
CTXφ Replication Depends on the Histone-Like HU Protein and the UvrD Helicase
One of the major strategies to prevent Cholera epidemics is the development of oral vaccines based on live attenuated Vibrio cholerae strains. The most promising vaccine strains have been obtained by deletion of the cholera toxin genes, which are harboured in the genome of an integrated phage, CTXϕ. However, they can re-acquire the cholera toxin genes when re-infected by CTXϕ or by hybrid phages between CTXϕ and other vibrio phages, which raised safety concerns about their use. Here, we developed a screening strategy to identify non-essential host factors implicated in CTXϕ replication. We show that the histone-like HU protein and the UvrD helicase are both absolutely required for its replication. We further show that they are essential for the replication of VGJϕ, a representative member of a family of phages that can form hybrids with CTXϕ. Accordingly, we demonstrate that the disruption of the two subunits of HU and/or of UvrD prevents infection of the V. cholerae by CTXϕ and VGJϕ. In addition, we show that it limits CTXϕ horizontal transmission. Taken together, these results indicate that HU - and/or UvrD - cells are promising candidates for the development of safer live attenuated cholera vaccine.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005256
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005256
Summary
One of the major strategies to prevent Cholera epidemics is the development of oral vaccines based on live attenuated Vibrio cholerae strains. The most promising vaccine strains have been obtained by deletion of the cholera toxin genes, which are harboured in the genome of an integrated phage, CTXϕ. However, they can re-acquire the cholera toxin genes when re-infected by CTXϕ or by hybrid phages between CTXϕ and other vibrio phages, which raised safety concerns about their use. Here, we developed a screening strategy to identify non-essential host factors implicated in CTXϕ replication. We show that the histone-like HU protein and the UvrD helicase are both absolutely required for its replication. We further show that they are essential for the replication of VGJϕ, a representative member of a family of phages that can form hybrids with CTXϕ. Accordingly, we demonstrate that the disruption of the two subunits of HU and/or of UvrD prevents infection of the V. cholerae by CTXϕ and VGJϕ. In addition, we show that it limits CTXϕ horizontal transmission. Taken together, these results indicate that HU - and/or UvrD - cells are promising candidates for the development of safer live attenuated cholera vaccine.
Introduction
Cholera remains a major health problem in many part of the developing world, with an estimation of 2.8 million cases and 100 000 to 200 000 deaths each year [1]. The agent of the cholera, the Vibrio cholerae bacterium, is found in briny waters all over the world [2]. However, most V. cholerae strains are not pathogenic or only cause local outbreaks of gastroenteritis. Pathogenicity depends on the acquisition of several virulence factors, of which the cholera toxin (CT) and the toxin-coregulated pilus (TCP) are considered the most significant. CT causes a voluminous watery diarrhoea, which is responsible for the high rate of death associated with cholera and its epidemic propagation [3], while TCP is required for colonization of the small intestine [4]. The cholera toxin genes, ctxAB, are encoded in the genome of a lysogenic filamentous phage, CTXϕ [5]. The genomic characterization of V. cholerae epidemic strains suggested that several independent toxigenic conversion events occurred in the history of cholera [6–8], which motivated studies on the life cycle of CTXϕ.
The amplification of the phage genome by rolling-circle replication (RCR) is central to this life cycle (Fig 1): once delivered in the cytoplasm of the cell via interactions with TCP and the TolQRA cell division proteins (Fig 1, (1)) [5,9], the circular single-stranded DNA (ssDNA) genome of CTXϕ is converted into a double stranded DNA (dsDNA) replicative form by the host machinery, which permits its RCR amplification and the production of new phage particles [10,11] (Fig 1, (2), (3) and (4)). In addition to phage particle production, RCR participates in the vertical transmission of ctxAB in the lineage of infected cells (Fig 1). However, vertical transmission is also assured by the integration of CTXϕ into the genome of its host [5] (Fig 1). CTXϕ exploits a chromosomally encoded site-specific recombination (Xer) machinery for integration [12,13] (Fig 1). The Xer machinery normally serves to resolve dimers of the circular chromosomes by the addition of a crossover at a specific site, dif [14,15]. In V. cholerae, as in most bacteria, it consists of two tyrosine recombinases, XerC and XerD. The attachment site of the phage, attPCTX, consists in the stem of a hairpin of its single stranded DNA genome [16,17] (Fig 1). XerC catalyses the formation of a Holliday Junction (HJ) between attPCTX and the dif site of one or the other of the two circular chromosomes of V. cholerae [16,17] (Fig 1 (6)). Replication converts the HJ intermediate into product [16–18]. The process is facilitated by EndoIII, a host-encoded base excision repair enzyme, which inhibits XerC catalysis once the HJ has been formed [18] (Fig 1, (7)). Nevertheless, the integration of non-replicative forms of CTXϕ is inefficient [18]. In contrast, the integration of replicative forms is very efficient and almost always leads to multiple tandem insertions, which suggests that it occurs after several rounds of amplification of the phage genome by RCR [18,19] (Fig 1). Multiple tandem insertions are permitted because a functional dif site is re-created on the right side of the prophage [13] (Fig 1). Tandem insertions are crucial for the life cycle of CTXϕ because the Xer recombination site on the left side of the prophage is masked in the dsDNA from of the prophage, which impedes excision [16] (Fig 1). Production of new free copies of the phage genome then depends on a process analogous to RCR between tandem prophage copies [11] (Fig 1, (8)).

CTXϕ RCR depends on a single phage-encoded protein, RstA (Fig 1). RstA production is under the control of the host SOS response [20] (Fig 1, (5)) and of a phage-encoded repressor, RstR [21]. RstA is an HUH endonuclease [22]. It creates a 5′-phosphotyrosine intermediate and a free 3′-OH at a specific cleavage site of the replicative form of CTXϕ, ori(+), to prime replication (Fig 1). The rest of the process is driven by the host machinery [21]. Host factors implicated in the replication of the E. coli filamentous phages are either essential, such as DNA polymerase III, or crucial to the proliferation of the cells, such as the Rep helicase [10,19]. However, marked differences in the life cycles of CTXϕ and of the proto-typical filamentous phages of enterobacteriaceae, including its ability to integrate into the genome of its host, the control exerted by the host SOS response on RstA production [20] and the requirement for a host-encoded protein for CTXϕ particle secretion [23], suggested that it might not be so for CTXϕ. Here, we screened for non-essential host factors involved in CTXϕ replication. We thus found that the histone-like protein HU [24] was essential for CTXϕ replication because it was necessary for RstA to introduce a nick in the phage genome at ori(+). We further found that in place of Rep, CTXϕ exploited UvrD, a DNA helicase mainly involved in DNA repair [25]. Finally, we found that HU and UvrD were implicated in the replication of other Vibrio filamentous phages, such a VGJϕ.
Results
Screening strategy
We previously described a colorimetric assay to monitor IMEX integration events in V. cholerae [17]. In brief, the dif site of the largest of the two chromosomes harboured by the V. cholerae N16961 El Tor strain, dif1, was inserted in the coding region of the Escherichia coli lacZ gene in such a manner as not to perturb β-galactosidase production. The lacZ::dif1 allele was inserted in place of the normal dif1 site of a N16961 El Tor strain in which the endogenous lacZ gene was deleted (Fig 2A). This strain forms blue colonies on X-gal media. However, 100% of the colonies obtained after the delivery of a truncated form of the El Tor variant of CTXϕ, RS2, which is fully functional in replication and integration, were white or contained large white sectors around a blue star shaped centre on X-gal plates (Fig 2B, panel (i) and (ii)). We previously used this property to search for non-essential host factors implicated in the integration of CTXϕ by transposition mutagenesis (Fig 2B, panel (iii), [18]). During the course of this first screen, we noted that fully white colonies represented a very limited fraction of the total colonies, confirming the importance of ssDNA amplification by RCR for the integration process (Fig 2B, panel (i)). It suggested that the assay could be used in a second screen to identify non-essential host factors involved in RCR (Fig 2B, panel (iv)). To this end, we cloned RS2 on a pSC101 plasmid that harboured a spectinomycin resistance gene and that could be delivered by conjugation (Fig 2A). By using a temperature-sensitive version of the pSC101 origin of replication, we could distinguish if the absence of integration was due to the disruption of host factors implicated in RCR or in the integration process (Fig 2B, panel (iii) and (iv)). As a control, we verified that conjugation of the pSC101-RS2 hybrid in ∆xerC cells yielded fully blue colonies at 30°C and 42°C. We also verified that disruption of RstA, which abolishes RCR, led to fully blue colonies at 30°C that couldn’t grow at 42°C.

We implemented the screen in two independent mariner transposition libraries of the lacZ::dif1 reporter strain. Conjugants were selected on plates supplemented with spectinomycin and X-gal at 30°C. We screened over 40 000 clones. Only 6 of them were both fully blue on X-gal plates and thermo-sensitive. All of them carried a transposon insertion in the VC1919 ORF of the V. cholerae genome (Fig 2C). Sequence analysis revealed that they corresponded to at least three independent transposition insertion events (Fig 2C).
HU is essential for CTXϕ replication
In E. coli, HU is composed of two subunits, HUα and HUβ, which are encoded by hupA and hupB, respectively [24]. The major form of HU is a heterodimer of HUα and HUβ, but HUα homo-dimers and HUβ homo-dimers are also formed. VC1919 encodes for a homologue of the β subunit of E. coli HU, HUβ. A homologue of the α subunit of E. coli HU, HU α, is encoded by VC0273. We engineered His-tag versions of the two gene products under their native promoters and showed that they were produced at the same level at 37°C and 42°C (S1 Fig). We purified the recombinant proteins and showed that they bound DNA with similar affinities (S2 Fig). These results suggested that VC0273 and VC1919 were the V. cholerae orthologs of E. coli hupA and hupB.
To confirm the results of our screen, we delivered a version of RS2 marked with a chloramphenicol resistance gene in a ΔhupB ΔxerC strain by conjugation. Note that, contrary to pSC101-RS2, this version of the phage does not contain a functional plasmid origin of replication. Because of the absence of XerC, RS2 cannot integrate in this strain and vertical transmission of chloramphenicol resistance to daughter cells entirely depends on RS2 RCR. In agreement with the results of our screen, no colonies were obtained on selection plates at 42°C (Fig 3A). Colonies were obtained at 37°C (Fig 3B), but they failed to propagate when re-streaked at 42°C (Fig 3C). To further determine the potential role of HU in CTXϕ replication, we engineered a ∆hupA ∆xerC strain and a ∆hupAB ∆xerC strain. The deletion of hupA did not affect the maintenance of RS2 at 37°C (Fig 3B and 3D) and 42°C (Fig 3A and 3C). However, no colonies were obtained when RS2 was delivered in the ∆hupAB ∆xerC strain whether at 42°C or 37°C (Fig 3A and 3C). Ectopic production of HUα or HUβ in ∆hupAB ∆xerC cells restored colony formation at 37°C, excluding any polar effect of the two deletions (Fig 3E). Taken together, these results suggested that HU was essential for CTXϕ replication, that HUα homo-dimers were sufficient to maintain the RF of the CTXϕ genome at 37°C but that HUβ homo-dimers and/or HUαβ hetero-dimers were absolutely required at 42°C.

The single deletion of hupB is sufficient to limit CTXϕ vertical and horizontal transmission
In order to gain a quantitative measure of the importance of HUα and HUβ in the CTXϕ replication process, we used quantitative PCR to monitor the number of RS2 ssDNA and dsDNA copies per genome equivalent in ∆hupA ∆xerC and ∆hupB ∆xerC cells that were grown under selection pressure at 37°C. The deletion of hupA had no visible effect on the relative number of RS2 copies, whether ssDNA or dsDNA (Fig 4A). In contrast, the deletion of hupB induced a 40% reduction in the number of RS2 copies per genome (Fig 4A). As the total number of RS2 copies per genome equivalent was now lower than 1, we suspected that the deletion of hupB would increase the instability of RS2 at 37°C even though it did not compromise colony formation on selection plates at this temperature. Indeed, a 100-fold reduction in the number of colony forming units was observed in ∆hupB ∆xerC cells compared to ∆hupA ∆xerC or ∆xerC cells after 5 hours of growth without selection pressure (Fig 4B). Because it limited the number of copies of the ssDNA CTXϕ genome, we further suspected that the deletion of hupB would also prevent RS2 integration. Indeed, we observed a 5-fold reduction in the integration efficiency of RS2 in ∆hupB lacZ::dif1 cells compared to lacZ::dif1 cells (Fig 4C). A weaker, yet significant, decrease in RS2 integration was also observed in ∆hupA lacZ::dif1 cells (Fig 4C). No decrease in the frequency of integration of a non-replicative plasmid harbouring attPCTX was observed in ∆hupA, ∆hupB and ∆hupAB, excluding any participation of HU in the integration process per se (Fig 4D). Finally, we suspected that the deletion of hupB might also prevent the production of phage particles by limiting the amount of ssDNA available for packaging. Indeed, a 1000-fold less phage particles were produced in ∆hupB ∆xerC cells than in ∆xerC cells (Fig 4E). Taken together, these results suggested that the deletion of hupB could by itself limit CTXϕ vertical transmission via lysogenic conversion and limit horizontal transmission via the production of new viral particles.

CTXϕ relies on UvrD for RCR
RCR of the proto-typical filamentous phages of E. coli depends on Rep, a helicase that is implicated in the replication of their host genome [26]. The E. coli Rep protein is not essential but its deletion leads to a severe growth defect [27,28]. The genome of V. cholerae encodes for a homologue of E. coli Rep. We found that it was not essential but that its deletion led to a severe growth defect, suggesting functional homology with E. coli Rep (S3 Fig). However, the deletion of V. cholerae Rep impeded neither the maintenance of RS2 in ∆xerC cells (Fig 5A) nor its integration (Fig 5B), suggesting that it was not implicated in CTXϕ RCR.

Some RCR plasmids of Gram+ bacteria replicate in E. coli using the UvrD DNA helicase [29]. The E. coli UvrD protein plays essential roles in methyl-directed mismatch repair and nucleotide excision repair of DNA [30]. It is also involved in clearing and restarting stalled replication forks [31–33]. It is under the control of two promoters: one is constitutive while the other is governed by LexA, which leads to a 3 to 6-fold overproduction of UvrD during SOS [34,35] (S4A Fig). E. coli UvrD is not essential and its deletion does not affect cell proliferation under normal growth conditions. The genome of V. cholerae encodes a homologue of E. coli UvrD. Its deletion did not affect cell proliferation (S4 Fig) but made them hyper sensitive to UV (S4B Fig). Inspection of the upstream region of the gene suggested the presence of two promoters, with a putative lexA-binding site overlapping the -10 box of one of them (S4A Fig). Correspondingly, introduction of a non-cleavable allele of lexA led to a 3-fold decrease in the expression of the gene (S4C Fig) while disruption of RecA or of the lexA box increased its expression (S4D Fig). Taken together, these results suggested that this gene was the functional homologue of E. coli uvrD and we wondered if its product was involved in CTXϕ RCR. Consistent with this view, deletion of V. cholerae uvrD almost abolished the maintenance of RS2 in ∆xerC cells (Fig 5A). Ectopic production of V. cholerae UvrD under an arabinose promoter on a plasmid restored colony formation, excluding any polar effect of the deletion (Fig 5B). The deletion of V. cholerae uvrD also led to over a 1000-fold drop in the frequency of integration of RS2 in XerC+ cells (Fig 5C). The few colonies that were obtained were fully white or only displayed a pinpoint blue dot at their centre, further indicating that integration occurred immediately after entry into the cell (Fig 5D). Taken together, these results suggested that CTXϕ relied on the UvrD helicase for RCR.
Nicking of ori(+) depends on HU
There are three different steps in RCR: (i) addition of a nick at ori(+) to prime replication; (ii) displacement of the old (+) ssDNA copy of the genome and synthesis of a new one; (iii) termination of replication and re-circularization of the old (+) ssDNA genome copy. HU could be involved in any of these steps. By definition, UvrD was expected to be only involved in the second step. To investigate whether HU and UvrD were involved in the first step of RCR, total genomic DNA was extracted from V. cholerae cells 3 hours after conjugation of RS2 and the presence of a nick at ori(+) was revealed by primer extension (Fig 6A and 6B). In wild-type cells, we observed a strong signal consistent with the introduction of a nick between the guanine and the thymine bases of the apical loop of the second hairpin of CTXϕ ori(+) (Fig 6B). The position of the observed nick fitted with previous genetic analysis of the cleavage position of RstA [36]. Nick formation was entirely suppressed when HU was deleted, suggesting that HU was essential for the activity of RstA (Fig 6B). In contrast, the deletion of UvrD did not affect nick formation, suggesting that UvrD was not implicated in RCR initiation.

One concern regarding our screening procedure was that we did not recover any transposition event in the uvrD gene even though it is not essential in V. cholerae. However, we found that pSC101-RS2 is not able to propagate in ∆uvrD ∆xerC V. cholerae cells even at the permissive temperature (Fig 6D). We then hypothesized that replication forks originating from the pSC101 origin would generate fatal double strand breaks when they reached a nicked ori(+), which could explain why the pSC101-RS2 hybrid failed to propagate in ΔuvrD ΔxerC cells (Fig 6C). In agreement with this hypothesis, deletion of HU or inactivation of RstA restored the propagation of the pSC101-RS2 hybrid in ΔuvrD ΔxerC cells (Fig 6D). There was little or no production of RS2 ssDNA in such cells, further illustrating the importance of HU for RCR (Fig 6E).
Role of HU and UvrD in the RCR of other V. cholerae phages
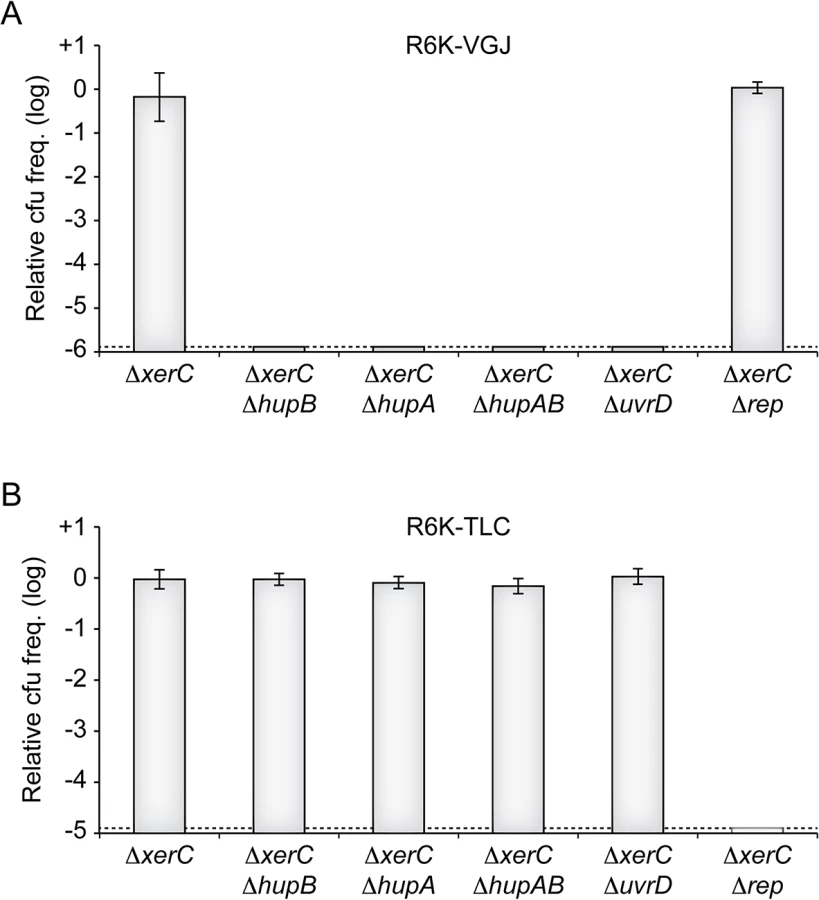
Ecological interactions between CTXϕ and several other filamentous phages and their satellites drives the continuous and rapid emergence of new epidemic variants of V. cholerae [13,15]. Foremost among the phages implicated in those interactions are RS1, which encodes for an anti-repressor [37,38], VGJϕ, which participates in the horizontal spreading of CTXϕ via the formation of CTX-VGJϕ hybrids [39,40], and TLCϕ, which is almost always found integrated before CTXϕ prophages in clinical isolates and which can lead to their excision [41–43]. We could easily predict that RS1 depended on HU and UvrD for replication, because it is essentially identical to RS2. To determine if VGJϕ and TLCJϕ might also depend on HU and UvrD, we conjugated a R6K suicide vector harbouring the replicative region of VGJϕ (R6K-VGJ) and a R6K suicide vector harbouring the replicative region of the satellite phage TLCϕ (R6K-TLC) in ∆xerC cells in which hupA, hupB, uvrD or rep were disrupted (Fig 7). No colonies were obtained when R6K-VGJ was conjugated in hupA or hupB mutants, suggesting that the HUαβ heterodimer was vital to VGJϕ RCR (Fig 7). R6K-VGJ also failed to be propagated in ∆uvrD cells, suggesting that UvrD was required for VGJϕ RCR (Fig 7). In contrast, ∆hupAB cells and ∆uvrD cells seemed to fully support TLCϕ replication (Fig 7). Finally, R6K-TLC was not maintained in ∆rep ∆xerC cells, suggesting that TLCϕ RCR depended on Rep (Fig 7).
Discussion
We developed a screening strategy to identify non-essential V. cholerae host factors involved in CTXϕ replication. We thus found that contrary to the proto-typical filamentous phages so far studied, the histone-like HU protein was absolutely necessary for RstA to prime RCR of the phage genome (Fig 2, 3 and 6). In addition, we showed that CTXϕ exploited UvrD, a helicase normally implicated in DNA repair, rather than Rep, the helicase normally associated to replication (Fig 5). Finally, we showed that a member of another family of vibrio filamentous phages, VGJϕ, also exploited HU and UvrD for RCR, demonstrating that CTXϕ is not an exception (Fig 7).
A role for HU in RCR
HU is a major component of the bacterial nucleoid, which binds dsDNA without any apparent specificity and with a low affinity but which recognizes with a higher affinity defined DNA structures and repair intermediates [44–46]. In E. coli, HU is involved in the initiation of chromosome replication [47–49]. However, it is not essential for survival: IHF, a protein belonging to the same family of DNA-binding proteins, can substitute for initiation of replication at oriC [50]. Likewise, deletion of hupAB does not compromise cell viability in V. cholerae, possibly because its genome encode for a homologue of IHF.
As far as we know, no reports exist on the implication of HU in the life circle of any other filamentous phages than CTXϕ and VGJϕ. HU was shown to be essential for replication of Mini-F and Mini-P plasmids [51]. However, these plasmids replicate by a theta system. In this case, HU bind to the origin without sequence-specificity and help to melt the origin to initiate replication [52]. Interestingly, it was observed in Salmonella typhimurium that replication of a Mini-F plasmid was strongly affected in a ∆hupB mutant, totally deficient in a ∆hupAB double mutant, but only mildly affected in a ∆hupA mutant [53]. This is remarkably similar to what we have observed in the case of CTXϕ and a similar role of HU in the initiation of replication should not be discarded. More interestingly, however, it was reported that HU played an essential role in the replication of pKYM, a plasmid from the Gram- bacterium Shigella sonei [54]. A shared characteristic of proto-typical filamentous phages and of most RCR plasmids is a very simple (+) origin of replication: Ff coliphages contain an approximately 36bp replication origin [55]; the Gram+ pC194 and pT181 plasmids harbour a small 55bp and 70bp origin, respectively [56,57]. None of these mobile elements require accessory proteins for the initiator protein nicking activity. In contrast, pKYM and CTXϕ (+) origins of replication are more complex. The (+) origin of replication of pKYM is 173bp long. It contains a core region corresponding to the RepK initiator binding-site and a downstream enhancer region. HU was shown to specifically recognize this enhancer region and assist in the binding of RepK [54]. CTXϕ ori(+) is 167bp long and contains several inverted repeat sequences upstream and downstream of the RstA cleavage site with the potential to form stem-loops [36]. It is therefore possible that HU helps CTXϕ replication by helping the binding of RstA and/or promoting its endonuclease activity. A weaker binding affinity and/or tighter control of the VGJϕ HUH endonuclease might explain why the two HU subunits are absolutely essential for this phage. Future biochemical work will need to clarify the exact mechanism of action of HU on RstA activity.
Implication of UvrD in RCR
Rep and UvrD are members of the SF1 family of helicases and share approximately 40% similarity [58]. They both unwind DNA in the 3’ – 5’ direction [59,60]. Despite the structural and functional similarities between Rep and UvrD, the physiological roles of the two helicases are well distinct. Rep is constitutively expressed in E. coli, where it is implicated in chromosome replication: it directly interacts with the replicative helicase DnaB and helps remove nucleoproteins complex in front of replication forks [61,62]. Rep is also implicated in the restart of stalled replication forks [63]. As a result, Δrep E. coli mutants display a 50–60% reduction in their replication rate [27,28]. Nevertheless, Rep is not essential. On the contrary, UvrD is overexpressed during the SOS response in E. coli and its role seems to be mainly limited to DNA repair: its activity is involved in MutHLS-dependent mismatch DNA repair [64] and UvrABC-dependent nucleotide excision repair [65]. UvrD also helps dismantle RecA filaments from ssDNA, which prevents unwanted recombination [66]. Finally, UvrD can promote the movement of the replisome along protein-bound DNA and participate in the restart of replication forks [62]. Nevertheless, its deletion does not directly affect replication fork progression in E. coli [61]. Consistent with its role in replication fork progression, Rep was shown to be critical for phage RCR in E. coli, including ϕX174 and the Ff family of filamentous phages [26]. In contrast, we found that CTXϕ and VGJϕ both exploited UvrD for RCR. As far as we know, this is the first time that UvrD has been shown to participate in the replication of a phage genome. A single SF1 helicase, PcrA, is encoded in the genome of Gram+ bacteria instead of Rep and UvrD. RCR of plasmids from Gram+ bacteria relies on PcrA. However, some of them can replicate in E. coli using UvrD [29]. In addition, UvrD was shown to be implicated in the RCR of pKYM [67]. Together, these results suggest that RCR depends on an activity common to Rep and UvrD, raising the question as to why these two helicases are not interchangeable, similarly to PcrA and UvrD. It is tempting to speculate that exploitation of UvrD or Rep is determined by the ability of the initiator protein to directly interact with one or the other of the two accessories helicases. In agreement with this hypothesis, the initiator protein of CTXϕ and VGJϕ share structural similarities with the initiator protein of the Gram+ plasmids that exploit UvrD to replicate in E. coli (pfam02486). In contrast, the initiator protein of TLCϕ shares sequence and structural similarities with the initiator protein of the E. coli proto-typical filamentous phages (pfam05144 and pfam05155). Future work will be directed at investigating the exact nature of the interaction between UvrD and RstA.
Considerations for the biosafety of live-attenuated vaccine cells
In 2011, recognizing that cholera was not sufficiently addressed despite its prevalence in epidemic forms in both endemic and non endemic areas, the World Health Assembly called for a comprehensive approach to cholera control, including the development of oral cholera vaccines (http://www.who.int/wer). The most promising live attenuated V. cholerae vaccine strains have been obtained by the deletion of one or both of the cholera toxin genes, ctxAB [68–71]. However, the possibility that such strains could be re-infected when in the intestinal track raised safety concerns about their use in a vaccine since they could promote the apparition of cholera symptoms in previously asymptomatic individuals and participate in the spreading of CTXϕ in the environment (S5A Fig). The concomitant deletion of the dif site of Chr1 in these strains only partially prevents ctxAB reacquisition since some phage variant can target the dif site of Chr2 [17] and does not block RCR amplification of the phage genome.
Several possibilities exist to limit the risk of re-acquisition of the genes and their further spreading. A simple way to block the delivery of the genome of CTXϕ could be to delete the production of its receptors at the cell surface, TCP and TolQRA. However, TCP is essential for intestinal colonization and hence immunogenicity [4]. TolQRA is part of the cell division machinery and is critical for the outer membrane stability of Gram- bacteria and their resistance to extra-cytoplasmic stress [72–76]. A simple way to limit further spreading of CTXϕ particles could be to block their secretion by deleting EspD [23]. However, EspD appears to be essential in V. cholerae [23]. As a result, the only valid vaccine cell protection strategy proposed to date was based on the observation that production of RstR from a resident CTXϕ prophage provided immunity against secondary infections by blocking initial rounds of RCR [77] (S5B Fig). However, this strategy has several limitations. First, several CTXϕ variants exist that harbour different RstR repressors cross-immunity is not assured among them [78] (S5B Fig). Thus, this strategy is limited to known CTXϕ repressor variants, with each repressor providing immunity against secondary infections by phages encoding the same repressor (S5C Fig). Second, CTXϕ interacts with other Integrative Mobile Element exploiting Xer (IMEX). Two of them, the RS1 satellite phage and fs2, harbour an anti-repressor, RstC [37,79] (Fig 7B). Third, hybrid phage formation between CTXϕ and other IMEXs, such a VGJϕ, can circumvent both the requirement for TCP expression and repressor immunity [39,40,80,81] (Fig 7B). Fourth, tandem CTXϕ genomes can be transduced by lytic phages, such as CP-T1 [82]. Finally, production of RstR does not affect the efficiency of the RCR process once it has been established, which permits production of new phage particles and further spreading of CTXϕ (S5D Fig).
Here, we showed that the deletion of hupB impedes ctxAB re-acquisition by CTX-VGJϕ hybrid infection and dramatically reduces CTXϕ production when its genome has been acquired by other horizontal transfer mechanisms (S5C Fig). Therefore, we think that the deletion of hupB would considerably increase the safety of RstR-producing vaccine cells. Moreover, we found that HU and UvrD were both essential for CTXϕ and VGJϕ replication, that their deletion compromised the ability of CTXϕ to integrate into the genome of its host and blocked the secretion of CTXϕ particles. HU is not essential for the proliferation of V. cholerae but we cannot discard a possible impaired colonization of the HU null mutants. However, Salmonella enterica strains lacking hupA and/or hupB are known to trigger an effective immune response protecting against salmonellosis, suggesting that HU is probably not essential for intestine colonization [83]. Therefore, the deletion of hupA and hupB is a promising strategy for the development of safe live attenuated cholera vaccines. UvrD participates in DNA mismatch repair, many genes of which have been shown to be important for colon colonization [84]. However, in the case the deletion of uvrD affects colon colonization, mutating it in such a way as to compromise its role in RCR without affecting its DNA repair activities could offer a third strategy for the development of safe live attenuated cholera vaccines.
Materials and Methods
Strains, plasmids and oligonucleotides
Strains, plasmids and oligonucleotides used in this study are described in S1, S2 and S3 Tables, respectively. All V. cholerae strains were constructed by natural transformation. Engineered strains were confirmed by PCR and sequencing. Bacterial strains were grown on Luria-Bertani (LB) agar. Antibiotics were used at the following concentrations: ampicillin (Amp), 100 μg/mL; spectinomycin (Sp), 100 μg/mL; chloramphenicol (Cm), 34 μg/mL for E. coli and 3 μg/mL for V. cholerae; kanamycin (Kn), 50 μg/mL; Zeocin (Zeo), 100 μg/mL for E. coli and 1 μg/mL for V. cholerae and rifampicin (Rif), 100 μg/mL for E. coli and 2 μg/mL for V. cholerae. 0.2% arabinose was used to induce UvrD production from the pBAD24 vector.
Mariner transposon-mutagenesis, screening and mutant characterization
A mariner transposon-mutagenesis bank of a V. cholerae reporter strain was created as described [18]. The bank was conjugated with a spectinomycin resistant (SpecR) derivative of RS2 El Tor containing a thermosensitive (TS) origin of replication (pSC101-RS2). Individual colonies were selected on X-Gal, IPTG and spectinomycin plates after 48 h of growth at 30°C. Fully blue colonies were selected and re-streaked in parallel at 30°C and 42°C. TS clones were cured from pSC101-RS2 by overnight growth in the absence of antibiotic and their phenotype was corroborated by re-conjugation with the same plasmid. The insertion was mapped by direct sequencing of the DNA flanking the point of insertion of the mariner transposons, which was amplified by arbitrary-random PCR [85].
Conjugation assay
E. coli β2163 meso-diaminopimelic acid (DAP) auxotroph donors and V. cholerae recipients were grown to 0.3 at OD600nm. Bacteria were pelleted by centrifugation, re-suspended in 50 μL and mixed at a 1 : 10 ratio, dropped onto sterile filter paper on top of an LB-agar plate supplemented with DAP and incubated for 3 h. Conjugants were selected for the plasmid antibiotic resistance and DAP prototrophy. To monitor integration, conjugants were spread on plates containing X-gal and incubated at 37°C overnight. Conjugants carrying a TS origin of replication were re-covered at 30°C.
Assay of CTXϕ infection efficiency and phage production
Strains harbouring kanamycin-marked CTXϕ were used as donors. Eighty microliters of filtered supernatant containing CTX-Kn particles was mixed with 20 μl of recipients strains that had been grown in AKI media to induce TCP expression [86]. The mix was incubated 20 min at 37°C to allow infection and then plated on LB to determine the number of potential recipients and LB supplemented with kanamycin to determine the number of infected cells. The frequency of infection was determined by the ration of KnR cells and the total number of recipients.
Q-PCR analysis
Total DNA was purified using the GenElute Bacterial Genomic DNA Kit from Sigma. Samples were analysed using a LightCycler FastStart DNA masterSYBR Green I system from Roche. Reactions were run in triplicate using a LightCycler 480 instrument (Roche). Primer 2690 and 2704, which amplify a specific 150 bp fragment inside rstA gene, were used for phage DNA quantification. Data were normalized with the bacterial chromosome using primers 768 and 769, which amplify a 150 bp fragment within the matP gene. For single strand DNA quantification, total DNA was digested 3 hours with ScaI to remove phage dsDNA. There is a cleavage site for ScaI within the phage fragment used for the analysis. Relative copy number of ssDNA was calculated as follows: 2 x e1Cp_digested/e2Cp_chromosome, in which e represents the amplification efficiency of the primers pairs used. A factor of 2 was used to normalize the ssDNA of the phage with the dsDNA of the chromosome. The analysis was run out in parallel without prior digestion, which permitted to calculate the relative copy number of dsDNA as follows: (e1Cp_undigested-e1Cp_digested)/e2Cp_chromosome.
SDS-page and western blot
Bacterial lysates were electrophoresed on 12% SDS-page gel. HUα or HUβ with a C-terminal 6xHis tag were analysed by western blot with a primary anti-4His mouse monoclonal antibody (Invitrogen) and a secondary anti-mouse IgG antibody coupled to peroxidase (Pierce). ECL Western Blotting Substrate (Pierce) was used to detect the reaction on a LAS-3000 Luminescent Analyser (Fujifilm).
Sequencing gel and nick detection
For nick detection, pBS66 was conjugated to the strain of interest and then total DNA was purified directly from the conjugation assay. After digestion with NotI, we performed a primer extension reaction using as a primer the 1269 oligonucleotide that had been labelled with γ-[32P] ATP. The sequence ladder was prepared using pBS66 purified from E. coli, in which CTXϕ does not replicate, and the fmol DNA Cycle Sequencing System (Promega).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ali M, Lopez AL, You YA, Kim YE, Sah B, Maskery B, et al. The global burden of cholera. Bull World Health Organ. 2012;90 : 209–218A. doi: 10.2471/BLT.11.093427 22461716
2. Sack DA, Sack RB, Nair GB, Siddique A. Cholera. The Lancet. 2004;363 : 223–233. doi: 10.1016/S0140-6736(03)15328-7 14738797
3. Kaper JB, Morris JG, Levine MM. Cholera. Clin Microbiol Rev. 1995;8 : 48–86. 7704895
4. Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168 : 1487–1492. doi: 10.1084/jem.168.4.1487 2902187
5. Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272 : 1910–4. 8658163
6. Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for multiple waves of global transmission in the seventh cholera pandemic. Nature. 2011;477 : 462–5. nature10392 [pii] doi: 10.1038/nature10392 21866102
7. Chun J, Grim CJ, Ha0073an NA, Lee JH, Choi SY, Haley BJ, et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci U A. 2009;106 : 15442–7. 0907787106 [pii] doi: 10.1073/pnas.0907787106 19720995
8. Kim EJ, Lee D, Moon SH, Lee CH, Kim SJ, Lee JH, et al. Molecular Insights Into the Evolutionary Pathway of Vibrio cholerae O1 Atypical El Tor Variants. PLoS Pathog. 2014;10: e1004384. doi: 10.1371/journal.ppat.1004384 25233006
9. Heilpern AJ, Waldor MK. CTXφ Infection of Vibrio cholerae Requires the tolQRA Gene Products. J Bacteriol. 2000;182 : 1739–1747. doi: 10.1128/JB.182.6.1739–1747.2000 10692381
10. Khan SA. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid. 2005;53 : 126–36. 15737400
11. Moyer KE, Kimsey HH, Waldor MK. Evidence for a rolling-circle mechanism of phage DNA synthesis from both replicative and integrated forms of CTXphi. Mol Microbiol. 2001;41 : 311–23. 11489120
12. Huber KE, Waldor MK. Filamentous phage integration requires the host recombinases XerC and XerD. Nature. 2002;417 : 656–9. 12050668
13. Das B, Martínez E, Midonet C, Barre F-X. Integrative mobile elements exploiting Xer recombination. Trends Microbiol. 2013;21 : 23–30. doi: 10.1016/j.tim.2012.10.003 23127381
14. Val M-E, Kennedy SP, El Karoui M, Bonne L, Chevalier F, Barre F-X. FtsK-dependent dimer resolution on multiple chromosomes in the pathogen Vibrio cholerae. PLoS Genet. 2008;4. doi: 10.1371/journal.pgen.1000201
15. Midonet C, Barre F - X. Xer Site Specific Recombination: Promoting vertical and horizontal transmission of genetic information. Mobile DNA III. ASM press. Waschington: Nancy Craig;
16. Val M-E, Bouvier M, Campos J, Sherratt D, Cornet F, Mazel D, et al. The Single-Stranded Genome of Phage CTX Is the Form Used for Integration into the Genome of Vibrio cholerae. Mol Cell. 2005;19 : 559–566. doi: 10.1016/j.molcel.2005.07.002 16109379
17. Das B, Bischerour J, Val M-E, Barre F-X. Molecular keys of the tropism of integration of the cholera toxin phage. Proc Natl Acad Sci. 2010;107 : 4377–4382. doi: 10.1073/pnas.0910212107 20133778
18. Bischerour J, Spangenberg C, Barre F-X. Holliday junction affinity of the base excision repair factor Endo III contributes to cholera toxin phage integration. EMBO J. 2012;31 : 3757–3767. doi: 10.1038/emboj.2012.219 22863778
19. Rakonjac J, Bennett NJ, Spagnuolo J, Gagic D, Russel M. Filamentous bacteriophage: biology, phage display and nanotechnology applications. Curr Issues Mol Biol. 2011;13 : 51–76. 21502666
20. Quinones M, Kimsey HH, Waldor MK. LexA cleavage is required for CTX prophage induction. Mol Cell. 2005;17 : 291–300. 15664197
21. Waldor MK, Rubin EJ, Pearson GDN, Kimsey H, Mekalanos JJ. Regulation, replication, and integration functions of the Vibrio cholerae CTXφ are encoded by region RS2. Mol Microbiol. 1997;24 : 917–926. doi: 10.1046/j.1365-2958.1997.3911758.x 9220000
22. Chandler M, Cruz F de la, Dyda F, Hickman AB, Moncalian G, Ton-Hoang B. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol. 2013;11. doi: 10.1038/nrmicro3067
23. Davis BM, Lawson EH, Sandkvist M, Ali A, Sozhamannan S, Waldor MK. Convergence of the Secretory Pathways for Cholera Toxin and the Filamentous Phage, CTXϕ. Science. 2000;288 : 333–335. doi: 10.1126/science.288.5464.333 10764646
24. Grove A. Functional evolution of bacterial histone-like HU proteins. Curr Issues Mol Biol. 2011;13 : 1–12. 20484776
25. Dillingham MS. Superfamily I helicases as modular components of DNA-processing machines. Biochem Soc Trans. 2011;39 : 413–423. doi: 10.1042/BST0390413 21428912
26. Takahashi s, Hours C, Iwaya M, Lane H E D, Denhardt D T. The Escherichia Coli Rep Gene in the Single-Stranded DNA Phages. Denhardt D.T., Dressler D.H., and Ray D.S. (eds). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press,. pp. 393–400.
27. Colasanti J, Denhardt DT. The Escherichia coli rep mutation. X. Consequences of increased and decreased Rep protein levels. Mol Gen Genet MGG. 1987;209 : 382–390. 2959842
28. Lane HE, Denhardt DT. The rep mutation. IV. Slower movement of replication forks in Escherichia coli rep strains. J Mol Biol. 1975;97 : 99–112. 1100854
29. Bruand C, Ehrlich SD. UvrD-dependent replication of rolling-circle plasmids in Escherichia coli. Mol Microbiol. 2000;35 : 204–210. doi: 10.1046/j.1365-2958.2000.01700.x 10632890
30. Caron PR, Kushner SR, Grossman L. Involvement of helicase II (uvrD gene product) and DNA polymerase I in excision mediated by the uvrABC protein complex. Proc Natl Acad Sci. 1985;82 : 4925–4929. 3161077
31. Bidnenko V, Lestini R, Michel B. The Escherichia coli UvrD helicase is essential for Tus removal during recombination-dependent replication restart from Ter sites. Mol Microbiol. 2006;62 : 382–396. doi: 10.1111/j.1365-2958.2006.05382.x 17020578
32. Florés M-J, Sanchez N, Michel B. A fork-clearing role for UvrD. Mol Microbiol. 2005;57 : 1664–1675. doi: 10.1111/j.1365-2958.2005.04753.x 16135232
33. Lestini R, Michel B. UvrD controls the access of recombination proteins to blocked replication forks. EMBO J. 2007;26 : 3804–3814. doi: 10.1038/sj.emboj.7601804 17641684
34. Arthur HM, Eastlake PB. Transcriptional control of the uvrD gene of Escherichia coli. Gene. 1983;25 : 309–316. 6319240
35. Finch P, Emmerson PT. Nucleotide sequence of the regulatory region of the uvrD gene of Escherichia coli. Gene. 1983;25 : 317–323. 6319241
36. Moyer KE, Kimsey HH, Waldor MK. Evidence for a rolling-circle mechanism of phage DNA synthesis from both replicative and integrated forms of CTXφ. Mol Microbiol. 2001;41 : 311–323. doi: 10.1046/j.1365-2958.2001.02517.x 11489120
37. Davis BM, Kimsey HH, Kane AV, Waldor MK. A satellite phage-encoded antirepressor induces repressor aggregation and cholera toxin gene transfer. Embo J. 2002;21 : 4240–9. 12169626
38. Kamruzzaman M, Robins WP, Bari SMN, Nahar S, Mekalanos JJ, Faruque SM. RS1 Satellite Phage Promotes Diversity of Toxigenic Vibrio cholerae by Driving CTX Prophage Loss and Elimination of Lysogenic Immunity. Infect Immun. 2014;82 : 3636–3643. doi: 10.1128/IAI.01699-14 24935981
39. Campos J, Martínez E, Marrero K, Silva Y, Rodríguez BL, Suzarte E, et al. Novel Type of Specialized Transduction for CTXφ or Its Satellite Phage RS1 Mediated by Filamentous Phage VGJφ in Vibrio cholerae. J Bacteriol. 2003;185 : 7231–7240. doi: 10.1128/JB.185.24.7231–7240.2003 14645284
40. Das B, Bischerour J, Barre F-X. VGJɸ integration and excision mechanisms contribute to the genetic diversity of Vibrio cholerae epidemic strains. Proc Natl Acad Sci. 2011;108 : 2516–2521. doi: 10.1073/pnas.1017061108 21262799
41. Midonet C, Das B, Paly E, Barre F-X. XerD-mediated FtsK-independent integration of TLCϕ into the Vibrio cholerae genome. Proc Natl Acad Sci. 2014;111 : 16848–53. doi: 10.1073/pnas.1404047111 25385643
42. Hassan F, Kamruzzaman M, Mekalanos JJ, Faruque SM. Satellite phage TLCphi enables toxigenic conversion by CTX phage through dif site alteration. Nature. 2010;467 : 982–5. nature09469 [pii] doi: 10.1038/nature09469 20944629
43. Rubin EJ, Lin W, Mekalanos JJ, Waldor MK. Replication and integration of a Vibrio cholerae cryptic plasmid linked to the CTX prophage. Mol Microbiol. 1998;28 : 1247–54. 9680213
44. Castaing B, Zelwer C, Laval J, Boiteux S. HU Protein of Escherichia coli Binds Specifically to DNA That Contains Single-strand Breaks or Gaps. J Biol Chem. 1995;270 : 10291–10296. doi: 10.1074/jbc.270.17.10291 7730334
45. Kamashev D, Rouviere-Yaniv J. The histone-like protein HU binds specifically to DNA recombination and repair intermediates. EMBO J. 2000;19 : 6527–6535. doi: 10.1093/emboj/19.23.6527 11101525
46. Rouvière-Yaniv J, Gros F. Characterization of a novel, low-molecular-weight DNA-binding protein from Escherichia coli. Proc Natl Acad Sci U S A. 1975;72 : 3428–3432. 1103148
47. Ogawa T, Wada M, Kano Y, Imamoto F, Okazaki T. DNA replication in Escherichia coli mutants that lack protein HU. J Bacteriol. 1989;171 : 5672–5679. 2676987
48. Kano Y, Ogawa T, Ogura T, Hiraga S, Okazaki T, Imamoto F. Participation of the histone-like protein HU and of IHF in minichromosomal maintenance in Escherichia coli. Gene. 1991;103 : 25–30. 1879696
49. Dixon NE, Kornberg A. Protein HU in the enzymatic replication of the chromosomal origin of Escherichia coli. Proc Natl Acad Sci U S A. 1984;81 : 424–428. 6364143
50. Bramhill D, Kornberg A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell. 1988;52 : 743–755. doi: 10.1016/0092-8674(88)90412-6 2830993
51. Ogura T, Niki H, Kano Y, Imamoto F, Hiraga S. Maintenance of plasmids in HU and IHF mutants of Escherichia coli. Mol Gen Genet MGG. 1990;220 : 197–203. 2183003
52. Wada M, Kohno K, Imamoto F, Kano Y. Participation of hup gene product in ori2-dependent replication of fertility plasmid F. Gene. 1988;70 : 393–397. 3063607
53. Hillyard DR, Edlund M, Hughes KT, Marsh M, Higgins NP. Subunit-specific phenotypes of Salmonella typhimurium HU mutants. J Bacteriol. 1990;172 : 5402–5407. 2168381
54. Yasukawa H, Ozaki E, Nakahama K, Masamune Y. HU protein binding to the replication origin of the rolling-circle plasmid pKYM enhances DNA replication. Mol Gen Genet MGG. 1997;254 : 548–554. 9197414
55. Rasched I, Oberer E. Ff coliphages: structural and functional relationships. Microbiol Rev. 1986;50 : 401. 3540571
56. Gros MF, te Riele H, Ehrlich SD. Replication origin of a single-stranded DNA plasmid pC194. EMBO J. 1989;8 : 2711–2716. 2583127
57. Novick RP. Staphylococcal Plasmids and their Replication. Annu Rev Microbiol. 1989;43 : 537–563. doi: 10.1146/annurev.mi.43.100189.002541 2679362
58. Lohman TM, Bjornson KP. Mechanisms of Helicase-Catalyzed DNA Unwinding. Annu Rev Biochem. 1996;65 : 169–214. doi: 10.1146/annurev.bi.65.070196.001125 8811178
59. Yarranton GT, Gefter ML. Enzyme-catalyzed DNA unwinding: Studies on Escherichia coli rep protein. Proc Natl Acad Sci U S A. 1979;76 : 1658–1662. 221901
60. Matson SW. Escherichia coli helicase II (urvD gene product) translocates unidirectionally in a 3’ to 5’ direction. J Biol Chem. 1986;261 : 10169–10175. 2942537
61. Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, et al. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell. 2009;36 : 654–666. doi: 10.1016/j.molcel.2009.11.009 19941825
62. Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29 : 145–157. doi: 10.1038/emboj.2009.308 19851282
63. Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16 : 430–438. 9029161
64. Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA Mismatch Repair: Functions and Mechanisms. Chem Rev. 2006;106 : 302–323. doi: 10.1021/cr0404794 16464007
65. Reardon JT, Sancar A. Nucleotide Excision Repair. In: Moldave Kivie, editor. Progress in Nucleic Acid Research and Molecular Biology. Academic Press; 2005. pp. 183–235. Available: http://www.sciencedirect.com/science/article/pii/S0079660304790042 16096029
66. Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, et al. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24 : 180–189. doi: 10.1038/sj.emboj.7600485 15565170
67. Yasukawa H, Hase T, Sakai A, Masamune Y. Rolling-circle replication of the plasmid pKYM isolated from a gram-negative bacterium. Proc Natl Acad Sci U S A. 1991;88 : 10282–10286. 1835091
68. Mekalanos JJ, Swartz DJ, Pearson GD, Harford N, Groyne F, de Wilde M. Cholera toxin genes: nucleotide sequence, deletion analysis and vaccine development. Nature. 1983;306 : 551–557. 6646234
69. Kaper JB, Lockman H, Baldini MM, Levine MM. Recombinant nontoxinogenic Vibrio cholerae strains as attenuated cholera vaccine candidates. Nature. 1984;308 : 655–658. 6324005
70. Valle E, Ledón T, Cedré B, Campos J, Valmaseda T, Rodríguez B, et al. Construction and Characterization of a Nonproliferative El Tor Cholera Vaccine Candidate Derived from Strain 638. Infect Immun. 2000;68 : 6411–6418. doi: 10.1128/IAI.68.11.6411–6418.2000 11035753
71. Liang W, Wang S, Yu F, Zhang L, Qi G, Liu Y, et al. Construction and Evaluation of a Safe, Live, Oral Vibrio cholerae Vaccine Candidate, IEM108. Infect Immun. 2003;71 : 5498–5504. doi: 10.1128/IAI.71.10.5498–5504.2003 14500467
72. Dubuisson J-F, Vianney A, Hugouvieux-Cotte-Pattat N, Lazzaroni JC. Tol-Pal proteins are critical cell envelope components of Erwinia chrysanthemi affecting cell morphology and virulence. Microbiology. 2005;151 : 3337–3347. doi: 10.1099/mic.0.28237–0 16207916
73. Gerding MA, Ogata Y, Pecora ND, Niki H, De Boer PAJ. The trans-envelope Tol–Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol. 2007;63 : 1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x 17233825
74. Vinés ED, Marolda CL, Balachandran A, Valvano MA. Defective O-Antigen Polymerization in tolA and pal Mutants of Escherichia coli in Response to Extracytoplasmic Stress. J Bacteriol. 2005;187 : 3359–3368. doi: 10.1128/JB.187.10.3359–3368.2005 15866920
75. Llamas MA, Ramos JL, Rodríguez-Herva JJ. Mutations in Each of the tol Genes ofPseudomonas putida Reveal that They Are Critical for Maintenance of Outer Membrane Stability. J Bacteriol. 2000;182 : 4764–4772. doi: 10.1128/JB.182.17.4764–4772.2000 10940016
76. Prouty AM, Velkinburgh JCV, Gunn JS. Salmonella enterica Serovar Typhimurium Resistance to Bile: Identification and Characterization of the tolQRA Cluster. J Bacteriol. 2002;184 : 1270–1276. doi: 10.1128/JB.184.5.1270–1276.2002 11844755
77. Kimsey HH, Waldor MK. CTXφ immunity: Application in the development of cholera vaccines. Proc Natl Acad Sci. 1998;95 : 7035–7039. 9618534
78. Raychoudhuri A, Patra T, Ghosh K, Ramamurthy T, Nandy RK, Takeda Y, et al. Classical ctxB in Vibrio cholerae O1, Kolkata, India. Emerg Infect Dis. 2009;15 : 131–2. doi: 10.3201/eid1501.080543 19116078
79. Nguyen DT, Nguyen BM, Tran HH, Ngo TC, Le TH, Nguyen HT, et al. Filamentous vibriophage fs2 encoding the rstC gene integrates into the same chromosomal region as the CTX phage [corrected]. FEMS Microbiol Lett. 2008;284 : 225–30. FML1200 [pii] doi: 10.1111/j.1574-6968.2008.01200.x 18503544
80. Campos J, Martínez E, Izquierdo Y, Fando R. VEJφ, a novel filamentous phage of Vibrio cholerae able to transduce the cholera toxin genes. Microbiology. 2010;156 : 108–115. doi: 10.1099/mic.0.032235–0 19833774
81. Campos J, Martínez E, Suzarte E, Rodríguez BL, Marrero K, Silva Y, et al. VGJφ, a Novel Filamentous Phage of Vibrio cholerae, Integrates into the Same Chromosomal Site as CTXφ. J Bacteriol. 2003;185 : 5685–5696. doi: 10.1128/JB.185.19.5685–5696.2003 13129939
82. Udden SM, Zahid MS, Biswas K, Ahmad QS, Cravioto A, Nair GB, et al. Acquisition of classical CTX prophage from Vibrio cholerae O141 by El Tor strains aided by lytic phages and chitin-induced competence. Proc Natl Acad Sci U A. 2008;105 : 11951–6. 0805560105 [pii] doi: 10.1073/pnas.0805560105 18689675
83. Method for constructing an attenuated mutant strain of pathogenic bacteria, vaccine, vaccine vector and use of said vaccine [Internet]. Available: http://www.google.com/patents/WO2012142684A1
84. Davies BW, Bogard RW, Dupes NM, Gerstenfeld TAI, Simmons LA, Mekalanos JJ. DNA Damage and Reactive Nitrogen Species are Barriers to Vibrio cholerae Colonization of the Infant Mouse Intestine. PLoS Pathog. 2011;7. doi: 10.1371/journal.ppat.1001295
85. O’Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol. 1998;28 : 449–61. 9632250
86. Iwanaga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol Immunol. 1986;30 : 1075–1083. 3543624
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy