire aberrant H3K4me3 during early embryogenesis in Jarid1b knockout embryos. H3K4me3 accumulates as embryonic development proceeds, leading to increased expression of neural master regulators like Pax6 and Otx2 in Jarid1b knockout brains. Taken together, these results suggest that Jarid1b regulates mouse development by protecting developmental genes from inappropriate acquisition of active histone modifications.
Published in the journal:
. PLoS Genet 9(4): e32767. doi:10.1371/journal.pgen.1003461
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003461
Summary
Embryonic development is tightly regulated by transcription factors and chromatin-associated proteins. H3K4me3 is associated with active transcription and H3K27me3 with gene repression, while the combination of both keeps genes required for development in a plastic state. Here we show that deletion of the H3K4me2/3 histone demethylase Jarid1b (Kdm5b/Plu1) results in major neonatal lethality due to respiratory failure. Jarid1b knockout embryos have several neural defects including disorganized cranial nerves, defects in eye development, and increased incidences of exencephaly. Moreover, in line with an overlap of Jarid1b and Polycomb target genes, Jarid1b knockout embryos display homeotic skeletal transformations typical for Polycomb mutants, supporting a functional interplay between Polycomb proteins and Jarid1b. To understand how Jarid1b regulates mouse development, we performed a genome-wide analysis of histone modifications, which demonstrated that normally inactive genes encoding developmental regulators acquire aberrant H3K4me3 during early embryogenesis in Jarid1b knockout embryos. H3K4me3 accumulates as embryonic development proceeds, leading to increased expression of neural master regulators like Pax6 and Otx2 in Jarid1b knockout brains. Taken together, these results suggest that Jarid1b regulates mouse development by protecting developmental genes from inappropriate acquisition of active histone modifications.
Introduction
Embryonic development is characterized by a coordinated program of proliferation and differentiation that is tightly regulated by transcription factors and chromatin-associated proteins. As embryonic cells differentiate, certain genes are activated while others are repressed, resulting in a unique pattern of gene expression in each cell type.
Histone H3 lysine 4 tri-methylation (H3K4me3) localizes to transcription start sites with high levels present at actively transcribed genes [1], [2], even though H3K4me3 at promoters is not a definite indication for transcriptional activity [3]. Methylation of H3K4 is catalyzed by a family of 10 histone methyltransferases in mammals [4]. Five of these are members of the Trithorax group of proteins that were first described in Drosophila to be required for maintenance of Hox gene expression by counteracting Polycomb-mediated repression. In Mll1 and Mll2 mutant mice, target genes are properly activated but expression fails to be maintained leading to embryonic lethality [5], [6]. In addition, H3K4 histone methyltransferases function in hematopoiesis [7], [8] and neurogenesis [9].
H3K4me3 is found in a constant balance with Polycomb-mediated repressive H3K27me3. Presence of both H3K4me3 and H3K27me3 at promoters is referred to as bivalency [10]. The category of bivalent genes is enriched in developmental regulators and is particularly abundant in embryonic stem cells (ESCs) that have the potential for several lineage choices [11]. Moreover, Polycomb proteins repress non-lineage specific gene expression, thereby ensuring developmental potency of embryonic and tissue stem cells during lineage specification, differentiation and development (reviewed in [12]). Polycomb proteins are classified into two separate complexes referred to as Polycomb repressive complex 2 (PRC2), which mediates H3K27me3, and PRC1, which catalyzes mono-ubiquitylation of H2A (H2AK119ub1) [13], [14]. Classical models propose a sequential mechanism in which H3K27me3 creates a binding site for PRC1 leading to further repression [14], [15], even though emerging studies suggest that Polycomb function is more complex [16]–[18].
While histone methylation was initially viewed as a stable modification, the discovery of histone demethylating enzymes has changed this paradigm [19]. Demethylation of H3K4me3 is catalyzed by the JARID1 (KDM5) family, which in mammals has four members: JARID1A, JARID1B, JARID1C and JARID1D [20]. The Drosophila JARID1 homologue LID (Little imaginal discs) is required for normal development [21], and the C. elegans homologue RBR-2 (retinoblastoma binding protein related 2) regulates vulva formation and lifespan [22], [23]. Mice mutant for Jarid1a are viable, displaying only mild phenotypes in hematopoiesis and behavior [24]. A recent report suggests that Jarid1b mutant mice are embryonic lethal between E4.5 and E7.5 [25]. The molecular mechanisms underlying this phenotype were not addressed. In contrast, others obtained viable Jarid1b mutant mice [26]. However, the requirement of Jarid1b for the differentiation of ESCs along the neural lineage [27], [28] suggests that Jarid1b may function in mouse development. In humans, JARID1B is highly expressed in several types of cancer, and it was shown to regulate proliferation of breast cancer cells and a slow cycling population of melanoma cells that promotes prolonged tumor growth (reviewed in [20]).
While the role of Jarid1b in mice remains controversial [25], [26], an understanding of its in vivo function is essential to direct future studies evaluating JARID1B as a potential drug target in cancer therapy. Jarid1b expression has been reported in various tissues during mouse embryogenesis whereas its expression becomes restricted in adults [29]. Here we report the first detailed analysis of the contribution of Jarid1b to mouse development. We show that Jarid1b is required for the proper development of several neural systems in the mouse and address the mechanisms underlying the observed defects.
Results
Knockout of Jarid1b leads to post-natal lethality in the majority of pups
To characterize the function of Jarid1b during mouse development, we generated constitutive Jarid1b knockout mice. Conditionally targeted Jarid1b mice containing a lacZ-Neo-reporter cassette flanked by FRT sites and in which Jarid1b exon 6 is flanked by loxP sites [28] were crossed with mice constitutively expressing Flp and Cre recombinase to obtain Jarid1b+/− mice. Jarid1b+/− mice were further intercrossed to generate Jarid1b−/− mice. Instead of the expected 25 percent of knockout mice, we only obtained 9.3 percent of adult Jarid1b knockouts (Figure 1A), suggesting that Jarid1b−/− mice are sub-viable. Analysis of early and late embryos from Jarid1b+/− intercrosses showed expected ratios while an increased number of Jarid1b knockouts was present among pups found dead during the first day after birth (Figure 1A), indicating that this might be the critical time for survival.
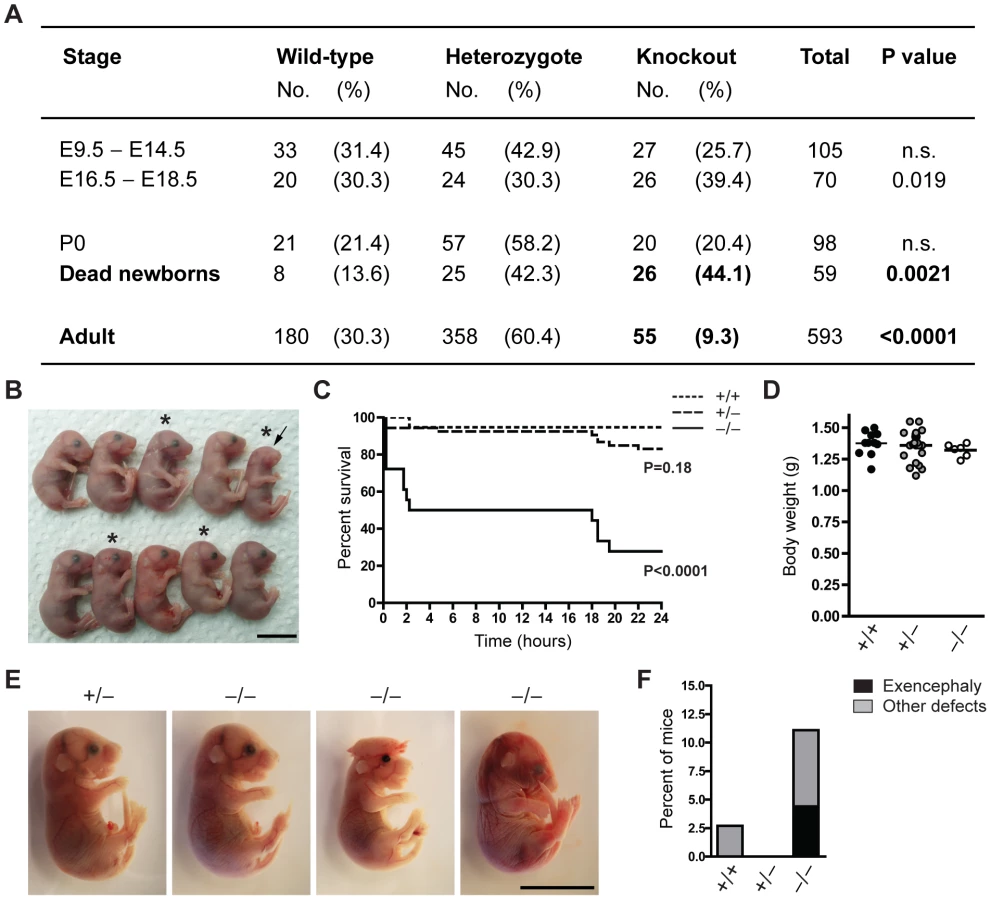
We have previously shown that conditional deletion of Jarid1b using this construct in vitro results in complete loss of Jarid1b protein and no generation of truncated or alternatively spliced variants [28]. Loss of Jarid1b in vivo was confirmed in all Jarid1b−/− embryos tested (see examples in Figure S1A and S1B), indicating that partial survival of Jarid1b knockouts in not due to incomplete deletion. Moreover, expression of other Jarid1 family members is unchanged both in vitro [28] and in vivo (Figure S1C).
To determine more precisely when Jarid1b−/− pups die, we performed caesarean deliveries and closely monitored the pups (Figure 1B). While approximately 95 percent of wild-type pups survive, we found that 50 percent of the knockouts die within the first two hours after delivery and another approximately 20 percent die after 14 to 24 hours (Figure 1C). All pups that survive the first day, develop normally until adulthood. Interestingly, survival of Jarid1b+/− pups is also slightly, even though not significantly, reduced during the first day. While most of the Jarid1b knockouts are grossly normal and not generally growth retarded (Figure 1D), we observed an increased incidence of developmental defects like exencephaly and eye defects among Jarid1b knockouts (Figure 1B, 1E and 1F). Taken together, loss of Jarid1b leads to major neonatal lethality of which only a small fraction can be explained by severe morphological abnormalities.
Respiratory function is not properly established in Jarid1b knockout pups
There is a large spectrum of physiological systems whose defects can challenge neonatal survival including those affecting parturition, breathing, suckling and neonatal homeostasis [30]. The first extrauterine challenge for neonates is breathing and since the majority of Jarid1b−/− pups die immediately after birth, we studied the respiratory system in more detail. Analysis of lungs from E18.5 fetuses revealed a normal size and weight (3.34±0.26 versus 3.45±0.44 percent body weight in heterozygotes versus knockouts, respectively) as well as a normal lobulation pattern (data not shown). Next, we isolated lungs from Jarid1b−/− newborns that had died within 2 hours after delivery and had either not shown any sign of breathing or exhibited gasping respiration (Figure 2A). While the wild-type lung showed saccular inflation, knockout lungs were compact and poorly inflated visible both from gross appearance and histology (Figure 2B and 2C), suggesting that Jarid1b−/− neonates die due to an inability to establish normal breathing. Moreover, preterm (E18.5) Jarid1b−/− lungs were abnormally compact compared to controls (Figure 2D), which might indicate a failure of prenatal breathing activity [31].
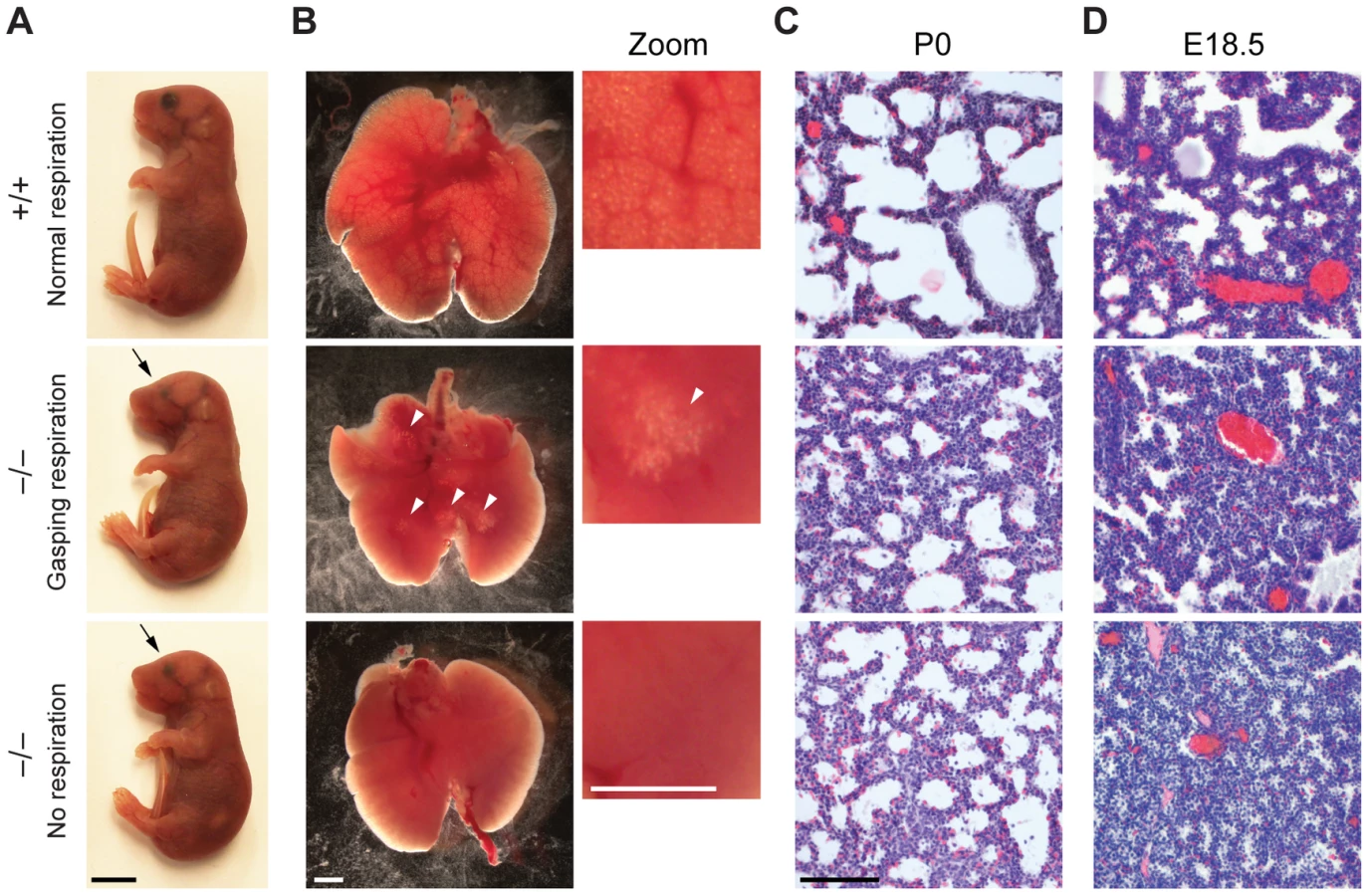
Respiratory failure might be caused by delayed lung maturation characterized by reduced surfactant expression [32]. Therefore, we analyzed expression of surfactant proteins (Sftpa1, Sftpb, Sftpc and Sftpd) in Jarid1b knockout mice (Figure S2A). None of the four surfactants was reduced in Jarid1b knockout lungs at E18.5, suggesting that respiratory failure is not due to pulmonary immaturity. In agreement with this, intrauterine administration of dexamethasone, a glucocorticoid that induces fetal lung maturation [33], did not improve survival of Jarid1b knockout pups (Figure S2B).
We also examined other physiological systems that are required for neonatal survival including the rib cage, diaphragm, craniofacial appearance and the palate as well as the cardiovascular system [30], but did not detect any abnormalities in the Jarid1b knockouts (Figure S3A–S3E). We conclude that while the lungs, skeletal and cardiovascular systems are properly developed, Jarid1b−/− neonates are unable to reliably establish respiratory function.
Central respiratory rhythmogenesis is intact despite abnormal breathing in knockouts
Immediate breathing after birth is also dependent on brainstem rhythmogenic and pattern forming neural circuits that develop before birth [34]. We therefore isolated brains from neonates after caesarean delivery, but found no gross abnormalities or differences in size of Jarid1b−/− brains compared to controls (Figure S3F and S3G). Essential rhythmogenic networks regulating breathing are located in the brainstem. Therefore, we recorded spontaneous C3–C5 nerve activity in an in vitro brainstem-spinal cord preparation from E18.5 embryos. Surprisingly, given the respiratory defects in newborn Jarid1b knockouts, central respiratory rhythmogenesis was unperturbed in Jarid1b−/− embryos (Figure S3H).
To monitor neurological reflexes of newborn Jarid1b−/− pups, we tested their response to pinching stimuli [35]. As opposed to control neonates, Jarid1b mutants only weakly reacted to a tail pinch (Figure S3I), suggesting that Jarid1b newborns show motosensory deficits characterized by hyporesponsiveness.
Cranial and spinal nerves are disorganized in Jarid1b knockout embryos
These results together with our previous in vitro data showing that Jarid1b is required for the differentiation of ESCs along the neural lineage [28] prompted us to analyze the development of neural systems in more detail in Jarid1b−/− embryos. As a first step we analyzed cranial nerves, a pair of 12 nerves that are essential for sensory and motor functions and reside in the mid - and hindbrain [36]. Defects in cranial nerve development may compromise neonatal survival. Cranial and spinal nerves can be visualized by whole-mount immunostaining at E10.5 using an anti-neurofilament antibody. Comparison of Jarid1b−/− embryos with controls revealed that while all nerve pairs are present, several cranial and spinal nerves are dysmorphic in the Jarid1b knockouts (Figure 3A). We used an arbitrary scoring system to quantify the differences between genotypes and found that Jarid1b knockouts are significantly affected while slight defects are already detectable in heterozygotes compared to wild-type (Figure 3B). Cranial nerves are involved in a diverse range of functions including movement of the eye, innervation of muscles of mastication, facial expression and tongue, and in transmitting information from chemoreceptors to the respiratory center [36], 37, and thus, defects in cranial nerve development may be relevant to reduced survival of Jarid1b knockouts. For example, the hypoglossal nerve (XII), which is dysmorphic in Jarid1b knockouts, innervates the muscles of the tongue, crucial for upper airway aperture during breathing.

Next, we analyzed Jarid1b expression during the time of mouse development when cranial nerves are specified. From embryonic day 8, the hindbrain becomes transiently partitioned along the anterior-posterior (AP) axis in a series of 8 rhombomeres that influence the spatial distribution of neuronal types [34]. Using the lacZ-Neo-reporter cassette present in the targeting construct [28], we observed high ubiquitous expression of Jarid1b in embryonic but not extraembryonic tissues at E8.5 (Figure S4). Moreover, in agreement with previous reports [38], at E12.5 and E14.5, Jarid1b expression was observed in several neural tissues including the fore - and hindbrain, neural retina, spinal cord and dorsal root ganglia as well as other tissues (Figures S5 and S6), indicating that Jarid1b could be involved in the development of several organs.
Cranial nerve development is imparted by genes involved in AP patterning and rhombomere specification, neuronal determination or survival and axonal migration [37]. Compartmentalization of the hindbrain, and in particular rhombomeres 3 and 4, have emerged as territories for the maintenance of breathing frequency after birth [34]. Rhombomeres are characterized by specific patterns of Hox gene, Krox20 (Egr2) and Kreisler (Mafb) expression, leading us to analyze expression of these genes by RNA in situ hybridization in Jarid1b−/− embryos. However, we did not observe any defects in the hindbrain patterning of E8.75 embryos (Figure 3C), suggesting that other mechanisms are responsible for spinal nerve abnormalities in Jarid1b−/− embryos.
Eye development is frequently disturbed in Jarid1b knockout embryos
In addition to sporadic cases of exencephaly, we frequently observed defects in eye development in Jarid1b−/− embryos and pups (Figure 4A–4D). In the most severe cases, eyes were completely absent (anophthalmia; Figure 4C). Other embryos exhibited microphthalmia (Figure 4B and 4D) or an incomplete closure of the optic fissure (Figure 4A and 4D). Moreover, after birth, the eyelid was often found open in Jarid1b−/− pups while it was closed in control mice at this time (Figure 4C). Altogether, externally visible eye defects were observed in approximately 22 percent of Jarid1b−/− embryos and pups (Figure 4E), but never in the Jarid1b knockouts that survive to adulthood. Histological analysis of two microphthalmic Jarid1b−/− eyes at E18.5 revealed a misfolding of the neural retina and a much smaller lense (Figure 4F and 4G). To test whether Jarid1b is expressed in the developing eye, we performed β-galactosidase stainings on sections of E12.5 and E14.5 eyes from targeted Jarid1b embryos (Figure 4H). At both stages, Jarid1b is specifically expressed in the inner layer of the neural retina, which contains retinal ganglion cells. Thus, Jarid1b seems required for the proper development of a mouse neurosensory organ, the eye.

Jarid1b knockout embryos display homeotic transformations of the skeleton
We have previously shown that Jarid1b binds to the transcription start sites of many developmental regulators in mouse ESCs, many of which are also bound by Polycomb group proteins [28]. Therefore, we speculated that Jarid1b might also regulate Polycomb target genes in vivo. Hox genes represent classical Polycomb targets and their misexpression in Polycomb mouse mutants results in transformations of the axial skeleton [39], [40].
To investigate whether such transformations are also present in Jarid1b mutants, we stained skeletal preparations of E17.5 embryos to visualize cartilage and bone. While we did not observe any defects in the anterior region of the vertebral column (occipito-cervico-thoracic region), we found a transformation of the 26th vertebra, which is supposed to be the last lumbar vertebra (L6) into the first sacral vertebrae (S1) (Figure 5A and 5B). Moreover, we also observed a transformation of the 34th vertebra (Figure 5B and Figure S7). Thus, Jarid1b−/− embryos display posterior transformations of the skeleton, which similar to Polycomb mutants are not completely penetrant [39], [40].

H3K4me3 is increased at normally repressed genes in early knockout embryos
To identify genes in addition to the Hox genes that might be misregulated in Jarid1b−/− embryos, we focused on an early embryonic stage (E8.5) where morphological defects were not yet observed. We expected that several of the phenotypes observed in the Jarid1b mutants arise from misspecification events early in development, as genes involved in eye specification, neural tube closure and hindbrain patterning start to be expressed from E8.0 [41], [42].
First, we performed chromatin immunoprecipitation (ChIP) followed by sequencing (seq) of head regions of E8.5 embryos (Figure S8A) for H3K4me3 and H3K27me3 to identify genes that change their chromatin state and thus might become misregulated in Jarid1b−/− embryos. By this analysis, we identified 492 peaks with increased H3K4me3 levels in Jarid1b knockouts versus heterozygotes, whereas only 27 peaks were detected in the reverse comparison (Figure S8B). Representative examples of loci with increased H3K4me3 in the knockouts as well as loci with unchanged chromatin states are shown in Figure 6A and 6B, respectively. The results were validated in an independent experiment by ChIP-qPCR showing that the differences in H3K4me3 are reproducible (Figure 6C). Comparison of genes with increased H3K4me3 in knockout embryos with all genes revealed an enrichment of repressed (H3K27me3 positive) and bivalent (H3K4me3/H3K27me3 positive) genes among genes with increased H3K4me3 (Figure 6D and Figure S8C), suggesting that aberrant active histone marks accumulate mainly at genes that are usually not actively transcribed. Gene ontology analysis of genes with increased H3K4me3 in the Jarid1b−/− embryos identified regulators of transcription and development including genes involved in ectoderm, nervous system and skeletal development as significantly overrepresented (Figure 6E and Figure S8D). To identify genes that are directly bound and regulated by Jarid1b, we also attempted ChIP experiments for Jarid1b in E8.5 embryos but unfortunately the results were of low quality due to very limited amounts of starting material. Instead, we compared genes with elevated H3K4me3 in Jarid1b−/− embryos with genes bound by Jarid1b in ESCs [28] and found that approximately one quarter was bound by Jarid1b in ESCs (Figure S8E). Thus, it is likely that some of the genes with increased H3K4me3 are also Jarid1b targets during early mouse development.

Next, we performed gene expression analysis of mRNA isolated from E8.5 Jarid1b heterozygotes and knockouts. Except for Jarid1b, we did not identify any genes that were more than 2-fold changed in the knockouts (Figure S8F). We validated a number of genes by RT-qPCR and confirmed that Jarid1b was not expressed in the knockouts, whereas Jarid1a and Jarid1c as well as L1cam and Pax2 remained unchanged (Figure S8G). Taken together, while we detected increased levels of H3K4me3 at a number of developmental regulators early in embryogenesis, these chromatin changes do not translate into detectable global transcriptional changes at this stage of development.
Increased levels of H3K4me3 in Jarid1b knockout embryos during development
Deletion of Jarid1b in ESCs leads to a global increase in H3K4me3, while global H3K4me3 levels remain unchanged in Jarid1b depleted neural stem cells isolated from E12.5 embryos [28]. Likewise, depletion of JARID1B in MCF7 cells [43] or depletion of Jarid1a in mouse embryonic fibroblasts [24] did not result in a global elevation of H3K4me3. To analyze the effect of Jarid1b depletion in vivo, we prepared protein extracts from different stages of embryos. We confirmed lack of Jarid1b protein in all knockout embryos analyzed (Figure 7A). While we detected little change in H3K4me3 by immunoblotting in heads of E12.5 (data not shown) and E14.5 Jarid1b−/− embryos, global H3K4me3 levels were strongly increased in heads of late (E17.5) Jarid1b−/− embryos and in forebrains of Jarid1b−/− newborns (Figure 7A). These results suggest that H3K4me3 accumulates in Jarid1b knockouts as embryonic development proceeds, while H3K4me3 levels remain fairly constant during normal fetal development (Figure S9).

Increased H3K4me3 at transcription start sites in brains of newborn Jarid1b knockouts
Next, we wanted to know at which classes of genes H3K4me3 accumulates in brains of newborn mice. Since the brain is a complex and heterogeneous organ, we first determined whether Jarid1b expression is limited to specific regions at this stage of brain development. However, β-galactosidase stainings on sections of brains from newborns revealed high overall expression of Jarid1b (Figure 7B). In addition, RT-qPCR analysis showed similar expression of Jarid1b in fore - and hindbrain, which is reduced in heterozygotes and lost in knockouts (Figure 7C). Thus, we divided the brain into forebrain and hindbrain for ChIP experiments and selected a number of genes that represent different chromatin states (Figure 7D–7G and S10). We observed increased H3K4me3 at repressed (H3K27me3-positive) genes, including Otx2, Pax9, HoxB5 and Hesx1, and at active (H3K4me3-positive) genes (Sema5b), but not at unmodified genes in P0 forebrains. Some bivalent genes, for example Pax6, showed increased H3K4me3 and slightly reduced H3K27me3, while others remained unchanged (e.g. Neurod2). Similar results were obtained in independent ChIP experiments using forebrain or hindbrain. These data suggest that H3K4me3 is increased at transcription start sites in late stages of brain development.
To test which genes are directly bound by Jarid1b, we also performed ChIP for Jarid1b (Figure 7D–7G and Figure S10). We detected Jarid1b binding at transcription start sites of bivalent (Pax6) and H3K4me3-positive (Sema5b) genes, which is in agreement with our previous findings in ESCs [28]. Moreover, we detected low levels of Jarid1b binding (2 - to 4-fold above background) at several of the H3K27me3-positive loci with increased H3K4me3 in the Jarid1b knockouts (Otx2, Pax9, Hoxb5), suggesting that elevated levels of H3K4me3 at many of these loci are due to a loss of direct association of Jarid1b.
Increased expression of several key transcription factors in Jarid1b knockouts
To determine whether changes in chromatin modifications are accompanied by differences in expression, we performed RT-qPCR in P0 brains of controls and Jarid1b knockouts (Figure 7D–7F and Figure S10). We detected increased levels of the transcription factor Otx2 in forebrains of Jarid1b−/− newborns. Furthermore, in line with a shifted balance of H3K4me3 versus H3K27me3, expression of the neural master regulator Pax6 was increased in Jarid1b−/− P0 brains. In contrast, expression of actively transcribed genes, like Sema5b, was unchanged despite higher levels of H3K4me3. We conclude that Jarid1b mutants accumulate higher levels of H3K4me3 and show increased expression of genes important for regulating embryonic development.
Next, we analyzed at which stage between E8.5 and P0 changes in gene expression arise in Jarid1b knockouts. While we did not detect transcriptional changes at E8.5, expression of Otx2, Pax6 and Sema5b was increased in heads of E12.5 Jarid1b knockout embryos compared to controls (Figure S11A). Since the transcription factor Pax6 controls the balance between neural stem cell (NSC) self-renewal and neurogenesis [44], we tested whether deletion of Jarid1b affected this balance. Sorting of NSCs and neuronal progenitor cells (NPs) from E12.5 brains (Figure S11B) revealed a slight (but not significant) increase in NSCs in Jarid1b knockouts and no change in NPs. Similarly, global levels of neuron and astrocyte markers remained unchanged in P0 brains (Figure S9B), which is in agreement with normal gross morphology of Jarid1b knockout brains (Figure S3F). Thus, the detectable changes in gene expression observed in Jarid1b knockout mice does not appear to be a result of abnormal numbers of NSCs or NPs.
Finally, we tested whether increased expression of Otx2 and Pax6 correlated with survival. However, as shown in Figure S11C, we did not detect a significant difference in expression of Otx2 and Pax6 in brains of newborns that were alive 2 hours after caesarean delivery versus newborns that died immediately. In contrast, the expression of Otx2 was significantly higher in the adult brain of surviving knockout animals as compared to wild type (Figure S11D), suggesting that transcriptional regulation by Jarid1b is not restricted to embryogenesis only, but affects selected genes rather than global transcription.
Discussion
Embryonic development is regulated by transcription factors as well as chromatin-mediated processes resulting in tissue-specific gene expression. Here, we show that the histone demethylase Jarid1b is required for faithful mouse embryonic development (see model in Figure S12). Deletion of Jarid1b results in major neonatal lethality caused by an inability of the newborn mice to establish breathing. Jarid1b mutant embryos display a number of defects related to neural systems, including the misorganization of cranial and spinal nerves as well as increased incidence of exencephaly that might contribute to neonatal lethality. Respiratory rhythmogenic circuits in the brainstem of Jarid1b mutant embryos appear intact since a spontaneous motor output on cervical nerves was observed under in vitro conditions. In agreement, Krox20 and Kreisler, essential genes involved in specification of respiratory-related rhombomeres, are also not affected in mutant embryos. Thus, we speculate that the breathing problems of Jarid1b mutant neonates may stem from either compromised pattern forming circuits controlling airway patency, or an inability of the rhythmic motor output to reach respiratory muscles, caused by defects in cranial and spinal nerve development. Several other organs important after birth appeared undisturbed. However, the spectrum of physiological systems required for neonatal survival is large [30] and we cannot exclude that there are other subtle defects that manifest in secondary physiological problems interfering with survival of Jarid1b knockouts.
Previous in vitro studies of ESCs with either reduced [28] or increased [27] levels of Jarid1b have reported a role for Jarid1b during differentiation of ESCs into neurons. This raises the question of why Jarid1b is specifically required in neural systems. During embryogenesis, Jarid1b is expressed in several neural organs including the brain, spinal cord and eye, but also in a number of other systems (this study, [38]). Moreover, in ESCs, Jarid1b is targeted to transcription start sites of genes that regulate development, including genes involved in neurogenesis and ectoderm development [28]. Thus, a combination of tissue-specific expression and target gene selectivity might explain neural-specific phenotypes. It should be noted, however, that other systems are also affected by Jarid1b depletion, exemplified by homeotic transformation of the skeleton (this study) or slightly reduced expression of meso - and endodermal markers during embryoid body differentiation of ESCs [28]. Interestingly, knockdown of Jarid1b in the retina of newborn mice leads to abnormal morphology of rod photoreceptor cells and misregulation of rod-expressed genes [45], supporting a role for Jarid1b in neuronal cells of the eye. In addition, other Jarid1 family members have reported functions in behavior and/or neurulation, suggesting that these processes are susceptible to changes in H3K4 methylation. Jarid1a knockout mice display abnormal clasping of the hindlimbs [24], while mutations of human JARID1C occur in patients with X-linked mental retardation [46]. Knockout of Jarid1c in the mouse results in embryonic lethality due to defects in neurulation and cardiogenesis [47]. Taken together, while several Jarid1 family members are involved in the control of neural systems, they may regulate different aspects of development and cannot fully compensate for the absence of other Jarid1 members resulting in gene-specific phenotypes.
To determine how Jarid1b contributes to the regulation of mouse development, we analyzed global as well as gene-specific histone methylation levels in Jarid1b−/− embryos. Consistent with the previously reported catalytic activity of Jarid1b [23], [43], H3K4me3 was increased around transcription start sites of developmental regulators already early during mouse development, particularly at genes that are normally in a repressed or poised state. At this early stage of development, we could not detect any global changes in gene expression levels, but we cannot exclude that expression of some genes might be affected in a subset of cells as the E8.5 mouse embryo is composed of many distinct cell layers. For example, the development of the eye initiates at E8.0 with the evagination of the optic pit from a subset of cells in the diencephalon [41]. In analogy, increased H3K4me3 at transcription start sites may reflect small increases in many cells of the early embryo or result from large increases in a subset of cells. Increased H3K4me3 around transcription start sites may render the associated genes more susceptible to later activation, especially during developmental time windows or in specific cell types where additional signaling molecules create a competent transcriptional state.
As embryonic development proceeds, a global increase in H3K4me3 becomes detectable. Locally, similar to early embryos, increased H3K4me3 is present at the transcription start sites of genes in Jarid1b−/− brains. Even though altered H3K4me3 per se may not be sufficient to induce transcriptional changes or cell fate conversions [48], it may have functional consequences when prompt responses to signaling events are required or for the fine control of steady state transcript levels [49]. Indeed, we detected increased expression of Pax6 and Otx2, two master regulators of eye and neural development (reviewed in [50], [51]), in forebrains of Jarid1b−/− pups. For both Pax6 and Otx2, it was shown that not only deletion but also overexpression affect eye and neural lineage development [44], [52], [53].
Binding of Jarid1b itself was found at H3K4me3-positive genes (both active and bivalent), which is in agreement with previous ChIP-seq data [28], [54]. Interestingly, the overlap between Jarid1b and Polycomb target genes was functionally supported in this study by the observation that Jarid1b knockout embryos show homeotic transformations of the skeleton, which is a hallmark of Polycomb mutant mice. Moreover, the observation that Jarid1b is bound to several repressed genes that are marked by aberrant H3K4me3 in the knockouts, suggests that Jarid1b is directly required to prevent aberrant accumulation of active chromatin modifications at developmental regulators during embryogenesis.
Most of the phenotypes that we observed in Jarid1b knockouts occurred with incomplete penetrance. This is not uncommon and has been reported for other histone demethylases [55] but also transcription factors [56]. The penetrance is often affected by the genetic background of the mice [50], [55]. This might also explain the difference in survival of our knockout mice compared to a previous study [25]. To test this hypothesis, we crossed our Jarid1b mutant mice that were derived on a C57BL/6 background into a mixed C57BL/6/129 genetic background (Figure S13). On the mixed background, 40% of knockouts die after birth, compared to 70% on a C57BL/6 background. Besides, we observed a similar frequency of exencephaly and a reduced response of the newborns to pinching stimuli, while no eye phenotypes were detected on the mixed background. These results suggest that different genetic background of mice strains used, could partly explain the divergence of obtained results.
Moreover, many disease-causing mutations only have detrimental defects in a subset of individuals, and phenotypic discordance remains even in the absence of genetic and environmental variation. It was shown that feedback induction of genes with related functions differs across individuals leading to a buffering of stochastic developmental failure through redundancy [57]. In the Jarid1b mutants, we did not observe upregulation of other Jarid1 members at the transcript level. However, we cannot exclude that protein levels are increased, or that these proteins are preferentially recruited to Jarid1b target sites to compensate for lack of Jarid1b. In addition, systematic analysis of transcript levels and their correlation with phenotypes has shown that variability in gene expression underlies incomplete penetrance [58]. It was proposed that fluctuations in gene expression can be controlled by wild-type developmental networks. In contrast to transcription factors that can induce cell fate conversions, chromatin modification are thought to rather fine tune transcription. In Jarid1b mutant embryos, both repressed and bivalent genes acquire increased levels of H3K4me3, which might render these genes more susceptible to unscheduled activation. Indeed, we observed increased expression of Pax6 and Otx2 in newborn knockout brains. Raj et al. [58] propose a model in which expression must surpass a threshold during a window of development. The same might be true for histone modifications. In plants, progressive increase in H3K27me3 was shown to be capable of switching a bistable epigenetic state of an individual locus [59]. While global levels of H3K4me3 are unchanged during early embryogenesis, H3K4me3 accumulates in the Jarid1b knockouts as embryonic development proceeds. For some genes or in specific cell types, this might lead to a switch in the balance of active versus repressive histone modifications, and if this coincides with a developmental window of transcriptional potency, it might affect phenotypic outcomes. Moreover, since Jarid1b binds to a large number of target genes [28], it could be expected that a wide range of phenotypes with varying severity is observed in Jarid1b knockout embryos.
The functions of histone demethylases in vivo are starting to emerge, however, in many cases the mechanisms of their action remain to be elucidated. Here, we present a detailed analysis of the role of the H3K4me2/3 demethylase Jarid1b during mouse development. In the adult organism, Jarid1b expression is also observed but it becomes more restricted. Since high expression of Jarid1b is detected during meiosis and in adult testis [29] as well as in several types of cancer [26], Jarid1b has been proposed to belong to the family of testis-cancer antigens [60]. In future studies, it will be very interesting to characterize the function of Jarid1b in adult mice as Jarid1b presents a potential drug target for anti-cancer therapies and an understanding of its in vivo role will help to guide targeting efforts.
Materials and Methods
Animals
The derivation of targeted (Jarid1bNeo/+) and conditional (Jarid1bF/F) Jarid1b mice has been previously described [28]. Conditional Jarid1b mice were crossed with Cmv-cre transgenic mice [61] to obtain heterozygous mice, which were further inter-crossed to generate Jarid1b knockouts. Jarid1b mice were maintained on a C57BL/6 background, unless otherwise stated. All mouse work was approved by the Danish Animal Ethical Committee (“Dyreforsøgstilsynet”).
For cesarean deliveries, timed matings were setup using Jarid1b+/− mice to generate experimental pups. Jarid1b+/+ mice were used for foster mothers. Pregnant Jarid1b+/− females were injected subcutaneously with 100 µl of Promon (50 mg/ml, Boheringer Ingelheim) at E16.5 and E18.5 to prevent natural birth. Pups were delivered at E19.5 by caesarean, massaged gently to stimulate breathing and placed on a 37°C warm plate during initial examination. Pups were then placed with a foster mother and examined regularly during the first 24 hours after delivery. Dexamethasone (Sigma) or saline control was administered subcutaneous (0.4 mg/kg) to pregnant females at E17.5 and E18.5 [33].
Histology
Histological analysis was performed according to standard procedures. Briefly, embryos or tissues were fixed over night in 4% buffered formaldehyde and subsequently incubated in baths of buffered formaldehyde, 96% ethanol, 99% ethanol, xylene and paraffin. Paraffin blocks were cut into 8–10 µm sections on a microtome (Microm HM355S). Sections were deparaffinised, stained with hematoxylin and eosin, dehydrated and mounted using VectaMount (Vector).
Nerve recordings in brainstem–spinal cord preparations
The neuraxis in caesarean delivered E18.5 embryos was removed by dissection in an ice cold, oxygenated (95% O2, 5% CO2) solution containing 250 mM glycerol, 3 mM KCl, 5 mM KH2PO4, 36 mM NaHCO3, 10 mM D-(+)-glucose, 2 mM MgSO4 and 0.7 mM CaCl2. Brainstem-spinal cord preparations, which contained the entire brainstem and the cervical part of the spinal cord, were placed in a 2 ml recording chamber with a temperature of 29°C and was constantly superfused at a rate of 2 ml/min with preheated oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid solution (ACSF). The ACSF solution contained 130 mM NaCl, 5.4 mM KCl, 0.8 mM KH2PO4, 26 mM NaHCO3, 30 mM D-(+)-glucose, 1 mM MgCl2 and 0.8 mM CaCl2. Glass-pipette suction electrodes (tip-diameter of 40–160 µm, A-M Systems, Carlsborg, USA) were placed on C3, C4, or C5 rootlets to record spontaneous respiratory-related nerve-activity. Nerve potentials were amplified by a custom-built nerve amplifier (×50,000), filtered at DC-2 KHz, and digitized (2.5 KHz) by a PCI–6289, M Series A/D-board (National Instruments, Austin, USA) controlled by Igor Pro (Wavemetrics, Lake Oswego, USA) software.
Beta-galactosidase stainings
For whole-mount beta-galactosidase stainings, embryos were fixed in PBS with 0.25% glutaraldehyde for 10–30 min, washed in PBS and stained with PBS containing 0.02% NP40/IGEPAL, 0.01% sodium deoxycholic acid, 2 mM MgCl2, 20 mM Tris-HCl pH 7.4, 5 mM Potassium ferrocyanide and 5 mM Potassium ferricyanide until desired colour intensity. After post-fixation in 4% paraformaldehyde over night at 4°C, embryos were passed through a glycerol gradient incubating several days at each concentration, and finally stored in 100% glycerol.
For beta-galactosidase stainings of cryo-sections, embryos were fixed in 0.2% paraformaldehyde over night at 4°C, incubated in PBS with 2 mM MgCl2 and 30% sucrose over night at 4°C, embedded in OCT (TissueTek, Sakura) and stored at −80°C. Samples were cut into 8 µm sections on a cryostat (Leica CM3050), post-fixed in 0.2% paraformaldehyde for 10 min on ice, washed in PBS with 2 mM MgCl2, incubated in PBS with 2 mM MgCl2, 0.01% deoxycholic acid and 0.02% NP40 for 20 min on ice, and stained as described above. Sections were washed, counter-stained with eosin, passed through an ethanol gradient, incubated in xylene and mounted in VectaMount (Vector).
In situ hybridisation
Whole-mount in situ hybridization of mouse embryos was performed as previously described [62].
Neurofilament staining
Embryos were isolated at E10.5, fixed in 4% paraformaldehyde for 2 hours and stored in 100% methanol at −20°C. After bleaching with methanol/H2O2, embryos were rehydrated, blocked in PBS with 2% milk and 0.1% triton (PBSMT) and stained over night at 4°C with anti-neurofilament antibody (Developmental Studies Hybridoma Bank, 2H3, 1∶50). Embryos were washed in PBSMT, and incubated over night at 4°C with peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, 111-035-146, 1∶500). After several washes in PBSMT, embryos were incubated in 0.3 mg/ml DAB (Sigma) in 0.5% NiCl2 for 30 min, H2O2 was added to a concentration of 0.0003% and the embryos incubated until the desired colour intensity was obtained. Finally, embryos were dehydrated through a methanol gradient and cleared in 1∶2 benzyl alcohol∶benzyl benzoate in glass containers.
Skeletal preparations
For skeletal preparation, embryos were isolated at E17.5, eviscerated and the skin removed. Embryos were fixed over night in 100% ethanol, rinsed in 95% ethanol and stained over night in 0.15 mg/ml Alcian Blue in 95% ethanol containing 20% glacial acidic acid. Embryos were washed in 95% ethanol, cleared in 1% KOH for 4 hours and stained with 50 mg/l Alizarin Red in 1% KOH over night at 4°C. Final clearing of embryos was performed in 1% KOH for 3 hours and through a gradient of 1% KOH/glycerol until final storage in 100% glycerol.
Antibodies
Antibodies used in this study include anti-Jarid1b (DAIN) [28], anti-H3K4me3 (Cell Signaling, C42D8), anti-H3K27me3 (Cell Signaling, D18C8), anti-H3 (Abcam, 1791), anti-H4 (Millipore, 05-858), anti-ß-III-tubulin (Sigma, T8660), anti-Glial Fibrillary Acidic Protein (GFAP) (DakoCytomation, Z0334), anti-Vinculin (Sigma, V9131) and anti-ß-tubulin (Santa Cruz, sc-9104).
Flow cytometry
Whole brain from E12.5 was dissociated, filtered, resuspended in PBS with 5% FBS and stained for 20 min on ice using the following antibodies: anti-Prominin-1-biotin (MACS Miltenyi Biotec, 130-092-441), anti-CD-15-FITC (BD Biosciences 332778), anti-A2B5-APC (MACS Miltenyi Biotec, 130-093-582) and anti-CD24 (BD Biosciences, 553262) [63]. Cells were washed, incubated with secondary fluorescent-conjugated antibodies for 20 min on ice, washed again and resuspended in buffer for viability dye staining containing 7AAD (BioLegend 420404). Cells were analyzed on a FACS Aria flow cytometer (BD Biosciences). Single viable cells were gated into the following populations: Neural progenitors: CD15 low, Prominin low, CD24 high; Neural stem cells: CD15 high, Prominin high, CD24 low.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) and ChIP-sequencing were performed as previously described [28]. MACS2 [64] was used to identify regions with increased histone methylation. For E8.5 embryos, five heads of embryos with 3–8 somites were pooled per ChIP, corresponding to approximately 100,000 cells in total. For ChIP and expression analysis of P0 brains, single brains were divided into fore - and hindbrain.
Base-calling and demultiplexing of raw sequencing data was performed using the standard Illumina pipeline (CASAVA, version 1.8.2) followed by alignment to the mouse genome (mm9 assembly) with bowtie (version 0.12.7) [65] using the following parameters: -S -m 1 mm9. Samtools (version 0.1.18) [66] and bedtools (version 0.1.18) [67] were applied for conversion of files between alignment formats. Furthermore, reads were extended to a total length of 250 bp (estimated DNA fragement size) in the 3′ direction. Various command-line utilities from UCSC (http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/) were used to generate normalized bigwig track files for viewing in the UCSC genome browser. The files were normalized to tags per million after removing duplicate reads. Peak calling was performed in MACS2 [64] using the following settings: –broad -f BAM -g mm.
Gene expression analysis
Gene expression analysis was previously described [28]. For RNA isolation of E8.5 embryos, the RNAeasy Microkit (Qiagen) was used. For microarray analysis, four heads of E8.5 embryos were pooled per sample and three biological replicates analyzed. RNA was hybridized on mouse Gene 1.0 ST arrays by the RH Microarray Center at Rigshospitalet, Copenhagen, following Affymetrix procedures. Microarray data was analyzed using Gene Array Analyzer software [68] with default settings, P-value<0.05 and a log2 fold change of +/−1. The ChIP-seq and microarray data have been submitted to the Gene Expression Omnibus (GEO) database (GSE41174). Primer sequences are provided in Table S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BarskiA, CuddapahS, CuiK, RohT-Y, SchonesDE, et al. (2007) High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 129 : 823–837.
2. SchübelerD, MacAlpineDM, ScalzoD, WirbelauerC, KooperbergC, et al. (2004) The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes & Development 18 : 1263–1271.
3. GuentherMG, LevineSS, BoyerLA, JaenischR, YoungRA (2007) A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130 : 77–88.
4. HublitzP, AlbertM, PetersAHFM (2009) Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol 53 : 335–354.
5. YuBD, HessJL, HorningSE, BrownGA, KorsmeyerSJ (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378 : 505–508.
6. GlaserS, SchaftJ, LubitzS, VinterstenK, van der HoevenF, et al. (2006) Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 133 : 1423–1432.
7. YagiH, DeguchiK, AonoA, TaniY, KishimotoT, et al. (1998) Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 92 : 108–117.
8. ErnstP, FisherJK, AveryW, WadeS, FoyD, et al. (2004) Definitive hematopoiesis requires the mixed-lineage leukemia gene. Developmental Cell 6 : 437–443.
9. LimDA, HuangY-C, SwigutT, MirickAL, Garcia-VerdugoJM, et al. (2009) Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458 : 529–533.
10. BernsteinBE, MikkelsenTS, XieX, KamalM, HuebertDJ, et al. (2006) A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 125 : 315–326.
11. PietersenAM, van LohuizenM (2008) Stem cell regulation by polycomb repressors: postponing commitment. Current Opinion in Cell Biology 20 : 201–207.
12. MargueronR, ReinbergD (2011) The Polycomb complex PRC2 and its mark in life. Nature 469 : 343–349.
13. CaoR, WangL, WangH, XiaL, Erdjument-BromageH, et al. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298 : 1039–1043.
14. CaoR, TsukadaY-I, ZhangY (2005) Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Molecular Cell 20 : 845–854.
15. WangL, BrownJL, CaoR, ZhangY, KassisJA, et al. (2004) Hierarchical recruitment of polycomb group silencing complexes. Molecular Cell 14 : 637–646.
16. SchoeftnerS, SenguptaAK, KubicekS, MechtlerK, SpahnL, et al. (2006) Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. The EMBO Journal 25 : 3110–3122.
17. PuschendorfM, TerranovaR, BoutsmaE, MaoX, IsonoK-I, et al. (2008) PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat Genet 40 : 411–420.
18. TavaresL, DimitrovaE, OxleyD, WebsterJ, PootR, et al. (2012) RYBP-PRC1 Complexes Mediate H2A Ubiquitylation at Polycomb Target Sites Independently of PRC2 and H3K27me3. Cell 148 : 664–678.
19. ShiY, LanF, MatsonC, MulliganP, WhetstineJR, et al. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119 : 941–953.
20. KooistraSM, HelinK (2012) Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13 : 297–311.
21. GildeaJJ, LopezR, ShearnA (2000) A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics 156 : 645–663.
22. GreerEL, MauresTJ, UcarD, HauswirthAG, ManciniE, et al. (2011) Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479 : 365–371.
23. ChristensenJ, AggerK, CloosPAC, PasiniD, RoseS, et al. (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128 : 1063–1076.
24. KloseRJ, YanQ, TothovaZ, YamaneK, Erdjument-BromageH, et al. (2007) The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128 : 889–900.
25. CatchpoleS, Spencer-DeneB, HallD, SantangeloS, RosewellI, et al. (2011) PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol 38 : 1267–1277.
26. BlairLP, CaoJ, ZouMR, SayeghJ, YanQ (2011) Epigenetic Regulation by Lysine Demethylase 5 (KDM5) Enzymes in Cancer. Cancers 3 : 1383–1404.
27. DeyBK, StalkerL, SchnerchA, BhatiaM, Taylor-PapidimitriouJ, et al. (2008) The Histone Demethylase KDM5b/JARID1b Plays a Role in Cell Fate Decisions by Blocking Terminal Differentiation. Molecular and Cellular Biology 28 : 5312–5327.
28. SchmitzSU, AlbertM, MalatestaM, MoreyL, JohansenJV, et al. (2011) Jarid1b targets genes regulating development and is involved in neural differentiation. The EMBO Journal 30 : 4586–4600.
29. MadsenB, TarsounasM, BurchellJM, HallD, PoulsomR, et al. (2003) PLU-1, a transcriptional repressor and putative testis-cancer antigen, has a specific expression and localisation pattern during meiosis. Chromosoma 112 : 124–132.
30. TurgeonB, MelocheS (2009) Interpreting Neonatal Lethal Phenotypes in Mouse Mutants: Insights Into Gene Function and Human Diseases. Physiological Reviews 89 : 1–26.
31. MoseleyAE, LieskeSP, WetzelRK, JamesPF, HeS, et al. (2003) The Na,K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J Biol Chem 278 : 5317–5324.
32. WhitsettJA, WeaverTE (2002) Hydrophobic surfactant proteins in lung function and disease. N Engl J Med 347 : 2141–2148.
33. KlingerS, TurgeonB, LévesqueK, WoodGA, Aagaard-TilleryKM, et al. (2009) Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc Natl Acad Sci USA 106 : 16710–16715.
34. BordayC, ChatonnetF, Thoby-BrissonM, ChampagnatJ, FortinG (2005) Neural tube patterning by Krox20 and emergence of a respiratory control. Respiratory Physiology & Neurobiology 149 : 63–72.
35. GomezaJ, HülsmannS, OhnoK, EulenburgV, SzökeK, et al. (2003) Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron 40 : 785–796.
36. GuthrieS (2007) Patterning and axon guidance of cranial motor neurons. Nat Rev Neurosci 8 : 859–871.
37. CordesSP (2001) Molecular genetics of cranial nerve development in mouse. Nat Rev Neurosci 2 : 611–623.
38. MadsenB, Spencer-DeneB, PoulsomR, HallD, LuPJ, et al. (2002) Characterisation and developmental expression of mouse Plu-1, a homologue of a human nuclear protein (PLU-1) which is specifically up-regulated in breast cancer. Mech Dev 119 (Suppl 1) S239–S246.
39. AkasakaT, KannoM, BallingR, MiezaMA, TaniguchiM, et al. (1996) A role for mel-18, a Polycomb group-related vertebrate gene, during theanteroposterior specification of the axial skeleton. Development 122 : 1513–1522.
40. del Mar LorenteM, Marcos-GutiérrezC, PérezC, SchoorlemmerJ, RamírezA, et al. (2000) Loss - and gain-of-function mutations show a polycomb group function for Ring1A in mice. Development 127 : 5093–5100.
41. ChowRL, LangRA (2001) Early eye development in vertebrates. Annu Rev Cell Dev Biol 17 : 255–296.
42. AlexanderT, NolteC, KrumlaufR (2009) HoxGenes and Segmentation of the Hindbrain and Axial Skeleton. Annu Rev Cell Dev Biol 25 : 431–456.
43. YamaneK, TateishiK, KloseRJ, FangJ, FabrizioLA, et al. (2007) PLU-1 Is an H3K4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Molecular Cell 25 : 801–812.
44. SansomSN, GriffithsDS, FaedoA, KleinjanD-J, RuanY, et al. (2009) The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet 5: e1000511 doi:10.1371/journal.pgen.1000511.
45. HaoH, KimDS, KlockeB, JohnsonKR, CuiK, et al. (2012) Transcriptional regulation of rod photoreceptor homeostasis revealed by in vivo NRL targetome analysis. PLoS Genet 8: e1002649 doi:10.1371/journal.pgen.1002649.
46. JensenLR, AmendeM, GurokU, MoserB, GimmelV, et al. (2005) Mutations in the JARID1C Gene, Which Is Involved in Transcriptional Regulation and Chromatin Remodeling, Cause X-Linked Mental Retardation. The American Journal of Human Genetics 76 : 227–236.
47. CoxBJ, VollmerM, TamplinO, LuM, BiecheleS, et al. (2010) Phenotypic annotation of the mouse X chromosome. Genome Res 20 : 1154–1164.
48. JiangH, ShuklaA, WangX, ChenW-Y, BernsteinBE, et al. (2011) Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 144 : 513–525.
49. El-SamadH, MadhaniHD (2011) Can a Systems Perspective Help Us Appreciate the Biological Meaning of Small Effects? Developmental Cell 21 : 11–13.
50. HeverAM, WilliamsonKA, van HeyningenV (2006) Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet 69 : 459–470.
51. OsumiN, ShinoharaH, Numayama-TsurutaK, MaekawaM (2008) Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells 26 : 1663–1672.
52. SchedlA, RossA, LeeM, EngelkampD, RashbassP, et al. (1996) Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell 86 : 71–82.
53. WorthamM, JinG, SunJL, BignerDD, HeY, et al. (2012) Aberrant Otx2 expression enhances migration and induces ectopic proliferation of hindbrain neuronal progenitor cells. PLoS ONE 7: e36211 doi:10.1371/journal.pone.0036211.
54. Lloret-Llinares M, Pérez-Lluch S, Rossell D, Morán T, Ponsa-Cobas J, et al.. (2012) dKDM5/LID regulates H3K4me3 dynamics at the transcription-start site (TSS) of actively transcribed developmental genes. Nucleic Acids Res.
55. FukudaT, TokunagaA, SakamotoR, YoshidaN (2011) Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly. Molecular and Cellular Neuroscience 46 : 614–624.
56. HarrisMJ, JuriloffDM (2010) An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Research Part A: Clinical and Molecular Teratology 88 : 653–669.
57. BurgaA, CasanuevaMO, LehnerB (2011) Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature 480 : 250–253.
58. RajA, RifkinSA, AndersenE, van OudenaardenA (2010) Variability in gene expression underlies incomplete penetrance. Nature 463 : 913–918.
59. AngelA, SongJ, DeanC, HowardM (2011) A Polycomb-based switch underlying quantitative epigenetic memory. Nature 476 : 105–108.
60. SimpsonAJG, CaballeroOL, JungbluthA, ChenY-T, OldLJ (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5 : 615–625.
61. SchwenkF, BaronU, RajewskyK (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23 : 5080–5081.
62. VitobelloA, FerrettiE, LampeX, VilainN, DucretS, et al. (2011) Hox and Pbx Factors Control Retinoic Acid Synthesis during Hindbrain Segmentation. Developmental Cell 20 : 469–482.
63. PanchisionDM, ChenH-L, PistollatoF, PapiniD, NiH-T, et al. (2007) Optimized flow cytometric analysis of central nervous system tissue reveals novel functional relationships among cells expressing CD133, CD15, and CD24. Stem Cells 25 : 1560–1570.
64. ZhangY, LiuT, MeyerCA, EeckhouteJ, JohnsonDS, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137.
65. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
66. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
67. QuinlanAR, HallIM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 : 841–842.
68. GellertP, TeranishiM, JennichesK, De GaspariP, JohnD, et al. (2012) Gene Array Analyzer: alternative usage of gene arrays to study alternative splicing events. Nucleic Acids Res 40 : 2414–2425.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The G4 Genome
- Neutral Genomic Microevolution of a Recently Emerged Pathogen, Serovar Agona
- The Histone Demethylase Jarid1b Ensures Faithful Mouse Development by Protecting Developmental Genes from Aberrant H3K4me3
- The Tissue-Specific RNA Binding Protein T-STAR Controls Regional Splicing Patterns of Pre-mRNAs in the Brain
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy