
Cooperative Action of Cdk1/cyclin B and SIRT1 Is Required for Mitotic Repression of rRNA Synthesis
In metazoans, transcription is arrested during mitosis. Previous studies have established that mitotic repression of cellular transcription is mediated by Cdk1/cyclin B-dependent phosphorylation of basal transcription factors that nucleate transcription complex formation. Repression of rDNA transcription at the onset of mitosis is brought about by inactivation of the TBP-containing transcription factor SL1 by Cdk1/cyclin B-dependent phosphorylation of the TAFI110 subunit, which impairs the interaction with UBF and the assembly of pre-initiation complexes. Here we show that hCdc14B, the phosphatase that regulates Cdk1/cyclin B activity and progression through mitosis, promotes reactivation of rDNA transcription by dephosphorylating TAFI110. In addition, the NAD+-dependent deacetylase SIRT1 becomes transiently enriched in nucleoli at the onset of mitosis. SIRT1 deacetylates TAFI68, another subunit of SL1, hypoacetylation of TAFI68 destabilizing SL1 binding to the rDNA promoter and impairing transcription complex assembly. The results reveal that modulation of SL1 activity by reversible acetylation of TAFI68 and phosphorylation of TAFI110 are key modifications that mediate oscillation of rDNA transcription during cell cycle progression.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005246
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005246
Summary
In metazoans, transcription is arrested during mitosis. Previous studies have established that mitotic repression of cellular transcription is mediated by Cdk1/cyclin B-dependent phosphorylation of basal transcription factors that nucleate transcription complex formation. Repression of rDNA transcription at the onset of mitosis is brought about by inactivation of the TBP-containing transcription factor SL1 by Cdk1/cyclin B-dependent phosphorylation of the TAFI110 subunit, which impairs the interaction with UBF and the assembly of pre-initiation complexes. Here we show that hCdc14B, the phosphatase that regulates Cdk1/cyclin B activity and progression through mitosis, promotes reactivation of rDNA transcription by dephosphorylating TAFI110. In addition, the NAD+-dependent deacetylase SIRT1 becomes transiently enriched in nucleoli at the onset of mitosis. SIRT1 deacetylates TAFI68, another subunit of SL1, hypoacetylation of TAFI68 destabilizing SL1 binding to the rDNA promoter and impairing transcription complex assembly. The results reveal that modulation of SL1 activity by reversible acetylation of TAFI68 and phosphorylation of TAFI110 are key modifications that mediate oscillation of rDNA transcription during cell cycle progression.
Introduction
Posttranslational modification of transcription factors is critical for cell cycle progression in a unidirectional and reversible manner. Cell cycle-dependent oscillation of transcriptional activity is governed by a complex network of regulatory proteins and signaling pathways that respond to various intra - and extracellular stimuli by influencing the activity and tertiary structure of proteins, controlling subcellular distribution, and regulating interactions with other proteins. Global repression of gene expression starts at prophase and is accompanied by release of most transcriptional regulators from mitotic chromatin [1–3, 4]. Mitotic switch-off of cellular transcription involves inactivation of key components of the transcription machinery. For class II genes, components of the basal transcription apparatus are inactivated by mitotic phosphorylation, including TAF subunits of TFIID [4, 5], the cdk7 subunit of TFIIH [6, 7] and the heptapeptide repeats of the carboxy-terminal domain (CTD) of RNA polymerase II [8]. For class III genes, inactivation of TFIIIB causes repression of RNA polymerase III (Pol III) transcription [9–11].
With regard to transcription by RNA polymerase I (Pol I), the nucleolar structure undergoes extensive changes at the onset of mitosis, and rDNA transcription ceases between pro-metaphase and telophase [3]. While most nucleolar proteins disperse throughout the mitotic cell after breakdown of the nuclear envelope, some components of the Pol I transcription machinery, including UBF and TTF-I, remain associated with nucleolus organizer regions (NORs) to bookmark active rDNA repeats [11, 12]. Consistent with post-translational modification of basal transcription factors controlling cell cycle-dependent fluctuations of gene expression, mitotic silencing and reactivation of rDNA transcription upon mitotic exit has been shown to be governed by reversible phosphorylation of the promoter selectivity factor SL1 [13]. SL1 is a multiprotein complex comprising the TATA-box binding protein (TBP) and five TBP-associated factors (TAFIs), TAFI110, TAFI68, TAFI48, TAFI41, and TAFI12 [14–17]. At the onset of mitosis, Cdk1/cyclin B, the kinase that triggers early mitotic events, e.g. chromosome condensation, nuclear envelope breakdown and spindle pole assembly, phosphorylates TAFI110. This phosphorylation impairs the interaction between SL1 and UBF, thus attenuating the assembly of pre-initiation complexes at the rDNA promoter [13, 18]. Upon exit from mitosis, rDNA transcription is restored, yet the mechanisms that restore transcriptional activity are poorly characterized [19].
In this study, we have investigated the molecular mechanisms that cause reversible mitotic inactivation of SL1 at the onset of mitosis and relieve transcriptional silencing at the end of mitosis. Consistent with prior studies showing that the phosphatase hCdc14B regulates progression through mitosis by counteracting mitotic phosphorylation by Cdk1/cyclin B [20], hCdc14B dephosphorylates TAFI110, thus promoting its reactivation as cells exit mitosis. Notably, though phosphorylation of TAFI110 by Cdk1/cyclin B is necessary, alone it is not sufficient for mitotic inactivation of rDNA transcription. Previous studies have established that another SL1 subunit, TAFI68, is acetylated by PCAF, acetylation of TAFI68 stabilizing binding of SL1 to the rDNA promoter [21]. Here we show that deacetylation of TAFI68 by the NAD+-dependent deacetylase SIRT1 is also crucial for mitotic inactivation of SL1. SIRT1 becomes enriched in nucleoli at the onset of mitosis and deacetylates TAFI68, which in turn weakens SL1 binding to rDNA and impairs transcription complex assembly. Thus both phosphorylation of TAFI110 by Cdk1/cyclin B and deacetylation of TAFI68 by SIRT1 are required for repression of Pol I transcription during mitosis. The finding that both Cdk1 and SIRT1 modulate the activity of SL1 underscores the functional significance of reversible modification of SL1 in linking cycle progression to regulation of rDNA transcription.
Results
The phosphatase Cdc14B counteracts mitotic phosphorylation of TAFI110
In asynchronous cells, TAFI110 (TAF1C) is constitutively phosphorylated at two tryptic peptides (labeled a and b in Fig 1A). In mitotic cells, a third peptide (labeled c) is phosphorylated by Cdk1/cyclin B, phosphorylation of peptide c correlating with mitotic inactivation of SL1 and transcriptional repression [13]. Phosphoamino acid analysis using 2-dimensional electrophoresis along with amino acid standards of phosphorylated serine, threonine and tyrosine showed that Cdk1/cyclin B phosphorylates TAFI110 at threonine, phosphorylation being reduced by the Cdk inhibitor roscovitine (S1A Fig). Two-dimensional phosphopeptide mapping experiments revealed that peptide c co-migrates with a synthetic peptide (SQQHpTPVLSSQPLR) that is phosphorylated at threonine 852 (Fig 1A and S1B Fig), suggesting that phosphorylation of T852 is causally involved in mitotic inactivation of SL1. Sequence alignment revealed that T852 as well as adjacent amino acids are conserved in TAFI110 from different vertebrates (S1C Fig). Regarding the phosphatase that counteracts mitotic phosphorylation of T852, we hypothesized that hCdc14B, the phosphatase that regulates Cdk1/cyclin B activity and progression through mitosis in mammals [20], could remove the inhibitory phosphate from T852. Indeed, recombinant hCdc14B dephosphorylated peptide c comprising T852, but not peptides a or b (Fig 1A, right). In vitro protein pull-down assays showed specific association of hCdc14B with hTAFI110 (Fig 1B). Moreover, co-immunoprecipitation experiments demonstrate that hCdc14B interacts with hTAFI110 in vivo (Fig 1C), supporting that hCdc14B is the phosphatase that removes the inhibitory phosphate from TAFI110. These results identify hTAFI110 as a novel substrate of hCdc14B, revealing that hCdc14B counteracts Cdk1/cyclin B-mediated phosphorylation of SL1.
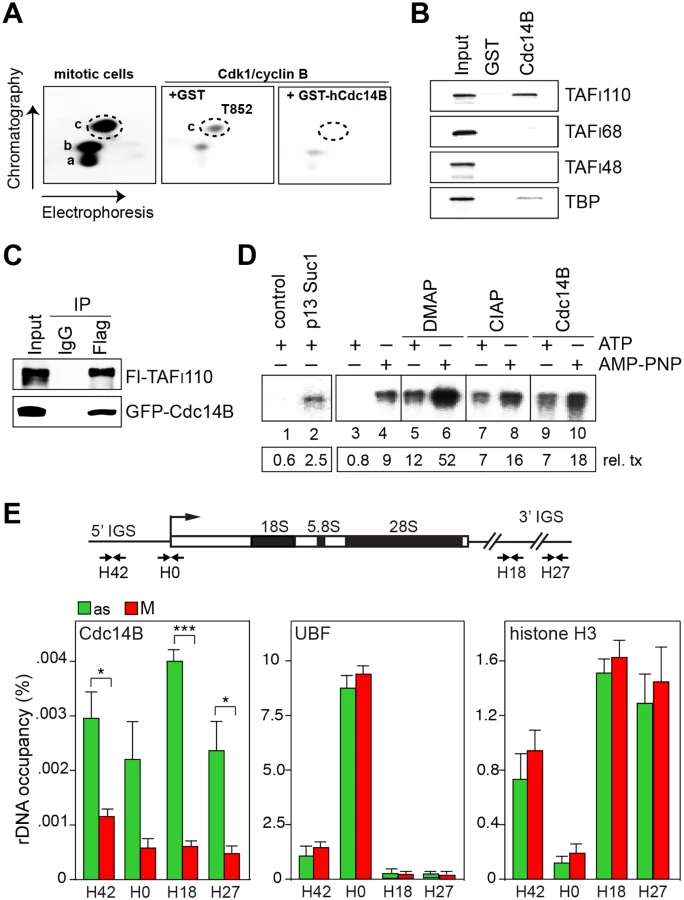
To examine whether hCdc14B is capable to overcome mitotic repression of Pol I transcription, we performed in vitro transcription assays using extracts from M-phase cells. As reported before [18, 19], extracts from mitotic cells are transcriptionally inactive (S1D Fig). Transcriptional repression was relieved if extracts were depleted from Cdk1/cyclin B by bead-bound p13suc1, underscoring the importance of Cdk1/cyclin B in mitotic repression of Pol I transcription (Fig 1D, lanes 1, 2). Transcriptional activity was also restored if transcription was performed in conditions that prevent Cdk1/cyclin B-dependent phosphorylation and thus inactivation of SL1. If ATP was replaced by the non-hydrolysable ATP analogue AMP-PNP (adenylyl-imidotriphosphate) or by inclusion of the kinase inhibitor DMAP (6-dimethylaminopurine), transcriptional repression was relieved (Fig 1D, lanes 3–10 and S1E Fig). Addition of calf intestine alkaline phosphatase (CIAP) or Cdc14B relieved transcriptional repression in ATP-containing reactions, reinforcing that Cdc14B-dependent dephosphorylation of TAFI110 at T852 reactivates Pol I transcription at the exit from mitosis.
Nucleoli disassemble during mitosis and many nucleolar proteins are released into the cytoplasm [3]. However, UBF remains bound to rDNA, thus bookmarking rDNA for resumption of transcription upon mitotic exit (Fig 1E and S1F Fig; see also ref. [22]). Both in yeast and mammals, Cdc14B is sequestered in nucleoli during interphase and activated both during mitosis and DNA damage upon release from nucleolar chromatin [20, 23]. In accord with these observations, we found that in asynchronous cells hCdc14B was preferentially bound to intergenic spacer sequences separating individual rDNA repeats (5’ - and 3’-IGS). Binding of hCdc14B to rDNA was abrogated in mitotic cells (Fig 1E, S1F and S1G Fig), reinforcing that Cdc14B is inactivated during interphase by confinement to the intergenic spacer, and is released from nucleolar chromatin during mitosis. Significantly, UBF and histone H3 remained associated with rDNA in M-phase cells. Together, the results suggest that release from rDNA enables hCdc14B to dephosphorylate SL1, a step that is required for resumption of rDNA transcription when cells re-enter the cell cycle.
Phosphorylation of T852 is not sufficient for mitotic inactivation of SL1
To prove that hCdc14B-dependent dephosphorylation of SL1 is required to activate rDNA transcription at the exit from mitosis, we assayed the activity of immunopurified SL1 in a reconstituted transcription system. SL1 from asynchronous cells stimulated transcription up to 8-fold (Fig 2A, lane 3), while the same amount of SL1 from mitotic cells was inactive (Fig 2A, lane 4). The activity of mitotic SL1 was restored by addition of hCdc14B, demonstrating that dephosphorylation of T852 by hCdc14B relieves Cdk1/cyclin-dependent mitotic inactivation of SL1 (Fig 2A, lanes 5, 6).

If Cdk1/cyclin B-mediated phosphorylation of TAFI110 is the only mechanism that inactivates SL1 and represses Pol I transcription during mitosis, then mutation of T852 should prevent mitotic inactivation of SL1. To test this, we established HeLa cell lines that stably overexpress Flag-tagged wildtype TAFI110 or a mutant in which T852 has been replaced by alanine (TAFI110/T852A). Both wildtype and mutant TAFI110 assembled into proper TBP-TAF complexes, indicating that replacement of T852 by alanine does not affect the interaction of TAFI110 with other SL1 subunits (Fig 2B). Moreover, both wildtype and mutant TAFI110 bound with similar efficiency to the rDNA promoter, supporting that phosphorylation of T852 has no impact on DNA binding (Fig 2C). To further corroborate these results, we compared rDNA occupancy of Pol I, UBF and SL1 in synchronized HeLa cells expressing Flag-TAFI110 or Flag-TAFI110/T852A. While UBF remained associated with rDNA throughout the cell cycle [22, 24], binding of SL1 and Pol I was decreased in mitotic cells (Fig 2D). Decreased rDNA occupancy of Pol I and SL1 was observed in mitotic cells regardless of whether wildtype or mutant TAFI110 were overexpressed, indicating that the assembly of Pol I pre-initiation complexes was impaired both in cells overexpressing wildtype TAFI110 or mutant TAFI110/T852A.
To confirm that rDNA transcription is switched-off during mitosis in cells expressing mutant TAFI110, we pulse-labeled nascent RNA with FUrd and visualized nascent transcripts that co-localize with UBF at mitotic NORs (Fig 2E). Similar amounts of FU-labeled transcripts were synthesized in interphase cells, regardless of whether wildtype or mutant TAFI110 was expressed. Surprisingly, though the level of ectopic TAFI110/T852A was about 2–3 fold higher than that of endogenous TAFI110 (S2A Fig), no nascent transcripts were visible in mitotic cells expressing mutant TAFI110/T852A. The observation that the phosphorylation-deficient mutant repressed rDNA transcription as efficiently as wildtype TAFI110 reveals that phosphorylation of TAFI110 by Cdk1/cyclin B is necessary but not sufficient for mitotic repression of rDNA transcription.
TAFI68 is hypoacetylated during mitosis
As phosphorylation of TAFI110 was not sufficient for mitotic inactivation of SL1, we reasoned that other posttranslational modifications contribute to inactivation of SL1 at the entry into mitosis. Previous work has established that TAFI68 (TAF1B), a TBP-associated factor that is structurally related to the general transcription factor TFIIB [25], is acetylated by the histone acetyltransferase PCAF at two lysine residues, K438 and K443 [26]. Acetylation was shown to augment the DNA-binding activity of TAFI68 and activate rDNA transcription [21]. Mutation of both lysine residues abolished PCAF-dependent acetylation of TAFI68 (S3A and S3B Fig). The correlation between acetylation and DNA binding efficiency of TAFI68 implies that deacetylation of TAFI68 impairs transcription complex assembly. In a previous study we have shown that PCAF-dependent acetylation of TAFI68 was counteracted by SIRT1 [21], the founding member of the Sirtuin family of NAD+-dependent histone deacetylases. In support of SIRT1 interacting with SL1, immobilized SIRT1 associated with TAFI110, TAFI68 and TBP, subunits of the SL1 complex (Fig 3A, top). Consistently, endogenous SIRT1 was co-immunoprecipitated with ectopic TAFI68 (Fig 3A, bottom). No association of TAFI68 was observed with either SIRT6 or SIRT7, highlighting the specificity of TAFI68 binding to SIRT1 (Fig 3B). This result reveals that the two nucle(ol)ar Sirtuins SIRT1 and SIRT7 serve opposing functions, SIRT7 activating rDNA transcription in cycling cells [27, 28], while deacetylation of TAFI68 by SIRT1 being required for mitotic repression of rDNA transcription. Accordingly, acetylation of TAFI68 was decreased in prometaphase - compared to G1/S-arrested cells, supporting that cell cycle-dependent fluctuations of SL1 acetylation are involved in mitotic repression of rRNA synthesis (Fig 3C). Consistent with SIRT1 targeting TAFI68, in vitro deacetylation of TAFI68 by SIRT1 required the presence of NAD+ (Fig 3D). Likewise, in vivo acetylation of TAFI68 was increased if cells were treated with nicotinamide (NAM), a competitive inhibitor of NAD+-dependent deacetylases (Fig 3E). Knockdown of SIRT1 further increased acetylation, proving that SIRT1 rather than another member of the Sirtuin family deacetylates TAFI68.

Next, we examined whether SIRT1-dependent deacetylation of TAFI68 contributes to mitotic inactivation of SL1. For this, we compared rDNA occupancy of SL1 (TBP, TAFI110 and TAFI68), Pol I (RPA116) and UBF in prometaphase cells expressing wildtype or mutant TAFI110 (Fig 3F). As expected, rDNA occupancy of UBF was comparable in both cell lines and was not affected by NAM-dependent inhibition of SIRT1 activity. In contrast, treatment with NAM similarly increased binding of SL1 regardless of whether cells expressed wildtype or mutant TAFI110. This indicates that deacetylation of TAFI68 by SIRT1 rather than phosphorylation of TAFI110 weakens the association of SL1 with the rDNA promoter, which leads to partial displacement of SL1 from rDNA in early mitosis. Consistent with acetylation of TAFI68 being required for binding of SL1 and transcription complex formation, rDNA promoter occupancy of Pol I was elevated in NAM-treated mitotic TAFI110/T852A cells, indicating that under these conditions transcriptional repression was partially relieved. Thus, unphosphorylated TAFI110 and acetylated TAFI68 are required for the assembly of productive Pol I transcription initiation complexes. Together, these results imply that mitotic repression of Pol I transcription is brought about by a dual mechanism. Deacetylation of TAFI68 by SIRT1 weakens the association of SL1 with rDNA, while phosphorylation of TAFI110 by Cdk1/cyclin B impairs the interaction with UBF, leading to mitotic repression of, rDNA transcription.
Inhibition of SIRT1 bypasses mitotic repression of Pol I transcription
In asynchronous cells the majority of SIRT1 resides in the nucleoplasm and is excluded from nucleoli. In prophase cells, however, SIRT1 transiently localizes in nucleoli, co-staining with UBF and Pol I (Fig 4A, dashed circles). Prophase cells have an intact nuclear envelope but show chromosome condensation and are positive for the mitotic histone mark H3-pSer10. Consistent with UBF bookmarking mitotic NORs [22, 29], UBF remained bound to NORs throughout mitosis. Co-localization of SIRT1 and UBF was confined to prophase and was not detected at later stages of mitosis (Fig 4B). Enrichment of SIRT1 at NORs preceded repression of Pol I transcription, monitored by visualization of NOR-associated nascent RNAs at different stages of mitosis. Consistent with Pol I transcription being switched-off during mitosis, no nascent RNA was detected at UBF-specific foci from prometaphase to telophase (Fig 4C).

Next we monitored nascent pre-rRNA levels in mitotic HeLa cells expressing Flag - TAFI110/WT or TAFI110/T852A in the absence or presence of the Sirtuin inhibitor NAM. The rationale of this experiment was to find out whether mitotic inactivation of SL1 would be relieved and pre-rRNA synthesis restored if both phosphorylation of T852 and deacetylation of TAFI68 were prevented. Immunofluorescence analysis of fluorouridine (FUrd)-labeled RNA revealed low but significant levels of nascent transcripts at mitotic NORs in NAM-treated cells that express TAFI110/T852A (Fig 4D). In contrast, there was no nascent RNA visible at mitotic NORs of HeLa cells expressing wildtype hTAFI110, regardless of whether the cells were treated with NAM or not. The finding that SL1 was not inactivated if both Cdk1/cyclin B-dependent phosphorylation of hTAFI110 and SIRT1-dependent deacetylation of TAFI68 were prevented highlight the functional significance of both activities in mitotic repression of Pol I transcription.
Discussion
At mitosis, there is substantial reorganization of chromosomal architecture as cells prepare to exit the G2-phase of the cell cycle and enter the prophase of mitosis. This reorganization of nuclear structure is accompanied by a global shut-off of transcriptional activity. Transcription by all three classes of nuclear DNA-dependent RNA polymerases stops by mid-prophase and resumes in late telophase [1, 2]. Mitotic repression of rDNA transcription correlates with perturbation of nucleolar structure and dispersion of most nucleolar proteins. However, basal factors required for transcription initiation are maintained on metaphase chromosomes [3, 12, 24, 30], thus marking rRNA genes for rapid assembly of pre-initiation complexes and resumption of rRNA synthesis in G1-phase. Mitotic repression of Pol I transcription is brought about by phosphorylation of TAFI110, the large subunit of the basal Pol I-specific transcription factor SL1 by Cdk1/cyclin B [13, 18]. Phosphorylation of TAFI110 at threonine 852 impairs the capability of SL1 to interact with UBF, thereby abrogating the assembly of transcription-competent initiation complexes [13]. Here we show that phosphorylation of TAFI110 at threonine 852 is counteracted by the phosphatase Cdc14B, which regulates progression through mitosis [20]. Cdc14B is sequestered during interphase in the nucleolus by association with intergenic spacer sequences that separate individual rDNA transcription units. At prometaphase, Cdc14B is released from rDNA, allowing dephosphorylation of TAFI110 and resumption of rRNA synthesis in early G1-phase [19]. These results suggested that Cdc14B-dependent dephosphorylation of TAFI110 is the molecular switch that reactivates SL1 at the exit from mitosis. Surprisingly, however, transcription was also repressed in mitotic cells that express the phosphorylation-deficient mutant TAFI110/T852A, indicating that phosphorylation of T852 by Cdk1/cyclin B is not the only mechanism that inactivates SL1 during mitosis.
In addition to phosphorylation of TAFI110, repression of rDNA transcription upon entry into mitosis involves deacetylation of another SL1 subunit, TAFI68, by the NAD+-dependent deacetylase SIRT1. TAFI68 is acetylated by the histone acetyltransferase PCAF, acetylation promoting the association of SL1 with the rDNA promoter [21]. The functional significance of TAFI68 acetylation, however, remained obscure. We found that mitotic repression of transcription was alleviated in the presence of nicotinamide, a competitive inhibitor of NAD+-dependent deacetylases. Moreover, PCAF-dependent acetylation of TAFI68 was counteracted by SIRT1, which is transiently enriched in nucleoli at prophase. This identifies TAFI68 as novel substrate of SIRT1, SIRT1-dependent deacetylation of SL1 reinforcing mitotic shut-off of Pol I transcription. In addition, SIRT1 is known to deacetylate the euchromatic histone mark H4K16Ac, and to facilitate loading of histone H1 and the condensin I complex, which promotes facultative heterochromatin formation and thereby contributes to chromosome integrity and stability during mitosis [31–33]. Our finding that SIRT1 deacetylates TAFI68 is in accord with numerous studies demonstrating that the deacetylase activity of SIRT1 targets histones, chromatin regulators, and a world of nonhistone substrates, including metabolic enzymes, transcription factors, cytoskeleton proteins, and many others [34]. Like other transcription factors, such as p53, acetylation of TAFI68 increases site-specific DNA binding activity. Accordingly, deacetylation by SIRT1 weakens the association of SL1 with rDNA [21]. Nucleolar enrichment of SIRT1 and decreased rDNA transcription correlates with increased dynamics of the Pol I transcription machinery at mitotic NORs determined by FRAP measurements [24, 35]. Thus, cells regulate rDNA transcription complex formation by reversible acetylation of TAFI68, acetylation of TAFI68 increasing and deacetylation by SIRT1 decreasing DNA binding of SL1. At the entry into mitosis two posttranslational modifications, i.e., phosphorylation of TAFI110 and deacetylation of TAFI68, inactivate SL1, thereby attenuating pre-initiation complex formation (Fig 5). These results reveal that fine-tuned reversible acetylation and phosphorylation of TAFIs is an effective means to regulate SL1 activity and mediate fluctuations of Pol I transcription during cell cycle progression.

Materials and Methods
Cell culture, transfections, and cell treatments
Cells cultured according to standard conditions (ATCC) were transfected with Fugene6, Lipofectamin 2000 (Invitrogen), or calcium phosphate. Clonal HeLa cell lines that stably express Flag-tagged wildtype hTAFI110 or mutant hTAFI110/T852A were selected in the presence of G418 (750 μg/ml). To knockdown SIRT1, cells transfected with specific shRNA expression plasmids (Sigma) were selected in the presence of puromycin (1 μg/ml) and analyzed after 5–6 days. HeLa and U2OS cells were synchronized at G1/S with thymidine (2 mM, 23 h), released for 8 h, and arrested in prometaphase with nocodazole (80 ng/ml). To inhibit SIRT1 activity, cells were treated for 5 h with 5–10 mM nicotinamide (NAM).
Plasmids and antibodies
Plasmids encoding SIRT1, SIRT6, SIRT7, hCdc14B, TBP, and individual TAFIs have been described [15, 20, 21, 27, 28]. cDNA encoding hTAFI110 (accession number NM_005679) was cloned into pRc/CMV (Invitrogen). Threonine at position 852 of hTAFI110, and lysine residues at position 438 and 443 of hTAFI68 were converted into alanine or arginine by site-directed mutagenesis. Primers used for PCR-mediated mutagenesis are listed in S2 Table. Antibodies against UBF [36], RPA194 and RPA116 [37, 38] and TAFIs [15] have been described. Anti-TBP antibodies (clone 3G3) were provided by Laszlo Tora. Antibodies against the Flag epitope (F3165), actin, BrdU (BU-33) were from Sigma, anti-acetylated lysine from Cell Signaling (9441), anti-UBF (sc-13125) and anti PCAF (H-369) from Santa Cruz, anti-SIRT1 (07–131), anti-histone H3-pSer10 (06–570) from Millipore, anti-GFP from Abcam, anti-histone H3 from Diagenode and anti-Cdc14B from Zymed.
Immunocytochemistry and nuclear run-on assays
Immunofluorescence was performed as described [39]. The secondary antibodies were conjugated to Cy2, Cy3, or FITC (Dianova), Alexa Fluor 488 or Alexa Fluor 555 (Molecular Probes). To visualize nascent RNAs, cells grown on poly-L-lysine coated coverslips were labeled with 2 mM fluorouridine (FUrd) for 20 min, fixed with 2% paraformaldehyde, permeabilized with methanol, incubated with the respective antibodies and stained with fluorophore-coupled secondary antibodies. DNA was stained with Hoechst 33342.
Microscopy, image acquisition and quantitative analysis
Images were visualized with a Zeiss microscope (Axiophot) using a 40×1.3 oil immersion Plan-Neofluar magnifying objective (Carl Zeiss), captured with a device camera (DS-Qi1Mc; Nikon) and processed with NIS-Elements software (version BR 3.10; Nikon). Images were quantified images using ImageJ and calculated as described [40]. Confocal laser scanning microscopy (CLSM) was done with LSM META 510 (Zeiss). Immunofluorescence images were quantified using ImageJ as described [40].
Expression and purification of recombinant proteins
HEK293T or HeLa cells expressing epitope-tagged proteins were lysed in buffer AM-600 (600 mM KCl, 20 mM Tris-HCl [pH 7.9], 5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 0.5 mM DTT) supplemented with 0.1% NP-40, protease inhibitors (Roche Complete, PMSF), and HDAC inhibitors (500 nM TSA, 5 mM sodium butyrate, 10 mM nicotinamide). Lysates were incubated for 4 h at 4°C with mouse or rabbit IgGs bound to protein A/G-agarose (Roche, Dianova) or with M2 anti-Flag beads (Sigma). After washing in lysis buffer, tagged proteins were eluted in buffer AM-300 supplemented with 0.1% NP-40 and Flag-peptide (20 μg/100 μl). GST-tagged proteins were expressed in E. coli and affinity-purified on Glutathione-Sepharose 4B (GE healthcare). Flag-tagged UBF was isolated from Sf9 cells [36]. GFP-tagged proteins were bound to GFP-Trap (ChromoTek) in buffer AM-300 containing 0.1% NP-40, Roche complete and HDAC inhibitors. Flag-tagged UBF and PCAF were isolated from baculovirus-infected Sf9 cells as described [36, 21].
Protein pull-down experiments and co-immunoprecipitation
2 μg of GST or GST-tagged proteins immobilized on Glutathione-Sepharose 4B were incubated with in vitro synthesized 35S-labeled proteins for 4 h at 4°C in 120 mM KCl, 20 mM Tris-HCl [pH 7.9], 5 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 0.2% NP-40 and protease inhibitors (Roche Complete, PMSF). After washing, bead-bound proteins were analyzed by SDS-PAGE and PhosphorImaging. Endogenous SL1 was immunopurified from HeLa nuclear extracts with anti-TBP antibody (clone 3G3) to Dynabeads, mouse IgGs were used as control [15]. To monitor the interaction of Flag-tagged hTAFI68 with GFP-tagged Sirtuins, cells were lysed in buffer containing 300 mM KCl, 20 mM Tris-HCl [pH 7.9], 5 mM MgCl2, 0.2 mM EDTA, 0.5% Triton X-100 and protease inhibitors (Roche Complete, 0.5 mM PMSF). After capture on GFP-trap beads (4 h, 4°C), associated hTAFI68 was monitored on Western blots. To examine the association of TAFI110 with Cdc14B, Flag-tagged hTAFI110 co-expressed with GFP-hCdc14B in HEK293T cells was bound to M2-agarose in the same buffer containing 120 mM KCl, and co-purified GFP-hCdc14B was visualized on immunoblots. Co-immunoprecipitation of Flag-tagged hTAFI110 with endogenous TAFI68 and TBP was performed in the presence of 250 mM KCl.
In vitro transcription assays
In vitro transcription reactions contained 40 ng of linearized plasmid pHrP2 comprising human rDNA sequences from -410 to +378 (with respect to the transcription start site) and 40 μg of extract from HeLa cells [13]. Depletion of Cdk1/cyclin B from mitotic extracts with immobilized p13suc1 has been described [18]. SL1 was immunopurified from extracts with anti-TBP (3G3) antibodies immobilized on anti-mouse IgG Dynabeads. Flag-tagged UBF was immunopurified from baculovirus-infected Sf9 cells [37]. The SL1-responsive reconstituted transcription system contained 5 μl of Pol I (MonoS fraction), 3 μl of TIF-IA (Q-Sepharose fraction) [41], and 10 ng of UBF. After incubation for 1 h at 30°C, transcripts were purified and analyzed by gel electrophoresis and PhosphorImaging.
Chromatin immunoprecipitation (ChIP) assays
Cells were fixed with 1% formaldehyde (10 min, RT), quenched with 0.125 M glycine, and sonicated to yield 250–500 bp DNA fragments. After 5-fold dilution with IP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.0], 167 mM NaCl) and preclearing with protein A/G Sepharose, lysates were incubated overnight with the respective antibodies, and protein-DNA complexes were captured on protein A/G Sepharose followed by washes in low salt buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 5 mM MgCl2, 1% Triton X-100), high salt buffer containing 500 mM NaCl, followed by LiCl buffer (250 mM LiCl, 10 mM Tris-HCl [pH 8.0], 5 mM EDTA, 0.5% Na-deoxycholate, 0.5% Triton X-100) and TE buffer. After reversal of the crosslink and proteinase K digestion, DNA was purified and analyzed by qPCR (Roche LightCycler480). Precipitated DNA was calculated as the percentage of DNA in the immunoprecipitates compared to input DNA. The primers used for PCR are listed in S1 Table.
Phosphopeptide mapping of TAFI110
Recombinant TAFI110 was immunopurified from HEK293T cells overexpressing FLAG-tagged TAFI110 and radiolabeled in vitro by incubation with extracts from mitotic HeLa cells or immobilized Cdk1/cyclin B in the presence of 32P-ATP. After digestion overnight at 37°C with trypsin (5 μg, Promega, sequencing grade) in 50 mM ammonium bicarbonate and lyophilisation, the peptides were resolved on cellulose thin-layer plates by electrophoresis for 25 min at 1000V in 1% (w/v) ammonium carbonate (pH 8.9), followed by ascending chromatography in a buffer containing 62.5% isobutyric acid, 1.9% n-butanol, 4.8% pyridine, and 2.9% acetic acid [13, 41]. Phosphoamino acid analysis was performed according to Boyle et al. [42].
Supporting Information
Zdroje
1. Taylor JH. Nucleic acid synthesis in relation to the cell division cycle. Ann N Y Acad Sci. 1960;90 : 409–421. 13775619
2. Prescott DM, Bender MA. Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp Cell Res. 1962;26 : 260–268. 14488623
3. Hernandez-Verdun D. Assembly and disassembly of the nucleolus during the cell cycle. Nucleus. 2011;2 : 189–194. doi: 10.4161/nucl.2.3.16246 21818412
4. Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin. 2013;6 : 6. doi: 10.1186/1756-8935-6-6 23547918
5. Segil N, Guermah M, Hoffmann A, Roeder RG, Heintz N. Mitotic regulation of TFIID: inhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev. 1996;10 : 2389–2400. 8843192
6. Long JJ, Leresche A, Kriwacki RW, Gottesfeld JM. Repression of TFIIH transcriptional activity and TFIIH-associated cdk7 kinase activity at mitosis. Mol Cell Biol. 1998;18 : 1467–1476. 9488463
7. Akoulitchev S, Reinberg D. The molecular mechanism of mitotic inhibition of TFIIH is mediated by phosphorylation of CDK7. Genes Dev. 1998;12 : 3541–3550. 9832506
8. Cisek LJ, Corden JL. Phosphorylation of RNA polymerase by the murine homologue of the cell-cycle control protein cdc2. Nature. 1989;339 : 679–684. 2662013
9. Hartl P, Gottesfeld J, Forbes DJ. Mitotic repression of transcription in vitro. J Cell Biol. 1993;120 : 13–24.
10. Gottesfeld JM, Wolf J, Dang T, Forbes DJ, Hartl P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science. 1994;263 : 81–84. 8272869
11. White RJ, Gottlieb TM, Downes CS, Jackson SP. Cell cycle regulation of RNA polymerase III transcription. Mol Cell Biol. 1995;15 : 6653–6662. 8524230
12. Sirri V, Roussel P, Hernandez-Verdun D. The mitotically phosphorylated form of the transcription termination factor TTF-1 is associated with the repressed rDNA transcription machinery. J Cell Sci. 1999;112 : 3259–3268. 10504331
13. Heix J, Vente A, Voit R, Budde A, Michaelidis TM, Grummt I. Mitotic silencing of human rRNA synthesis: inactivation of the promoter selectivity factor SL1 by cdc2/cyclin B-mediated phosphorylation. EMBO J. 1998;17 : 7373–7381. 9857193
14. Zomerdijk JC, Beckmann H, Comai L, Tjian R. Assembly of transcriptionally active RNA polymerase I initiation factor SL1 from recombinant subunits. Science. 1994;266 : 2015–2018. 7801130
15. Heix J, Zomerdijk JCBM, Ravanpay A, Tjian R, Grummt I. Cloning of murine RNA polymerase I-specific TAF factors: conserved interactions between the subunits of the species-specific transcription initiation factor TIF-IB/SL1. Proc Natl Acad Sci USA. 1997;94 : 1733–1738. 9050847
16. Denissov S, van Driel M, Voit R, Hekkelman M, Hulsen T, Hernandez N, et al. Identification of novel functional TBP-binding sites and general factor repertoires. EMBO J. 2007;26 : 944–954. 17268553
17. Gorski JJ, Pathak S, Panov K, Kasciukovic T, Panova T, Russell J, et al. A novel TBP-associated factor of SL1 functions in RNA polymerase I transcription. EMBO J. 2007;26 : 1560–1568. 17318177
18. Kuhn A, Vente A, Doree M, Grummt I. Mitotic phosphorylation of the TBP-containing factor SL1 represses ribosomal gene transcription. J Mol Biol. 1998;284 : 1–5. 9811537
19. Klein J, Grummt I. Cell cycle-dependent regulation of RNA polymerase I transcription: the nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc Natl Acad Sci USA. 1999;96 : 6096–6101. 10339547
20. Tumurbaatar I, Cizmecioglu O, Hoffmann I, Grummt I, Voit R. Human Cdc14B promotes progression through mitosis by dephosphorylating Cdc25 and regulating Cdk1/cyclin B activity. PLoS One. 2011;6: e14711. doi: 10.1371/journal.pone.0014711 21379580
21. Muth V, Nadaud S, Grummt I, Voit R. Acetylation of TAFI68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J. 2001;20 : 1353–1362. 11250901
22. Mais C, Wright JE, Prieto JL, Raggett SL, McStay B. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev. 2005;19 : 50–94. 15598984
23. Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134 : 256–267. doi: 10.1016/j.cell.2008.05.043 18662541
24. Chen D, Dundr M, Wang C, Leung A, Lamond A, Misteli T, et al. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J Cell Biol. 2005;168 : 41–54. 15623580
25. Naidu S, Friedrich JK, Russell J, Zomerdijk JCBM. TAF1B is a TFIIB-like component of the basal transcription machinery for RNA polymerase I. Science. 2011;333 : 1640–1642. doi: 10.1126/science.1207656 21921199
26. Choudhary C, Kumar C, Gnad F, Nielsen M, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325 : 834–840. doi: 10.1126/science.1175371 19608861
27. Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006;20 : 1075–1080. 16618798
28. Chen S, Santiago-Reichelt M, Seiler J, Felbel K, Grummt, I, Voit R. Repression of RNA polymerase I transcription upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol Cell. 2013;52 : 303–313 doi: 10.1016/j.molcel.2013.10.010 24207024
29. Grob A, Colleran C, McStay B. Construction of synthetic nucleoli in human cells reveals how a major functional nuclear domain is formed and propagated through cell division. Genes Dev. 2014;28 : 220–230. doi: 10.1101/gad.234591.113 24449107
30. Zatsepina OV, Voit R, Grummt I, Spring H, Semenov MV, Trendelenburg MF. The RNA polymerase I-specific transcription initiation factor UBF is associated with transcriptionally active and inactive ribosomal genes. Chromosoma. 1993;102 : 599–611. 8306821
31. Vaquero A, Scher M, Erdjument-Bromage H, Tempst P, Serrano L, Reinberg D. SIRT1 regulates the histone methyl-transferase during heterochromatin formation. Nature 2007;450 : 440–444. 18004385
32. Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16 : 93–105. 15469825
33. Fatoba ST, Okorokov AL. Human SIRT1 associates with mitotic chromatin and contributes to chromosomal condensation. Cell Cycle. 2011;10 : 2317–2322. 21636977
34. Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrin Metab. 2014;3 : 138–145.
35. Gorski SA, Snyder SK, John S, Grummt I, Misteli T. Modulation of RNA polymerase assembly in transcriptional regulation. Mol Cell. 2008;30 : 486–497. doi: 10.1016/j.molcel.2008.04.021 18498750
36. Voit R, Hoffmann M, Grummt I. Phosphorylation by G1-specific cdk-cyclin complexes activates the nucleolar transcription factor UBF. EMBO J. 1999;18 : 1891–1899. 10202152
37. Seither P, Zatsepina O, Hoffmann M, Grummt I. Constitutive and strong association of PAF53 with RNA polymerase I. Chromosoma 1997;106 : 216–225. 9254723
38. Percipalle P, Fomproix N, Cavellán E, Voit R, Reimer G, Krüger T, et al. The chromatin remodeling complex WSTF-SNF2h interacts with nuclear myosin 1 and has a role in RNA polymerase I transcription. EMBO Rep. 2006;7 : 525–530. 16514417
39. Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, et al. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA. 2009;106 : 17781–17786. doi: 10.1073/pnas.0909873106 19815529
40. Gavet O, Pines J. Progressive activation of cyclin B1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010;18 : 533–543. doi: 10.1016/j.devcel.2010.02.013 20412769
41. Schnapp A, Grummt I. Transcription complex formation at the mouse rDNA promoter involves the stepwise association of four transcription factors and RNA polymerase. J Biol Chem. 1991;266 : 24588–24595. 1761556
42. Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;210 : 110–149.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy