
Post-transcriptional Regulation of Keratinocyte Progenitor Cell Expansion, Differentiation and Hair Follicle Regression by
Up to 60% people suffer from hair loss throughout their lifetime. Hair growth undergoes recurrent cycling of growth, regression, and resting phases with a defined periodicity. The main cause of human hair loss is due to the premature transition from growth to regression. Understanding of the molecular basis underlying hair regression is important to elucidate the mechanisms of hair loss. Here, we demonstrated that miR-22, a highly conserved microRNA, is critical for the transition from growth to regression of the hair follicle. Importantly, miR-22 could be a novel target for therapeutic therapy of hair loss disorders.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005253
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005253
Summary
Up to 60% people suffer from hair loss throughout their lifetime. Hair growth undergoes recurrent cycling of growth, regression, and resting phases with a defined periodicity. The main cause of human hair loss is due to the premature transition from growth to regression. Understanding of the molecular basis underlying hair regression is important to elucidate the mechanisms of hair loss. Here, we demonstrated that miR-22, a highly conserved microRNA, is critical for the transition from growth to regression of the hair follicle. Importantly, miR-22 could be a novel target for therapeutic therapy of hair loss disorders.
Introduction
Hair follicles undergo recurrent cycles of growth (anagen), regression (catagen), and resting (telogen) phases with a defined periodicity [1,2]. Abnormal regulation of genes associated with hair cycling may cause several types of human hair growth disorders [3]. For example, male pattern baldness is due to the premature transition from anagen-to-catagen induced by androgens [4]. The hair follicle contains epithelial cells of the outer root sheath (ORS), matrix, the inner root sheath and hair shaft, and mesenchymal cells of dermal papilla [1,5]. The interaction between epithelial and mesenchymal cells is required for proper hair development and follicle cycling [2]. At the onset of growth (anagen), the dermal papilla is at the proximal end of the follicle, in close proximity to the hair stem cell niche known as the follicle bulge [6]. Signals from the condensed dermal papilla induce proliferation of hair bulge stem cells, resulting in the downward extension of the hair germ where it ultimately envelops the dermal papilla and triggers hair matrix formation. Transit-amplifying matrix cells proliferate rapidly in response to signals from the dermal papilla after which they terminally differentiate to form the inner root sheath and hair shaft. Several important signaling pathways including Wnt and BMP play an important role in hair differentiation [7]. Further, a host of transcription factors promote hair differentiation during anagen including Gata3 [8], Cutl1 [9], Lef1 [10], Dlx3 [11], Msx2 [12], Foxn1 [13], TCF3 [14] and Hoxc13 [15]. In contrast to anagen, catagen is the physiological involution of the hair follicle. The hair follicle rapidly degenerates and shortens until it is again adjacent to the hair follicle stem cell reservoir in the bulge region. This process may be triggered by a variety of stimuli, including apoptosis and loss of supportive growth factor signaling needed to maintain cell proliferation and differentiation during anagen [16]. While some apoptotic triggers promoting catagen have been defined, including Bcl2/Bax [17], p53 [18], p57 [19], and the transforming growth factors, TGF-β1 and TGF-β2 [20,21], little is known about how the stimulatory signals that drive anagen are terminated upon entry to catagen and maintained during telogen.
MicroRNAs play important roles in many biological processes including hair follicle development [22], and hundreds of microRNAs are expressed in the skin [23,24]. Global ablation of microRNA activity through genetic deletion of microRNA processing enzymes, DGCR8, Drosha, and Dicer demonstrated that microRNAs are critical for both embryonic and adult hair follicle development [23,25,26]. Conditional deletion of Drosha and Dicer during anagen causes failure of catagen and follicular degradation [25], indicating a critical role of microRNAs in the transition of anagen to catagen. However, the specific microRNAs involved in this process are unknown. Recently, numerous reports have documented the functional contribution of several specific microRNAs to hair development, including miR-24 [27], miR-125b [28], miR-31 [24], and miR-205 [29], and thus a microRNA network governing hair follicle development is beginning to emerge.
The function of miR-22 in hair follicles is of particular interest, as microRNA profiling revealed that miR-22 expression markedly increases during the catagen stage, reaching peak expression at the transition to telogen [24], suggesting a potential role for miR-22 in hair follicle involution. Here we utilize miR-22 gain - and loss-of-function mouse models to investigate the physiological role of miR-22 in hair cycling. We demonstrate that miR-22 is an important post-transcriptional regulator that governs exit from anagen and maintenance of the hair follicle in telogen through inhibition of a transcriptional program driving keratinocyte proliferation, differentiation, and hair shaft assembly.
Results
miR-22 expression pattern during hair cycling
To elucidate the role of miR-22 in hair follicle development, we initially confirmed published findings of miR-22 upregulation during catagen [24]. miR-22 is markedly upregulated during catagen and reaches maximal expression in telogen (S1A Fig), suggesting a potential role of miR-22 in hair involution. Further, in-situ hybridization at successive time points during anagen, catagen and telogen phases revealed that miR-22 expressing cells are located in the ORS and hair matrix but not in the IRS at postnatal day 11 (P11, mid-anagen) (Fig 1). At P17 (catagen), miR-22 is strongly expressed throughout the hair follicle including both ORS and IRS, with the in-situ signal becoming weaker in the hair matrix (Fig 1). At P21 (telogen), miR-22 is strongly expressed in the ORS (Fig 1). At P30, the mid-anagen phase of the second hair cycle, miR-22 is again mainly expressed in the upper ORS (S1B and S1C Fig). We observed miR-22 signal in a few cells of hair matrix in close proximity of the dermal papilla (S1C Fig). In older animals, hair follicles reside much longer in telogen [30]. Thus, we examined the miR-22 expression level in the skin of 18 month-old mice. As expected, miR-22 expression increases in the aged skin (S1D Fig), consistent with a potential function for miR-22 as a regulator of follicle regression.

miR-22 induction causes hair loss in vivo
To determine the functional consequences of miR-22 activity in hair follicles, we generated mice in which miR-22 overexpression could be induced in the ORS of hair follicles with temporal specificity. MiR-22 was placed under the control of a tetracycline regulatory element (TRE) (Fig 2A) and TRE-miR-22 transgenic mice were generated. TRE-miR-22 mice were crossed with K14-rtTA transgenic mice, in which the Keratin 14 (K14) promoter that is active in the basal layer of the epidermis and the ORS of the hair follicle controls expression of the reverse tetracycline transactivator (rtTA). After induction with Doxycycline (Dox), K14-rtTA/TRE-miR-22 double transgenic (DTG) mice exhibited robust miR-22 expression in the basal layer of the epidermis and ORS of the hair follicle (S1B Fig). Expression analysis demonstrated strong miR-22 induction in DTG mice, compared to TRE-miR22 and K14-rtTA controls (Fig 2B).
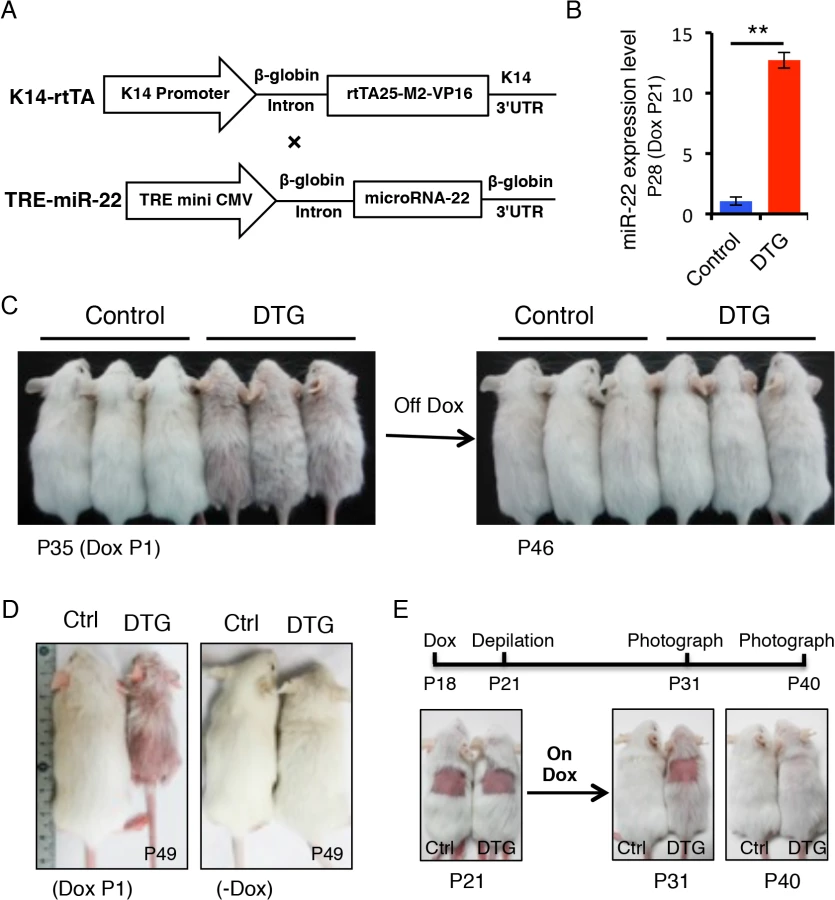
We initially induced miR-22 expression at postnatal day 1 during embryonic anagen stage. Strikingly, Dox-treated DTG mice began to exhibit premature hair loss 35 days after induction (Fig 2C). Within 11 days after Dox withdrawal at P35, there was a full recovery from the hair loss phenotype (Fig 2C), demonstrating that the effect of miR-22 induction on hair growth is reversible. When DTG mice were maintained on Dox, hair loss became more pronounced at P49 during the telogen phase, while hair growth of the control mice was normal and morphologically indistinguishable from wildtype littermates (Fig 2D). The hair loss persisted as long as the mice were maintained on Dox. In addition to the hair loss phenotype, we also found that the DTG mice were smaller in size compared to the control littermates (Fig 2C and 2D). Because the K14 promoter is also active in the esophagus and stomach, the reduction in body size is most likely due to digestion defects caused by miR-22 induction.
To further explore the effect of miR-22 on hair morphorgenesis, we induced miR-22 expression at embryonic day 15 when the hair placode begins to form. miR-22 induction resulted in obvious thinning of the hair coat in DTG pups at P12 (S2A Fig), indicating that miR-22 induction represses hair morphorgenesis. Surprisingly, the DTG pups died at around P17. We next initiated a synchronous hair growth cycle at P21 by depilating the dorsal hair concomitant to miR-22 induction. External hair regrowth was observed in littermate controls 10 days post-depilation, but was absent in the depilated DTG skin (Fig 2E). Similar findings were observed at P65 when both control and DTG mice were treated with Dox at P53 and depilated at P56 (S2B Fig). Taken together, our data reveal that miR-22 overexpression is sufficient to repress hair morphorgenesis and development in vivo.
miR-22 induction is sufficient to promote anagen-to-catagen transition of hair follicles
To investigate how miR-22 overexpression impairs hair development, we studied the progression of hair cycling by analyzing histology of DTG hair follicles at successive time points after Dox induction. Both DTG and control mice were treated with Dox at P1. At P9, the DTG follicles were morphologically normal, suggesting a completion of hair follicle morphogenesis (S2C Fig). Abnormal hair follicles were clearly observed in the DTG backskin by P16. While the hair follicles of control littermates were in early catagen at P16, the DTG follicles had already entered late catagen, characterized by the narrow hair bulb and shortened hair length (Fig 3A and 3B and S2D Fig). This indicates that miR-22 induction promotes the anagen-to-catagen transition. Both control and DTG follicles can normally enter telogen at P21, although the DTG follicles appeared slightly abnormal to be considered a typical telogen follicle (S2C Fig). By P26, control follicles entered early anagen, while the DTG follicles remained in telogen (Fig 3A and S2D Fig), suggesting that miR-22 induction retards the telogen-to-anagen transition. By P30 and 35, control follicles entered anagen and formed enlarged hair bulbs with differentiated hair shafts. While the DTG follicles also formed hair bulbs, these hair bulbs were smaller in size. The extent of downward growth in DTG follicles was reduced (Fig 3A and 3B), and the DTG follicles had no keratinized medulla (Fig 3A). By P43 and P46, the control follicles shortened and entered late catagen. While DTG hair follicles also entered catagen, characterized by narrow hair bulbs, the DTG follicles failed to further regress and shorten (S2C Fig). Most likely, the hair cycle is arrested in a mid-catagen phase, which may be the major cause of hair loss at this stage. As this phenomenon is contrary to the promotion of the anagen-to-catagen transition in the first hair cycle, we asked whether short-term miR-22 induction would also result in an arrest of the hair cycle at this stage. We induced miR-22 expression in mid-anagen at P27, and collected samples at P32 in late anagen. Control hair follicles were morphologically in late anagen (Fig 3C), while the DTG follicles had prematurely entered mid-catagen, characterized by narrow hair bulbs and short follicles (Fig 3C).

We next induced miR-22 expression at P32 in late anagen, and collected samples at P37 of early catagen. We found that miR-22 induction markedly promoted progression to catagen (Fig 3C). These findings demonstrate that miR-22 induction promotes the transition from anagen to catagen, as well as progression through catagen. To further confirm this, we examined expression of a number of genes normally expressed in catagen at P16 (early catagen), in response to 16 days of Dox induction (S2E Fig). Expression of MMP11, K16, TGFβ2, SPINK12 are markedly upregulated in DTG backskin (S2E Fig), supporting a function for miR-22 in promoting the anagen-to-catagen transition. To test this idea that miR-22 represses the telogen-to-anagen transition, we induced miR-22 expression at P18 and collected samples at P26. The control hair follicles normally entered anagen, while DTG follicles were much delayed (Fig 3C). The data further supported that miR-22 retards the telogen-to-anagen transition.
In addition to defective follicular development, the DTG epidermis became markedly thickened and was hyperproliferative by P30, with increased numbers of Ki67 positive cells and P63 positive cells (S3 Fig), however differentiation of DTG epidermis appeared unaffected (S3 Fig).
miR-22 is required for proper anagen-to-catagen transition
The TRE-miR-22 mouse model demonstrates the sufficiency of miR-22 in promoting the anagen-to-catagen transition, but does not address a physiological requirement in this process. We thus examined histology of miR-22 knockout (KO) hair follicles at successive time points. No significant difference was found in the KO follicles at P1 (Fig 3D), however by P9 hair follicle length in the KO was significantly longer than in the controls, although KO follicles appeared morphologically normal (Fig 3D and S2G Fig). By P18, the defects in KO follicles became more pronounced. Control follicles had entered late catagen at this time point, however KO follicles were delayed in catagen entry, evidenced by a large hair bulb (Fig 3D and S2F Fig). By P21, control follicles uniformly had entered telogen, while up to 15% late catagen-stage follicles remained in the KO backskin, indicating a delay in hair development (Fig 3D and S2G Fig). By P26 both control and KO hair follicles entered a new anagen, however the penetrance of downward growth was increased in the KO follicles (Fig 3D and S2G Fig). Overall, the miR-22 KO phenotype is in direct opposition to that of miR-22 gain of function. Taken together, these data point to an important physiological role for miR-22 in promoting the anagen-to-catagen transition and in proper telogen maintenance in hair follicles.
miR-22 induction inhibits keratinocyte expansion and differentiation, and promotes apoptosis
To understand how miR-22 gain of function induces hair loss, we asked whether sustained miR-22 expression regulates cell proliferation and differentiation in the hair follicle. By P30, control follicles are in mid-anagen, with large hair bulbs and terminally differentiated hair shafts. Both DTG and control follicles were morphologically similar at P30, after 30 days of Dox administration (Fig 3A). Subsequently, the hair loss phenotype manifests in DTG mice after P30 (Fig 2C). Thus, we chose to examine proliferation and differentiation at P30, the onset of phenotypic manifestation. The number of proliferative cells was significantly reduced in the DTG matrix (Fig 4A), suggesting that miR-22 represses cell proliferation. The hair loss defect could arise from an impairment of matrix cells in producing differentiated lineages. To test this possibility, we first examined the proliferative expansion of matrix cells. P13 mice were pulsed by a single injection of BrdU and analyzed 11 hours later. In control follicles, BrdU labeled cells could readily be observed in the hair precortex. In contrast, upward movement of BrdU+ cells into the DTG precotex was impaired (Fig 4B), suggesting defects in both proliferation and migration. To further pursue this, we treated DTG mice with Dox at P18, depilated hair at P21, and assessed new hair generation at P27 after 11-hour BrdU pulse. As expected, BrdU labeled cells were present in the precortex of control follicles (S4A Fig). In contrast, the DTG precortex lacked the BrdU labeled cells (S4A Fig). These data support a model in which miR-22 inhibits the proliferative expansion and subsequent differentiation of keratinocyte progenitors.

To investigate the regulation of miR-22 on keratinocyte differentiation, we first examined transcription factors that are important for keratinocyte differentiation. Lef1 is a transcription factor that promotes the differentiation of hair stem cells [31]. Examination of Lef1 expression in the DTG hair follicles revealed a decrease in Lef1 positive cells (Fig 4C and S4B Fig). In contrast, an increase in Lef1 positive cells was observed in the miR-22 KO mice at P18 (Fig 4C and S4B Fig). The transcription factor Gata-3 plays an essential role in the differentiation of IRS progenitor cells [8], and Gata-3 signals were negative in DTG follicles (Fig 4D). In contrast, Gata-3 positive cells increased in the miR-22 KO follicles (Fig 4D and S4B Fig). The data further suggest that miR-22 represses keratinocyte differentiation. We next examined markers of terminal keratinocyte differentiation. AE13 is a hair cortex marker, which is used to detect cortical keratins. While the follicles of control littermates were AE13 positive, the DTG follicles completely lacked AE13 reactivity (Fig 4E), suggesting that miR-22 induction inhibits hair cortex formation. Expression of the IRS and medulla tricohyalins marker AE15 was detected in both of these sites in control follicles. In contrast, AE15 immunoreactivity was reduced in the IRS of DTG follicles relative to control follicles, and DTG follicles lacked AE15 positive medulla tricohyalins (Fig 4E). These results demonstrate that miR-22 overexpression inhibits differentiation of HF keratinocytes.
Involution of the hair follicle during catagen is driven by apoptosis [16], and thus we examined cleaved Caspase 3 in both DTG and miR-22 KO hair follicles. DTG hair follicles at P16 and P31 after Dox induction at P1 showed an increase in apoptotic events, whereas control follicles have no apoptotic cells at these stages (Fig 4F). In contrast, miR-22 KO follicles have significantly fewer apoptotic cells than the control follicles at P18 (Fig 4G and S4B Fig). Thus, these data demonstrate that miR-22 also induces apoptosis of hair follicle keratinocytes, which may further contribute to the hair loss phenotype in DTG mice.
miR-22 induction represses colony forming capacity of hair follicle stem cells and hair neogenesis
The failed proliferative expansion of follicles observed upon miR-22 overexpression indicates that miR-22 may affect stem cell self-renewal and commitment. We examined the Sox9+ bulge stem cell compartment and observed no obvious difference in the frequency of hair follicle stem cells (HFSCs) in the DTG follicles (S5A Fig). Flow cytometric profiles further confirmed that the DTG and control hair follicles contained equivalent numbers of CD49f+, CD34+ HFSCs (S5B Fig). We next isolated HFSCs from control and DTG skin and compared their colony forming capacity in vitro. Strikingly, the number of large holoclones was significantly reduced in DTG HFSCs (Fig 5A and 5B), indicating that miR-22 overexpression compromises colony forming capacity of hair follicle stem cells. This further explains why the DTG hair bulb is smaller in size at P30 (Fig 3A).
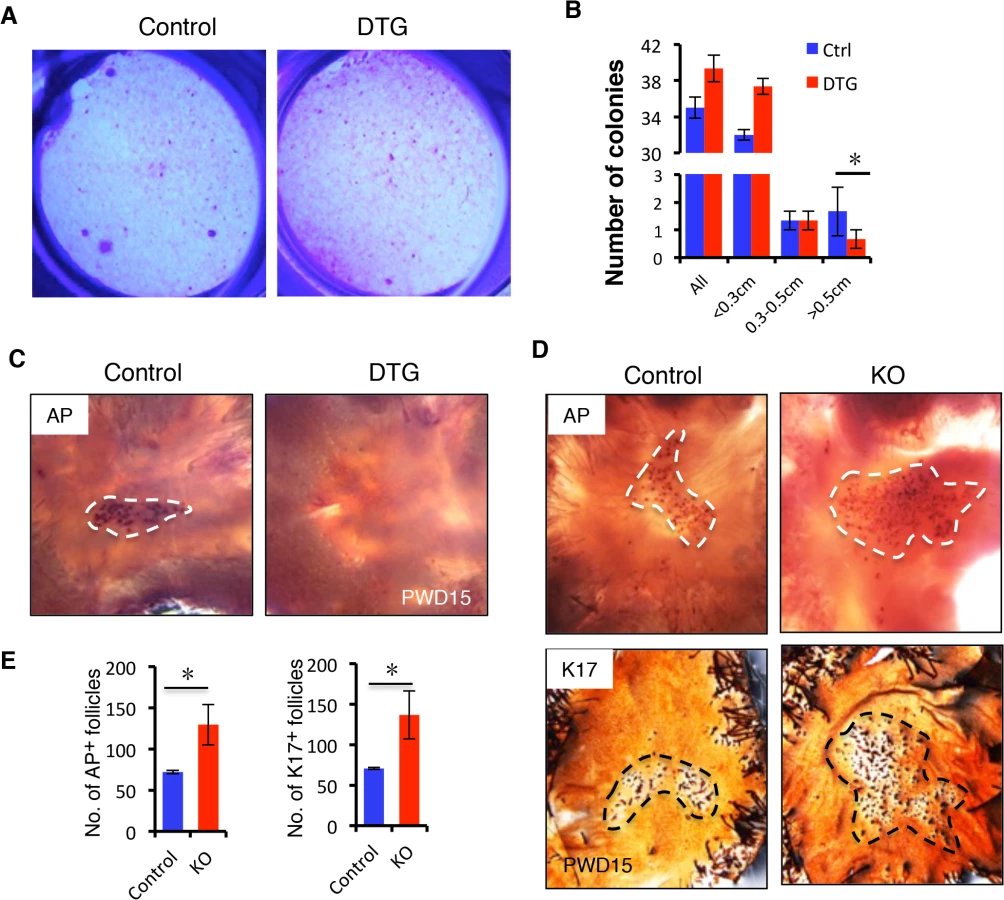
We next asked whether miR-22 induction also inhibits hair follicle neogenesis following 1 cm2 skin wounding (S5C Fig). We induced miR-22 expression at the time of wounding and collected samples at 15 days post-wounding (PWD). Interestingly, while a certain number of hair follicles regenerated de novo within the wound center of control mice, hair follicle neogenesis was completely abrogated in DTG mice (Fig 5C). In contrast, we found a significant increase number of newly formed hair follicles in miR-22 KO mice 15 days post-wounding (Fig 5D and 5E). Taken together, these findings provide evidence that miR-22 represses colony forming capacity of HFSCs and hair follicle neogenesis.
miR-22 is a critical negative regulator of the keratinocyte differentiation gene expression program during catagen and telogen
To gain mechanistic insights into the molecular events underlying transitions from anagen to catagen, to telogen, we analyzed the transcriptome profiles of catagen (mid-catagen via late anagen) and telogen (telogen via mid-catagen) [32]. The gene ontology (GO) categories of genes downregulated in the catagen profiles include non-membrane bounded organelle, Keratin, cell cycle, and negative regulation of apoptosis, consistent with an inhibition of proliferation, abrogation of differentiation-related gene expression (keratins), loss of hair shaft production (as hair shaft assembly occurs in a non-membrane bound organelle), and increased apoptosis (S7A Fig) that occurs in the transition from anagen to catagen. We also analyzed downregulated functions in the telogen transcriptome profiles. These also include Keratin, non-membrane bounded organelle, and negative regulation of apoptosis, and epithelial development (S7B Fig). Collectively, these findings suggested that the programs driving proliferation, survival, and keratin-mediated hair shaft assembly are silenced in the catagen-to-telogen transition.
To understand the molecular mechanisms of how miR-22 contributes to the termination of the hair cycle, we performed transcriptome profiling on control and DTG backskin after 7 days of Dox treatment (P21-P28) (S6 Fig). We identified 305 downregulated genes and 137 upregulated genes with a significance q<0.05 and a fold change >1.5. Enriched downregulated GO clusters upon miR-22 induction included keratin, non-membrane bounded organelle, hair cycle, melanin metabolism process and negative regulation of apoptosis (Fig 6A). These gene expression patterns are in good agreement with our phenotypic observations of decreased differentiation and increased apoptosis resulting from ectopic induction of miR-22 (Figs 2–4), and in agreement with functions normally downregulated upon transition from anagen to catagen and telogen, suggesting that miR-22 may be a critical regulator of this transition.

Anti-apoptotic genes that are suppressed by miR-22 induction include GCLG, Msx2, Foxc1, Sgk3, Adamts20, MITF, and Vnn1 (Fig 6B) [33–37]. The 305 genes downregulated by miR-22 exhibited considerable overlap with genes downregulated in telogen (153/305, 50.16%) and catagen (131/305, 42.95%) (Fig 6C; S1 Table and S2 Table). Remarkably, among the 153 genes commonly suppressed during telogen and by miR-22 induction were 42 keratin genes (27.45%) (S7C Fig). We confirmed the downregualtion of K32, K35 and K85 by qRT-PCR (Fig 6D). Interestingly, there are 74 genes commonly downregulated by miR-22 induction, during catagen, and during telogen. These genes include 27 keratin genes and the transcriptional regulators of keratinocyte differentiation Dlx3, Foxn1 and Msx2 (Fig 6C and S3 Table). These findings suggest that miR-22 directly represses a transcriptional program governing hair shaft assembly and keratinocyte differentiation.
The much smaller set of genes upregulated by miR-22 induction (137) had little in common with the Catagen (5 genes) and Telogen (4 genes) signatures (S8 Fig), further suggesting that the phenotypic consequences of miR-22 activation are through transcript repression.
Multiple hair differentiation regulators are miR-22 direct targets
To shed light on how miR-22 negatively regulates keratinocyte phenotypic gene expression, we initially identified all downregulated transcription factors in the DTG transcriptome profiles. Included were Dlx3, Msx2, Hoxc13, Foxn1, Nfe2I3, Elf5, Smad6, Lef1, Dlx4 and MITF, mostly known regulators of keratinocyte differentiation (Fig 7A). Among them, Dlx3, Hoxc13, Foxn1 and Lef1 are capable of directly controlling keratin gene expression [11] [10,13,15]. Identification of miR-22 binding sites using TargetScan indicated that 3’UTR of transcription factors Dlx3, Hoxc13, Foxn1, Nfe2I3 and Elf5 are direct miR-22 targets (S9A Fig). The secreted BMP antagonist Sostdc1, which promotes migration of DP progenitors [38], also contains miR-22 binding sites and is significantly downregulated in DTG transcriptome profiles (S9A Fig). In addition, Foxc1, Sgk3, and MITF contain miR-22 binding sites in their 3’UTR (S6B Fig), which are downregulated anti-apoptotic genes [33–35]. We focused on the transcription factors that directly regulate keratin gene expression (Dlx3, Foxn1, Hoxc13) and the BMP antagonist Sostdc1 for functional validation as miR-22 targets. Q-PCR results confirmed that Dlx3, Hoxc13, Foxn1 and Sostdc1 mRNA levels were markedly reduced in the DTG skin (S7D Fig), and significantly upregulated in miR-22 KO skin (Fig 7B). Immunofluorescence and Western blotting confirmed that Dlx3, Foxn1 and Sostdc1 protein levels are greatly deceased in DTG follicles, however Hoxc13 protein levels did not appear significantly altered (Fig 7C and 7D). To further test whether 3’UTRs of these genes are direct targets of miR-22, we constructed luciferase reporters for the Dlx3, Hoxc13, Foxn1 and Sostdc1 3’UTRs, as well as reporter constructs in which the predicted miR-22 binding sites were mutated (S9C Fig). miR-22 mimics significantly repressed wild type Dlx3, Hoxc13, Foxn1 and Sostdc1 3’UTR reporter activity, but had no significant influence on the mutant reporters (Fig 7E). These results further demonstrate that miR-22 directly regulates the Dlx3, Hoxc13, Foxn1 and Sostdc1 3’UTRs.

Taken together, our findings demonstrate that miR-22 acts as a pleiotropic regulator of a transcriptional program that regulates hair follicle stem and progenitor cell expansion, migration, and subsequent differentiation and hair shaft formation.
Discussion
In this study we demonstrate that miR-22 promotes hair follicle involution through negative post-transcriptional regulation of keratinocyte progenitor expansion, differentiation and hair shaft assembly in the catagen and telogen phases. The fact that miR-22 overexpression promotes anagen-to-catagen transition and maintains hair follicles in telogen, coupled with the observed delayed entry to catagen from anagen and accelerated transition from telogen to anagen upon deletion of miR-22, point to an important role of miR-22 in controlling termination of the hair cycle. MiR-22 appears to promote the anagen-to-catagen transition through increasing apoptosis and repressing master regulators of keratinocyte differentiation (Fig 8).

During hair cycling, the HF undergoes dynamic changes in morphology [39]. In the anagen phase, the TA matrix cells undergo proliferation and differentiation to form the hair shaft. The hair shaft is a highly keratinized product of the hair follicle, formed by type I and type II keratins, which generate intermediate filaments in the cytoplasm and contribute to cytoskeleton formation [39]. The transition from anagen, to catagen, and to telogen is a physiological involution, with HF rapidly degenerating and shortening [7]. GO analysis of catagen and telogen transcriptome profiles revealed that keratin genes are continuously downregulated from catagen to telogen phases, supporting the idea that the involution requires the repression of keratinocyte differentiation and hair shaft assembly [16]. Until now, the molecular regulators terminating hair shaft generation and promoting follicle involution were largely unknown. In the current study we identify miR-22 as a critical post-transcriptional regulator repressing keratinocyte differentiation and promoting transition from anagen to catagen and maintaining telogen phases.
Mir-22 represses a number of transcription factors that promote keratinocyte differentiation and hair shaft formation, including Dlx3, Msx2, Hoxc13, Foxn1, Nfe2I3, Elf5, Smad6, Lef1, Dlx4 and MITF in the DTG skin. Further, we identified miR-22 binding sites in the 3’UTR of Nfe2I3, Elf5, Dlx3, Hoxc13 and Foxn1. Our results demonstrated that miR-22 directly regulates the expression of Dlx3, Hoxc13 and Foxn1 using luciferase activity assay (Fig 7F). Thus, miR-22 may directly repress these transcription factors to control keratin gene expression and keratinocyte differentiation. Among them, a core of transcription factors including Dlx3, Foxn1, Hoxc13 and Lef1 can directly bind to and activate the promoters of hair keratin genes including K32, K35, and K37 [10,11,13]. Consistent with our model, the miR-22 target Dlx3 is expressed at its highest levels in anagen, with expression decreasing in catagen and terminating in telogen [11]. Thus, the Dlx3 expression pattern is inversely correlated to that of miR-22, supporting this notion that miR-22 repression of Dlx3 contributes to HF involution. Consistent with this notion, deletion of Dlx3 leads to hair loss by directly repressing hair keratin gene expression [11]. Dlx3 can directly bind to the promoter of K32, K35 and Hoxc13 to activate gene expression [11]. The Dlx3 KO phenotype resembles the miR-22 overexpression mouse phenotype, indicating that Dlx3 is a functionally important target of miR-22 during hair cycling. Foxn1 is another direct target of miR-22. Foxn1 knockout mice were initially utilized as a nude mouse model [40]. The nude phenotype is predominantly due to the impaired hair shaft formation caused by reduction of hair keratin gene expression [13]. Like Dlx3, Foxn1 is capable of directly regulating keratin gene expression as a transcription factor or it can act indirectly as a co-activator [13]. Another phenotype of Foxn1 KO mice is defective differentiation of thymic epithelia [41]. Consistently, we found that thymic development is impaired in the miR-22 overexpressing DTG transgenic mice (S10 Fig), providing further support that Foxn1 is a functionally important target of miR-22. Consistent with the downregulation of keratinocyte differentiation-related transcription factors, 58 keratin genes are markedly reduced in the miR-22 overexpressing mice. Thus, miR-22 plays an important role in hair follicle keratinocyte differentiation during hair cycling.
Global deletion of microRNA processing enzymes Dicer1 and Drosha during anagen lead to failed catagen entry and impaired follicle degradation, revealing an essential function of microRNAs in HF regression [25]. Loss of apoptosis might account for this failed follicle degradation, however Dicer1 and Drosha mutant hair matrix cells exhibit a slight increase in apoptosis, suggesting that apoptosis-independent hair regression mechanisms are defective in Dicer1 and Drosha mutant follicles. In the miR-22 KO mice, we observed elongated anagen follicles and delayed entry into catagen; phenotypes consistent to with the Dicer1 and Drosha-null follicles. Thus, miR-22 mediated repression of keratinocyte differentiation may partially account for the Dicer1 and Drosha mutant phenotype. However, the miR-22 KO phenotype is less severe than that observed Dicer1 or Drosha KO mice, suggesting that other microRNAs contribute to HF regression from anagen, to catagen, and then to telogen. Numerous reports have documented the functions of specific microRNAs on hair development [24,27–29]. Among them, miR-24 and miR-125b have expression patterns similar to that of miR-22 during hair cycling [24]: sharp upregulation at catagen and maximal expression at telogen phase. Consistent with miR-22, ectopic expression of miR-24 in the ORS of hair follicles resulted in hair loss with severe defects in hair follicle morphogenesis. miR-24 repressed hair keratinocyte differentiation by negatively regulating TCF3, an important upstream regulator of hair differentiation [14,27]. miR-125b is another well-characterized microRNA in hair development. Similarly to miR-22, miR-125b overexpression leads to hair loss by repressing hair differentiation. miR-125b directly represses the vitamin D receptor, which plays an important role in hair differentiation [28]. Thus, miR-22, miR-24 and miR-125b might synergistically function to promote hair regression by repressing a keratinocyte differentiation regulatory program in the transition into catagen and telogen. This phenomenon may in fact be more broad. In addition to miR-24, miR-125b, numerous microRNAs including miR-29a, miR-27a, miR-27b, miR-30a, miR-152 and miR-143 also exhibit the same expression pattern as miR-22 [24]. Thus, future studies are warranted to determine the combinatorial effects of this suite of microRNAs in hair cycle transitions.
Another important finding in the current study is that miR-22 inhibits hair follicle stem cell colony formation capacity and hair neogenesis. Interestingly, the effects on HFSCs were also observed for miR-24 and miR-125b. MiR-24 overexpression leads to depletion of HFSCs [27], suggesting a role of miR-24 in promoting HFSC commitment and loss of self-renewal. Thus, while these microRNAs are important in regulating HFSC self-renewal, they likely have unique functions.
It is well known that catagen is an apoptosis-driven physiological involution. A variety of signaling pathways inducing apoptosis have been identified in the process of hair regression during catagen [16]. These regulators include c-Kit, Bcl-2, hairless, survivin, Bax, p53, Dkk1, and transforming growth factors, TGF-1 and TGF-2 [16]. In this study we show that miR-22 promote apoptosis in hair follicles. Several negative regulators of apoptosis were downregulated in the miR-22 overexpressing mice including ADAMTS20, Foxc1, GCLC, MITF, SGK3, Msx2, Vnn1. Among them, Foxc1, MITF, and SGK3 contain miR-22 binding sites. miR-22 might directly repress these transcripts to activate apoptotic signaling pathways. Thus, our findings suggest that a post-transcriptional mechanism regulating apoptosis might be involved in hair regression.
Interestingly, it has recently been reported that miR-22 is strongly induced in response to testosterone treatment [42,43]. Male pattern baldness is due to the premature transition from anagen-to-catagen induced by androgens [3]. Thus, this raises the hypothesis that miR-22 might function in the pathogenesis of Androgenic alopecia (AGA).
Materials and Methods
Ethics statement
This study was performed in strict accordance with the recommendations in the Regulations of Beijing Laboratory Animal Management. The protocol was approved by the Institutional Animal Care and Use Committee of China Agricultural University (approval number: SKLAB-2011-04-03).
Mice
To generate TRE-miR-22 mice, the mmu-miR-22 coding sequence was amplified by PCR from mouse genomic DNA (Forward Primer: 5’ - GTGGATCCGCTAGA GCCAGGTTTG - 3’ and Reverse primer: 5’ - GCTCTAGAATTTCTTCCCACTGCC -3’), cloned into BamH I/XbaI sites of the pTRE2 plasmid (Clontech) to generate pTRE-miR-22 construct. The TRE-miR-22 transgenic mice were generated using standard protocols. K14-rtTA transgenic mice (#007678) and miR-22-/- mice (#018155) were purchased from the Jackson Laboratory and backcrossed into the FVB strain.
Microarray and data analysis
For mRNA microarrays, total RNA samples of dorsal skin were extracted from P28 DTG and control mice (Dox since P21) using TRIzol reagent (Invitrogen). Then the RNA samples were submitted to the Beijing CapitalBio Corporation and analyzed on Affymetrix mouse 430_2 arrays. The data were analyzed using Significant Analysis of Microarray (SAM) software to identify the differentially expressed genes. Differentially expressed transcripts were defined by a fold-change (FC) ≥1.5 and a q-value <5%. The transcriptome profiles of catagen and telogen are published in [32]. The differentially expressed genes were identified with a significance P value < 0.05 and an FC of 1.5. We utilized DAVID Functional Annotation Bioinformatics Microarray Analysis software to analyze gene ontology (GO) based on the KEGG pathway database.
Histology, immunochemistry and immunofluorescence
Immunochemistry and immunofluorescence were performed as described previously [44]. Skin samples were fixed in 4% PFA, paraffin-embedded 5-μm sections were stained with hematoxylin and eosin (H&E). For immunofluorescence staining, paraffin sections were microwave pretreated, and incubated with primary antibodies, then incubated with secondary antibodies (Invitrogen) and counterstained with DAPI in mounting media. The following antibodies and dilutions were used: K14 (rabbit, 1 : 1000, Covance), K14 (mouse, 1 : 500, Abcam),K5 (rabbit, 1 : 1000, Covance), Sox9 (rabbit, 1 : 100, Santa Cruz), AE13(mouse, 1 : 100, Abcam), AE15 (mouse, 1 : 100, Abcam), BrdU (rat, 1 : 200, Abcam), Cleaved Caspase-3 (rabbit, 1 : 100, Cell Signaling), Gata-3 (mouse, 1 : 100, Santa Cruz), K17 (rabbit, 1 : 100, Abcam), Foxn1 (mouse, 1 : 100, Santa Cruz), Dlx3 (rabbit, 1 : 100, Santa Cruz), Sostdc1 (rabbit, 1 : 100, Abcam), Hoxc13 (mouse, 1 : 100, Sigma), Lef1 (rabbit, 1 : 100, Cell Signaling), Ki67 (mouse, 1 : 100,Novacastra), Bmpr1a (rabbit, 1 : 100, Abcam), Msx2 (rabbit, 1 : 200, Santa Cruz), k15 (mouse, 1 : 150, Vector Labs), CD49f (rat,1 : 100,BD), p63 (mouse, 1 : 200, Santa Cruz), K10 (rabbit, 1 : 1000, Covance), Loricrin (rabbit, 1 : 500, Covance), K1 (rabbit, 1 : 500, Covance).
Wounding and whole-mount hair follicle neogenesis assay
The wounding and wound-induced hair neogenesis analyses were performed as described [45]. The mice were anaesthetized with ketamine/xylazine. A 1 cm2 full thickness excision of skin was made on the mid back of 3 week-old mice. To detect newly developing hair follicles in the wound, we incubated the skin in 20 mM EDTA in PBS at 37°C overnight. Then, we gently peeled the epidermal portion off under a dissecting microscope, fixed the epidermis in 4% PFA for 1 h, rinsed with PBS, blocked with 3% H2O2, and performed immunostaining for KRT17 in 1.5 ml eppendorf tubes. We fixed the dermis in acetone at 4°C overnight, rinsed it with PBS, and then incubated with NBT/BCIP (Roche) for 30 min at 37°C. We stopped the reaction with 20 mM EDTA in PBS.
FACS
Subcutaneous fat was removed by a scalpel and the whole skin was placed dermis side down in Trypsin (GIBCO) at 4°C O/N. Single-cell suspensions were obtained by scraping the skin to remove the epidermis and HFs from the dermis. The cells were washed with PBS containing 5% of fetal bovine serum (FBS), then filtered through 70 μm cell strainers, followed by 40 μm. Cell suspensions were incubated with the appropriate antibodies for 90 min on ice. The following antibodies were used: Alpha6-PE (1 : 500, eBioscience), CD34-eFluor660 (1 : 100, eBioscience). DAPI was used to exclude dead cells. Cell isolations were performed on MoFlo XDP sorters equipped with Summit 5.2 software (Beckman Coulter).
Colony-forming assay
The colony-forming assay was performed according to previously reported method [46]. 3,000 viable cells per well were plated onto mitomycin-treated 3T3 fibroblasts in E-media supplemented with 15% serum and 0.3 mM calcium using a six-well plate. After 14 days in vitro, to visualize colony number and morphology, cells were stained with 1% Rhodamine B (Sigma). The growth medium was changed every second day. To induce terminal differentiation, serum was reduced to 5%, and calcium was raised to 1.5 mM.
miR-22 in situ hybridizations
miR-22 in situ hybridizations were performed based on the previously described protocol [47]. Double DIG-labeled miR-22 and scrambled LNA probes (Exiqon) were hybridized at 55°C. In situ signals were detected by staining with Anti-Digoxigenin-AP antibody (Roche) and developing using BM purple substrate (Roche).
Quantitative PCR
Total RNA was isolated from dorsal skin using the mirVanaTM RNA Isolation kit following manufacturer’s instructions (Ambion). Each RNA sample was reverse transcribed with the M-MLV Reverse Transcriptase (Sigma) using Oligo (dT) primers. Real-time PCR was performed with the LightCycler 480 SYBR Green I master mix on a LightCycler 480 real-time PCR system (Roche). For microRNA expression, mature miR-22 was quantified using TaqMan microRNA assays according to the manufacturer’s instructions. U6 snRNA was used as an internal control (Applied Biosystems). Q-PCR primers were listed in S4 Table.
Dual luciferase activity assays
To generate reporter constructs for luciferase assays, segments of about 650 bp in length containing predicted miRNA target sites in the 3’UTRs of Dlx3, Sostdc1,Hoxc13 and Foxn1 were cloned into the psiCHECK-2 vector (Promega) between the XhoI or SgfI and NotI sites immediately downstream of the Renilla luciferase gene. To generate reporters with mutant 3’UTRs, six nucleotides (GCAGCT) in the target site complementary to the position 2–7 of the Mir22 seed region were mutated to TAGATC by a QuikChange Site-Directed Mutagenesis kit according to the manufacturer’s protocol (Stratagene).
293T cells were seeded in 96-well plates one day before transfection. Ten nanograms of each reporter construct was co-transfected with miR-22 mimic or a negative control at a final concentration of 50 nM into 293T cells using Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). After 24h, firefly and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system according to the manufacturer’s instructions (Promega).
The primers used for amplifying 3’-UTR of candidate target genes of miR-22 were listed in S4 Table.
Western blot
P24 Dorsal skin (Dox since P21) was homogenized in ice-cold lysis buffer (Beyotime, China) with 1% PMSF (Phenylmethylsulfonyl fluoride). Samples were centrifuged at 10,000g for 20 min at 4℃ and the protein concentration of the resulting supernatant was determined by the BCA (bicinchoninic acid) protein assay kit (Beyotime, China). Proteins (30μg) were separated by SDS-PAGE electrophoresis and subsequently transferred to PVDF membranes. Membranes were blocked with 5% nonfat dry milk in incubation buffer and incubated with primary antibodies against Tubulin, Dlx3, Sostdc1, Hoxc13 and Foxn1. Bound antibody was detected with peroxidase-linked secondary antibody and a chemiluminescence detection system.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Millar SE (2002) Molecular mechanisms regulating hair follicle development. J Invest Dermatol 118 : 216–225. 11841536
2. Botchkarev VA, Kishimoto J (2003) Molecular control of epithelial-mesenchymal interactions during hair follicle cycling. J Investig Dermatol Symp Proc 8 : 46–55. 12894994
3. Duverger O, Morasso MI (2014) To grow or not to grow: hair morphogenesis and human genetic hair disorders. Semin Cell Dev Biol 25–26 : 22–33. doi: 10.1016/j.semcdb.2014.06.001 24977333
4. Fiuraskova M, Kucerova R, Kolar Z (2003) Pathobiology of androgenetic alopecia. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 147 : 37–41. 15034603
5. Muller-Rover S, Handjiski B, van der Veen C, Eichmuller S, Foitzik K, et al. (2001) A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol 117 : 3–15. 11442744
6. Morgan BA (2014) The dermal papilla: an instructive niche for epithelial stem and progenitor cells in development and regeneration of the hair follicle. Cold Spring Harb Perspect Med 4: a015180. doi: 10.1101/cshperspect.a015180 24985131
7. Lee J, Tumbar T (2012) Hairy tale of signaling in hair follicle development and cycling. Semin Cell Dev Biol 23 : 906–916. doi: 10.1016/j.semcdb.2012.08.003 22939761
8. Kaufman CK, Zhou P, Pasolli HA, Rendl M, Bolotin D, et al. (2003) GATA-3: an unexpected regulator of cell lineage determination in skin. Genes Dev 17 : 2108–2122. 12923059
9. Ellis T, Gambardella L, Horcher M, Tschanz S, Capol J, et al. (2001) The transcriptional repressor CDP (Cutl1) is essential for epithelial cell differentiation of the lung and the hair follicle. Genes Dev 15 : 2307–2319. 11544187
10. Zhou P, Byrne C, Jacobs J, Fuchs E (1995) Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev 9 : 700–713. 7537238
11. Hwang J, Mehrani T, Millar SE, Morasso MI (2008) Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development 135 : 3149–3159. doi: 10.1242/dev.022202 18684741
12. Ma L, Liu J, Wu T, Plikus M, Jiang TX, et al. (2003) 'Cyclic alopecia' in Msx2 mutants: defects in hair cycling and hair shaft differentiation. Development 130 : 379–389. 12466204
13. Schlake T, Schorpp M, Maul-Pavicic A, Malashenko AM, Boehm T (2000) Forkhead/winged-helix transcription factor Whn regulates hair keratin gene expression: molecular analysis of the nude skin phenotype. Dev Dyn 217 : 368–376. 10767081
14. Merrill BJ, Gat U, DasGupta R, Fuchs E (2001) Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev 15 : 1688–1705. 11445543
15. Jave-Suarez LF, Winter H, Langbein L, Rogers MA, Schweizer J (2002) HOXC13 is involved in the regulation of human hair keratin gene expression. J Biol Chem 277 : 3718–3726. 11714694
16. Botchkareva NV, Ahluwalia G, Shander D (2006) Apoptosis in the hair follicle. J Invest Dermatol 126 : 258–264. 16418734
17. Lindner G, Botchkarev VA, Botchkareva NV, Ling G, van der Veen C, et al. (1997) Analysis of apoptosis during hair follicle regression (catagen). Am J Pathol 151 : 1601–1617. 9403711
18. Botchkarev VA, Komarova EA, Siebenhaar F, Botchkareva NV, Sharov AA, et al. (2001) p53 Involvement in the control of murine hair follicle regression. Am J Pathol 158 : 1913–1919. 11395365
19. Botchkarev VA, Botchkareva NV, Albers KM, Chen LH, Welker P, et al. (2000) A role for p75 neurotrophin receptor in the control of apoptosis-driven hair follicle regression. FASEB J 14 : 1931–1942. 11023977
20. Inui S, Fukuzato Y, Nakajima T, Yoshikawa K, Itami S (2003) Identification of androgen-inducible TGF-beta1 derived from dermal papilla cells as a key mediator in androgenetic alopecia. J Investig Dermatol Symp Proc 8 : 69–71. 12894997
21. Hibino T, Nishiyama T (2004) Role of TGF-beta2 in the human hair cycle. J Dermatol Sci 35 : 9–18. 15194142
22. Botchkareva NV (2012) MicroRNA/mRNA regulatory networks in the control of skin development and regeneration. Cell Cycle 11 : 468–474. doi: 10.4161/cc.11.3.19058 22262186
23. Andl T, Murchison EP, Liu F, Zhang Y, Yunta-Gonzalez M, et al. (2006) The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr Biol 16 : 1041–1049. 16682203
24. Mardaryev AN, Ahmed MI, Vlahov NV, Fessing MY, Gill JH, et al. (2010) Micro-RNA-31 controls hair cycle-associated changes in gene expression programs of the skin and hair follicle. FASEB J 24 : 3869–3881. doi: 10.1096/fj.10-160663 20522784
25. Teta M, Choi YS, Okegbe T, Wong G, Tam OH, et al. (2012) Inducible deletion of epidermal Dicer and Drosha reveals multiple functions for miRNAs in postnatal skin. Development 139 : 1405–1416. doi: 10.1242/dev.070920 22434867
26. Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, et al. (2009) DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci U S A 106 : 498–502. doi: 10.1073/pnas.0810766105 19114655
27. Amelio I, Lena AM, Bonanno E, Melino G, Candi E (2013) miR-24 affects hair follicle morphogenesis targeting Tcf-3. Cell Death Dis 4: e922. doi: 10.1038/cddis.2013.426 24232098
28. Zhang L, Stokes N, Polak L, Fuchs E (2011) Specific microRNAs are preferentially expressed by skin stem cells to balance self-renewal and early lineage commitment. Cell Stem Cell 8 : 294–308. doi: 10.1016/j.stem.2011.01.014 21362569
29. Wang D, Zhang Z, O'Loughlin E, Wang L, Fan X, et al. (2013) MicroRNA-205 controls neonatal expansion of skin stem cells by modulating the PI(3)K pathway. Nat Cell Biol 15 : 1153–1163. doi: 10.1038/ncb2827 23974039
30. Keyes BE, Segal JP, Heller E, Lien WH, Chang CY, et al. (2013) Nfatc1 orchestrates aging in hair follicle stem cells. Proc Natl Acad Sci U S A 110: E4950–4959. doi: 10.1073/pnas.1320301110 24282298
31. Zhang Y, Yu J, Shi C, Huang Y, Wang Y, et al. (2013) Lef1 contributes to the differentiation of bulge stem cells by nuclear translocation and cross-talk with the Notch signaling pathway. Int J Med Sci 10 : 738–746. doi: 10.7150/ijms.5693 23630438
32. Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, et al. (2009) Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet 5: e1000573. doi: 10.1371/journal.pgen.1000573 19629164
33. Hoshino Y, Katsuno Y, Ehata S, Miyazono K (2011) Autocrine TGF-beta protects breast cancer cells from apoptosis through reduction of BH3-only protein, Bim. J Biochem 149 : 55–65. doi: 10.1093/jb/mvq114 20880961
34. Wang Y, Zhou D, Phung S, Masri S, Smith D, et al. (2011) SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol 25 : 72–82. doi: 10.1210/me.2010-0294 21084382
35. Chen YS, Lee SM, Lin CC, Liu CY (2014) Hispolon decreases melanin production and induces apoptosis in melanoma cells through the downregulation of tyrosinase and microphthalmia-associated transcription factor (MITF) expressions and the activation of caspase-3, -8 and -9. Int J Mol Sci 15 : 1201–1215. doi: 10.3390/ijms15011201 24445257
36. Gomes WA, Kessler JA (2001) Msx-2 and p21 mediate the pro-apoptotic but not the anti-proliferative effects of BMP4 on cultured sympathetic neuroblasts. Dev Biol 237 : 212–221. 11518517
37. Silver DL, Hou L, Somerville R, Young ME, Apte SS, et al. (2008) The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet 4: e1000003. doi: 10.1371/journal.pgen.1000003 18454205
38. Clavel C, Grisanti L, Zemla R, Rezza A, Barros R, et al. (2012) Sox2 in the dermal papilla niche controls hair growth by fine-tuning BMP signaling in differentiating hair shaft progenitors. Dev Cell 23 : 981–994. doi: 10.1016/j.devcel.2012.10.013 23153495
39. Shimomura Y, Christiano AM (2010) Biology and genetics of hair. Annu Rev Genomics Hum Genet 11 : 109–132. doi: 10.1146/annurev-genom-021610-131501 20590427
40. Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T (1994) New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature 372 : 103–107. 7969402
41. Nehls M, Kyewski B, Messerle M, Waldschutz R, Schuddekopf K, et al. (1996) Two genetically separable steps in the differentiation of thymic epithelium. Science 272 : 886–889. 8629026
42. Delic D, Grosser C, Dkhil M, Al-Quraishy S, Wunderlich F (2010) Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids 75 : 998–1004. doi: 10.1016/j.steroids.2010.06.010 20600201
43. Wang WL, Chatterjee N, Chittur SV, Welsh J, Tenniswood MP (2011) Effects of 1alpha,25 dihydroxyvitamin D3 and testosterone on miRNA and mRNA expression in LNCaP cells. Mol Cancer 10 : 58. doi: 10.1186/1476-4598-10-58 21592394
44. Yu Z, Lin KK, Bhandari A, Spencer JA, Xu X, et al. (2006) The Grainyhead-like epithelial transactivator Get-1/Grhl3 regulates epidermal terminal differentiation and interacts functionally with LMO4. Dev Biol 299 : 122–136. 16949565
45. Ito M, Yang Z, Andl T, Cui C, Kim N, et al. (2007) Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 447 : 316–320. 17507982
46. Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, et al. (2008) Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet 40 : 1291–1299. doi: 10.1038/ng.239 18849992
47. Jorgensen S, Baker A, Moller S, Nielsen BS (2010) Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods 52 : 375–381. doi: 10.1016/j.ymeth.2010.07.002 20621190
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy