
Characterization of TCF21 Downstream Target Regions Identifies a Transcriptional Network Linking Multiple Independent Coronary Artery Disease Loci
While coronary artery disease (CAD) is due in part to environmental and metabolic factors, about half of the risk is genetically predetermined. Genome-wide association studies in human populations have identified approximately 150 sites in the genome that appear to be associated with CAD. The mechanisms by which mutations in these regions are responsible for predisposition to CAD remain largely unknown. To begin to explore how disease-specific gene sequences and disease gene function promotes pathology, we have mapped the loci and genes that are downstream of the transcription factor TCF21, which is strongly associated with CAD. By identifying genes that are regulated by TCF21 we have been able to link together multiple other CAD associated genes and begin to identify the critical molecular processes that mediate atherosclerosis in the blood vessel wall and contribute to the genesis of ischemic cardiovascular events.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005202
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005202
Summary
While coronary artery disease (CAD) is due in part to environmental and metabolic factors, about half of the risk is genetically predetermined. Genome-wide association studies in human populations have identified approximately 150 sites in the genome that appear to be associated with CAD. The mechanisms by which mutations in these regions are responsible for predisposition to CAD remain largely unknown. To begin to explore how disease-specific gene sequences and disease gene function promotes pathology, we have mapped the loci and genes that are downstream of the transcription factor TCF21, which is strongly associated with CAD. By identifying genes that are regulated by TCF21 we have been able to link together multiple other CAD associated genes and begin to identify the critical molecular processes that mediate atherosclerosis in the blood vessel wall and contribute to the genesis of ischemic cardiovascular events.
Introduction
Recent large-scale GWAS have identified 46 genome-wide significant CAD loci and a further 104 independent variants associated at a 5% false discovery rate (FDR), yet the biological and disease-relevant mechanisms for these associations remain largely unknown [1]. It is estimated that at least two-thirds of the disease loci contain causal genes that are not related to known cardiovascular risk factors such as diabetes and lipid metabolism, suggesting that they are involved in disease promoting processes in the blood vessel wall. Thus the great promise of these genetic findings is the elucidation of atherosclerosis disease pathways, and further investigation of mechanisms by which genes in disease loci work together to regulate cellular and molecular functions that are involved in disease risk is sorely needed. Among the significant loci, a small subset of genes encode transcription factors (TFs) which are likely to impact disease risk by regulating disease relevant genes and possibly other CAD associated genes. Further study of downstream targets of these TFs, employing well established genome-wide methods, would be expected to provide biological insights through links to established pathways and to identify informative relationships among other apparently independent causal CAD loci.
Tcf21 is a member of the basic helix-loop-helix (bHLH) TF family and is critical for the development of a number of cell types during embryogenesis of the heart, lung, kidney, and spleen [2–5]. Tcf21 is expressed in mesodermal cells in the proepicardial organ that give rise to coronary artery smooth muscle cells (SMC) and loss of Tcf21 results in increased expression of smooth muscle markers by cells on the heart surface consistent with premature SMC differentiation [6]. Knockout animals also exhibit a dramatic failure of cardiac fibroblast development suggesting a role for Tcf21 in the fate decisions of a precursor cell for SMC and cardiac fibroblast lineages [2,6]. These data are consistent with the hypothesis that early expression of Tcf21 is important for expansion of the SMC compartment of the coronary circulation, with persistent Tcf21 expression being required for cardiac fibroblast development [2,6].
To better understand the cellular functions of TCF21 in the SMC lineage, and to gain insights into how such functions might contribute to CAD risk, we performed chromatin immunoprecipitation coupled with high throughput sequencing (ChIP-Seq), examined the downstream target loci and genes that harbor TCF21 binding sites, and employed bioinformatic and experimental approaches to investigate how the target genes work together to mediate the risk of CAD. Pathway analysis of downstream target genes revealed that TCF21 regulates cell-cell and cell-matrix interactions as well as growth factor signaling pathways. Also, we found TCF21 target regions to be over-represented among CAD associated loci, and that genes in these regions assemble into pathways that mediate fundamental processes such as cell cycle, chromatin remodeling, and growth factor signaling. Taken together, these studies elucidate disease-associated genes and pathways that lie downstream of TCF21, and show how SMC related processes may be responsible for a substantial portion of the genetic risk for CAD.
Results
ChIP-Seq studies identify TCF21 core target loci and genes
Our primary interest in these studies was the transcriptional network of TCF21-regulated genes contributing to the development of human CAD. Because of the known role of TCF21 in the embryonic development of coronary vascular SMC, we undertook these ChIP-Seq experiments in primary cultured human coronary artery SMC (HCASMC). Furthermore, we selected culture conditions that maintain these cells in the synthetic, undifferentiated state that most closely reflects the disease phenotype [7]. Two polyclonal antibodies raised against peptides representing different epitopes of TCF21 and previously validated by the manufacturers were employed in these studies. ChIP-Seq was performed with both antibodies (Ab1 and Ab2), with two replicates per antibody and an IgG control condition. We then followed best practices for computational analysis of sequence data as put forth by the ENCODE project, including genome alignment, peak calling, and replicate consolidation using the Irreproducible Discovery Rate (IDR) method to identify high confidence peaks for each antibody [8].
ChIP-Seq using Ab1 identified 10,523 peaks while Ab2 identified 4,900 peaks that largely overlapped with those identified by Ab1. These two sets of peaks were within 50 kb of 12,226 and 7,150 genes, respectively (Table 1). To better understand the disparity between the numbers of DNA regions immunoprecipitated by the two antibodies, we characterized the distribution and relationship between the two sets of peaks identified. The spatial distribution of each peak set was investigated by graphing the distance between peaks and the transcription start site of the nearest gene (S1A and S1B Fig). These distributions were nearly identical for the two peak sets. In each case, peaks were distributed primarily within 100 kb of the transcription start sites, with 90% of peaks being found within this interval. The similarity between antibody peak localization was further demonstrated by relating the peak coordinates to structural gene features (S1C and S1D Fig). The pattern of distribution for peaks associated with both antibodies revealed the majority of binding sites were located within intronic and intergenic regions, with a significant number of peaks also being found within the promoter and exonic regions and a very small number of peaks mapping to transcript untranslated sequences.

Next, we investigated the overlap in genomic regions represented by peaks associated with each antibody precipitation. Results of this analysis also revealed a high degree of overlap between the two peak sets, with all but 72 of the 4900 peaks identified by Ab2 sharing one or more basepairs with peaks identified by Ab1 (Fig 1A). Visualization of peaks with the IGV browser provided further evidence of extensive overlap of peaks, although Ab2 frequently showed decreased peak size compared to Ab1 (Fig 1C). Shown here are TCF21 peak regions in three genes that have been identified as replicated CAD GWAS loci, IL6R, SH2B3, and SMG6. Due to this overlap, as well as the similarities in peak binding patterns described above, we intersected the two datasets to refine the number of peaks to those identified by both antibodies (Ab_shared, Fig 1B) and, unless otherwise noted, employed this data set for the analyses presented below.

ChIP-Seq peaks are located in regions of open chromatin and identify genes differentially regulated by TCF21
A number of approaches were employed to validate results obtained with ChIP-Seq. First, we investigated the overlap between TCF21 peaks and regions of open chromatin as defined for HCASMC by the assay of transposase accessible chromatin high-throughput sequencing (ATAC-Seq) performed in this laboratory [9] and with ENCODE data for human aortic smooth muscle cells (HAoSMC) as identified by DNase hypersensitivity assay. We found that the TCF21 ChIP-Seq peaks significantly overlap with ATAC-Seq signals (P<1.0e-300, fold enrichment = 1.85; S1 Table and Fig 1C). Similarly, the TCF21 ChIP-Seq peaks also significantly overlap with the HAoSMC DHS signals (P<1.0e-300, fold enrichment = 4.29; S1 Table and Fig 1C). Second, we performed technical replication with ChIP employing separately isolated chromatin from HCASMC derived from a different donor, with PCR primers flanking a number of TCF21 peaks. In these studies, the chosen genes showed 4 - to 90-fold enrichment compared to a select non-target region (S2 Fig).
To investigate whether genes with binding peaks are directly regulated by TCF21, we took advantage of existing co-expression networks to investigate overlap between ChIP-Seq identified target genes and those genes that track with TCF21 gene expression. We retrieved 2705 co-expression modules, each containing highly co-regulated genes, derived from 108 coexpression networks constructed from many different tissues in human and mouse populations [10–19]. We then performed enrichment analysis between the co-expression modules and genes associated with Ab_shared peaks using Fisher’s exact test. The target genes tended to form coexpression modules, and 296 such modules from 10 coexpression networks were significantly enriched with TCF21 target genes at FDR<0.01 (S2 Table). Vascular endothelial cell and vascular disease related adipose tissue coexpression networks were most strongly associated with TCF21 target genes [20]. Taken together, these data provide evidence that expression of target genes is highly coordinated by TCF21 and that identified peaks functionally regulate target gene expression.
Pathway analyses suggest that TCF21 regulates growth factor signaling as well as cell-cell and cell-matrix interactions
To investigate the molecular and cellular processes downstream of TCF21 and possible mechanisms of disease association, studies were conducted to look for over-representation of TCF21 target genes among well annotated regulatory pathways. Here, we analyzed the common peak set (Ab_shared) with the Genomic Regions Enrichment of Annotations Tool (GREAT) [21]. GREAT assigned genes to peaks and queried a number of functional databases with the resulting gene list (Table 2). Evaluation of gene ontology (GO) Molecular Function, GO Biological Process, PANTHER Pathway, and Pathway Commons databases identified terms related to growth factor signaling (“platelet growth factor (PDGF) receptor binding/signaling,” “vascular endothelial cell growth factor (VEGF) signaling”), cell-matrix interactions (“integrin binding,” “cell adhesion”), matrix biology (“extracellular matrix structural constituent”), actin contractile function (“actin filament-based processes,” “actin cytoskeleton”). Mouse phenotype database terms included “abnormal cardiovascular system physiology”, “abnormal blood circulation”, and “abnormal blood vessel morphology”. Importantly, MSigDB Predicted Promoter Motifs ontology identified enrichment among the TCF21 target genes for those with JUN family member binding sites in their promoter regions.

TCF21 peaks contain the CAGCTG E-box motif as well as an activator protein-1 (AP-1)-like motif that mediates JUN factor binding
The availability of sequence information across a large number of TF binding sites allowed identification of the canonical binding sequence for TCF21. We employed the HOMER and MEME-ChIP algorithms for this task, investigating de novo TF motif enrichment within TCF21 peaks shared between Ab1 and Ab2 [22,23]. This analysis identified the nucleotide sequence CAGCTG in 67.2% of peaks (P = 1e-1010) (Fig 2A). This sequence matched the CANNTG sequence that is the common E-box binding motif used by bHLH factors, and is identical to the E-box motif that is known to mediate binding of bHLH partners of TCF21, including TCF12 [24]. An additional E-box motif (CATCTG) was found in 66.7% of peaks (P = 1e-627), and identified as identical to the motif recognized by bHLH factor Olig2, likely representing a second motif that is recognized by TCF21. Interestingly, an additional enriched TF binding motif was also identified in approximately 30% of peaks, corresponding to the bZIP motif TGA(G/C)TCA (P = 1e-336) that is known to bind the AP-1 family of TFs. Other motifs of interest included those that mediate binding of TEAD and CEBP transcription factor families. Graphing the distribution of these motifs in comparison to the summits of TCF21 peaks suggested that AP-1 and possibly ATF1 factors bind in a bimodal pattern flanking TCF21, suggesting a possible steric binding relationship between TCF21 and these bZip factors (Fig 2B). These motifs likely mediate binding of TFs that cooperate with TCF21 to direct transcriptional programs associated with target genes, as has been characterized for other TFs [25].

Follow-up ChIP-Seq studies verify over-representation of JUN family member binding at TCF21 target genes
We have previously shown that JUN and other AP-1-related transcription factors transactivate TCF21, and disruption of this pathway by disease-associated allelic variation in the related binding site may account in part for the CAD susceptibility observed at this locus [27]. To explore whether JUN factors may also bind in association with TCF21 to co-regulate target genes in arterial smooth muscle cells, we performed ChIP-Seq in HCASMC for JUN and JUND. Samples were processed, sequences aligned to the genome, and peaks called with the same algorithms as for the TCF21 experiments. We quantified the overlap of TCF21 and JUN/JUND binding regions against a background of all regions of open chromatin, with the analyses employing both ATAC-Seq study of HCASMC and DHS study of HAoSMC to define this background. For the analysis with ATAC-Seq regions we found significant overlap of TCF21 with JUN and JUND binding sites (P<4.12e-215, fold enrichment = 2.84), and employing the same methods with HAoSMC DHS regions as background, TCF21 overlap with JUN and JUND peaks remained significant (P<1.79e-183, fold enrichment = 2.21).
Example genomic regions with overlap of TCF21, JUN and JUND binding are shown for the developmental WT1 gene and the developmental growth factor PDGFRB gene (Fig 2C). This and other labs have shown that WT1 regulates TCF21 [27,28], but TCF21 binding in the WT1 locus as demonstrated here is novel and provides support for a bidirectional regulatory interaction between these important developmental factors. Collectively, these analyses reveal common genome-wide binding patterns between TCF21, JUN and JUND, providing strong evidence for the coordinated binding of TCF21 and JUN family members in HCASMC. Taken together with previously published data showing that JUN factors are upstream regulators of TCF21 transcription, these results suggest a compelling functional link between these TF pathways.
Functional SNPs mapped to genes targeted by TCF21 are significantly enriched among CAD GWAS SNPs
To look for more functional relationships between TCF21 target gene SNPs and those associated with other CAD genes, further analyses were conducted employing regulatory SNPs (eQTLS) which have been identified through studies in a variety of tissues investigating the genetic basis of gene expression [11,16,29–31]. We retrieved eQTLs from liver, brain, blood, human aortic endothelial cells (HAEC), adipose tissues and collected all the eQTLs/functional SNPs mapped to specific target genes [11–16,29,31–33]. eQTLs/functional SNPs mapped to genes targeted by TCF21 were significantly enriched among SNPs with low CARDIoGRAM GWAS P-values (P<0.01) (fold enrichment 1.78 to 2.42, P = 1.24e-12 to 3.69e-95 in all eSNP sets tested; Table 3). Additionally, we obtained all the functional SNPs from RegulomeDB (http://www.regulomedb.org/, based on ENCODE) and evaluated them in the context of their functional annotation. Functional SNPs for TCF21 target genes as defined by the RegulomeDB Category I (i.e., SNPs with highest level of evidence that they have functional influence on genes) showed the highest fold enrichment for SNPs with CAD GWAS P-values < 0.01 (Category I fold enrichment 2.06; P = 1.04e-155). Category II SNPs (SNPs with less functional evidence than Category I) also showed highly significant enrichment for low P-values (fold enrichment 1.42; P = 2.60e-40) (Table 3). All analyses were controlled for LD, with SNPs possessing r2>0.3 removed.

TCF21 target genes show significant over-representation among GWAS genes associated with CAD
We noted previously that a number of the CAD loci that have reached genome wide significance contain TCF21 peaks (Fig 1C), and reasoned that since TCF21 is a transcription factor one mechanism for its disease association might be through regulation of these other CAD loci. We were thus interested to perform an enrichment analysis with GWAS data to test the hypothesis that TCF21 affects CAD by modulating a larger than expected number of CAD-related genes. Significant enrichment of TCF21 targets among CAD loci would support this hypothesis. To investigate this possibility, we took two complementary approaches to look for over-representation of TCF21 binding regions among CAD GWAS loci. The first approach was based on gene-level overlap by assessing enrichment of TCF21 target genes among candidate genes in the CHD GWAS loci. The second approach was based on SNP-level linkage information by evaluating whether the average linkage disequilibrium (LD) between TCF21 peak SNPs and CAD GWAS loci SNPs is greater than expected by chance. To test the specificity of TCF21 target regions to CAD, we also included additional phenotypes for comparison. The traits/phenotypes that we investigated with both analyses included: i) coronary artery disease phenotypes, ii) risk factors that are known to be associated with CAD, iii) non-atherosclerotic vascular diseases that are not specifically associated with CAD, iv) primarily inflammatory disease phenotypes that are known to involve molecular pathways that are also linked to CAD, v) disease phenotypes related to tissues where TCF21 is known to not be expressed and which were predicted negative controls. Also, we focused analyses on traits with the largest number of associated variants, with the goal of strengthening the statistical analysis, and primarily employed traits/phenotypes with at least 20 associated variants. Phenotypes or traits that passed P<0.05 from both methods were deemed significant, yielding a combined cutoff of P<0.0025. Considering ~20 disease sets were tested, this combined statistical cutoff is equivalent to a Bonferroni-corrected P<0.05.
In the first analysis, we investigated over-representation of TCF21 binding region genes among CAD locus genes. As a preliminary analysis, to test for possible confounding, we tested whether TCF21 binding is by chance more likely to be near genes mapped for various GWAS phenotypes by running an enrichment analysis between genes linked to TCF21 binding sites and all GWAS genes from the GWAS Catalog [34]. As shown in S3 Table, although there is statistically significant over-representation of Ab2 and Ab_Shared binding site genes among CAD GWAS genes, the fold enrichments are all close to 1, indicating very minor enrichment of overall GWAS signals among the TCF21 targets. We thus assigned genes to TCF21 peaks employing a distance metric of 50 kb (S4 Table), compiled a list of candidate genes for each phenotype/trait, and tested for enrichment of TCF21 target genes among disease/trait candidate genes. Enrichment analyses for chosen phenotypes/traits were conducted using all GWAS genes as background to correct for the slight over-representation of GWAS genes among those that are TCF21 targets. In addition, to correct for any potential bias in the large numbers of GWAS genes for certain traits such as height and CAD, we implemented a permutation strategy by generating 1000 random GWAS gene sets of matching size for each trait to derive permutation-based enrichment P-values. The methodology for this permutation-based analysis is provided in the Methods section.
Employing genes in the GWAS catalog associated with CAD phenotypes, enrichment was found for TCF21 target genes among CAD genes compared to a background of all GWAS genes (CAD, 1.34-fold enrichment, permutation P = 0.014) and these results did not change substantially with exclusion of lipid trait genes (CAD no lipid, 1.34-fold enrichment, permutation P = 0.03) (Table 4). When CARDIoGRAM+C4D data was included in the analysis (CAD extended) the fold enrichment increased to 1.51 (permutation P<1.0e-03) and again this did not change substantially with removal of lipid trait variation (CAD extended no lipid, 1.53-fold enrichment, permutation P<1.0e-03). We also found that the candidate sets of GWAS genes associated with the CAD related trait platelet number and a disease phenotype related to a dysfunctional immune system, inflammatory bowel disease (IBD), also showed a high degree of enrichment for TCF21 target genes among the GWAS gene sets. At Bonferroni corrected P<0.05 (raw P<0.0022) in this test, height, CAD extended, CAD extended no lipid, IBD, and platelet phenotypes reached statistical significance. Importantly, we found little evidence of enrichment of TCF21 target genes among GWAS candidate genes for risk factors blood pressure, lipids, and glucometabolic related traits.

In a second type of analysis conducted at the SNP level, we investigated whether common variants in regions targeted by TCF21 binding tend to demonstrate higher LD with SNPs associated with CAD by GWAS. Such a link would provide additional evidence for the involvement of TCF21 in the genetic pathways that contribute to CAD risk and serve as a complementary approach to the gene-based analysis described above. After pruning SNPs associated with both the CAD loci and TCF21 peaks for LD, we investigated whether SNPs in the TCF21 binding sites were in higher than expected LD with CAD-associated genetic variants compared to random SNPs. A similar analysis was done for other GWAS phenotypes to test the specificity to CAD. For each trait-associated SNP set, permutation analysis was utilized to generate distributions of average r2 using 10,000 random sets of TCF21-GWAS SNP pairs, and statistical significance was assigned to those categories where fewer than 5% of permutations produced an average r2 greater than or equal to the true data. Results from this analysis showed that SNPs in TCF21 peaks have significantly greater LD than expected by chance with SNPs for CAD related phenotypes: CAD (permutation P = 0.0209) and for CAD Extended (permutation P = 0.0086) that analyzed GWAS SNPs plus those from CARDIoGRAM+C4D (Table 5 and Fig 3). With these analyses, the CAD categories without lipid variants were marginally more significant than CAD categories including lipid loci. Interestingly, the greatest enrichment was found for non-CAD phenotypes, including height and IBD as found for the gene enrichment analysis, as well as schizophrenia. With Bonferroni correction for multiple testing these three non-CAD phenotypes reached statistical significance of P<0.05. However, when considering the consistent phenotypes between this test and the previous gene level enrichment analysis for GWAS phenotypes, the CAD phenotypes CAD, CAD extended, CAD nolipid, CAD extended nolipid, as well as height, IBD, and platelet number were found to be significant at P<0.05 in both tests, yielding a combined P<0.0025 which is equivalent to Bonferroni-corrected P<0.05.


TCF21 target CAD loci are enriched for genes involved in vascular wall biological processes
To investigate possible functional relationships among the TCF21 target CAD loci, we evaluated the degree of representation of these genes in annotated functional pathways. For this assessment, we used the genes assigned to TCF21 peaks with the DAVID algorithm. We first investigated the pathways that were identified individually for the TCF21 target CAD loci and the full list of CAD loci, quantifying enrichment of genes in loci with those gene ontology (GO) terms annotated for the “biological processes” category. For the TCF21 target CAD genes (57% of the total CAD genes), the top terms that reached significance were disease ontology terms related to migration, including “regulation of cell migration,” “regulation of locomotion,” as well as those related to cell division, “regulation of proliferation,” and metabolism, “regulation of phosphorous metabolic and signaling processes,” regulation of protein amino acid phosphorylation” (S5 Table). Only 3 of the top 20 terms were related to lipids or lipid metabolism. By contrast, when all of the CAD genes were employed in the analysis, there was a preponderance of top terms related to lipid metabolism, “lipid homeostasis,” “cholesterol transport,” and “cholesterol efflux,” with 17 of the top 20 GO terms related to lipids (S6 Table). Also, we performed the analysis with TCF21 target CAD genes being analyzed with the background of all CAD genes (S7 Table). Although this resulted in a decreased number of significant results, this analysis identified cellular traits, including “actin filament based processes,” and “actin cytoskeleton organization,” which represent a smooth muscle signature, and “transcription factor binding,” “intracellular signaling cascade,” and “regulation of DNA binding,” which are all highly suggestive of fundamental cell signaling functions of smooth muscle cells in the vessel wall, possibly related to disease-related processes.
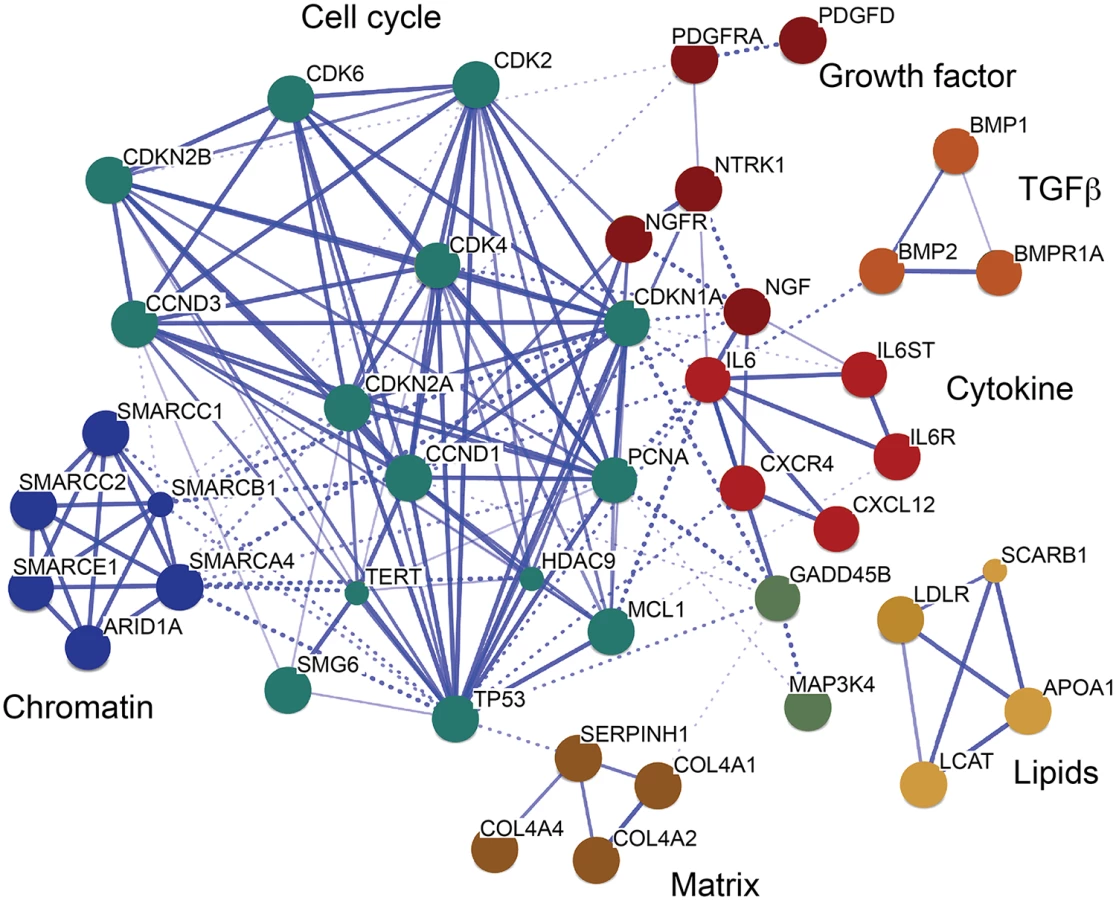
Further, we investigated the interaction of TCF21 peak CAD genes with other genes with the STRING algorithm that integrates protein-protein interaction, text mining and genomic data to link candidate genes into a related network [35]. For this analysis, we employed STRING to generate a network seeded with CHD genes within 50 kb of a TCF21 peak, and then further modified that gene set by including related interacting genes and removing non-connected genes. The resulting network showed clusters of genes associated with cell cycle regulation, extracellular matrix, lipid metabolism, cytokine signaling and growth factor signaling, suggesting that these processes that are relevant to SMC biology are involved in the pathogenesis of CAD (Fig 4).
Discussion
We focused ChIP-Seq experiments on smooth muscle cells based on previous findings demonstrating the role of TCF21 in coronary vascular development during embryogenesis and its relevance for the CAD phenotype. During development, TCF21 is expressed in SMC progenitors but not in endothelial or other cell types present in vascular disease lesions [2,6]. Vascular SMC response to growth factor signaling is increasingly recognized as an important component of the vascular response to injury where atherosclerotic disease progression is characterized by SMC undergoing phenotypic modulation, changing from a quiescent cell expressing contractile lineage markers to a proliferative, migratory, matrix synthesizing cell that is characteristic of the disease state [7]. In order to better understand the role of TCF21 in CAD progression, we utilized an in vitro model of undifferentiated SMC phenotype based on serum stimulation for the studies presented here [36]. Analysis of TCF21 peaks revealed enrichment for a number of pathways related to the known vascular developmental role of TCF21, as well as growth factor stimulation and signaling, cell-matrix interactions, matrix composition, and actomyosin contractile function. The query of relevant mouse databases revealed enrichment within terms related to vascular phenotypes, including “abnormal blood vessel morphology” and “abnormal blood circulation.” These data suggest that TCF21 directly regulates genes centrally involved in pathways that mediate pathologic SMC processes. Given the association of TCF21 with CAD, our findings thus provide novel evidence supporting the role of SMC in the etiology of disease processes within the vascular wall.
We provide evidence for cooperation of TCF21 with other transcription factors through the finding of other TF binding motifs in the ChIP-Seq peak sequences. We found evidence for enrichment of JUN (AP-1) and several other bZip factor binding motifs, including CEBP and ATF1, in the TCF21 peaks. There was also enrichment for binding sites recognizing the TEAD family of transcription factors that have been linked to signaling through the Hippo pathway that regulates developmental and cancer related cellular processes [37]. We found the identification of JUN related binding sites particularly interesting, but given that these AP-1 motifs are among those that have been found to be commonly enriched in datasets for which they were not the targeted transcription factor [38], we were compelled to perform ChIP-Seq studies for JUN and JUND in the HCASMC. These studies confirmed the in silico prediction, revealing significant enrichment of binding sites found for both of these TFs in genes that also contain TCF21 peaks. For a number of genes, TCF21 and JUN factors were found to bind in close proximity with evidence for JUN factor binding flanking the TCF21 binding site. As noted above, the pathway analysis of TCF21 target genes has identified enrichment for signaling through growth factor binding pathways, including PDGF. PDGF signaling related to smooth muscle cell proliferation is known to involve JUN TFs [39], and our previous studies have shown that PDGF and JUN are upstream regulators of TCF21 expression [27]. These findings, when coupled with the observation that TCF21 promotes cellular proliferation and dedifferentiation in skeletal muscle cells [40], provide new insight into the relationship between TCF21 and certain growth factor pathways involving AP-1, and suggest that TCF21 is both regulated by and cooperates with this TF family.
These ChIP-Seq studies have identified a number of other CAD associated loci and genes that are bound by TCF21, identifying a disease related transcriptional network. To investigate the hypothesis that TCF21 targets a greater than expected number of CAD-related genes we performed enrichment analysis at both the gene and SNP level with GWAS data. These studies identified a greater than expected number of TCF21 targets among CAD GWAS genes, and showed that TCF21 peak region SNPs are in greater linkage disequilibrium with CAD GWAS SNPS than expected. Since the ChIPseq data was generated in HCASMC, a cell type found in the coronary artery, the results of CAD loci enrichment is consistent with the hypothesis that TCF21 regulates a greater than expected number of CAD genes in the vessel wall and that its effect on disease risk may be due in part to its regulation of the network of TCF21 target CAD associated genes. Consistent with TCF21 CAD target genes being active primarily in the vascular wall, we saw no difference in enrichment between the CAD analyses with and without lipid genes or SNPs being included in the analysis, and no enrichment was found for vascular risk factor traits “LDL cholesterol” and “Total cholesterol.” The enrichment for “Platelets” is not surprising given the known association of platelet number with vascular disease events, and this association could explain some portion of the TCF21 risk for CAD [41–43]. Human genetic epistasis studies are required to validate these putative gene-gene interactions between TCF21 and the other CAD genes in the network, but further study of the functional links among validated TCF21 target CAD genes should provide an opportunity for further dissecting the molecular basis of disease risk.
Surprisingly, both the gene - and SNP-level enrichment GWAS analyses also found TCF21 target genes to be over-represented among those associated with height and immune related IBD. Although SMC would seem unlikely to be involved in the height and IBD phenotypes, we speculate that enrichment of TCF21 target genes within these other phenotypes is real and reflects overlapping biology between the physiology/pathophysiology of these phenotypes and that of CAD. For instance, short stature has been repeatedly linked to an elevated risk for CAD, suggesting the presence of a substantial overlap in the biological pathways that determine height as well as the risk of CAD [44–46]. In support of a causal link between shorter stature and higher risk of CAD, a recent Mendelian Randomization study found a graded relationship between a genetic risk score of 180 height raising alleles identified by the GIANT consortium and the risk of CAD [47,48]. Furthermore, pathway analysis of genes near or within the height loci identified enrichment of several canonical pathways involved in both growth-development and atherosclerosis [48]. It is also not surprising to observe enrichment for immune related pathways that mediate IBD given inflammation has been extensively linked to atherosclerosis at an experimental level [49–51]. These findings are also consistent with our pathway analyses of TCF21 related CAD genes (S7 Table). We note that the genes that contribute to the enrichment in these phenotypes are unlikely to be regulated by TCF21 given TCF21 is not associated with these phenotypes in GWAS. Instead, we suspect this enrichment reflects functional roles of related networks in traits that involve multiple other tissues.
Our analyses provide multiple lines of circumstantial evidence supporting a role for the identified TCF21 related transcriptional network in the etiology of CAD. First, the TCF21 target genes are highly enriched for molecular processes with established relationships to CAD, such as growth factor binding, cell adhesion, vascular development, inflammation, and PDGF signaling. Second, the TCF21 target genes are enriched for established CAD genes identified through GWAS. Third, the functional SNPs of TCF21 target genes are enriched for, and tend to be in significant LD with, SNPs associated with CAD in GWAS. Fourth, the finding that the TCF21 transcriptional network is enriched in other traits is consistent with a biological program that contributes to a number of disease-relevant tissue-specific cellular processes. However, further work is required to firmly establish our hypothesis that the TCF21 transcriptional gene network contributes to CAD risk. If TCF21 binding to other CAD loci is important for the disease risk mechanism, one may be able to identify epistatic effects between polymorphisms in or near TCF21 and other CAD SNPs in the TCF21 target regions. At an experimental level it will be important to link TCF21 binding sites in target loci to function of the causal variant and the causal gene, and to investigate how TCF21 binding may regulate the causal mechanisms at other CAD loci. Finally, it will be important to functionally assess the predicted TCF21 gene network by perturbation in vitro and in vivo with experimental cell culture and animal models. Taken together, these approaches may provide significant new insights into how a disease associated transcription factor may work through other disease-associated loci to modulate disease risk.
Methods
HCASMC culture and TCF21 knockdown
Human primary Coronary Artery Smooth Muscle Cells (HCASMCs, #CC-2583, Lot No. 200212) were purchased from Lonza (Allendale, NJ, USA) and cultured in SmGM-2 Smooth Muscle Growth Medium-2 including hEGF, insulin, hFGF-B and FBS, but without antibiotics (Lonza, #CC-3182) for 3 passages. Technical replicate samples were cultured from the same stock vial but harvested 24h apart. The crosslinked nuclei of all replicates were thereafter processed in parallel.
TCF21 chromatin immunoprecipitation and next generation sequencing (ChIP-Seq)
Ten million HCASMC cells per condition were crosslinked for 10 min with 1% formaldehyde. Cells were lysed for 10 min on ice (10 mM Tris pH8.0, 10 mM NaCl, 0.2% NP-40). Nuclei were then lysed for 10 min on ice (50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS). Crosslinked chromatin was sheared for 3x5 min by sonication using a Bioruptor (Diagenode, Denville, NJ, USA) and pre-cleared with 10 μ g anti-rabbit IgG pre-immune serum (DAKO, Carpintera, CA, USA, #X0903, Lot. No. 25509) using protein G sepharose beads (Roche Diagnostics, Indianapolis, IN, USA, #11 243 233 001). TCF21 ChIP was performed overnight in IP dilution buffer (20 mM Tris pH 8.1, 2 mM EDTA, 150 mM NaCl, 1% Triton X100, 0.01% SDS), using 14 μ g anti-TCF21 (Sigma-Aldrich, St. Louis, MA, USA, #HPA013189, Lot. No. R02939 (Ab1) or Abcam, Cambridge, UK, #ab49475, Lot No. 153899 (Ab2)) or anti-rabbit IgG respectively. JUN and JUND ChIP was performed with Santa Cruz Biotechnology sc-1694 and sc-74 respectively. Beads were washed twice with buffer 1 (20 mM Tris pH 8.1, 2 mM EDTA, 50 mM NaCl, 1% Triton X100, 0.1% SDS), once with buffer 2 (10 mM Tris pH 8.1, 1 mM EDTA, 250 mM LiCl, 1% NP40, 1% sodium deoxycholate monohydrate) and twice with TE buffer. Bound chromatin was eluted from the beads twice with 100 mM NaHCO3 containing 1% SDS. After reverse-crosslinking, RNaseA and proteinase K digestion, chromatin was cleaned up using the Qiagen PCR purification kit (Qiagen, Crawley, UK).
Follow-up confirmatory ChIP was performed with immunoprecipitated and reverse-crosslinked chromatin from HCASMC that was prepared as described above. Quantitative real-time PCR (ViiA 7, Life Technologies) was performed with primers specific for ChIP-Seq peak sequences using SYBR Green (Applied Biosystems) assays and fold enrichment was calculated by measuring the delta Ct—delta Ct IgG. Melting curve analysis was also performed for each ChIP primer pair.
Original data has been deposited at GEO, accession number: GSE61369.
ChIP-Seq peak identification and target gene mapping
Sequence data were aligned to the reference genome (hg19) using BWA [52]. In the case of TCF21, we identified peaks present in both biological replicates of each of the two antibody precipitations by using Irreproducible Discovery Rate analysis, or IDR, a method extensively used by the ENCODE project [8]. Here, we followed the workflow developed by ENCODE, using MACSv2 to identify peaks against an IgG ChIP-Seq control [8]. Parameters for peak calling and IDR thresholds were set to values recommended by the ENCODE pipeline. We analyzed each antibody composite dataset individually, and also intersected these two datasets to create a dataset with minimal number of peaks and minimal peak length (Fig 1). In the case of JUN and JUND, we used MACSv2 to call peaks against an IgG control using default parameters and selected the top ten thousand peaks (ranked by Q-value) for subsequent analysis. We utilized the Genomic Regions Enrichment of Annotations Tool (GREAT) [21] to analyze the detected peaks of each TF, with the parameter “Association rule settings” set to single nearest gene within 50 kb. We also independently identified genes within 50 kb of TF binding sites using the BEDTools software suite [53]. Genes obtained from each method were designated TF target genes.
Assessment of co-expression among the predicted TCF21 targets
To more broadly assess the co-expression patterns among the TCF21 target genes, we retrieved 2,705 co-expression modules derived from 108 coexpression networks constructed from many different tissues in human and mouse populations [10–19]. The enrichment analysis between the co-expression modules and the target genes was evaluated using Fisher’s exact test.
ATAC-sequencing
ATAC-Seq was performed with slight modifications to published protocol [9]. Briefly, human coronary artery smooth muscle cells (passage 5–6) were cultured in normal media until about 75% confluence. Approximately 5x10e4 fresh cells were collected by centrifugation at 500 x g and washed twice with cold 1X phosphate buffered saline. Nuclei were extracted with cold lysis buffer containing 10mM Tris-HCl, pH7.4, 10mM NaCL, 3mM MgCl2, 0.1% IGEPAL and nuclei were resuspended in transposition reaction buffer containing Tn5 transposases (Illumina Nextera). Transposition reactions were incubated at 37 C for 30 minutes, followed by DNA purification using Zymo DNA Clean-up and Concentration Kit. Libraries were initially PCR amplified using Nextera barcodes and High Fidelity polymerase (NEB). The number of cycles was empirically determined from an aliquot of the PCR mix, by calculating the Ct value at 25% maximum Rn for each library preparation. The final amplified library was again purified using the Zymo DNA Clean-up and Concentration kit, and the DNA was evaluated by TBE gel electrophoresis and quantitated using Bioanalyzer, nanodrop, and quantitative PCR (KAPA Biosystems). Libraries were multiplexed and paired-end 50bp next generation sequencing was performed using an Illumina HiSeq 2500. Raw FastQ files were evaluated using a modification of the FastQC pipeline to generate per base and per sequence level summary statistics. Libraries that achieved consistent high quality scores from this tool were aligned to the human genome (hg19) using Bowtie2 and transposase sensitive regions were called using MACS, and bigwig files were generated [9].
De novo and known motif enrichment
To discover DNA binding motifs enriched among TCF21 peaks, HOMER findMotifsGenome.pl script was used to search for the known TRANSFAC motifs and to create de novo motifs from 4852 TCF21 ChIP-Seq binding regions (Ab_Shared regions) [22]. All software parameters were set to default values, with the addition of the “-size given” command to define the width of each peak from the data rather than a constant value. Motifs discovered by HOMER were validated with MEME-ChIP [23] with a maximum motif length of 10. The motifs identified by MEME-ChIP were further compared to the binding motifs of known transcription factors. Density plots of motifs were created as follows. TCF21 summits were defined using MACS with default parameters for the collection of 4852 TCF21 ChIP-Seq binding regions. Motif distribution plots were made using HOMER annotatePeaks.pl script and centering on the locations of TCF21 summits. TRANSFAC matrices for the top HOMER known-motif outputs (TCF12, OLIG2, AP-1, unknown-NANOG, CEBP, TEAD4, ATF1) were used for the scan of TCF21 summit locations in the human GRCh37/hg19 genome. Scanning for motifs was performed using annotatePeaks.pl with the following parameters: hg19-size 2000-hist 20.
TCF21, ATAC-Seq and HAoSMC DHS peak overlap analysis
We retrieved the HAoSMC DHS data from the ENCODE project and selected peaks with P<0.05 as true HAoSMC DHS signal (n = 121731). The overlaps among TCF21 binding regions, ATAC-Seq, and HAoSMC DHS peaks were evaluated through one-sided Fisher exact test. In the overlap between TCF21 ChIP-Seq and ATAC-Seq peaks, we only focused on the ATAC-Seq signals with high-confidence fold-enrichment (mfold>200).
TCF21 and JUN/JUND peak overlap analysis
We tested co-activity between TCF21 and the AP-1 factors JUN and JUND by evaluating the level of overlap (using BEDTools) between ChIP-Seq binding regions corresponding to each TF. Peaks from each dataset were first reduced to those that overlap with open chromatin as identified with our HCASMC ATAC-Seq or ENCODE HAoSMC DHS data. In each analysis, a one-sided Fisher’s exact test was used to determine whether the observed peak overlap between TCF21 and JUN or between TCF21 and JUND was statistically greater when compared against a background of all HCASMC ATAC-Seq or HAoSMC DHS peaks.
Analysis of enrichment of association of functional SNPs in TCF21 target loci among functional SNPs of CAD loci
We utilized the publicly available summary level association results (~2.5 million HapMap SNPs) of the CARDIoGRAM consortium involving 22,233 CAD cases and 64,762 controls to further analyze the relevance of the TCF21 target genes to CAD [54]. We explored whether the subset of functional SNPs located in TCF21 target genes had a higher proportion of low P-values of association (P<0.01) with CAD when compared to the proportion of low P-values observed for all functional SNPs examined in CARDIoGRAM, using a Fisher's exact test. The distribution of the P-values of these two sets of SNPs was also tested using a one-sided Kolmogorov-Smirnov (KS) test. For this analysis, we identified 189,002 functional SNPs (eSNPs) through eQTL experiments involving liver, brain, blood, human aortic endothelial cells (HAEC), and adipose tissues [11–16,29,31–33,55,56]. In the second approach, we identified 1,201,481 potentially functional SNPs through the ENCODE-based RegulomeDB (http://www.regulomedb.org/). Functional SNPs from RegulomeDB were further classified into various categories based on multiple measures of functional implication according to the database, with category 1 SNPs having the highest number of independent functional measures. TCF21 target genes were first mapped to their associated eSNPs or RegulomeDB SNPs and then compared with the corresponding background functional SNPs of all genes for enrichment of low P-value associations with CAD. For both analyses, LD structure was addressed by pruning the eQTL and ENCODE SNPs. This was done by removing those with r2 > 0.3 to derive independent SNPs.
Analysis of enrichment for CAD related and other sets of GWAS genes among TCF21 targets
To investigate possible over-representation of TCF21 binding regions among CHD GWAS loci, we conducted analyses at both the gene and variant level, looking for enrichment of TCF21 target genes among CHD GWAS loci, and for enrichment of average linkage disequilibrium between TCF21 peak SNPs and GWAS loci SNPs.
We first assessed whether there was a spurious association of TCF21 target genes with CAD due to confounding, by looking for an over-representation of these genes among those associated with GWAS loci genes in general. For this analysis, we downloaded the NHGRI GWAS catalog and extracted candidate GWAS genes for all diseases or phenotypes curated in the catalog. Enrichment of candidate GWAS genes among the TCF21 target genes located in the TCF21 peaks was estimated using one-sided Fisher’s exact test. We then looked for an over-representation of CAD candidate genes discovered thorough recent GWAS studies among genes targeted by TCF21. The list of CAD candidate genes we considered (“CAD”) was also collated from the NHGRI GWAS catalog and includes all genes assigned to a SNP in the catalog for phenotypes relevant to CAD including "coronary heart disease", "myocardial infarction", and "coronary artery calcification". For a second category (“CAD extended”) this list was supplemented with genes identified through the CARDIoGRAMplusC4D study, which derive from analysis of Metabochip data and for this reason have not been included in the GWAS catalog [1,34]. Lipid metabolism genes were removed from each of these categories to create lists “CAD no lipid” and “CAD extended no lipid.” We compared the proportion of CAD genes observed among all GWAS catalog phenotypes to the proportion of CAD genes observed among the subset of TCF21 target genes using one-tailed Fisher’s exact test.
For both GWAS enrichment methods we also included additional phenotypes as described in the Result section to test the specificity of CAD signal enrichment in TCF21 targets. These phenotypes included cholesterol measures related to CAD (LDL cholesterol, total cholesterol, triglycerides), diseases known to be associated with dysfunction of the arterial vessel wall or myocardial infarction (aneurysmal conditions involving either the root, the thoracic, or the abdominal aorta, or intracranial vessels (aneurysm), migraine, high blood pressure, platelet number, and Kawasaki's disease), height, which has been associated with CAD, glucometabolic related phenotypes insulin resistance (insulin resistance measures), β-cell function (B function glucose), diseases associated with a dysfunctional immune system (IBD), systemic lupus erythematosus, celiac disease), neuropsychiatric disorders (schizophrenia and Parkinson’s disease), breast cancer and macular degeneration which are well represented in the GWAS catalog but unlikely to be associated with CAD.
To correct for any potential bias in the large numbers of GWAS genes for certain traits such as height and CAD, we implemented a permutation strategy by generating 1000 random GWAS gene sets of matching size for each trait to derive permutation-based enrichment P-values. For example, there are 414 GWAS genes for height, and we generated 1000 gene sets each containing 414 randomly selected GWAS genes from the pool of 8277 GWAS genes in the GWAS catalog. In addition to the enrichment P-value obtained with Fisher’s exact test, (Ptrait) for the observed GWAS gene set of each trait we also calculated 1000 enrichment P-values from the 1000 random gene sets (Prandom1, Prandom2,…, Prandom1000). Ptrait was then compared against the 1000 P-values from the random gene sets to derive a permutation-based enrichment P-value as defined by (the number of Prandom from the random sets that are smaller than Ptrait)/1000. This was done for each of the CAD phenotypes and each additional phenotypes/traits to correct for the different numbers of GWAS genes between phenotypes.
For the TCF21-GWAS LD analysis, we developed a custom pipeline to assess the linkage between variants in TCF21 peaks and variants associated with disease by GWAS. The same GWAS traits/phenotypes were investigated in this analysis as for the gene enrichment analysis. SNPs were identified from the GWAS catalog, where we excluded any GWAS studies that did not contain a cohort of European descent. TCF21 peak SNPs were defined as those 1000 Genomes phase 1 variants that are polymorphic in Europeans and overlap with a TCF21 peak [57]. We used data from the HapMap project (April 2009 release #27) restricted to the CEU population, as well as in-house LD calculations among European SNPs in 1000 Genomes, to assign a linkage disequilibrium r2 value to TCF21—GWAS SNP pairs [58]. In the instance where HapMap and 1000 Genomes r2 values for a given SNP pair did not agree, we used the average of the two. Using custom software developed in the R language, we calculated the average of all r2 values between TCF21 peak—trait SNPs pairs, which was denoted as the “true average” for the association between TCF21 peak SNPs and that particular trait. We then computed 10,000 null averages, each time permuting the trait labels in the GWAS catalog and determining the r2 average between TCF21 peak SNPs and an equal number of SNPs now incorrectly associated with the trait of interest. In this analysis, if a TCF21 peak SNP was in LD with multiple GWAS SNPs, only the maximum r2 value of these associations was taken into calculating the r2 average for a given trait. This filter ensured that each TCF21 SNP mapped onto at most one GWAS trait SNP, although a GWAS trait SNP was allowed to map onto multiple TCF21 SNPs.
Pathway and network analysis of TCF21 target genes
For pathway analysis, the DAVID algorithm [59] was employed with Gene Ontology (GO) terms to annotate the functions of the TCF21 target genes in relation to “biological processes.” Enrichment analysis was carried out in DAVID using default settings. TCF21 target CAD genes and the full list of CAD genes were assessed individually and compared, and the TCF21 target CAD genes were also evaluated in the context of a background composed of all CAD loci genes. For network analysis of TCF21 target CAD genes, those loci associated with CAD at FDR< 0.05 were identified, and genes linked to these loci in the original publications by distance from the lead SNP, eQTL, or allele-specific expression were collated. This gene list was employed with the STRING algorithm [35] to search protein-protein interaction and other types of databases to develop a molecular interaction network.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. (2012) Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45 : 25–33. doi: 10.1038/ng.2480 23202125
2. Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, et al. (2012) The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139 : 2139–2149. doi: 10.1242/dev.079970 22573622
3. Hidai H, Bardales R, Goodwin R, Quertermous T, Quertermous EE (1998) Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech Dev 73 : 33–43. 9545526
4. Lu J, Chang P, Richardson JA, Gan L, Weiler H, et al. (2000) The basic helix-loop-helix transcription factor capsulin controls spleen organogenesis. Proc Natl Acad Sci U S A 97 : 9525–9530. 10944221
5. Quaggin SE, Schwartz L, Cui S, Igarashi P, Deimling J, et al. (1999) The basic-helix-loop-helix protein pod1 is critically important for kidney and lung organogenesis. Development 126 : 5771–5783. 10572052
6. Braitsch CM, Combs MD, Quaggin SE, Yutzey KE (2012) Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev Biol 368 : 345–357. doi: 10.1016/j.ydbio.2012.06.002 22687751
7. Gomez D, Owens GK (2012) Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95 : 156–164. doi: 10.1093/cvr/cvs115 22406749
8. Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, et al. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22 : 1813–1831. doi: 10.1101/gr.136184.111 22955991
9. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods doi: 10.1038/nmeth.2688
10. Derry JMJ, Zhong H, Molony C, MacNeil D, Guhathakurta D, et al. (2010) Identification of Genes and Networks Driving Cardiovascular and Metabolic Phenotypes in a Mouse F2 Intercross. Plos One 5: e14319. doi: 10.1371/journal.pone.0014319 21179467.
11. Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, et al. (2008) Genetics of gene expression and its effect on disease. Nature 452 : 423–428. doi: 10.1038/nature06758 18344981
12. Fehrmann RS, Jansen RC, Veldink JH, Westra HJ, Arends D, et al. (2011) Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet 7: e1002197. doi: 10.1371/journal.pgen.1002197 21829388
13. Greenawalt DM, Dobrin R, Chudin E, Hatoum IJ, Suver C, et al. (2011) A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res 21 : 1008–1016. doi: 10.1101/gr.112821.110 21602305
14. Nica AC, Parts L, Glass D, Nisbet J, Barrett A, et al. (2011) The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7: e1002003. doi: 10.1371/journal.pgen.1002003 21304890
15. Romanoski CE, Che N, Yin F, Mai N, Pouldar D, et al. (2011) Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res 109: e27–41. doi: 10.1161/CIRCRESAHA.111.241869 21737788
16. Schadt EE, Molony C, Chudin E, Hao K, Yang X, et al. (2008) Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107. doi: 10.1371/journal.pbio.0060107 18462017
17. Tu ZD, Keller MP, Zhang CS, Rabaglia ME, Greenawalt DM, et al. (2012) Integrative Analysis of a Cross-Loci Regulation Network Identifies App as a Gene Regulating Insulin Secretion from Pancreatic Islets. PLoS genetics 8: e1003107. doi: 10.1371/journal.pgen.1003107 23236292.
18. Wang SS, Schadt EE, Wang H, Wang XP, Ingram-Drake L, et al. (2007) Identification of pathways for atherosclerosis in mice—Integration of quantitative trait locus analysis and global gene expression data. Circulation research 101: E11–E30. 17641228
19. Yang X, Schadt EE, Wang S, Wang H, Arnold AP, et al. (2006) Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Research 16 : 995–1004. 16825664
20. Yang X, Peterson L, Thieringer R, Deignan JL, Wang X, et al. (2010) Identification and validation of genes affecting aortic lesions in mice. J Clin Invest 120 : 2414–2422. doi: 10.1172/JCI42742 20577049
21. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28 : 495–501. doi: 10.1038/nbt.1630 20436461
22. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38 : 576–589. doi: 10.1016/j.molcel.2010.05.004 20513432
23. Machanick P, Bailey TL (2011) MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27 : 1696–1697. doi: 10.1093/bioinformatics/btr189 21486936
24. Murre C, McCaw PS, Baltimore D (1989) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 56 : 777–783. 2493990
25. Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M (2008) Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4: e1000133. doi: 10.1371/journal.pgen.1000133 18654640
26. Miyagishi M, Nakajima T, Fukamizu A (2000) Molecular characterization of mesoderm-restricted basic helix-loop-helix protein, POD-1/Capsulin. Int J Mol Med 5 : 27–31. 10601570
27. Miller CL, Anderson DR, Kundu RK, Raiesdana A, Nurnberg ST, et al. (2013) Disease-Related Growth Factor and Embryonic Signaling Pathways Modulate an Enhancer of TCF21 Expression at the 6q23.2 Coronary Heart Disease Locus. PLoS Genet 9: e1003652. doi: 10.1371/journal.pgen.1003652 23874238
28. White JT, Zhang B, Cerqueira DM, Tran U, Wessely O (2010) Notch signaling, wt1 and foxc2 are key regulators of the podocyte gene regulatory network in Xenopus. Development 137 : 1863–1873. doi: 10.1242/dev.042887 20431116
29. Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, et al. (2009) Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325 : 1246–1250. doi: 10.1126/science.1174148 19644074
30. Nica AC, Montgomery SB, Dimas AS, Stranger BE, Beazley C, et al. (2010) Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 6: e1000895. doi: 10.1371/journal.pgen.1000895 20369022
31. Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, et al. (2012) Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8: e1002639. doi: 10.1371/journal.pgen.1002639 22532805
32. Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, et al. (2010) Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464 : 773–777. doi: 10.1038/nature08903 20220756
33. Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, et al. (2007) Population genomics of human gene expression. Nat Genet 39 : 1217–1224. 17873874
34. Welter D, MacArthur J, Morales J, Burdett T, Hall P, et al. (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42: D1001–1006. doi: 10.1093/nar/gkt1229 24316577
35. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, et al. (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41: D808–815. doi: 10.1093/nar/gks1094 23203871
36. Chamley-Campbell JH, Campbell GR, Ross R (1981) Phenotype-dependent response of cultured aortic smooth muscle to serum mitogens. J Cell Biol 89 : 379–383. 7251658
37. Zhao B, Li L, Guan KL (2010) Hippo signaling at a glance. Journal of cell science 123 : 4001–4006. doi: 10.1242/jcs.069070 21084559
38. Worsley Hunt R, Wasserman WW (2014) Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome Biol 15 : 412. doi: 10.1186/s13059-014-0412-4 25070602
39. Zhan Y, Kim S, Yasumoto H, Namba M, Miyazaki H, et al. (2002) Effects of dominant-negative c-Jun on platelet-derived growth factor-induced vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol 22 : 82–88. 11788465
40. Funato N, Ohyama K, Kuroda T, Nakamura M (2003) Basic helix-loop-helix transcription factor epicardin/capsulin/Pod-1 suppresses differentiation by negative regulation of transcription. J Biol Chem 278 : 7486–7493. 12493738
41. Kaplan S, Kaplan ST, Kiris A, Gedikli O (2014) Impact of initial platelet count on baseline angiographic finding and end-points in ST-elevation myocardial infarction referred for primary percutaneous coronary intervention. International journal of clinical and experimental medicine 7 : 1064–1070. 24955183
42. Goliasch G, Forster S, El-Hamid F, Sulzgruber P, Meyer N, et al. (2013) Platelet count predicts cardiovascular mortality in very elderly patients with myocardial infarction. European journal of clinical investigation 43 : 332–340. doi: 10.1111/eci.12049 23398046
43. Dangas GD, Claessen BE, Mehran R, Xu K, Fahy M, et al. (2012) Development and validation of a stent thrombosis risk score in patients with acute coronary syndromes. JACC Cardiovascular interventions 5 : 1097–1105. doi: 10.1016/j.jcin.2012.07.012 23174632
44. Paajanen TA, Oksala NK, Kuukasjarvi P, Karhunen PJ (2010) Short stature is associated with coronary heart disease: a systematic review of the literature and a meta-analysis. Eur Heart J 31 : 1802–1809. doi: 10.1093/eurheartj/ehq155 20530501
45. Rosenbush SW, Parker JM (2014) Height and heart disease. Reviews in cardiovascular medicine 15 : 102–108. 25051127
46. Emerging Risk Factors C (2012) Adult height and the risk of cause-specific death and vascular morbidity in 1 million people: individual participant meta-analysis. International journal of epidemiology 41 : 1419–1433. doi: 10.1093/ije/dys086 22825588
47. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, et al. (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467 : 832–838. doi: 10.1038/nature09410 20881960
48. Nelson CP, Hamby SE, Saleheen D, Hopewell JC, Zeng L, et al. (2015) Genetically-Determined Height and Coronary Artery Disease. New England Journal of Medicine In Press.
49. Makinen VP, Civelek M, Meng Q, Zhang B, Zhu J, et al. (2014) Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet 10: e1004502. doi: 10.1371/journal.pgen.1004502 25033284
50. Lees CW, Barrett JC, Parkes M, Satsangi J (2011) New IBD genetics: common pathways with other diseases. Gut 60 : 1739–1753. doi: 10.1136/gut.2009.199679 21300624
51. Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on A (2009) Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 54 : 2129–2138. doi: 10.1016/j.jacc.2009.09.009 19942084
52. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760. doi: 10.1093/bioinformatics/btp324 19451168
53. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 : 841–842. doi: 10.1093/bioinformatics/btq033 20110278
54. Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, et al. (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43 : 333–338. doi: 10.1038/ng.784 21378990
55. Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, et al. (2007) A genome-wide association study of global gene expression. Nature Genetics 39 : 1202–1207. 17873877
56. Duan S, Huang RS, Zhang W, Bleibel WK, Roe CA, et al. (2008) Genetic architecture of transcript-level variation in humans. American Journal of Human Genetics 82 : 1101–1113. doi: 10.1016/j.ajhg.2008.03.006 18439551
57. Consortium GP, Abecasis GR, Auton A, Brooks LD, DePristo MA, et al. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491 : 56–65. doi: 10.1038/nature11632 23128226
58. Consortium IH, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature 467 : 52–58. doi: 10.1038/nature09298 20811451
59. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57. doi: 10.1038/nprot.2008.211 19131956
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy