
Feeding and Fasting Signals Converge on the LKB1-SIK3 Pathway to Regulate Lipid Metabolism in
Liver kinase B1 (LKB1), a serine/threonine kinase, controls 14 different AMP-activated protein kinase (AMPK) family kinases, including salt-inducible kinase 3 (SIK3), suggesting that it plays a variety of roles. Using the fruit fly as an in vivo model system, we reveal that LKB1 kinase activity is critical for lipid storage and controls the lipolysis pathway in the fat body, which is equivalent to mammalian adipose and liver tissue. We find that the lipolytic defects of LKB1 mutants are rescued by the expression of constitutively active SIK3 in the fat body. We show that LKB1 and SIK3 regulate lipid storage by altering the gene expression of brummer, the Drosophila homolog of human adipose triglyceride lipase (ATGL), a critical lipolytic gene. We also identify that LKB1-SIK3 signaling controls the nuclear and cytosolic localization of the class IIa deacetylase HDAC4 via SIK3-dependent phosphorylation in feeding and fasting conditions, respectively. Collectively, these data suggest that the LKB1-SIK3-HDAC4 pathway plays a critical role in maintaining fly lipid homeostasis in response to dietary conditions.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005263
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005263
Summary
Liver kinase B1 (LKB1), a serine/threonine kinase, controls 14 different AMP-activated protein kinase (AMPK) family kinases, including salt-inducible kinase 3 (SIK3), suggesting that it plays a variety of roles. Using the fruit fly as an in vivo model system, we reveal that LKB1 kinase activity is critical for lipid storage and controls the lipolysis pathway in the fat body, which is equivalent to mammalian adipose and liver tissue. We find that the lipolytic defects of LKB1 mutants are rescued by the expression of constitutively active SIK3 in the fat body. We show that LKB1 and SIK3 regulate lipid storage by altering the gene expression of brummer, the Drosophila homolog of human adipose triglyceride lipase (ATGL), a critical lipolytic gene. We also identify that LKB1-SIK3 signaling controls the nuclear and cytosolic localization of the class IIa deacetylase HDAC4 via SIK3-dependent phosphorylation in feeding and fasting conditions, respectively. Collectively, these data suggest that the LKB1-SIK3-HDAC4 pathway plays a critical role in maintaining fly lipid homeostasis in response to dietary conditions.
Introduction
Perturbation of energy homeostasis either directly or indirectly causes human health problems such as obesity and type II diabetes [1]. Lipid stores are the major energy reserves in animals and are dynamically regulated by alternating between the lipogenesis and lipolysis cycles in response to food availability. Dissecting the regulatory mechanisms of lipid homeostasis is therefore essential to our understanding of how energy metabolism is maintained.
Drosophila has emerged as a useful genetic model organism for studying lipid homeostasis and energy metabolism [2]. Drosophila lipid reserves are mainly stored as triacylglycerol (TAG) in the fat body, the insect equivalent of mammalian adipose tissue. In addition, lipolytic factors are evolutionarily conserved between insects and mammals. Brummer (Bmm) is the Drosophila homolog of ATGL, a key regulator of lipolysis. bmm mutant flies are obese and display partial defects in lipid mobilization [3]. Furthermore, hormonal regulation of lipid metabolism is also highly conserved in Drosophila. Under starvation conditions, the primary role of AKH, the functional analogue of glucagon and β-adrenergic signaling in mammals [4,5], is to stimulate lipid mobilization by activating AKH receptor (AKHR) [6] and consequently inducing cAMP/PKA signaling in the fat body [7]. A report demonstrated that AKH acts in parallel with Bmm to regulate lipolysis and that AKHR mutation leads to obesity phenotypes and defects in fat mobilization [7]. However, bmm expression is hyperstimulated in starved AKHR mutants [7], implying the existence of an unknown regulatory mechanism between Bmm and AKHR in Drosophila.
LKB1 (liver kinase B1, also known as STK11) is a serine/threonine kinase that was first identified as a tumor suppressor gene associated with Peutz-Jeghers syndrome [8,9]. LKB1 phosphorylates and activates AMP-activated protein kinase (AMPK) in response to cellular energy status, thus controlling cell metabolism, cell structures, apoptosis, etc. [10–13]. Moreover, LKB1 is the master upstream protein kinase for 12 AMPK-related kinases, including salt-inducible kinases (SIKs) [14], suggesting that it plays diverse roles. Although the metabolic functions of AMPK have been highly studied, the in vivo functions of LKB1 and AMPK-related kinases in metabolism, including lipid homeostasis, are still largely unknown [15]. Recent reports showed that LKB1 is required for the growth and differentiation of white adipose tissue [16] and that SIK3 maintains lipid storage size in adipose tissues [17]. In addition, we and others determined that Drosophila SIK family kinases regulate lipid levels and starvation responses [18,19]. However, to further understand the roles and mechanisms of LKB1 signaling in lipid metabolism, proper genetic animal models are urgently required.
Here we demonstrate the role of LKB1 and its downstream SIK3 in the regulation of lipid homeostasis using Drosophila as an in vivo model system. We demonstrated that LKB1-activated SIK3 regulates the nucleocytoplasmic localization of HDAC4 to control lipolytic gene expression. We also identified that DILPs modulate SIK3 activity via Akt-dependent phosphorylation and the AKH pathway regulates LKB1 activity in phosphorylating SIK3 to control its lipolytic responses upon short-term fasting. Furthermore, we identified that AKH-independent signaling modulates the LKB1-SIK3-HDAC4 pathway upon prolonged fasting. Altogether, these studies showed that the LKB1-SIK3 signaling pathway plays a crucial regulatory role in maintaining lipid homeostasis in Drosophila.
Results and Discussion
Drosophila LKB1 mutants display reduced lipid storage in the fat body
LKB1 functions in a complex with two scaffolding proteins, STE20-related adaptor (STRAD) and mouse protein 25 (MO25) [20,21]. As the first step toward elucidation of the role of LKB1 in lipid metabolism, we demonstrated the gene expression of each component of the LKB1 complex in the fat body (Fig 1A), suggesting that Drosophila LKB1 forms the heterotrimeric complex when activated in tissues. Additionally, we characterized an LKB1-null mutant line, LKB1X5 [22], and found that these flies showed markedly decreased lipid storage compared to wild-type flies, despite having similar food intake and retaining expression of the lipogenic genes (SREBP, FAS, and ACC) (Figs 1B, 1C and S1A). However, expression of bmm and lipolysis activity were elevated in LKB1X5 mutants (Figs 1C and S1B, respectively). Moreover, transgenic expression of wild-type LKB1 with two different fat body drivers (FB-Gal4 and cg-Gal4) rescued the decreased lipid levels and increased bmm expression phenotypes of LKB1X5 mutants, whereas expression of the kinase-dead form of LKB1 (LKB1 K201I) did not (Figs 1E, 1F, S2A and S2B). Additionally, overexpression of LKB1 induced significant increases in the lipid levels and decreases in bmm expression in a dose-dependent manner (S3A and S3B Fig). The implication behind these observations is that LKB1 plays a critical role in lipid storage in Drosophila by regulating the lipolysis pathway in a kinase activity-dependent manner.
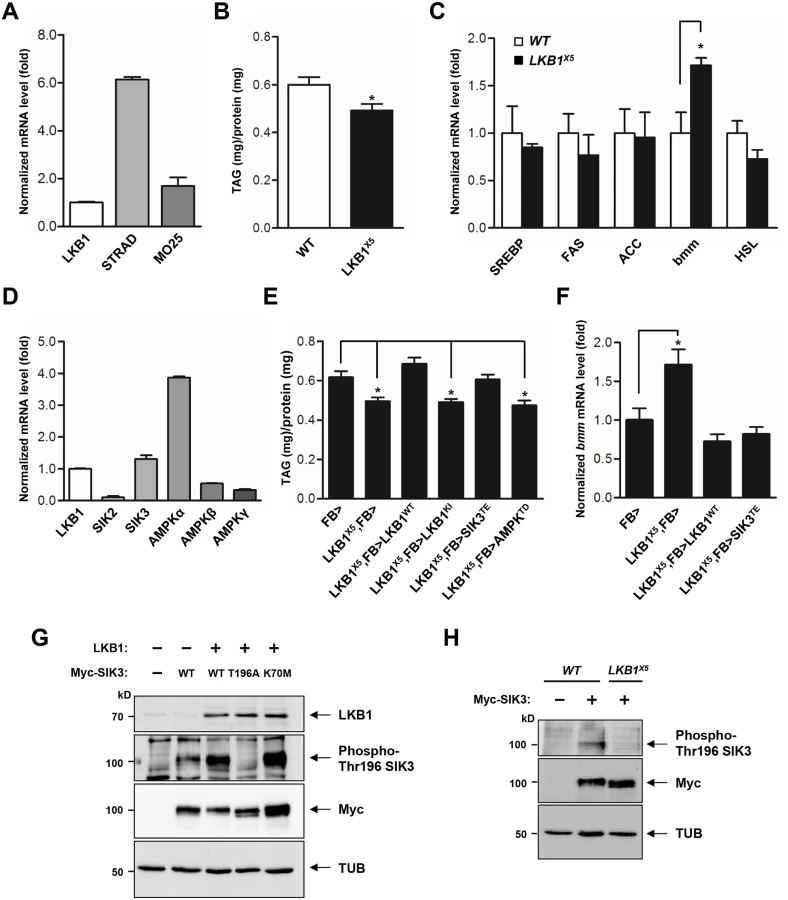
SIK3 is a critical target of LKB1 for controlling lipid storage in Drosophila
To identify the lipolytic target of LKB1 among AMPK-related kinases in Drosophila, we determined mRNA levels of SIKs and AMPK, which are heavily involved in various metabolic pathways. As shown in Fig 1D, SIK3 and AMPKalpha were more highly expressed in the fat body. Furthermore, transgenic expression of constitutively active SIK3 (SIK3 T196E) in the fat body rescued the lipid accumulation and bmm expression defects of LKB1-null mutants, whereas expression of constitutively active AMPK (AMPK T184D) or inactive SIK3 with a mutation in the LKB1 phosphorylation site (SIK3 T196A) failed to rescue the lipid levels of the null mutants (Figs 1E, 1F, S4A and S4B). These results clearly suggest a specific role for SIK3 in the LKB1-mediated regulation of lipid storage in the fat body of Drosophila. Supporting this conclusion, overexpression of LKB1 highly augmented the phosphorylation of conserved Thr196 in SIK3 (Fig 1G), but this phosphorylation was completely lost in LKB1X5 mutants (Fig 1H).
Drosophila SIK3 mutants display lipodystrophy
Drosophila SIK3, one of the AMPK-related kinases, shares considerable sequence homology with the kinase domain of mammalian SIK3 (Fig 2A). To assess the in vivo role of SIK3, SIK3 loss-of-function mutants were generated by mobilizing the EP-element from SIK3G7844 (Fig 2B). From 600 EP excision alleles, we generated SIK3Δ5–31 mutant, which lacks 2,476 bp (2R14578001~14580477) that encodes for the translation start site and the ATP-binding site of SIK3 (Fig 2B and 2C). Confirming that SIK3Δ5–31 is a null mutant, SIK3 mRNA was not detected in the mutant (Fig 2D). However, the internal gene (CG15071) in the coding region of SIK3 was not affected (Fig 2D).

SIK3Δ5–31 mutant flies died before the mid-pupal stage and showed a decreased survival rate (Fig 2E and 2F). The SIK3 null mutant also exhibited a lipodystrophic phenotype (Fig 2G), and FB-Gal4-driven EGFP expression further confirmed the lean fat body phenotype of SIK3Δ5–31 mutants compared to control flies (Fig 2H). Consistently, SIK3Δ5–31 mutant had decreased lipid stores despite having a similar food intake in the larval stage (Figs 2I and S1A, respectively). Surprisingly, the fat body-specific expression of exogenous wild-type SIK3 rescued the lethality of SIK3Δ5–31 mutant, while the expression of a kinase-dead SIK3 (SIK3 K70M) failed to rescue the mutant (Fig 2E and 2F). These results demonstrated that the phosphotransferase activity of SIK3 in the fat body is crucial for its function.
To further investigate the role of SIK3 in lipid metabolism, we analyzed bmm gene expression in SIK3Δ5–31 mutant. Expectedly, the mutant showed markedly increased expression of bmm and increased lipase activity (Figs 2J and S1B, respectively), a phenotype similar to the LKB1 null mutant. Transgenic expression of either wild-type (SIK3 WT) or constitutively active SIK3 (SIK3 T196E) in the fat body of SIK3Δ5–31 mutant resulted in full recovery of lipid levels and bmm expression compared to wild-type controls (Figs 2K, 2L, S2C and S2D). In contrast, expression of either inactive SIK3 harboring a mutation in the LKB1 phosphorylation site (SIK3 T196A) or a kinase-dead mutant (SIK3 K70M) failed to rescue the SIK3Δ5–31 mutant phenotypes (Fig 2K and2L, respectively). Therefore, the kinase activity of SIK3 controlled by LKB1 is critical for the lipid storage in Drosophila fat body.
The LKB1-SIK3 pathway is upstream of HDAC4, which regulates lipid storage and bmm expression
LKB1 and AMPK-related kinases play a major role in the inhibition of hepatic gluconeogenesis in response to high glucose levels via phosphorylation of the class IIa HDACs and the CREB co-activator CRTC [23–25]. To test whether HDAC4 or CRTC is involved in the LKB1 and SIK3 pathway, we analyzed the genetic interactions of LKB1 and SIK3 with HDAC4 and CRTC in Drosophila. We found that ablation of CRTC exacerbated the lethality of LKB1 and SIK3 null mutants (S5A and S5B Fig). However, strikingly, the loss of HDAC4 rescued the lethality of SIK3 null mutants, but did not affect the lethality of LKB1 null mutants (S6A–S6D Fig), suggesting that HDAC4 participates in LKB1-SIK3 signaling of Drosophila.
To evaluate whether HDAC4 is crucial for the regulation of lipid storage by LKB1 and SIK3, we expressed HDAC4 RNAi in the fat body of LKB1 and SIK3 mutants. Surprisingly, knockdown of HDAC4 in the fat body fully rescued the TAG levels and bmm gene expression of LKB1 and SIK3 null mutants (Fig 3A and 3B, respectively), indicating that HDAC4 is indeed a critical downstream target of LKB1 and SIK3 in lipid metabolism of Drosophila.

SIK3 induces the translocation of HDAC4 from the nucleus to the cytoplasm via phosphorylation
SIKs can regulate target gene expression by directly phosphorylating the class IIa HDACs and consequently inhibiting their translocation to the nucleus [26,27]. Expression of wild-type SIK3 (SIK3 WT) or constitutively active SIK3 (SIK3 T196E) augmented the phosphorylation of HDAC4 but not of the phosphorylation-defective HDAC4 (HDAC4 3A), demonstrating that SIK3 induces HDAC4 phosphorylation in Drosophila (Fig 3C). HDAC4 localized to both the cytoplasm and nuclei of larval fat body cells under feeding conditions, but localized mostly to the nucleus under fasting conditions (Fig 3D). However, HDAC4 accumulated in the nuclei of the fat body cells of LKB1 and SIK3 null mutants even under feeding conditions (Fig 3D). In addition, HDAC4 3A was retained in the nuclei of the fat body cells under both feeding and fasting conditions (Fig 3D). These results indicated that Drosophila SIK3, under the control of LKB1, phosphorylates HDAC4 in the fat body and regulates its nucleocytoplasmic localization in different dietary conditions.
The class IIa HDACs deacetylate and activate FOXO transcription factors [19,24], and the activated FOXO then induces ATGL/Bmm expression [19,28]. Overexpression of wild-type HDAC4 increased the mRNA levels of bmm (Fig 3E), indicating that HDAC4 regulates bmm gene expression in the fat body. Furthermore, overexpression of constitutively active SIK3 completely blocked the increased bmm expression induced by HDAC4 overexpression (Fig 3E), and bmm knockdown in the fat body blocked the decreases in TAG levels induced by LKB1 or SIK3 null mutation (Fig 3F and 3G). Altogether, these results suggested that the LKB1-SIK3 signaling pathway controls HDAC4-dependent Bmm activity in Drosophila fat body.
LKB1-SIK3-HDAC4 signaling acts downstream of the AKH pathway to control lipid homeostasis
Under fasting conditions, AKH activates the mobilization of fat body triglyceride by triggering AKHR and consequent activation of cAMP signaling in the fat body [7]. Consistently, we showed that AKHR mutation highly increased TAG levels (Fig 4A) and decreased bmm gene expression (Fig 4B). To determine the functional interaction between AKHR signaling and the LKB1-SIK3 signaling pathway, we crossed LKB1 or SIK3 null mutant flies with AKHR mutant flies. Interestingly, deletion of LKB1 or SIK3 reversed both the lipid accumulation and the reduced bmm expression phenotypes of AKHR mutant flies (Fig 4A and 4B, respectively), suggesting that the LKB1-SIK3 pathway likely acts downstream of AKHR. Furthermore, SIK3 Thr196 phosphorylation was reduced in both fasting and AKH overexpression conditions compared to that in feeding conditions (Fig 4C), supporting that the AKH pathway inhibits the kinase activity of LKB1 as shown by decreased SIK3 Thr196 phosphorylation.

On the basis of the observation that fasting induces the nuclear translocation of HDAC4, we also examined subcellular localization of HDAC4 in AKHR mutant flies. Intriguingly, HDAC4 in AKHR mutants localized to both the cytoplasm and the nucleus of larval fat body cells in 4 hr fasting condition compared to control (Figs 3D and 4D), suggesting that AKHR-dependent regulation is critical for HDAC4 localization. In addition, overexpression of the phosphorylation-defective HDAC4 by SIK3 (HDAC4 3A) suppressed the TAG levels and enhanced bmm expression in AKHR mutants (Fig 4E and 4F, respectively). Collectively, these results suggested that the LKB1-SIK3-HDAC4 pathway acts downstream of the AKH pathway to control lipolysis activity in Drosophila.
AKH-independent signaling modulates the LKB1-SIK3-HDAC4 pathway under prolonged fasting
We showed that AKHR-dependent regulation of LKB1-SIK3 activity is critical for HDAC4 nuclear localization in ~4 hr fasting condition (Fig 4D). However, Gronke et al. showed that bmm gene expression is stimulated in AKHR mutant flies in 6 hr fasting condition [7]. Notably, in contrast to 4 hr fasting condition (Fig 4D), HDAC4 accumulated in the nuclei of the fat body cells in AKHR mutant flies after prolonged fasting (~10 hr) (Fig 5A), indicating that there should be AKHR-independent HDAC4 regulation during prolonged fasting. Furthermore, knockdown of HDAC4 in the fat body blocked the increased bmm gene expression in AKHR mutant flies after 10 hr fasting (Fig 5B), indicating that AKHR-independent signaling promotes HDAC4 nuclear localization to induce bmm gene expression under prolonged fasting conditions. Interestingly, expression of constitutively active SIK3 blocked the prolonged fasting-induced nuclear localization of HDAC4 (Fig 5C), suggesting that LKB1-SIK3 activity is critical for bmm expression under prolonged fasting. Taken together, these results demonstrated that the LKB1-SIK3-HDAC4 pathway acts as the primary lipolytic signaling upon both short-term and prolonged fasting while AKH plays a major role only in short-term fasting. Thus, it is of particular interest to investigate novel signaling mechanisms regulating the LKB1-SIK3-HDAC4 pathway under prolonged fasting conditions.

Our study provides evidence that LKB1 is necessary for maintaining Drosophila lipid storage via the regulation of lipolysis through the activation of SIK3. Consistent with our results in Drosophila, adipose tissue-specific LKB1 knockout mice showed decreased serum triglycerides [16], and the basal lipogenesis activity of adipocytes was significantly lower in LKB1 hypomorphic mice [29]. Recently, SIK3 null mice were also found to display a malnourished phenotype with lipodystrophy and were resistant to high-fat diets [17]. Thus, the LKB1-SIK3 pathway is indeed an evolutionally conserved regulatory mechanism for lipid homeostasis.
LKB1 is ubiquitously expressed and constitutively active in mammalian cells [15], which raises the question of how dietary conditions change the activity of LKB1 and SIK3 to control lipid homeostasis. Our findings suggested that fasting and the AKH pathway inhibit LKB1 activity to regulate SIK3 Thr196 phosphorylation (Figs 4C and 6C). It is possible that fasting - and AKH-induced inhibition of LKB1 activity can be achieved by altered subcellular localization, protein conformation, stability, and/or protein-protein interactions of LKB1 and its associated proteins. Interestingly, in HEK-293 cells, fasting triggers autophosphorylation of human LKB1 at Thr336 [30] that corresponds to Thr460 in Drosophila LKB1 [31]. This phosphorylation promotes the protein-protein interaction between LKB1 and 14-3-3 proteins [30] and inhibits the ability of LKB1 for suppressing cell growth [31].
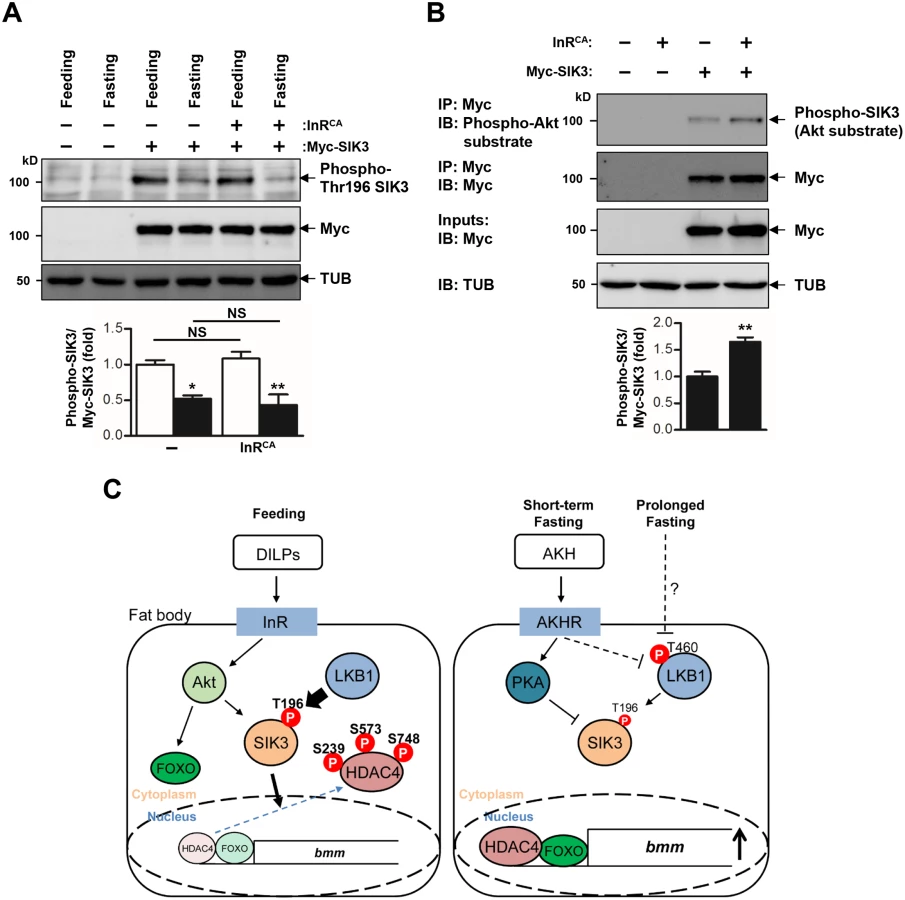
In addition, the AKH pathway activates cAMP/PKA signaling in Drosophila [7]. Mammalian PKA inhibits SIK activity by phosphorylating a conserved serine residue [32,33] that corresponds to Ser563 in Drosophila SIK3 [19], suggesting that the AKH pathway also controls SIK3 activity via PKA-dependent phosphorylation (Fig 6C). On the other hand, the Drosophila insulin-like peptides (DILPs) did not increase SIK3 Thr196 phosphorylation (Fig 6A), but induced Akt-mediated SIK3 phosphorylation (Fig 6B), suggesting that DILPs directly regulate SIK3 activity independently of affecting LKB1 activity [19,34] (Fig 6C). Interestingly, these Drosophila signaling circuits are highly similar to mammalian insulin and glucagon pathways in controlling lipid metabolism and storage, raising questions regarding whether the LKB1-SIK3-HDAC4 signaling pathway is also conserved in mammalian systems as a converging point between feeding and fasting signals to control lipid homeostasis.
Is SIK3 also involved in the modulation of other LKB1 functions, such as the regulation of cell polarity and mitosis? SIK3 null mutants showed normal epithelial polarity and mitosis (S7A and S7B Fig). Additionally, transgenic expression of constitutively active SIK3 (SIK3 T196E) failed to suppress the cell polarity and mitosis defects of LKB1 mutants (S8A and S8B Fig), suggesting that SIK3 does not participate in the regulation of cell polarity and mitosis by LKB1. In addition, both fat body-specific expression of LKB1 and ablation of HDAC4 failed to rescue the lethality of LKB1 null mutants (S6C Fig), indicating that LKB1 has SIK3/HDAC4-independent roles and additional targets in other tissues and developmental processes.
In summary, we have demonstrated that the LKB1-SIK3 pathway is important for maintaining lipid homeostasis in Drosophila. As alterations in lipolysis are closely associated with human obesity [35], future studies will be required to unravel the relationship between LKB1-SIK3-HDAC4 signaling and obesity-related metabolic diseases.
Materials and Methods
Fly strains
The following fly stocks were used in this study: LKB1X5, UAS-LKB1WT, and UAS-LKB1KI [22], HDAC4KG09091 (Bloomington #15159), UAS-HDAC4 RNAi (VDRC #20522), UAS-bmm RNAi (Bloomington #25926), UAS-InRCA (Bloomington #15159), cg-Gal4 (Bloomington #7011), hs-Gal4 (Bloomington #1799), UAS-2xEGFP (Bloomington #6874), UAS-HA-AMPKTD [12], CRTC25-3 [36], UAS-FLAG-HDAC4WT and UAS-FLAG-HDAC43A [19], AKHR1 [7], and FB-Gal4 [37]. SIK3Δ5–31 was generated by imprecise excision of SIK3G7844 line (KAIST Drosophila Library Facility, Daejeon, Korea). To generate UAS-SIK3 flies, SIK3 EST cDNA (Berkeley Drosophila Genome Project accession no. LD07105) was cloned into the Myc-tagged pUAST vector and microinjected into w1118 embryos. All flies were grown on food containing approximately 35 g cornmeal, 70 g dextrose, 5 g agar, 50 g dry active yeast (Ottogi, Inc., Korea), 4.6 ml propionic acid, and 7.3 ml Tegosept (100 g/l in ethanol) per liter at 25°C. All flies were backcrossed for a minimum of 6 generations into w1118 background.
Site-directed mutagenesis
The QuickChange kit (Stratagene) was used for site-directed mutagenesis. For generation of a kinase-dead mutant SIK3 (Lys70Met, SIK3K70M), 5’-CAAGACAAAGGTGGCCATCATGATCATAGACAAAACATGTC-3’ and 5’-GACATGTTTTGTCTATGATCATGATGGCCACCTTTGTCTTG-3’ primers were used. For generation of a SIK3 mutant non-phosphorylatable by LKB1 (Thr196Ala, SIK3 T196A), 5’-GGGTGCCACCTTAAAAGCTTGGTGTGGATCAC-3’ and 5’-GTGATCCACACCAAGCTTTTAAGGTGGCACCC-3’ primers were used. For generation of a SIK3 mutant mimicking LKB1-dependent phosphorylation (Thr196Glu, SIK3 T196E), 5’-GAGGGTGCCACCTTAAAAGAATGGTGTGGATCACCGCCC-3’ and 5’-GGGCGGTGATCCACACCATTCTTTTAAGGTGGCACCCTC-3’ primers were used.
qPCR analysis
Larvae were collected, and RNA was extracted using the RNeasy Mini Kit (QIAGEN). Total RNA (1 μg) was reverse-transcribed by M-MLV Reverse Transcriptase (Promega) to generate cDNA for quantitative real-time RT-PCR (Bio-Rad CFX96 Real-Time PCR detection system, SYBR Green) using a 500 nM primer concentration and 2 ng of cDNA template. The primers were used in S1 Table. The relative values were calculated using the ΔΔCt method via normalization to rp49 mRNA levels. Results were expressed in arbitrary units, with each control value as 1 unit.
Immunohistochemistry
Third instar larvae were dissected in Drosophila Ringer’s solution and fixed with 4% formaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature. After being washed with 0.1% Triton X-100 in PBS (PBST), the samples were blocked for 1 hr incubation at room temperature with 5% bovine serum albumin (BSA) in PBST. The samples were further incubated at 4°C for 16 hr with the indicated antibodies: anti-FLAG-M2 (Sigma, F1804), anti-Myc (Cell Signaling Technology, #2272), anti-aPKC (Santa Cruz, sc-216), and anti-PH3 (Millipore, 06–570). Following three washes with PBST, the samples were incubated with appropriate secondary antibodies (and with Hoechst 33258 used for staining DNA, if required) for 3 hr at room temperature. The samples were washed with PBST and mounted with 80% glycerol in PBS, then observed by a confocal microscope LSM710 (Zeiss).
Feeding assay
Feeding assay was performed according to previously described with minor modifications [38]. Blue food dye (Erioglaucine Disodium Salt, Sigma, #861146) was added at 1% (w/v) to fly food. Larvae were switched from normal food to blue-color food for 2 hr. After feeding, larvae were frozen immediately. Samples were homogenized in PBS buffer and centrifuged for 25 min at 13,200 rpm. The absorbance of the supernatant was measured at 625 nm using Infinite M200 spectrophotometer (Tecan).
Immunoblot analysis
Larvae were lysed in a lysis buffer (20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 5 mM EGTA, 150 mM NaCl, 20 mM NaF, 1% Triton X-100, 1 μg/ml leupeptin, and 1mM PMSF) for 30–60 min on ice. After centrifugation for 15 min at 13,200 rpm, supernatants were reserved for SDS-PAGE analysis, and proteins were then transferred to nitrocellulose membranes (GE Healthcare, #BA85). Membranes were incubated in a blocking solution (Tris-buffered saline (TBS) containing 0.1% Tween-20, 5% BSA) for 1hr. The primary antibodies used were anti-LKB1 [22], anti-phospho-Thr196 SIK3 [39], anti-phospho-Ser239 HDAC4 (Cell Signaling Technology, #3443), anti-phospho-Akt substrate (Cell Signaling Technology, #9614), anti-FLAG-M2 (Sigma, #F1804), anti-Myc (Cell Signaling Technology, #2272), anti-AKH (a gift from Dr. Veenstra), and anti-β-tubulin antibody (Developmental Studies Hybridoma Bank, E7). Protein detection was done using the LAS-4000 imaging system (Fujifilm), and densitometric analysis was performed using Multi Gauge 3.0 software.
Lipid measurement and lipase assay
TAG measurement was performed according to previously described methods using Free Glycerol Reagent (Sigma, #F6428) and Triglyceride Reagent (Sigma, #T2449) [40]. A standard curve was generated with a glycerol standard solution (Sigma, #G7793). Samples were assayed at 540 nm using Infinite M200 spectrophotometer (Tecan). In each homogenate, amounts of TAG (in mg) were normalized to those of protein (in mg) using Bradford protein assay (Bio-Rad). Lipase activity was determined according to the manufacturer’s instructions with QuantiChrom kit (BioAssay Systems, DLPS-100).
Survival rate analysis
The analysis for survival rate was performed as previously described [12]. The eggs from flies with the appropriate genotypes were laid on 60 mm dishes containing standard apple juice-agar with yeast paste for 4 hr. The hatched larvae were collected using selection markers and transferred to plates containing normal food media. The green balancer chromosome (CyO, Actin-GFP) was used to select the homozygous SIK3Δ5–31 mutant. The number of larvae was scored for viability at each developmental stage, and dead larvae were removed. At least 100 larvae were studied per genotype.
Statistical analysis
All quantitative data are analyzed using Student’s t tests or ANOVA with a post Tukey’s multiple comparison test, and P < 0.05 was considered statistically significant. Each experiment was repeated at least three times, data are presented as the average ± standard error of the mean (SEM). The P values given in the survival data are the result of a log rank test using GraphPad Prism 5 software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Kopelman PG (2000) Obesity as a medical problem. Nature 404 : 635–643. 10766250
2. Schlegel A, Stainier DY (2007) Lessons from "lower" organisms: what worms, flies, and zebrafish can teach us about human energy metabolism. PLoS Genet 3: e199. 18081423
3. Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, et al. (2005) Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab 1 : 323–330. 16054079
4. Kim SK, Rulifson EJ (2004) Conserved mechanisms of glucose sensing and regulation by Drosophila corpora cardiaca cells. Nature 431 : 316–320. 15372035
5. Lee G, Park JH (2004) Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics 167 : 311–323. 15166157
6. Staubli F, Jorgensen TJ, Cazzamali G, Williamson M, Lenz C, et al. (2002) Molecular identification of the insect adipokinetic hormone receptors. Proc Natl Acad Sci U S A 99 : 3446–3451. 11904407
7. Gronke S, Muller G, Hirsch J, Fellert S, Andreou A, et al. (2007) Dual lipolytic control of body fat storage and mobilization in Drosophila. PLoS Biol 5: e137. 17488184
8. Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, et al. (1998) A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391 : 184–187. 9428765
9. Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, et al. (1998) Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18 : 38–43. 9425897
10. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, et al. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101 : 3329–3335. 14985505
11. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, et al. (2003) Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2 : 28. 14511394
12. Lee JH, Koh H, Kim M, Kim Y, Lee SY, et al. (2007) Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447 : 1017–1020. 17486097
13. Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE (2007) LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol 177 : 387–392. 17470638
14. Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, et al. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23 : 833–843. 14976552
15. Alessi DR, Sakamoto K, Bayascas JR (2006) LKB1-dependent signaling pathways. Annu Rev Biochem 75 : 137–163. 16756488
16. Zhang W, Wang Q, Song P, Zou MH (2013) Liver kinase b1 is required for white adipose tissue growth and differentiation. Diabetes 62 : 2347–2358. doi: 10.2337/db12-1229 23396401
17. Uebi T, Itoh Y, Hatano O, Kumagai A, Sanosaka M, et al. (2012) Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS One 7: e37803. doi: 10.1371/journal.pone.0037803 22662228
18. Choi S, Kim W, Chung J (2011) Drosophila salt-inducible kinase (SIK) regulates starvation resistance through cAMP-response element-binding protein (CREB)-regulated transcription coactivator (CRTC). J Biol Chem 286 : 2658–2664. doi: 10.1074/jbc.C110.119222 21127058
19. Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, et al. (2011) A hormone-dependent module regulating energy balance. Cell 145 : 596–606. doi: 10.1016/j.cell.2011.04.013 21565616
20. Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, et al. (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J 22 : 3062–3072. 12805220
21. Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, et al. (2003) MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J 22 : 5102–5114. 14517248
22. Lee JH, Koh H, Kim M, Park J, Lee SY, et al. (2006) JNK pathway mediates apoptotic cell death induced by tumor suppressor LKB1 in Drosophila. Cell Death Differ 13 : 1110–1122. 16273080
23. Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, et al. (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437 : 1109–1111. 16148943
24. Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, et al. (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145 : 607–621. doi: 10.1016/j.cell.2011.03.043 21565617
25. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, et al. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310 : 1642–1646. 16308421
26. Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, et al. (2007) SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med 13 : 597–603. 17468767
27. van der Linden AM, Nolan KM, Sengupta P (2007) KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. EMBO J 26 : 358–370. 17170704
28. Chakrabarti P, Kandror KV (2009) FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem 284 : 13296–13300. doi: 10.1074/jbc.C800241200 19297333
29. Gormand A, Henriksson E, Strom K, Jensen TE, Sakamoto K, et al. (2011) Regulation of AMP-activated protein kinase by LKB1 and CaMKK in adipocytes. J Cell Biochem 112 : 1364–1375. doi: 10.1002/jcb.23053 21312243
30. Bai Y, Zhou T, Fu H, Sun H, Huang B (2012) 14-3-3 interacts with LKB1 via recognizing phosphorylated threonine 336 residue and suppresses LKB1 kinase function. FEBS Lett 586 : 1111–1119. doi: 10.1016/j.febslet.2012.03.018 22575644
31. Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, et al. (2002) Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J 362 : 481–490. 11853558
32. Okamoto M, Takemori H, Katoh Y (2004) Salt-inducible kinase in steroidogenesis and adipogenesis. Trends Endocrinol Metab 15 : 21–26. 14693422
33. Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, et al. (2004) The CREB coactivator TORC2 functions as a calcium - and cAMP-sensitive coincidence detector. Cell 119 : 61–74. 15454081
34. Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, et al. (2007) Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 449 : 366–369. 17805301
35. Langin D, Dicker A, Tavernier G, Hoffstedt J, Mairal A, et al. (2005) Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 54 : 3190–3197. 16249444
36. Wang B, Goode J, Best J, Meltzer J, Schilman PE, et al. (2008) The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metab 7 : 434–444. doi: 10.1016/j.cmet.2008.02.010 18460334
37. Gronke S, Beller M, Fellert S, Ramakrishnan H, Jackle H, et al. (2003) Control of fat storage by a Drosophila PAT domain protein. Curr Biol 13 : 603–606. 12676093
38. Bjordal M, Arquier N, Kniazeff J, Pin JP, Leopold P (2014) Sensing of amino acids in a dopaminergic circuitry promotes rejection of an incomplete diet in Drosophila. Cell 156 : 510–521. doi: 10.1016/j.cell.2013.12.024 24485457
39. Al-Hakim AK, Goransson O, Deak M, Toth R, Campbell DG, et al. (2005) 14-3-3 cooperates with LKB1 to regulate the activity and localization of QSK and SIK. J Cell Sci 118 : 5661–5673. 16306228
40. Palanker L, Tennessen JM, Lam G, Thummel CS (2009) Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab 9 : 228–239. doi: 10.1016/j.cmet.2009.01.009 19254568
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy