
Promotion of Bone Morphogenetic Protein Signaling by Tetraspanins and Glycosphingolipids
The bone morphogenetic protein (BMP) signaling pathway is required for multiple developmental processes during metazoan development. Various diseases, including cancer, can result from mis-regulation of the BMP pathway. Thus, it is critical to identify factors that ensure proper regulation of BMP signaling. Using the nematode C. elegans, we have devised a highly specific and sensitive genetic screen to identify new modulators in the BMP pathway. Through this screen, we identified three conserved tetraspanin molecules as novel factors that function to promote BMP signaling in a living organism. We further showed that these three tetraspanins likely form a complex and function together with glycosphingolipids to promote BMP signaling. Recent studies have implicated several tetraspanins in cancer initiation, progression and metastasis in mammals. Our findings suggest that the involvement of tetraspanins in cancer may partially be due to their function in modulating the activity of BMP signaling.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005221
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005221
Summary
The bone morphogenetic protein (BMP) signaling pathway is required for multiple developmental processes during metazoan development. Various diseases, including cancer, can result from mis-regulation of the BMP pathway. Thus, it is critical to identify factors that ensure proper regulation of BMP signaling. Using the nematode C. elegans, we have devised a highly specific and sensitive genetic screen to identify new modulators in the BMP pathway. Through this screen, we identified three conserved tetraspanin molecules as novel factors that function to promote BMP signaling in a living organism. We further showed that these three tetraspanins likely form a complex and function together with glycosphingolipids to promote BMP signaling. Recent studies have implicated several tetraspanins in cancer initiation, progression and metastasis in mammals. Our findings suggest that the involvement of tetraspanins in cancer may partially be due to their function in modulating the activity of BMP signaling.
Introduction
Bone morphogenetic proteins (BMPs) belong to the transforming growth factor β (TGFβ) superfamily of secreted polypeptides that regulate a variety of developmental and homeostatic processes [1, 2]. The TGFβ ligands are synthesized as precursor proteins that can be subsequently processed by proteases [3]. Active TGFβ ligands bind to a heterotetrameric receptor complex composed of type I and type II receptors, leading to the phosphorylation of the type I receptor by the type II receptor. The phosphorylated type I receptor then phosphorylates and activates the receptor-regulated Smads (R-Smads). The activated R-Smads form a complex with common-mediator Smads (Co-Smads) and enter the nucleus to regulate downstream gene expression. Malfunction of the TGFβ pathway can result in numerous somatic and hereditary disorders in humans, including various cancers, bone skeletal disorders, and cardiovascular diseases [4–7]. Multiple levels of regulation ensure proper spatiotemporal activity of TGFβ signaling in the correct cellular context [8–11]. Identifying factors involved in modulating the TGFβ pathway and determining their modes of action in vivo will not only provide valuable insights into our understanding of TGFβ signaling, but may also provide therapeutic targets for the many diseases caused by alterations in TGFβ signaling.
C. elegans, with its wealth of genetic and molecular tools and its well-defined cell lineage, provides an excellent model system to study the functions and modulation of TGFβ signaling during the development of a whole organism at single-cell resolution. There are at least three TGFβ-related pathways in C. elegans: one that controls dauer formation, one that regulates axon guidance and cell migration, and a third BMP-like “Sma/Mab” pathway that regulates body size and male tail formation, among its multiple functions [12]. The Sma/Mab pathway includes a BMP-like molecule DBL-1 [13, 14], the type I receptor SMA-6 [15], the type II receptor DAF-4 [16], the R-Smads SMA-2 and SMA-3, and the Co-Smad SMA-4 [17]. Loss-of-function mutations in any component of this pathway will cause small body size and male tail sensory ray formation defects [12].
We have previously shown that the Sma/Mab pathway also plays a role in patterning the C. elegans postembryonic mesoderm. The hermaphrodite postembryonic mesodermal M lineage arises from a single pluripotent precursor cell, the M mesoblast. During larval development, the M mesoblast divides to produce a dorsal lineage that gives rise to striated bodywall muscles (BWMs) and macrophage-like coelomocytes (CCs), as well as a ventral lineage that produces BWMs and the sex muscle precursor cells, the sex myoblasts (SMs) ([18]; Fig 1A and 1C and 1E). This dorsoventral asymmetry is regulated by the schnurri homolog sma-9 [19]. Mutations in sma-9 lead to a dorsal-to-ventral fate transformation in the M lineage ([20]; Fig 1B and 1D and 1F). We have shown that mutations in the core components of the Sma/Mab pathway (Fig 2A) do not cause any M lineage defect on their own, but they suppress the dorsoventral patterning defects of sma-9 mutants, suggesting that SMA-9 regulates M lineage dorsoventral patterning by antagonizing Sma/Mab signaling [20]. Using this sma-9 M lineage suppression phenotype (Fig 1A and 1C and 1E), we have recently identified two new modulators of the Sma/Mab pathway, the RGM protein DRAG-1 and the DCC/neogenin homolog UNC-40, which directly associate with each other to positively regulate Sma/Mab signaling [21, 22, Fig 2A]. We further showed that their functions in modulating BMP signaling are evolutionarily conserved [22, 23].


In this study, we describe our identification and analysis of additional sma-9 M lineage phenotype suppressors that function in modulating Sma/Mab signaling. One novel modulator is TSP-21, which belongs to a family of transmembrane molecules called tetraspanins [24]. Tetraspanins are a distinct family of integral membrane proteins that have four conserved transmembrane (TM) domains separated by a small extracellular loop (EC1), a small intracellular loop (IL) and a large extracellular loop (EC2). They are known to interact with each other to form homo - and hetero-oligomers, and organize membranes into the so-called tetraspanin-enriched microdomains that are also enriched in cholesterol and glycosphingolipids [24–27]. There are 33 tetraspanins in humans and 21 in the C. elegans genome. The in vivo functions of most of these tetraspanins are not well understood. Here we provide evidence that TSP-21, the C. elegans ortholog of human TSPAN4, TSPAN9 and CD53, is localized to the cell membrane and functions positively to regulate Sma/Mab signaling in the signal-receiving cells at the ligand-receptor level. We further show that two additional tetraspanins that belong to the C8 subfamily of tetraspanins, TSP-12 and TSP-14, also function to promote Sma/Mab signaling. TSP-12 and TSP-14 can physically interact with each other, with TSP-21, and with the type I receptor of the Sma/Mab pathway, SMA-6. In addition, we find that mutants defective in glycosphingolipid biosynthesis exhibit defects in Sma/Mab signaling. Collectively, our results provide in vivo evidence supporting the roles of tetraspanins and glycosphingolipid-enriched membrane microdomains in modulating BMP signaling. Finally, we provide evidence that like TSP-12 and TSP-14, which have been previously shown to function in promoting LIN-12/Notch signaling [28], TSP-21 also appears to function in LIN-12/Notch signaling in a cell type-specific manner.
Results
Mutations in the BMP-like Sma/Mab pathway specifically suppress the sma-9 M lineage phenotype
We have previously shown that mutations in all core components of the Sma/Mab pathway, but not the TGFβ-like dauer pathway, can suppress the M lineage phenotype of sma-9(0) mutants [20]. Here we show that null mutations in unc-129, which encodes a TGFβ-like molecule important for axon guidance [29], or null mutations in genes that regulate body size but do not function in the Sma/Mab pathway, such as the β-spectrin gene sma-1 [30], or the cuticle collagen gene lon-3 [31, 32], do not suppress the M lineage phenotype of sma-9(0) mutants (Table 1). Similarly, mutations in the Sma/Mab pathway do not suppress the M lineage defect of let-381(RNAi), which also leads to a dorsal-to-ventral fate transformation defect in the M lineage by inactivating the FoxF/FoxC transcription factor LET-381 ([33]; Table 1). In contrast, two deletion alleles of sma-10, which encodes a conserved, leucine-rich repeats - and immunoglobulin (Ig)-like domain (LRIG)-containing transmembrane protein that promotes Sma/Mab signaling in regulating body size [34, Fig 2A], do suppress the M lineage phenotype of sma-9(0) mutants (Table 1).

Motivated by the specificity of the BMP-like Sma/Mab pathway mutants in suppressing the sma-9 M lineage defect and our previous success from the sma-9 suppressor screen in the identification of evolutionarily conserved modulators of BMP signaling, such as DRAG-1/RGM and UNC-40/DCC/neogenin [21, 22], we performed a large-scale screen for sma-9 suppressors, named susm (suppressor of sma-9) mutations, with the aim of identifying additional modulators of BMP signaling (see Materials and Methods). Using a combination of linkage analysis, complementation tests and whole genome sequencing (see Materials and Methods), we identified the corresponding genes for 32 susm mutations. As shown in Table 2, our suppressor screen successfully and specifically identified mutations in all core members and known modulators of the Sma/Mab pathway. Intriguingly, we isolated lon-1(jj67) as a sma-9 suppressor and showed that an existing, strong loss-of-function allele, lon-1(e185), also suppresses the sma-9 M lineage phenotype (Tables 1 and 2). lon-1 encodes a member of the cysteine-rich secretory protein (CRISP) family of proteins and is known to function downstream of, and be negatively regulated by, the Sma/Mab pathway [13, 35, Fig 2A]. The suppression of the sma-9 M lineage phenotype by lon-1 mutations and the increased expression of the Sma/Mab responsive reporter RAD-SMAD [21] in lon-1(jj67) mutants (Fig 3B) suggest that LON-1 may exert feedback regulation on Sma/Mab signaling, rather than being strictly regulated by this pathway (Fig 2A). In addition to these factors known to function in Sma/Mab signaling, we also identified a novel factor defined by two non-complementing alleles, jj60 and jj77 (Table 2).


Mutations in the membrane protein tetraspanin-21 (TSP-21) are novel sma-9 suppressors
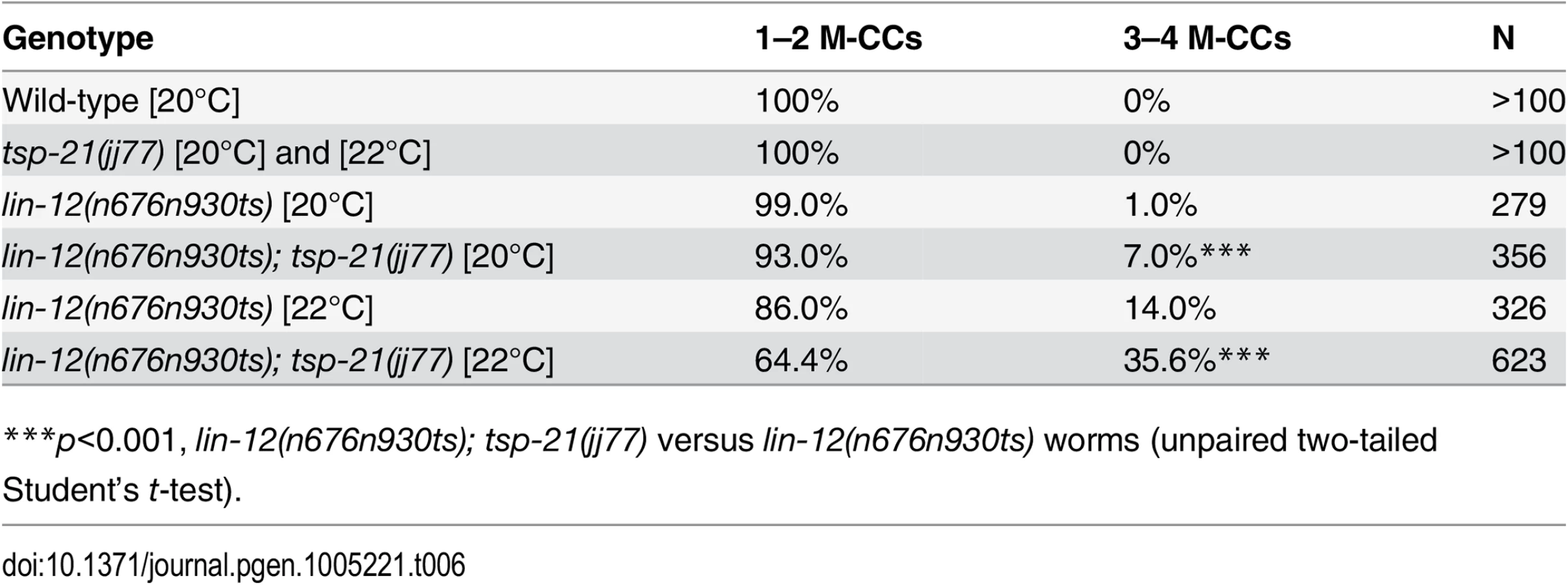
We performed whole genome sequencing (WGS) of the novel complementation group that includes jj60 and jj77 to identify the corresponding gene. Four lines of evidence indicate that the corresponding gene is tsp-21. 1) RNAi of tsp-21 suppressed the sma-9 M lineage phenotype (Table 3). 2) Both jj60 and jj77 contain molecular lesions in tsp-21 (Fig 4A): jj60 contains a G-to-A change in nucleotide 1827, resulting in a Glycine (G) to Glutamic acid (E) change of amino acid 109 (G109E). jj77 contains a T-to-A change in nucleotide 3327, resulting in a Valine (V) to Glutamic acid (E) change of amino acid 236 (V236E). jj77 also carries a 84bp deletion (between nucleotides 3202 and 3287) and a 6bp insertion (ATCTCT), resulting in a 13 amino acid deletion and 2 amino acid insertion between amino acids 209 and 223. 3) A DNA fragment containing the genomic sequence of tsp-21 (5kb upstream, entire coding region including introns, and 1.7kb downstream sequences for C17G1.8 in pJKL1005, Fig 4B) rescued the Susm phenotype of jj77 (Table 3). 4) A deletion allele of tsp-21 that we recently obtained, tm6269, exhibited defects similar to those of jj77 and jj60 animals (Fig 4B and Table 3).

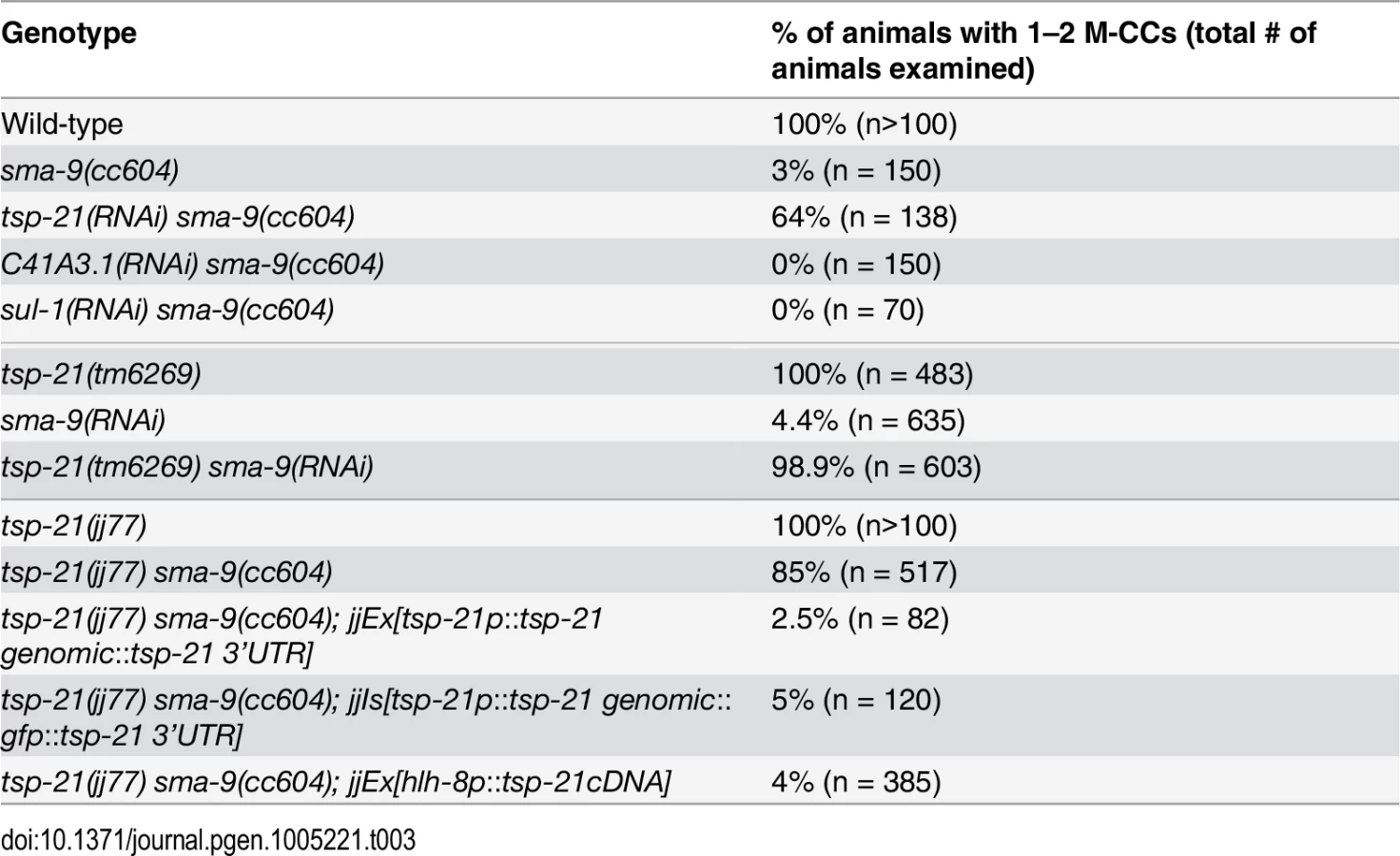
tsp-21 encodes a conserved but previously unstudied 301 amino acid transmembrane protein of the tetraspanin family, TSP-21 (Fig 4C). Based on the number of cysteine (C) residues in the large EC2 loop, tetraspanins can be classified into three groups, C4, C6 and C8 [36, 37]. TSP-21 belongs to the C6a group, with the following configuration of cysteine residues in EC2: CCG——CC——C——C (Fig 4C). The closest vertebrate homologs of TSP-21 are TSPAN4, TSPAN9 and CD53 (Figs 4C and S1). These proteins, except for CD53, share the conserved C6a configuration in the EC2 loop as well as conserved transmembrane (TM) domains (Fig 4C). The G109E mutation in jj60 affects the last residue of TM3 (Fig 4A and 4C), and likely results in the production of a partial loss-of-function TSP-21 protein. The deleted residues of TSP-21 in jj77 mutants include one of the six highly conserved cysteine residues in EC2 (Fig 4C). jj77 also contains a missense mutation in a conserved residue in TM4 (V236E, Fig 4), and is therefore likely a strong loss-of-function, or likely null, allele of tsp-21. We have therefore used jj77 for all our subsequent analyses.
TSP-21 positively modulates the Sma/Mab pathway at the level of the ligand-receptor complex
Because our sma-9 suppressor screen is highly specific in identifying components of the BMP-like Sma/Mab pathway, we examined tsp-21(jj77) mutants for any additional Sma/Mab signaling defects, such as body size, male tail patterning and expression of RAD-SMAD, a Sma/Mab signaling reporter that we previously generated [21]. tsp-21(jj77) animals are smaller than wild-type animals (Fig 2B–2E) and exhibited reduced RAD-SMAD reporter expression (Fig 3). Unlike mutants in core members of the Sma/Mab pathway, tsp-21(jj77) mutant males can mate and they do not exhibit any significant male tail patterning defects (based on examining 100 sides of tsp-21(jj77) male tails). These tsp-21 phenotypes are very similar to those exhibited by null mutants in two previously identified Sma/Mab pathway modulators, drag-1 and unc-40 [21, 22]. Furthermore, like mutants in other members of the Sma/Mab pathway [20–22], tsp-21(jj77) mutants exhibited no M lineage defects (Table 3). Finally, tsp-21(jj77) mutants showed no dauer defects, and tsp-21 exhibited no genetic interaction with daf-1 and daf-7, two genes functioning in the TGFβ-like dauer pathway ([12]; S1 Table). Collectively, these phenotypic analyses suggest that TSP-21 positively modulates the BMP-like Sma/Mab pathway, but does not appear to play a role in the TGFβ-like dauer pathway.
The smaller body size of tsp-21(jj77) mutants allowed us to use genetic epistasis to determine where in the Sma/Mab pathway TSP-21 functions. We generated double mutants between tsp-21(jj77) and null mutations in various Sma/Mab pathway components (Fig 2A) and measured their body sizes. As shown in Fig 2E, dbl-1(wk70); tsp-21(jj77) and sma-3(jj3); tsp-21(jj77) double mutants were as small as dbl-1(wk70) and sma-3(jj3) single mutants, respectively. These observations are consistent with TSP-21 functioning in the Sma/Mab pathway in regulating body size. lon-1(jj67); tsp-21(jj77) double mutants were as long as lon-1(jj67) single mutants, while lon-2(e678) tsp-21(jj77) double mutants showed intermediate body size between lon-2(e678) and tsp-21(jj77) single mutants (Fig 2E), suggesting that tsp-21 is likely to function upstream of lon-1, but in parallel to lon-2, in the Sma/Mab pathway. drag-1(jj4); tsp-21(jj77) and unc-40(e1430); tsp-21(jj77) double mutants, or drag-1(jj4) unc-40(e1430); tsp-21(jj77) triple mutants were significantly smaller than each respective single mutant (Fig 2E), which is consistent with tsp-21 functioning in parallel to drag-1 and unc-40. Taken together, these results indicate that TSP-21 functions at the ligand-receptor level to positively modulate Sma/Mab signaling.
TSP-21 is localized to the plasma membrane and functions in the hypodermis and M lineage, which are Sma/Mab signal-receiving cells
To determine how TSP-21 functions in the Sma/Mab pathway, we examined the expression and localization pattern of TSP-21. We first generated integrated transgenic lines carrying a translational TSP-21::GFP fusion (pJKL1004, see Materials and Methods) that contains the entire tsp-21 genomic region including 5kb 5’ sequences and 1.7kb 3’ sequences (Fig 4B). This translational fusion rescued the Susm phenotypes of tsp-21(jj77) mutants (Table 3). Subsequently we generated the same fusion in the endogenous tsp-21 locus via CRISPR-Cas9 mediated homologous recombination (see Materials and Methods). Both reporters showed that TSP-21::GFP is plasma membrane-localized and is expressed in a wide variety of somatic cell types, including the pharynx, intestine and hypodermis starting in embryos after the 100 cell stage (Fig 5A–5C at mid-embryogenesis) and peaking in L1 and L2 larvae (Fig 5D–5L). The TSP-21::GFP signal in these tissues decreases in late larval and adult stage animals. TSP-21::GFP is also present at the surface of M lineage cells from the 1-M stage to the 16-M stage (Fig 5S–5Z). In addition to expression in these tissues, the two TSP-21::GFP lines generated via the CRISPR-Cas9 system also showed GFP expression in the somatic gonad and vulva in L2-L4 larvae and adults (Fig 5M–5O), as well as in the rectal epithelium in L4 larvae (Fig 5P–5R). These observations suggest that the enhancer elements for tsp-21 expression in the somatic gonad, the vulva and the rectal epithelium lie outside of the 10.5kb tsp-21 genomic region included in pJKL1004.

We noticed that TSP-21::GFP is enriched in the basolateral side of intestinal cells while being absent from their apical sides (Fig 5G–5I). A similar localization pattern has been reported for the type I receptor SMA-6 and the type II receptor DAF-4 [38]. In addition, while TSP-21::GFP in the M lineage cells is primarily plasma membrane localized, there is also a significant intracellular distribution of TSP-21::GFP in the M mesoblast (Fig 5S and 5W). At present, the functional significance of either the asymmetric localization of TSP-21::GFP in intestinal cells or its intracellular localization in the M cell is not clear.
The Sma/Mab pathway is known to function in the hypodermal cells to regulate body size and in the M lineage to regulate M lineage development. We next tested whether TSP-21 functions in these cell types to exert its role in Sma/Mab signaling. Using cell-type-specific promoters to drive tsp-21 expression, we found that forced expression of tsp-21 cDNA in hypodermal cells, but not in pharyngeal or intestinal cells, rescued the small body size phenotype of tsp-21(jj77) mutants (Table 4). Similarly, forced expression of tsp-21 cDNA in the M lineage also rescued the Susm phenotype of tsp-21(jj77) mutants (Table 3). Thus TSP-21 functions autonomously in the signal-receiving cells to promote Sma/Mab signaling.

TSP-12 and TSP-14 function redundantly to promote Sma/Mab signaling
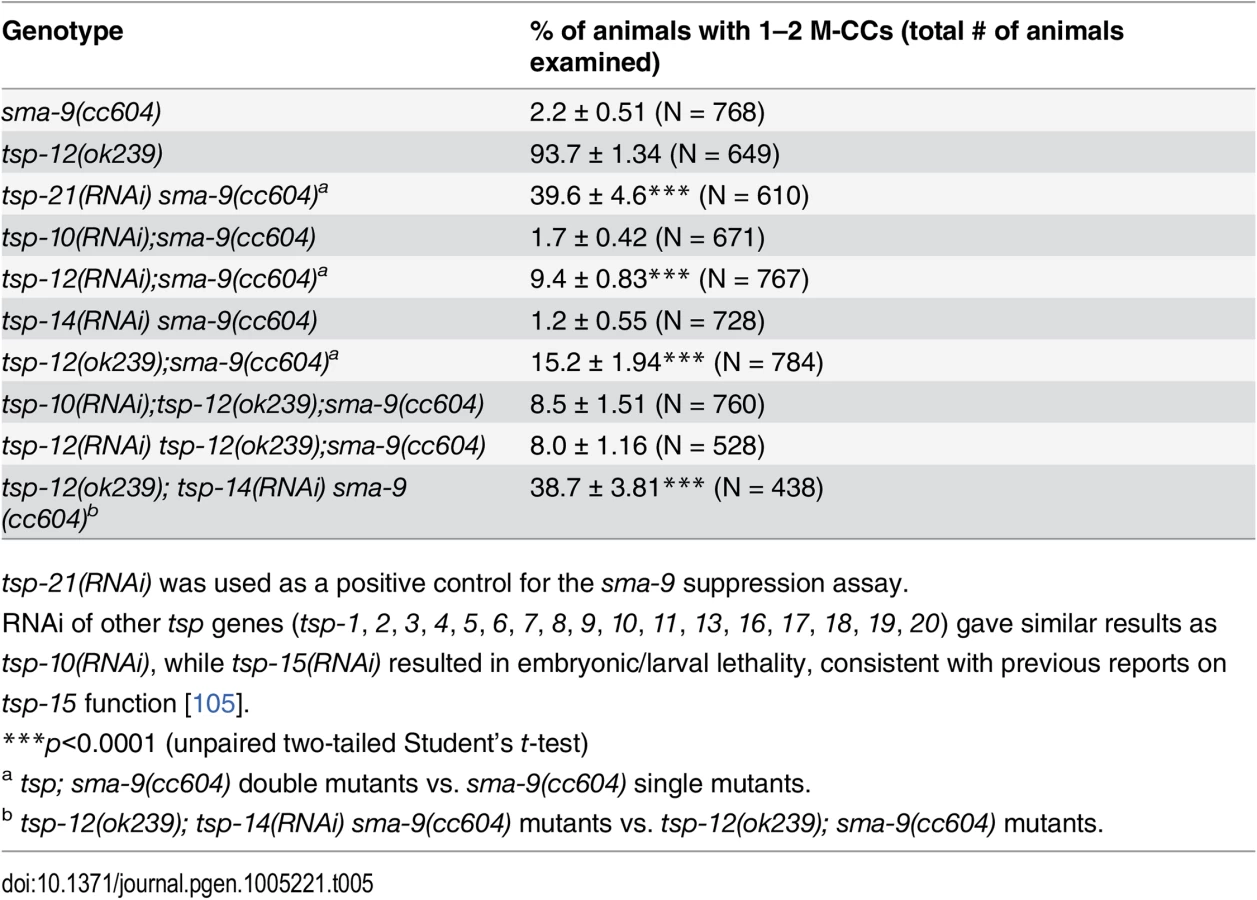
There are 21 tetraspanins in C. elegans. Our finding that TSP-21 functions in Sma/Mab signaling prompted us to ask whether other tetraspanins might also function in the Sma/Mab pathway. We therefore screened through the remaining 20 tetraspanin tsp genes by RNAi injection, testing whether any of them are involved in Sma/Mab signaling using the sma-9 suppression assay. Only tsp-12(RNAi) resulted in a low penetrance (9.4%, n = 767) Susm phenotype (Table 5). We then tested a deletion allele of tsp-12, ok239, and found that it also exhibited the Susm phenotype (Table 5), suggesting that the tetraspanin TSP-12 also plays a role in modulating Sma/Mab signaling. Dunn and colleagues [28] have previously reported that TSP-12 and TSP-14 function redundantly to promote Notch signaling. We asked whether tsp-14 and tsp-12 might also share a redundant role in the Sma/Mab pathway, and found that tsp-14(RNAi) enhanced the penetrance of the Susm phenotype of the tsp-12(ok239) mutation (Table 5). This effect appears to be specific since tsp-10(RNAi) failed to enhance the penetrance of the Susm phenotype of the tsp-12(ok239) mutation (Table 5). Thus, TSP-12 and TSP-14 also function redundantly to promote Sma/Mab signaling, in addition to their role in Notch signaling.
TSP-21 positively modulates LIN-12/Notch signaling in the M lineage
The dual functions of TSP-12 and TSP-14 in both the Notch and the Sma/Mab signaling pathways prompted us to examine whether TSP-21 also functions in the Notch signaling pathway. The LIN-12/Notch signaling pathway is known to function in the M lineage to promote the ventral fate: loss of LIN-12/Notch function results in a ventral-to-dorsal fate transformation in the M lineage, namely the loss of M-derived SMs and the gain of M-derived CCs ([39, 40]; S2 Fig). tsp-21(jj77) single mutants exhibit no M lineage defects. We therefore examined whether tsp-21(jj77) could enhance the M lineage defect of the lin-12 temperature sensitive, partial loss-of-function allele lin-12(n676n930ts) by scoring the number of M-derived CCs. As shown in Table 6, tsp-21(jj77) significantly enhanced the M lineage defect of lin-12(n676n930ts) at both 20°C and 22°C, suggesting that TSP-21 functions to promote LIN-12/Notch signaling in the M lineage. However, tsp-21(jj77) failed to enhance the sterility and embryonic lethality of bn18ts, a mutation in the second Notch receptor gene in C. elegans, glp-1 [41]. The lack of genetic interaction between tsp-21(jj77) and glp-1(bn18) is consistent with the absence of TSP-21::GFP expression in the germline and early embryo, as described above.
TSP-21 can form a complex with TSP-12 and TSP-14
Tetraspanins often associate with each other and with other membrane or membrane-associated proteins to organize membranes into tetraspanin-enriched microdomains [24–26]. Our finding that in addition to TSP-21, TSP-12 and TSP-14 also function in promoting Sma/Mab signaling suggested that these tetraspanins might interact with each other. We tested this hypothesis by using the mating-based split-ubiquitin system (mbSUS, [42]) in budding yeast. The mbSUS is based on the observation that a full-length ubiquitin can be reconstituted when the N-terminal ubiquitin domain (Nub) and the C-terminal ubiquitin domain (Cub) are brought into close proximity [43, 44]. This system can be used to identify potential interactions between full-length membrane proteins or between a membrane protein and a soluble protein: a mutant form of Nub, NubG, that has reduced affinity for Cub, can only reconstitute with Cub via two interacting proteins. The reconstituted ubiquitin will direct ubiquitin-specific proteases to liberate PLV (protein A, LexA and VP16) from Cub, which then enters the nucleus and activates transcription of reporter genes. We generated TSP-Cub fusions and Nub-TSP or TSP-Nub fusions (see Materials and Methods, and S2 Table for a list of the plasmids generated), and tested pairwise interactions among the three tetraspanins, as well as interactions between these tetraspanins and the type I and type II receptors SMA-6 and DAF-4, respectively. Results from these experiments are summarized in Fig 6. TSP-12-Cub appeared to auto-activate reporter expression, while the TSP-14-Cub was not detectable on western blots (see Materials and Methods). For the remaining three Cub fusions (TSP-21, SMA-6 and DAF-4), we found that TSP-21 can associate with itself, as well as with TSP-12 and TSP-14 (Fig 6). In addition, SMA-6 can associate with both TSP-12 and TSP-14, but not TSP-21. We also detected a very weak interaction between DAF-4 and TSP-14 (Fig 6). The use of multiple positive and negative controls in these experiments (see Materials and Methods, and Fig 6) indicated that the observed interactions are highly specific. For example, TSP-21 did not show any interaction with the C. elegans LKB homolog PAR-4 ([45]; Fig 6), or the plant potassium channel KAT1 ([42]; Fig 6), or with the C. elegans ABC transporter HMT-1 [46]. Except for the weak DAF-4-TSP-14 interaction, the other observed interactions all appeared to be particularly strong, as yeast growth on SC-Trp,-Leu,-Ade,-His,-Ura,-Met plates supplemented with 0.3mM of methionine was detectable only 2 days after streaking the mated yeast. Thus, TSP-21 can form both homo-oligomers and heteromeric complexes with TSP-12 and TSP-14. These findings are consistent with our genetic evidence that all three tetraspanins function to promote Sma/Mab signaling. The strong interactions between SMA-6 and TSP-12 and TSP-14 suggest that these tetraspanins might function by directly recruiting the receptor molecules to specific membrane microdomains.

Glycosphingolipids are required for promoting Sma/Mab signaling
Tetraspanin-enriched microdomains are also enriched in cholesterol and glycosphingolipids [24–26]. We therefore tested whether cholesterol and/or glycosphingolipids are required for Sma/Mab signal transduction. Our results suggest that Sma/Mab activity is influenced by glycosphingolipids but not cholesterol.
C. elegans worm survival requires exogenous cholesterol [47, 48]. In the lab, worms are normally fed with E. coli bacteria on agar plates supplemented with 5μg/mL cholesterol [49]. Using a method that can lead to nearly complete cholesterol depletion ([50, 51] and see Materials and Methods), we grew L1 or L4 worms on cholesterol-depleted plates and scored their phenotypes or their progeny’s phenotype, respectively, at the adult stage. We found no suppression of the M lineage phenotype when sma-9(cc604) worms were grown on cholesterol-depleted media, even though the worms were sterile, a known phenotype resulting from cholesterol depletion [47, 48]. Thus cholesterol does not seem to be essential for Sma/Mab signaling.
To determine the requirement of glycosphingolipids in Sma/Mab signaling, we generated double mutants between sma-9(cc604) and mutations that reduce or eliminate the activity of enzymes involved in glycosphingolipid biosynthesis [52], S3 Fig), and examined their Susm phenotype. As shown in Table 7, mutations in cgt-3 and bre-5 partially suppressed the sma-9 M lineage phenotype. cgt-3 encodes the ceramide glucosyltransferase that converts ceramide to glucosylceramide, a precursor of complex glycosphingolipids [53, 54]. Previous work has shown that CGT-3 is the major enzyme among the three worm CGT proteins [54]. We found that a deletion allele of cgt-3, ok2877, which deletes most of the coding exons of cgt-3, resulted in a late L1 or early L2 larval arrest and a partial suppression of the sma-9(cc604) M lineage defects (Table 7). cgt-3(ok2877) mutants exhibited additional defects in Sma/Mab signaling: the relative fluorescence intensity of the RAD-SMAD reporter in cgt-3(ok2877) mutants is only 58% of that in stage-matched wild-type animals (see Materials and Methods) and cgt-3(ok2877) mutants exhibited a smaller body size compared to stage-matched wild-type control animals (72% of wild-type body length, n = 23). We also observed a low penetrance of the Susm phenotype in ye17, an allele of bre-5 that encodes a β-1,3-galactosyltransferease involved in glycosphingolipid biosynthesis (Table 7, [55, 56]). Taken together, our data suggest that glycosphingolipids are required for Sma/Mab signaling. The lack of a Susm phenotype for the other mutations affecting glycosphingolipid biosynthesis (Table 7) may be because many of them are partial loss-of-function alleles, since null mutations in many of these genes result in lethality [52]. Alternatively, proper Sma/Mab signaling may require specific type(s) of glycosphingolipids.

cgt-3 is widely expressed in multiple cell types in C. elegans [53, 54]. We tested whether cgt-3, and therefore glycosphingolipids, are required in the signal-receiving cells for proper Sma/Mab signaling. Expression of cgt-3 in the M lineage using the hlh-8 promoter partially, but significantly, rescued the Susm phenotype of cgt-3(ok2877) mutants (Table 7), suggesting that proper Sma/Mab signaling requires glycosphingolipids in the signal-receiving cells. The lack of complete rescue suggests that glycosphingolipids are also required outside of the signal-receiving cells to promote Sma/Mab signaling.
During the course of our study, we observed that both cgt-3(ok2877) and bre-5(ye17) single mutants exhibited a low penetrance M lineage phenotype like that of a lin-12(lf) mutant: extra M-derived CCs due to the fate transformation of M-derived SMs to CCs ([39, 40]; Table 8 and S2 Fig). We further found that cgt-3(ok2877) enhanced the penetrance of the M lineage defects of a hypomorphic lin-12 temperature sensitive allele, n676n930, at a semi-permissive temperature (Table 8). These observations are consistent with previous findings by Katic and colleagues [57] showing that enzymes required for glycosphingolipid biosynthesis, such as BRE-5, are required for promoting LIN-12/Notch signaling. The requirement of glycosphingolipids in LIN-12/Notch signaling appears to be distinct from their requirement in Sma/Mab signaling. Mutations in the Sma/Mab pathway fully restore the sma-9(0) M lineage phenotype back to that of wild-type animals ([20–22, 40]; S2 Fig). However, lin-12(0); sma-9(0) double mutants exhibit a reversal of the M lineage dorsoventral polarity, so that the double mutants have 2 SMs born on the dorsal side and 2 M-derived CCs located on the ventral side ([39, 40]; S2 Fig). Careful examination of the position of the M-derived CCs in cgt-3(ok2877);sma-9(cc604) and bre-5(ye17);sma-9(cc604) mutants showed that a majority of the double mutant animals have their M-derived CCs located on the dorsal side (S3 Table), indicating a suppression rather than a reversal of polarity. Taken together, our results support the notion that glycosphingolipids are required for both LIN-12/Notch and Sma/Mab signaling.

Discussion
Tetraspanins play important roles in promoting BMP signaling
In this study, we identified TSP-21, a C6a class tetraspanin, as a key factor promoting the BMP-like Sma/Mab signaling in C. elegans. tsp-21 mutants exhibit small body size and Susm phenotypes similar to that shown by mutants in core Sma/Mab pathway components. The TSP-21 protein is localized to the plasma membrane, and tsp-21 is expressed and functions in the signal-receiving cells at the ligand-receptor level to promote Sma/Mab signaling. We found that among the remaining 20 C. elegans tetraspanins, TSP-12 and TSP-14 function redundantly to also promote Sma/Mab signaling. How do these three tetraspanins function to promote Sma/Mab signaling? We envision two possible, non-mutually exclusive, scenarios.
In the first scenario, the three tetraspanins might promote clustering of the receptor complexes or the ligand-receptor complexes to modulate Sma/Mab signaling. Tetraspanins are known to homo - and hetero-oligomerize to organize membranes into tetraspanin-enriched microdomains, which are also enriched in tetraspanin-associated proteins [24–26]. Previous work has shown that in mouse, TSPAN12 promotes Norrin/β-catenin signaling by enhancing clustering of the Norrin receptor FZD4 [58, 59]. In particular, TSPAN12 and Norrin can each enhance FZD4 clustering but work together cooperatively to further increase the clustering of the ligand-receptor complex to promote Norrin/β-catenin signaling [58]. We have shown that C. elegans TSP-12, -14 and -21 can interact with each other in yeast and that both TSP-12 and TSP-14 can interact with the type I receptor SMA-6. In addition, we found that glycosphingolipids, which are enriched in tetraspanin-enriched microdomains, are also required for proper Sma/Mab signaling. These findings suggest that TSP-21, TSP-12 and TSP-14 may function by recruiting the receptor complex, or the ligand-receptor complex, to glycosphingolipid-enriched membrane microdomains containing TSP-21-TSP-12-TSP-14, thereby increasing the local concentration of the receptors, or the ligand-receptor complexes, to promote Sma/Mab signaling (Fig 7). Supporting this model, SMA-6, DAF-4 and TSP-21 are all localized to the basolateral membranes of the polarized intestinal cells ([38]; this work). We envision that several previously identified positive modulators of the Sma/Mab pathway, including DRAG-1/RGM, UNC-40/neogenin, and SMA-10/LRIG, might be localized in these microdomains as well, as all three proteins are plasma membrane-localized, are expressed and function in the signal-receiving cells, and interact with the ligand and the receptors (for DRAG-1), or the receptors (for SMA-10), or with each other (for DRAG-1 and UNC-40) [21, 22, 34]. Further biochemical and cell biological experiments are needed to determine the presence and subcellular localization of TSP-21-TSP-12-TSP-14-containing membrane microdomains, whether the Sma/Mab pathway receptors and modulators are indeed localized to these microdomains, and what other factors are also present there.

Alternatively, but not mutually exclusively, the three tetraspanins might be involved in the trafficking of essential Sma/Mab pathway components. Tetraspanins have been found to be present in the plasma membrane or various types of intracellular membranous organelles, and multiple tetraspanins are known to regulate the processing and trafficking of associated proteins [60]. In C. elegans, TSP-12 and TSP-14 have previously been shown to function redundantly in promoting Notch signaling [28]. Their Drosophila and mammalian homologs, the TspanC8 tetraspanins, interact with the ADAM (a disintegrin and metalloprotease) protease ADAM10 to promote its maturation and trafficking to the cell surface, which in turn promotes Notch signaling [61–63]. TSP-12 and TSP-14 may function in a similar manner in promoting Sma/Mab signaling. Since both TSP-12 and TSP-14 can bind to the type I receptor SMA-6 in yeast, they may promote Sma/Mab signaling by regulating the trafficking of SMA-6 (Fig 7), and/or other players in the Sma/Mab pathway. Further work is needed to test this hypothesis. Since the role of TspanC8 tetraspanins in promoting Notch signaling is evolutionarily conserved [61–63], it will be interesting to determine whether the role of TspanC8 tetraspanins in modulating BMP signaling is also evolutionarily conserved, and whether these tetraspanins function in a similar manner in promoting both BMP and Notch signaling.
Using C. elegans as a model, Gleason and colleagues recently showed that the type I receptor SMA-6 and the type II receptor DAF-4 utilize distinct mechanisms for their intracellular recycling, providing physiological evidence supporting the roles of endocytosis and intracellular trafficking in regulating BMP signaling [38]. In light of the roles of multiple tetraspanins in regulating the processing and trafficking of associated proteins [60], our findings, together with that of Gleason and colleagues [38], highlight the usefulness of C. elegans as a model system in identifying cell biological mechanisms that regulate BMP signaling.
Possible links between tetraspanins, cancer and TGFβ or BMP signaling
The family of tetraspanin proteins is large: there are 21 tetraspanins in C. elegans and 33 tetraspanins in humans. Recent studies have implicated tetraspanins in multiple diseases and physiological processes in humans [60]. In particular, several tetraspanins, such as CD151 [64], TSPAN12 [65], and TSPAN8 [66], among others, have been implicated in cancer initiation, progression and metastasis in mammals. These and other tetraspanins have emerged as diagnostic and prognostic markers, and possible therapeutic targets, for tumor progression (for reviews, see [27, 67]). However, the mechanism by which the mis-regulation of these tetraspanins contributes to cancer is not fully understood [27, 67]. It is well known that mis-regulation of TGFβ signaling contributes to cancer initiation and progression [6, 68]. CD151 is the only tetraspanin whose role in cancer has been directly linked to altered TGFβ signaling [69]. Sadej and colleagues showed that CD151 is required for TGFβ1-induced proliferation and scattering of breast cancer cell line MDA-MB-231 through regulating TGFβ-induced p38 phosphorylation, rather than canonical TGFβ-induced Smad phosphorylation. Furthermore, this function of CD151 in TGFβ signaling requires its interaction with the integrins [69]. How CD151-integrin interaction regulates TGFβ-induced p38 phosphorylation is not clear. Recently a study on the tetraspanin-interacting protein EWI-2 indirectly implicates two other tetraspanins, CD9 and CD81, in regulating TGFβ signaling in melanoma growth and metastasis [70]. But the detailed mechanism on how these two tetraspanins regulate TGFβ signaling is not known.
We have provided a direct in vivo link between BMP signaling and three tetraspanins, TSP-21, TSP-12 and TSP-14, in living animals using C. elegans as a model. Our genetic epistasis results showed that TSP-21 acts through SMA-3, one of the R-Smads in the canonical BMP-like Sma/Mab signaling pathway (Fig 2E). Due to the embryonic arrest of null mutants in the C. elegans integrin genes, we could not determine whether the function of TSP-21 in Sma/Mab signaling is dependent on integrins. We have found that strong-loss-of function mutations in one of the two C. elegans genes encoding the α subunit of integrin, ina-1(gm39) and ina-1(gm144) [71], did not exhibit any Susm phenotype (n = 53 for gm39, and n = 109 for gm144). But we cannot rule out the possibility that in these mutants residual ina-1 function or function of pat-2, another gene encoding the α subunit of integrin [72] is sufficient to mediate Sma/Mab signaling.
TSP-21 is orthologous to human TSPAN4, TSPAN9 and CD53, but is much more distantly related to CD151 (whose C. elegans ortholog is TSP-17; S1 Fig). It is therefore possible that the differences between CD151 and TSP-21 in regulating TGFβ signaling are due to intrinsic biochemical differences between the two types of proteins. Alternatively, since TSP-21 regulates a BMP-like Sma/Mab signaling pathway, it is likely that tetraspanins can regulate both TGFβ signaling and BMP signaling, but via distinct downstream effectors. Interestingly, each of the three human orthologs of TSP-21 (TSPAN4, TSPAN9 and CD53), as well as two out of the six human orthologs of TSP-12 and TSP-14 (TSPAN10 and TSPAN33), are expressed at elevated levels in certain cancer cell lines or tumors [73–75] In addition, one human ortholog of TSP-12 and TSP-14 (TSPAN14) is genetically altered in non-small-cell lung cancer [76]. However, the functional significance of their overexpression or mutation in human cancers is not fully understood. We propose that the involvement of these tetraspanins in cancer may be partially due to their role in modulating the activity of TGFβ and/or BMP signaling.
Screening for sma-9 suppressor is a highly specific and sensitive method for identifying key players in the BMP-like Sma/Mab pathway in C. elegans
Previous genetic studies in C. elegans have led to the identification of key players in BMP signaling (for example, [16, 17]). A screen based on the body size phenotype has also been fruitful in identifying factors involved in modulating Sma/Mab signaling, such as SMA-10/LRIG [32] and LON-2/glypican [34, 77]. Potential modulators of the Sma/Mab pathway may also exist among a collection of mutants with a small body size phenotype [78]. However, it may be difficult to identify the genes for which mutations produce only a subtle effect on body size, such as tsp-21(jj77). Furthermore, since genes not functioning in the Sma/Mab pathway also regulate body size (for example, [30–32, 79]), not all mutations affecting body size will identify factors specifically functioning in the Sma/Mab pathway. The sma-9 suppressor screen appears to be a highly specific and sensitive means to identify new components of the Sma/Mab pathway: (1) Mutations in all (except for crm-1, see Table 1) previously identified Sma/Mab pathway members suppress the sma-9 M lineage phenotype (Table 1 and Table 2). In general, partial loss-of-function alleles for a given gene exhibited lower penetrance of the Susm phenotype compared to putative null alleles (Table 2), demonstrating that the suppression of the sma-9 M lineage phenotype is highly sensitive to altered levels of Sma/Mab signaling. (2) Mutations in other signaling pathways, such as the dauer pathway or the Wnt pathway, or mutations that exclusively affect body size without affecting Sma/Mab signaling, do not suppress the M lineage phenotype of the sma-9 mutant ([20]; Table 1). (3) Using this screen, we have identified three evolutionarily conserved modulators of the Sma/Mab pathway, DRAG-1/RGM [21], UNC-40/neogenin/DCC [22], and TSP-21/TSPAN4,9 (this study). Additional modulators of this pathway probably exist, as our screen has only recovered single alleles of several genes known to function in Sma/Mab signaling and is, therefore, unlikely to be genetically saturated (Table 2).
In summary, we have developed a highly specific and sensitive way to identify new modulators of the BMP pathway in C. elegans. This genetic approach has confirmed known regulators and identified novel players. Because of the high degree of conservation of the BMP pathway, the factors that we identify in our screen and the mode of their action that we decipher in C. elegans will be broadly relevant in understanding modulation of BMP signaling in other metazoans, including humans.
Materials and Methods
C. elegans strains
Strains were grown using standard culture conditions, as described by Brenner [49]. Analyses were performed at 20°C, unless otherwise noted. Cholesterol depletion conditions were following those described in Merris et al. [80] by replacing agar with agarose, and by growing bacteria OP50 and C. elegans worms on defined media, which contains 3.5mM Tris.HCl, 2mM Tris, 34mM NaCl, and 3.1g/L of ether-extracted peptone. Eggs or L4 hermaphrodite animals were placed on cholesterol-depleted plates and the resulting adult animals were scored for M lineage phenotypes.
The following mutations and integrated transgenes were used: Linkage group I (LG I): drag-1(jj4), arIs37(secreted CC::gfp), bre-4(ok3167), bre-5(ye17); LG II: sma-6(e1482), pod-2(ye60), cgt-3(ok2877)/mIn1[mIs14 dpy-10(e128)], jjIs2437[CXTim50.19[pCXT51(5*RLR::deleted pes-10p::gfp) + LiuFD61(mec-7p::rfp)], sptl-1(ok1693); LG III: daf-4(m63), daf-7(m62), sma-2(e502), sma-3(e491), sma-4(e729), lon-1(e185), cup-5(ar465), ina-1(gm144), ina-1(gm39), bre-2(ye31), bre-3(ye26), bre-3(ye28), lin-12(n676n930ts), hT2[qIs48], ccIs4438[intrinsic CC::gfp]; LG IV: daf-1(m40), daf-1(m213), fat-2(wa17), fat-3(wa22), fat-6(tm331), tsp-12(ok239), nT1[qIs51]; LG V: dbl-1(wk70), fat-7(wa36), sma-10(wk89), sma-10(ok2224), sma-1(ru18), lon-3(ct417), crm-1(tm2218), him-5(e1467), bre-1(ye4), bre-5(ye17), cgt-1(ok1045), acs-1(gk3066)V/nT1[qIs51]IV;V; LG X: lon-2(e678), tsp-21(tm6269), sma-9(cc604), jjIs2433[RAD-SMAD: CXTim50.1[pCXT51(5*RLR::deleted pes-10p::gfp) + LiuFD61(mec-7p::rfp)]].
tsp-21 and sma-9 are located 0.79 map unit apart from each other on the X chromosome. We therefore separated the tsp-21(jj77) mutation from sma-9(cc604) via recombination. Specifically, progeny from tsp-21(jj77) sma-9(cc604)/+ + heterozygous parents were scored for the number of CCs. Animals with 6 CCs (jj77 cc604/+ + or + +/+ + or jj77 cc604/jj77 +) were genotyped for jj77 homozygosity by PCR. jj77 cc604/jj77 + animals were selected and their progeny were further genotyped by sequencing the sma-9 gene in order to obtain jj77 +/jj77 + animals. Four independent recombinants were obtained, #570, #778, #898 and #954. Each recombinant was then outcrossed with N2 three more times before further phenotypic analysis. All four recombinants behaved similarly regarding body size, RAD-SMAD and male tail patterning phenotypes.
The lon-2(e678) tsp-21(jj77) double mutant was generated from a lon-2(e678) egl-15(n484)/tsp-21(jj77) heterozygous worms by identifying Lon-non-Egl recombinants, and scoring for the presence of the tsp-21(jj77) and the lon-2(e678) mutations by PCR genotyping.
RNAi
let-381(RNAi) was performed via feeding following the protocol described in [33]. Other RNAi experiments were performed by injection. In general, gene specific fragments were amplified using RNAi clones from the Ahringer library [81] or the Vidal library [82], or using N2 genomic DNA as template. dsRNAs were generated using the T7 Ribomax RNA Production System (Promega) and injected into gravid adult hermaphrodite animals of specific genotypes carrying CC::gfp. The resulting progeny were scored at the adult stage for the number of CCs.
Isolation and characterization of sma-9 suppressor mutations
arIs37(secreted CC::gfp) I; cup-5(ar465) III; sma-9(cc604) X animals lacking M-derived coelomocytes (having a total of 4 CCs) were treated with 50 mM ethyl methanesulfonate (EMS). Individual F1 animals were picked to 3F1s per plate and their combined F2 progeny were screened for the restoration of M lineage-derived coelomocytes (having a total of 5–6 CCs) by direct visual examination using a fluorescence stereomicroscope. Plates that segregated 5–25% of animals with 6 CCs were kept for further analysis, including determining whether the mutations bred true, the degree of suppression for each suppressor mutation when homozygous and whether the mutations are dominant or recessive.
By screening through 5,300 haploid genomes using the above method, we isolated 37 true-breeding sma-9 suppressors, named susm (suppressor of sma-9) mutations (jj49-jj85, Table 2). Four of these, jj68, jj80, jj81 and jj84 showed a relatively low degree of suppression (near 30%, Table 2), and were not further characterized in this work. jj58 might be a dominant mutation and was not further analyzed. All of the remaining susm alleles appear to be recessive, single locus mutations, although some suppressors exhibited partial dominance in their Susm phenotype (Tables 1 and 2). The suppressor mutations were then mapped to chromosome X or chromosome III based on their linkage to sma-9(cc604) X or to cup-5(ar465) III. Further complementation tests were carried out between each suppressor mutation and mutations in each known members of the Sma/Mab pathway, and between different suppressor mutations that did not affect known genes in the Sma/Mab pathway.
LW0214, which has arIs37(secreted CC::gfp) I and sma-9(cc604) X introgressed into the CB4856 Hawaiian strain by 6x backcrossing, was used for mapping the sma-9 suppressors via snip-SNP mapping [83] and whole genome sequencing (WGS) [84]. LW0214 was tested using a panel of SNP markers and subsequently by WGS, and found to contain CB4856 SNPs for all six chromosomes except for the following regions that still contain N2 SNP markers: chromosome I—from the left end to -12 and from +24 to the right end; chromosome II—from the left end to -18; and chromosome X—between +1.73 and +11. Snip-SNP markers used were described in Wicks et al. [83] and Davis et al. [85].
For the sma-9 suppressor mutations that appeared to affect known genes in the Sma/Mab pathway, either by complementation tests, or by whole genome sequencing (WGS, see below), their molecular lesions were identified by sequencing PCR products spanning the entire genomic regions of the corresponding genes, which include dbl-1, daf-4, sma-6, sma-2, sma-3, sma-4, lon-1, sma-10 and unc-40. For jj69 that contains a single base pair change in the upstream regulatory region of sma-6, a plasmid pJKL1060, which contains 3kb of upstream sequences, the genomic coding region and 2kb of downstream sequences of sma-6, was used to rescue the Susm phenotype of jj69.
Whole genome sequencing (WGS) and data analysis
Direct WGS of the homozygous suppressor mutant DNA was performed for some sma-9 suppressors. For others, the suppressors were simultaneously mapped and identified using the SNP-WGS method of Doitsidou et al. [84]. For the SNP-WGS method, each sma-9; suppressor mutant was crossed with LW0214, which has sma-9 introgressed into the polymorphic Hawaiian strain CB4856 (described above). Between 36 and 59 F2 progeny that were homozygous for both sma-9 and the suppressor mutation were collected. F3 generation worms from these F2 progeny were pooled for DNA extraction and library construction. Worm genomic DNA was prepared using the Qiagen Gentra Puregene Kit. 5μg of genomic DNA was used to prepare the sequencing library using the NEBNext DNA Sample Prep Master Mix Set 1. Single-end 50bp short-read (51 cycle) sequencing was performed on the HiSeq 2000 instrument (Illumina), yielding 38 ~ 78 million reads (20 ~ 41 fold coverage) per sample.
For direct WGS (jj58, jj60, jj61, jj71, jj77), data analysis was done using the MAQGene platform [86, 87] with the default setting. SNP variants on the X chromosome compared to the reference C. elegans genome ce6 W221 were analyzed. Genes with missense SNP variants in jj60 and jj77, but not in jj58 and jj71, were among the candidate genes that were targeted by RNAi for their ability to suppress the sma-9(cc604) M lineage defects by injection. These included C41A3.1, K09C4.8 and C17G1.8. Further PCR and sequencing confirmed the jj60 and jj77 mutations in C17G1.8 (tsp-21).
For mapping additional suppressors using either direct WGS (for jj2, jj5, jj7, jj50, jj52 and jj70) or SNP-WGS (for jj49, jj57, jj62, jj69, jj71, jj73, jj78 and jj83), sequence data were aligned to C. elegans reference genome version WS220 using BFAST [88] with default parameters. SNP calling was performed by SAMTOOLS [89]. A valid SNP call required a minimum read depth of three. ANNOVAR [90] was used for annotation of SNP coding potential. For SNP-WGS, Hawaiian SNPs were annotated with a custom Perl script. Scatter plots of heterozygous (0.2–0.7 fraction of total reads) Hawaiian SNPs were generated as chromosome position vs. fractional total graphs. Mapping intervals were defined by visual inspection for gaps (i.e., Hawaiian SNP fraction <0.2). Candidate suppressor genes were identified as homozygous (fraction >0.8), non-Hawaiian, nonsynonymous SNPs in the mapped interval. The SNPs in the identified suppressor genes were verified by PCR and sequencing.
jj61 was mapped via SNP-WGS to the region on the X chromosome where lon-2 is located. Direct inspection of the sequence reads around the lon-2 region showed that jj61 contains a large deletion (11.8kb) spanning the lon-2 region, which was subsequently verified by PCR and sequencing.
Plasmid constructs and transgenic lines
sma-6 reporter and rescuing constructs
pJKL840: sma-6p::nls::rfp::lacZ::unc-54 3’UTR
pJKL1048: sma-6(jj69)p::nls::rfp::lacZ::unc-54 3’UTR
pJKL1060: sma-6p::sma-6 rescuing construct
tsp-21 reporter constructs
pJKL1005: 5kb tsp-21p::tsp-21 genomic ORF::1.7kb tsp-21 3’UTR
pJKL1004: 5kb tsp-21p::tsp-21 genomic ORF::gfp::1.7kb tsp-21 3’UTR
pJKL998: 5kb tsp-21p::nls::gfp::lacZ::unc-54 3’UTR
pZL11: 5kb tsp-21p::tsp-21 genomic ORF (sgRNA target site modified)::gfp::1.7kb tsp-21 3’UTR
Constructs for tissue-specific expression of tsp-21
pJKL1015: tsp-21p::tsp-21 cDNA::tsp-21 3’UTR
pJKL1017: rol-6p::tsp-21 cDNA::tsp-21 3’UTR
pJKL1018: elt-3p::tsp-21 cDNA::tsp-21 3’UTR
pJKL1019: elt-2p::tsp-21 cDNA::tsp-21 3’UTR
pJKL1020: hlh-8p::tsp-21 cDNA::tsp-21 3’UTR
pJKL1021: myo-2p::tsp-21 cDNA::tsp-21 3’UTR
The full-length tsp-21 cDNA clone yk1449c02, which contains a SL1 trans-splice leader sequence, and full length 5’ and 3’ UTRs, was kindly provided by Dr. Yuji Kohara (National Institute of Genetics, Japan). A point mutation in the coding region of tsp-21 in yk1449c02 was corrected by site-directed mutagenesis to generate pJKL994. Transgenic animals were generated using the plasmid pRF4 or pJKL449 (myo-2p::gfp::unc-54 3’UTR) as markers. Integrated transgenic lines carrying pJKL1004[TSP-21::GFP] (jjIs3113 and jjIs3114) were generated using gamma-irradiation. pJKL840[sma-6p::nls::rfp::lacZ::unc-54 3’UTR] was used for co-localization of TSP-21::GFP and sma-6p::nls::rfp. pTAA1[hlh-8p::cgt-3.1a ORF::unc-54 3’UTR] was used to test for function of cgt-3 in the M lineage.
Generating TSP-21::GFP using CRISPR technology
The following mix of plasmid DNAs was injected into the N2 gravid adults: (1) a Cas9 expression plasmid pDD162 [91], (2) a tsp-21-specific sgRNA plasmid pZL10, which has GAAACTGACACGGTAGAAGATGG replacing the unc-119 sgRNA in plasmid 46169 [92], (3) the homologous repair template pZL11: 5kb tsp-21p::tsp-21 genomic ORF (sgRNA target site modified)::gfp::1.7kb tsp-21 3’UTR, (4) a co-injection marker pCFJ90[myo-2p::mCherry] [93]. GFP knock-in events were screened via PCR using a primer in GFP (ZL21: CGCATATCTTGGACGCCTAATTTG) and a primer in the tsp-21 3’ region outside of the sequences included in pZL11 (ZL22: TCCACACAATCTGCCCTTTCG). Single worm PCR of 250 F1s failed to detect any germline integration event. However, we checked the F2 generation for high transmission efficiency lines (myo-2::mCherry positive) and screened via PCR 5–10 F3 progeny from each of the three high transmission efficiency lines (>50%). One of the three transgenic lines gave us two homozygous GFP knock-in strains: LW3670: jj93(tsp-21::gfp) and LW3671: jj94(tsp-21::gfp).
RT-PCR
Total RNA was isolated from mixed-stage N2 or sma-6(jj69) worms using TRIzol Reagent (Invitrogen). Reverse transcription was performed with SuperScript III First-Strand Synthesis System (Invitrogen) following the manufacturer’s instructions. The primers used to detect the cDNAs of sma-6 and act-1 are: sma-6, MLF-34 and MLF-44; act-1, NMA-163 and NMA-164.
Body size measurement, RAD-SMAD reporter assay and dauer formation assay
Body size measurement and RAD-SMAD reporter assay were carried out as described in Tian et al. [22]. Dauer formation assay was carried out as described in Tian et al. [21]. Statistical analyses were performed using Microsoft Excel and GraphPad Prism (http://www.graphpad.com/scientific-software/prism/).
Fluorescence microscopy
GFP and RFP epifluorescence in transgenic animals was visualized either on a Leica DMRA2 compound microscope, where the images were captured by a Hamamatsu Orca-ER camera using the OPENLAB software, or on a Zeiss LSM 710 confocal microscope. Subsequent image analysis was performed using ImageJ and Photoshop CC.
Tetraspanin phylogenetic analysis
We identified tetraspanin homologs by running hmmsearch from HMMER 3.1b1 [94] with the hidden Markov model (HMM) profile for tetraspanins (PF00335.15) from PFAM 27.0 [95] against the reference proteome set of the Quest for Orthologs consortium ([96]; source URL, ftp:/ftp.ebi.ac.uk/pub/databases/reference_proteomes/QfO/QfO_release_2014_04.tar.gz). hmmsearch was run with the arguments '-E 1e-06—domE 1e-06—incE 1e-06—incdomE 1e-06-A [alignment]', which generated aligned regions of similarity to these core domains. Since the regions of homology were extracted from full-length proteins with an HMM, the specific residues extracted were generally a subset of the full protein; moreover, it was possible for two or more such regions to be independently extracted from a single protein chain, although this proved rare for tetraspanins.
These regions were then realigned with MAFFT v7.158b [97] in L-INS-i, its slowest and most reliable mode, using the arguments '—localpair—maxiterate 1000'. The resulting alignments were purged of poorly aligned members by first running trimal v1.4.rev15 [98] using the argument '-gt 0.5', and then running BMGE 1.1 [99] using the arguments '-t AA-h 1-g 0.5:1'. This purged the alignments of any columns in which over 50% of the columns' positions consisted of gaps rather than amino acid residues, and then any sequences in which over 50% of the residues were gapped, yielding global alignments that lacked excessive loops and gaps. From the filtered alignments, we computed protein maximum-likelihood phylogenies, with a WAG model of amino acid evolution [100] and with pseudocounts for gaps, via FastTree 2.1.7 [101], using the arguments '-pseudo-wag'. Confidence values for the branches of trees (ranging from 0.00 to 1.00) were automatically computed by FastTree with 1,000 internal replicates. We visualized the resulting trees with FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree). Branch lengths were measured in average substitutions (when comparing full sequences or their profiles) among non-gap positions in the aligned sequences, with distances derived from the BLOSUM45 matrix, a correction for multiple substitutions, and an allowed maximum of 3.0 substitutions per individual site [102].
Split-ubiquitin yeast two-hybrid analysis
The split-ubiquitin yeast two-hybrid experiments were carried out following the detailed method described in Grefen et al. [103]. The bait CubPLV and prey NubG constructs were generated via PCR and recombinational in vivo cloning in yeast [103]. The resulting fusion constructs were recovered from yeast and transformed into E. coli and confirmed by sequencing. The primers, cDNA templates, and the names of the resulting bait and prey constructs are summarized in S2 Table. The bait and prey constructs were transformed into the haploid yeast strains THY.AP4 (MATa) and THY.AP5 (MATα), respectively, and the resulting yeast strains were mated to generate diploid yeast cells carrying specific combinations of bait and prey constructs [103]. Interactions among each pair of bait and prey constructs were visualized by streaking diploid cells on SC-Trp,-Leu,-Ade,-His,-Ura,-Met plates that were supplemented with four different concentrations of methionine: 0mM, 0.075mM, 0.150mM and 0.300mM, respectively. Methionine can repress the expression of the CubPLV fusion, which is under the control of the Met-repressible MET25 promoter [42]. Growth was monitored for 2–9 days at 30°C.
The plasmids KAT-1-Cub-PLV, NubG-KAT-1 (in pNX33 vector) and KAT-1-NubG (in pXN21 vector) [42], HMT-1-Cub-PLV and HMT-1-NubG (in pXN21 vector) [46] were kindly provided by Sungjin Kim (Cornell University) and used as specificity controls. NubG fusions for PAR-4, a protein unexpected to interact with any of the proteins tested, was included as another control for specificity of the interactions. The empty NubG vector was used as a control to determine if any Cub-PLV fusions can auto-activate the reporters. The vector expressing soluble wild-type Nub (NubWT) was used as a control to indicate expression of the Cub-PLV fusion. Additional confirmation of expression of each fusion protein came from western blot analysis using rabbit polyclonal anti-VP16 antibodies (ab4808, Abcam, for CubPLV fusions) and monoclonal anti-HA antibodies (Clone 12CA5, Sigma, for NubG fusions).
Supporting Information
Zdroje
1. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003;113 : 685–700. 12809600
2. Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev Cell 2009;16 : 329–43. doi: 10.1016/j.devcel.2009.02.012 19289080
3. Constam DB. Regulation of TGFbeta and related signals by precursor processing. Semin Cell Dev Biol 2014;32 : 85–97. doi: 10.1016/j.semcdb.2014.01.008 24508081
4. Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 2008;1782 : 197–228. doi: 10.1016/j.bbadis.2008.01.006 18313409
5. Cai J, Pardali E, Sanchez-Duffhues G, ten Dijke P. BMP signaling in vascular diseases. FEBS Lett 2012;586 : 1993–2002. doi: 10.1016/j.febslet.2012.04.030 22710160
6. Massague J. TGFbeta in Cancer. Cell 2008;134 : 215–30. doi: 10.1016/j.cell.2008.07.001 18662538
7. Massague J. TGF-beta signaling in development and disease. FEBS Lett 2012;586 : 1833. doi: 10.1016/j.febslet.2012.05.030 22651913
8. Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development 2009;136 : 3699–714. doi: 10.1242/dev.030338 19855013
9. Umulis D, O'Connor MB, Blair SS. The extracellular regulation of bone morphogenetic protein signaling. Development 2009;136 : 3715–28. doi: 10.1242/dev.031534 19855014
10. Massague J. TGFbeta signaling in context. Nat Rev Mol Cell Biol 2012;13 : 616–30. doi: 10.1038/nrm3434 22992590
11. Ramel MC, Hill CS. Spatial regulation of BMP activity. FEBS Lett 2012;586 : 1929–41. doi: 10.1016/j.febslet.2012.02.035 22710177
12. Gumienny TL, Savage-Dunn C. TGF-beta signaling in C. elegans. WormBook 2013;:1–34.
13. Morita K, Flemming AJ, Sugihara Y, Modhii M, Suzuki Y, Yoshida S, et al. A Caenorhabditis elegans TGF-beta, DBL-1, controls the expression of LON-1, a PR-related protein, that regulates polyploidization and body length. EMBO J 2002;21 : 1063–73. 11867534
14. Suzuki Y, Yandell MD, Roy PJ, Krishna S, Savage-Dunn C, Ross RM, et al. A BMP homolog acts as a dose-dependent regulator of body size and male tail patterning in Caenorhabditis elegans. Development 1999;126 : 241–50. 9847238
15. Krishna S, Maduzia LL, Padgett RW. Specificity of TGFbeta signaling is conferred by distinct type I receptors and their associated SMAD proteins in Caenorhabditis elegans. Development 1999;126 : 251–60. 9847239
16. Estevez M, Attisano L, Wrana JL, Albert PS, Massague J, Riddle DL. The daf-4 gene encodes a bone morphogenetic protein receptor controlling C. elegans dauer larva development. Nature 1993;365 : 644–9. 8413626
17. Savage C, Das P, Finelli AL, Townsend SR, Sun CY, Baird SE, et al. Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc Natl Acad Sci U S A 1996;93 : 790–4. 8570636
18. Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 1977;56 : 110–56. 838129
19. Liang J, Lints R, Foehr ML, Tokarz R, Yu L, Emmons SW, et al. The Caenorhabditis elegans schnurri homolog sma-9 mediates stage - and cell type-specific responses to DBL-1 BMP-related signaling. Development 2003;130 : 6453–64. 14627718
20. Foehr ML, Lindy AS, Fairbank RC, Amin NM, Xu M, Yanowitz J, et al. An antagonistic role for the C. elegans Schnurri homolog SMA-9 in modulating TGFbeta signaling during mesodermal patterning. Development 2006;133 : 2887–96. 16790477
21. Tian C, Sen D, Shi H, Foehr ML, Plavskin Y, Vatamaniuk OK, et al. The RGM protein DRAG-1 positively regulates a BMP-like signaling pathway in Caenorhabditis elegans. Development 2010;137 : 2375–84. doi: 10.1242/dev.051615 20534671
22. Tian C, Shi H, Xiong S, Hu F, Xiong WC, Liu J. The neogenin/DCC homolog UNC-40 promotes BMP signaling via the RGM protein DRAG-1 in C. elegans. Development 2013;140 : 4070–80. doi: 10.1242/dev.099838 24004951
23. Tian C, Liu J. Repulsive guidance molecules (RGMs) and neogenin in bone morphogenetic protein (BMP) signaling. Mol Reprod Dev 2013;80 : 700–17. doi: 10.1002/mrd.22199 23740870
24. Hemler ME. Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 2005;6 : 801–11. 16314869
25. Charrin S, le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem J 2009;420 : 133–54. doi: 10.1042/BJ20082422 19426143
26. Yanez-Mo M, Barreiro O, Gordon-Alonso M, Sala-Valdes M, Sanchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol 2009;19 : 434–46. doi: 10.1016/j.tcb.2009.06.004 19709882
27. Zoller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer 2009;9 : 40–55. doi: 10.1038/nrc2543 19078974
28. Dunn CD, Sulis ML, Ferrando AA, Greenwald I. A conserved tetraspanin subfamily promotes Notch signaling in Caenorhabditis elegans and in human cells. Proc Natl Acad Sci U S A 2010;107 : 5907–12. doi: 10.1073/pnas.1001647107 20220101
29. Colavita A, Krishna S, Zheng H, Padgett RW, Culotti JG. Pioneer axon guidance by UNC-129, a C. elegans TGF-beta. Science 1998;281 : 706–9. 9685266
30. McKeown C, Praitis V, Austin J. sma-1 encodes a betaH-spectrin homolog required for Caenorhabditis elegans morphogenesis. Development 1998;125 : 2087–98. 9570773
31. Nystrom J, Shen ZZ, Aili M, Flemming AJ, Leroi A, Tuck S. Increased or decreased levels of Caenorhabditis elegans lon-3, a gene encoding a collagen, cause reciprocal changes in body length. Genetics 2002;161 : 83–97. 12019225
32. Suzuki Y, Morris GA, Han M, Wood WB. A cuticle collagen encoded by the lon-3 gene may be a target of TGF-beta signaling in determining Caenorhabditis elegans body shape. Genetics 2002;162 : 1631–9. 12524338
33. Amin NM, Shi H, Liu J. The FoxF/FoxC factor LET-381 directly regulates both cell fate specification and cell differentiation in C. elegans mesoderm development. Development 2010;137 : 1451–60. doi: 10.1242/dev.048496 20335356
34. Gumienny TL, Macneil L, Zimmerman CM, Wang H, Chin L, Wrana JL, et al. Caenorhabditis elegans SMA-10/LRIG is a conserved transmembrane protein that enhances bone morphogenetic protein signaling. PLoS Genet 2010;6:e1000963. doi: 10.1371/journal.pgen.1000963 20502686
35. Maduzia LL, Gumienny TL, Zimmerman CM, Wang H, Shetgiri P, Krishna S, et al. lon-1 regulates Caenorhabditis elegans body size downstream of the dbl-1 TGF beta signaling pathway. Dev Biol 2002;246 : 418–28. 12051826
36. DeSalle R, Mares R, Garcia-Espana A. Evolution of cysteine patterns in the large extracellular loop of tetraspanins from animals, fungi, plants and single-celled eukaryotes. Mol Phylogenet Evol 2010;56 : 486–91. doi: 10.1016/j.ympev.2010.02.015 20171294
37. Huang S, Tian H, Chen Z, Yu T, Xu A. The evolution of vertebrate tetraspanins: gene loss, retention, and massive positive selection after whole genome duplications. BMC Evol Biol 2010;10 : 306,2148-10-306. doi: 10.1186/1471-2148-10-306 20939927
38. Gleason RJ, Akintobi AM, Grant BD, Padgett RW. BMP signaling requires retromer-dependent recycling of the type I receptor. Proc Natl Acad Sci U S A 2014;111 : 2578–83. doi: 10.1073/pnas.1319947111 24550286
39. Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell 1983;34 : 435–44. 6616618
40. Foehr ML, Liu J. Dorsoventral patterning of the C. elegans postembryonic mesoderm requires both LIN-12/Notch and TGFbeta signaling. Dev Biol 2008;313 : 256–66. 18036582
41. Kodoyianni V, Maine EM, Kimble J. Molecular basis of loss-of-function mutations in the glp-1 gene of Caenorhabditis elegans. Mol Biol Cell 1992;3 : 1199–213. 1457827
42. Obrdlik P, El-Bakkoury M, Hamacher T, Cappellaro C, Vilarino C, Fleischer C, et al. K+ channel interactions detected by a genetic system optimized for systematic studies of membrane protein interactions. Proc Natl Acad Sci U S A 2004;101 : 12242–7. 15299147
43. Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci U S A 1994;91 : 10340–4. 7937952
44. Stagljar I, Korostensky C, Johnsson N, te Heesen S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc Natl Acad Sci U S A 1998;95 : 5187–92. 9560251
45. Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development 2000;127 : 1467–75. 10704392
46. Kim S, Selote DS, Vatamaniuk OK. The N-terminal extension domain of the C. elegans half-molecule ABC transporter, HMT-1, is required for protein-protein interactions and function. PLoS One 2010;5:e12938. doi: 10.1371/journal.pone.0012938 20886084
47. Kurzchalia TV, Ward S. Why do worms need cholesterol? Nat Cell Biol 2003;5 : 684–8. 12894170
48. Zhu H, Han M. Exploring developmental and physiological functions of Fatty Acid and lipid variants through worm and fly genetics. Annu Rev Genet 2014;48 : 119–48. doi: 10.1146/annurev-genet-041814-095928 25195508
49. Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974;77 : 71–94. 4366476
50. Yochem J, Tuck S, Greenwald I, Han M. A gp330/megalin-related protein is required in the major epidermis of Caenorhabditis elegans for completion of molting. Development 1999;126 : 597–606. 9876188
51. Crowder CM, Westover EJ, Kumar AS, Ostlund RE,Jr, Covey DF. Enantiospecificity of cholesterol function in vivo. J Biol Chem 2001;276 : 44369–72. 11598105
52. Zhang H, Abraham N, Khan LA, Hall DH, Fleming JT, Gobel V. Apicobasal domain identities of expanding tubular membranes depend on glycosphingolipid biosynthesis. Nat Cell Biol 2011;13 : 1189–201. doi: 10.1038/ncb2328 21926990
53. Marza E, Simonsen KT, Faergeman NJ, Lesa GM. Expression of ceramide glucosyltransferases, which are essential for glycosphingolipid synthesis, is only required in a small subset of C. elegans cells. J Cell Sci 2009;122 : 822–33. doi: 10.1242/jcs.042754 19240113
54. Nomura KH, Murata D, Hayashi Y, Dejima K, Mizuguchi S, Kage-Nakadai E, et al. Ceramide glucosyltransferase of the nematode Caenorhabditis elegans is involved in oocyte formation and in early embryonic cell division. Glycobiology 2011;21 : 834–48. doi: 10.1093/glycob/cwr019 21325339
55. Griffitts JS, Whitacre JL, Stevens DE, Aroian RV. Bt toxin resistance from loss of a putative carbohydrate-modifying enzyme. Science 2001;293 : 860–4. 11486087
56. Griffitts JS, Huffman DL, Whitacre JL, Barrows BD, Marroquin LD, Muller R, et al. Resistance to a bacterial toxin is mediated by removal of a conserved glycosylation pathway required for toxin-host interactions. J Biol Chem 2003;278 : 45594–602. 12944392
57. Katic I, Vallier LG, Greenwald I. New positive regulators of lin-12 activity in Caenorhabditis elegans include the BRE-5/Brainiac glycosphingolipid biosynthesis enzyme. Genetics 2005;171 : 1605–15. 16157663
58. Junge HJ, Yang S, Burton JB, et al. TSPAN12 regulates retinal vascular development by promoting Norrin - but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009;139 : 299–311. doi: 10.1016/j.cell.2009.07.048 19837033
59. Ke J, Harikumar KG, Erice C, Chen C, Gu X, Wang L, et al. Structure and function of Norrin in assembly and activation of a Frizzled 4-Lrp5/6 complex. Genes Dev 2013;27 : 2305–19. doi: 10.1101/gad.228544.113 24186977
60. Berditchevski R. Tetraspanins. Springer 2013;418. Available: http://link.springer.com/book/10.1007%2F978-94-007-6070-7.
61. Dornier E, Coumailleau F, Ottavi JF, Moretti J, Boucheix C, Mauduit P, et al. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J Cell Biol 2012;199 : 481–96. doi: 10.1083/jcb.201201133 23091066
62. Haining EJ, Yang J, Bailey RL, Khan K, Collier R, Tsai S, et al. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J Biol Chem 2012;287 : 39753–65. doi: 10.1074/jbc.M112.416503 23035126
63. Prox J, Willenbrock M, Weber S, Lehmann T, Schmidt-Arras D, Schwanbeck R, et al. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell Mol Life Sci 2012;69 : 2919–32. doi: 10.1007/s00018-012-0960-2 22446748
64. Sadej R, Grudowska A, Turczyk L, Kordek R, Romanska HM. CD151 in cancer progression and metastasis: a complex scenario. Lab Invest 2014;94 : 41–51. doi: 10.1038/labinvest.2013.136 24247563
65. Knoblich K, Wang HX, Sharma C, Fletcher AL, Turley SJ, Hemler ME. Tetraspanin TSPAN12 regulates tumor growth and metastasis and inhibits beta-catenin degradation. Cell Mol Life Sci 2014;71 : 1305–14. doi: 10.1007/s00018-013-1444-8 23955570
66. Berthier-Vergnes O, Kharbili ME, de la Fouchardiere A, Pointecouteau T, Verrando P, Wierinckx A, et al. Gene expression profiles of human melanoma cells with different invasive potential reveal TSPAN8 as a novel mediator of invasion. Br J Cancer 2011;104 : 155–65. doi: 10.1038/sj.bjc.6605994 21081927
67. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 2014;14 : 49–60. 24505619
68. Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res 2009;19 : 89–102. doi: 10.1038/cr.2008.316 19050696
69. Sadej R, Romanska H, Kavanagh D, et al. Tetraspanin CD151 regulates transforming growth factor beta signaling: implication in tumor metastasis. Cancer Res 2010;70 : 6059–70. doi: 10.1158/0008-5472.CAN-09-3497 20570898
70. Wang HX, Sharma C, Knoblich K, Granter SR, Hemler ME. EWI-2 negatively regulates TGF-beta signaling leading to altered melanoma growth and metastasis. Cell Res 2015;25 : 370–85. doi: 10.1038/cr.2015.17 25656846
71. Baum PD, Garriga G. Neuronal migrations and axon fasciculation are disrupted in ina-1 integrin mutants. Neuron 1997;19 : 51–62. 9247263
72. Williams BD, Waterston RH. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J Cell Biol 1994;124 : 475–90. 8106547
73. Li Y, Wang L, Qiu J, Da L, Tiollais P, Li Z, et al. Human tetraspanin transmembrane 4 superfamily member 4 or intestinal and liver tetraspan membrane protein is overexpressed in hepatocellular carcinoma and accelerates tumor cell growth. Acta Biochim Biophys Sin (Shanghai) 2012;44 : 224–32. doi: 10.1093/abbs/gmr124 22236579
74. Nowee ME, Snijders AM, Rockx DA, de Wit RM, Kosma VM, Hämäläinen K,et al. DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J Pathol 2007;213 : 46–55. 17668415
75. Ferrer M, Yunta M, Lazo PA. Pattern of expression of tetraspanin antigen genes in Burkitt lymphoma cell lines. Clin Exp Immunol 1998;113 : 346–52. 9737661
76. Bankovic J, Stojsic J, Jovanovic D, Andjelkovic T, Milinkovic V, Ruzdijic S, et al. Identification of genes associated with non-small-cell lung cancer promotion and progression. Lung Cancer 2010;67 : 151–9. doi: 10.1016/j.lungcan.2009.04.010 19473719
77. Gumienny TL, MacNeil LT, Wang H, de Bono M, Wrana JL, Padgett RW. Glypican LON-2 is a conserved negative regulator of BMP-like signaling in Caenorhabditis elegans. Curr Biol 2007;17 : 159–64. 17240342
78. Savage-Dunn C, Maduzia LL, Zimmerman CM, Roberts AF, Cohen S, Tokarz R, et al. Genetic screen for small body size mutants in C. elegans reveals many TGFbeta pathway components. Genesis 2003;35 : 239–47. 12717735
79. Hirose T, Nakano Y, Nagamatsu Y, Misumi T, Ohta H, Ohshima Y. Cyclic GMP-dependent protein kinase EGL-4 controls body size and lifespan in C elegans. Development 2003;130 : 1089–99. 12571101
80. Merris M, Wadsworth WG, Khamrai U, Bittman R, Chitwood DJ, Lenard J. Sterol effects and sites of sterol accumulation in Caenorhabditis elegans: developmental requirement for 4alpha-methyl sterols. J Lipid Res 2003;44 : 172–81. 12518036
81. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003;421 : 231–7. 12529635
82. Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 2004;14 : 2162–8. 15489339
83. Wicks SR, Yeh RT, Gish WR, Waterston RH, Plasterk RH. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet 2001;28 : 160–4. 11381264
84. Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 2010;5:e15435. doi: 10.1371/journal.pone.0015435 21079745
85. Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 2005;6 : 118. 16156901
86. Bigelow H, Doitsidou M, Sarin S, Hobert O. MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods 2009;6 : 549. doi: 10.1038/nmeth.f.260 19620971
87. Sarin S, Bertrand V, Bigelow H, Boyanov A, Doitsidou M, Poole RJ, et al. Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics 2010;185 : 417–30. doi: 10.1534/genetics.110.116319 20439776
88. Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One 2009;4:e7767. doi: 10.1371/journal.pone.0007767 19907642
89. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25 : 2078–9. doi: 10.1093/bioinformatics/btp352 19505943
90. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. doi: 10.1093/nar/gkq603 20601685
91. Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 2013;10 : 1028–34. doi: 10.1038/nmeth.2641 23995389
92. Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 2013;10 : 741–3. doi: 10.1038/nmeth.2532 23817069
93. Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, et al. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 2008;40 : 1375–83. doi: 10.1038/ng.248 18953339
94. Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform 2009;23 : 205–11. 20180275
95. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res 2014;42:D222–30. doi: 10.1093/nar/gkt1223 24288371
96. Sonnhammer EL, Gabaldon T, Sousa da Silva AW, Martin M, Robinson-Rechavi M, Boeckmann B, et al. Big data and other challenges in the quest for orthologs. Bioinformatics 2014;30 : 2993–8. doi: 10.1093/bioinformatics/btu492 25064571
97. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30 : 772–80. doi: 10.1093/molbev/mst010 23329690
98. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009;25 : 1972–3. doi: 10.1093/bioinformatics/btp348 19505945
99. Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol 2010;10 : 210,2148-10-210. doi: 10.1186/1471-2148-10-210 20626897
100. Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 2001;18 : 691–9. 11319253
101. Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 2010;5:e9490. doi: 10.1371/journal.pone.0009490 20224823
102. Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 2009;26 : 1641–50. doi: 10.1093/molbev/msp077 19377059
103. Grefen C, Lalonde S, Obrdlik P. Split-ubiquitin system for identifying protein-protein interactions in membrane and full-length proteins. Curr Protoc Neurosci 2007;Chapter 5:Unit 5.27.
104. Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23 : 2947–8. 17846036
105. Moribe H, Yochem J, Yamada H, Tabuse Y, Fujimoto T, Mekada E. Tetraspanin protein (TSP-15) is required for epidermal integrity in caenorhabditis elegans. J Cell Sci 2004;117(Pt 22): 5209–5220. 15454573
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy