
Parallel Gene Expression Differences between Low and High Latitude Populations of and .
While gene expression variation in natural populations is common, the population genetic processes responsible for the maintenance of this variation remain obscure. Here we study geographic differences in gene expression in recently established low and high latitude populations of two closely related species of Drosophila. We observe substantial parallelism in expression differences and expression plasticity between populations, which supports the idea that spatially varying selection correlated with latitude contributes to the maintenance of gene expression variation in these species. Comparison of inter-population sequence differentiation and expression differentiation suggests that cis-acting variants play a role in geographic expression differentiation.
Published in the journal:
. PLoS Genet 11(5): e32767. doi:10.1371/journal.pgen.1005184
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005184
Summary
While gene expression variation in natural populations is common, the population genetic processes responsible for the maintenance of this variation remain obscure. Here we study geographic differences in gene expression in recently established low and high latitude populations of two closely related species of Drosophila. We observe substantial parallelism in expression differences and expression plasticity between populations, which supports the idea that spatially varying selection correlated with latitude contributes to the maintenance of gene expression variation in these species. Comparison of inter-population sequence differentiation and expression differentiation suggests that cis-acting variants play a role in geographic expression differentiation.
Introduction
Parallel adaptive evolution in natural populations has been of longstanding interest to evolutionary biologists because its prevalence speaks to the repeatability of adaptive trajectories or mechanisms and in doing so, informs our understanding of the general “rules” of divergence under selection. Indeed, convergent phenotypic evolution is now typically considered prima facie evidence of adaptation [1]. Much empirical support for parallel adaptive evolution at both the phenotypic and molecular level comes from documentation of evolutionary changes on the relatively long timescale of interspecific divergence [1–5]. Alternatively, relatively few studies provide insight into the question of whether recent, potentially transient adaptive processes occurring within species, perhaps associated with spatially varying selection, are repeated in different species [6–10].
In Drosophila melanogaster, extensive phenotypic, cytological, and genomic investigations of latitudinal differentiation have revealed widespread signals of local adaptation in North America and Australia [11–15] following colonization out of an African ancestral range [11,16–18]. Less well understood is whether D. simulans, the sister species of D. melanogaster, also shows latitudinal differentiation, and if so, whether there is substantial overlap in the targets of spatially varying selection in the two species. D. simulans is hypothesized to have had an ancestral origin in Madagascar [19] and like D. melanogaster, only recently colonized Eurasia, North America and Australia [11]. The two species are broadly sympatric human commensals, and can often be simultaneously collected in the field. Like D. melanogaster, D. simulans exhibits high levels of nucleotide diversity and low levels of linkage disequilibrium, consistent with large population sizes [20,21]. Unlike D. melanogaster, which is polymorphic for a number of clinally varying paracentric chromosome inversions [22,23], D. simulans segregates no common chromosome inversions [24]. Despite this major cytological difference between the two species, their recent shared ancestry and general population genetic similarities suggest the possibility that their evolutionary response to recent colonization of novel habitats might be similar in magnitude and perhaps even overlap substantially in the genetic details. Interestingly, however, the relatively few studies of latitudinal variation in D. simulans provide little support for this expectation. For example, studies of protein electrophoretic variation and life-history traits indicate that D. simulans exhibits less geographic differentiation than does D. melanogaster [12,25,26]. Arthur et al. 2008 [27] revealed scant evidence for latitudinal differentiation in heat or cold tolerance in Australian populations of D. simulans, despite the fact that these traits are strongly correlated with latitude in D. melanogaster [28]. However, latitudinal clines in wing size in both D. simulans and D. melanogaster on multiple continents support the hypothesis that larger body size is favored at higher latitude [27,29–32]. A relatively unbiased and large-scale approach for generating hypotheses regarding the extent and biological basis of parallel latitudinal adaptation in the two species is characterization of gene expression variation.
Here we take this approach to ask whether there is substantial overlap of gene expression divergence associated with high and low latitude American population samples in both species. We also ask whether the geographic differentiation in the influence of temperature variation on gene expression is shared between the two species, which would support the idea that variation in gene expression plasticity is also shaped by spatially varying selection in the two species.
Results
Transcriptome Overview of Low and High Latitude D. melanogaster and D. simulans
To identify genes that are differentially expressed between low and high latitude populations, we characterized population variation in whole male transcriptomes of D. melanogaster and D. simulans at 21°C and 29°C using sympatric isofemale strains established from flies collected in Maine and Panama. We created three biological replicates for each species × population × temperature combination for a total of 24 libraries. Sequences were generated by 90 bp paired-end Illumina Hiseq. After filtering, we obtained 347.7 million paired reads for D. melanogaster and 359.4 million paired reads for D. simulans (S1 Table). Estimates of transcript abundance (Fragments Per Kilobase Of Exon Per Million Fragments Mapped, FPKM) were highly correlated across biological replicates (Pearson’s r correlation ~ 0.99 and 0.98 for D. melanogaster and D. simulans, respectively, P <2.2E-16, S1 Fig).
Differential Gene Expression in High vs. Low Latitude Populations
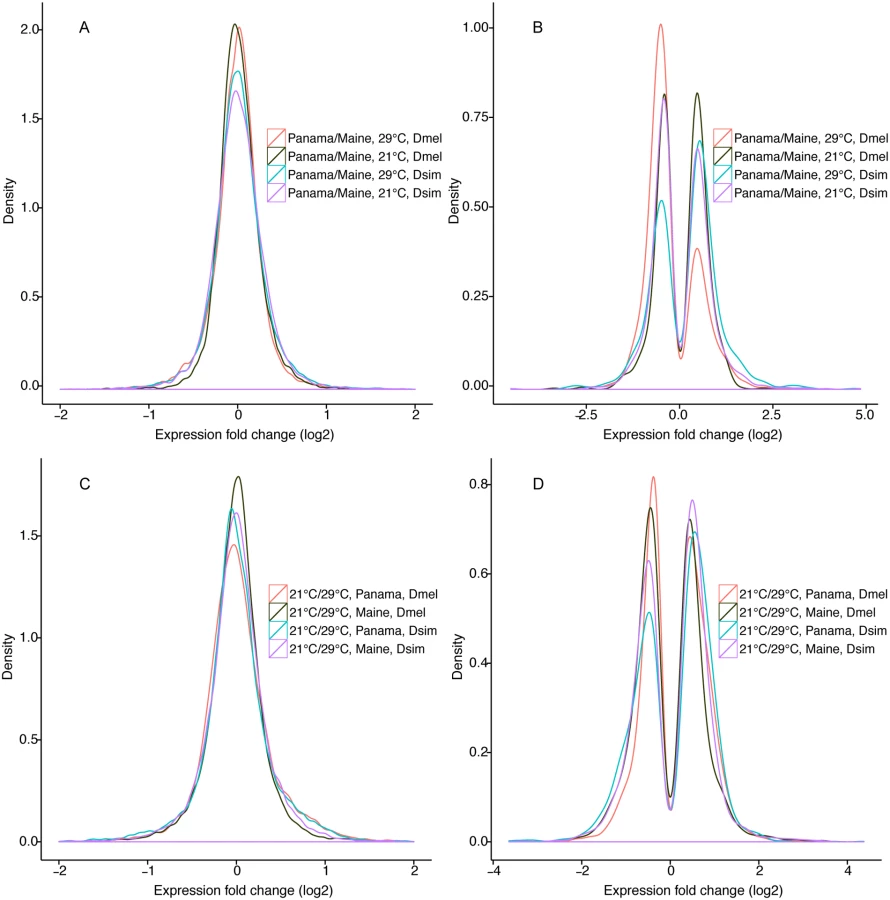
In D. melanogaster, 759 (5.4%) and 980 (6.9%) genes were significantly differentially expressed between populations (False discovery rate (FDR) <0.05) at 21°C and 29°C, respectively (Table 1, Fig 1A and 1B, S2 Fig, S2 Table). The magnitude of expression differences was significantly greater at 29°C than at 21°C. At 21°C, similar numbers of differentially expressed genes showed higher expression in Maine vs. lower expression in Maine (Fig 1B, dark green line, 396 vs. 363, χ2 test, P >0.05), while at 29°C most differentially expressed genes showed higher expression in Maine than in Panama (Fig 1B, red line, 681 vs. 299, χ2 test, P <0.001). Only 193 genes were differentially expressed between populations at both temperatures, supporting the idea that expression differences between these populations are not solely attributable to a similar generalized stress response for Panama at 21°C and Maine and 29°C. Moreover, GO enrichment analysis (below) provided no evidence that stress response pathway genes were enriched among differentially expressed genes.

We used DAVID [33] and GOTermFinder [34] analysis for functional category enrichment using all differentially expressed genes. At 21°C differentially expressed genes were enriched in response to toxic substance (P = 1.88E-07), response to insecticide (P = 7.92E-07), detection of visible light (P = 0.003), electron carrier activity (P = 0.001), and cytochrome P450s (P = 0.02) (S3 Table). Cytochrome P450 genes were previously inferred to be differentiated between low and high latitude populations in genomic analyses [23,35] and in expression analysis [36,37]. At 29°C differentially expressed genes were significantly enriched in multiple light-stimulus related functional categories, including phototransduction (P = 1.44E-11), detection of visible light (P = 2.66E-09), sensory perception of light stimulus (P = 6.48E-07) and transmission of nerve impulse (P = 0.002, S3 Table). At 29°C, 5 of the top 10 differentially expressed genes (ranked by adjusted P-value) were trp and trp-interacting inaF genes (inaF-A, inaF-B, inaF-C, and inaF-D; note that the inaF genes may not be independent as they are co-localized in a small genomic region. The fundamental role of TRP proteins in the light-induced calcium channel in Drosophila photoreceptors [38] further supports the idea that the visual system is influenced by spatially varying selection. Interestingly, previously published genomic data [15] from the USA provide evidence of two strongly differentiated trp non-synonymous SNPs as well as differentiated SNPs in the 5’-flanking region. Focusing on the top 50 differentially expressed protein-coding genes at 21°C and 29°C (S4 Table), eight (Cyp12d1-d, Cyp12d1-p, dy, dsf, CG15221, inaF-B, inaF-D, and CG7884) were differentially expressed at both temperatures; several of the genes showing the greatest expression differences between populations are associated with response to environmental stimulus (S4 Table). Of the 585 expressed annotated transcription factors (TFs), 22 (3.7%) and 39 (6.6%) were differentially expressed between populations (S5 Table) at 21°C and 29°C, respectively. These TFs include srp, which is associated with wing size variation in Australian D. melanogaster populations [39]. Interestingly, 13 of the 22 differentially expressed transcription factors, including eyeless (ey) and glass (gl), are associated with eye development and vision. This is potentially noteworthy given the observed high levels of expression differentiation for vision-related genes. Four transcription factors, pros, dsf, az2, and CG32006 were differentially expressed at both temperatures.
One possible explanation for differences in transcript abundance in whole males from different populations is the presence of population differences in relative size of different organs or tissues, though we are unaware of studies documenting such variation. To address this issue, albeit indirectly, we estimated tau [40,41], an index of tissue specificity, for each gene using FlyAtlas data [42]. We also determined for each expressed gene the tissue that showed the highest expression. If expression differences between populations were attributable primarily to geographic variation in relative sizes of certain organs or tissues, we would expect differentially expressed genes to be enriched with genes showing more tissue expression bias. However, we found no such enrichment for either somatic or testis expression (χ2 tests, all P >0.1). Moreover, we found no correlation between tau and fold change for all expressed genes or for significantly differentially expressed genes (Pearson’s R2 <0.01). Taken together, these results do not support the idea that high and low latitude population expression differences were driven by geographic differences in relative organ size.
Significantly differentiated genes were not neighbors more often than expected by chance (empirical P >0.1). Additionally, the median physical distance between differentially expressed genes was not significantly different from the expected value assuming such genes were randomly distributed on each chromosome arm (P >0.1, see Methods), which suggests that expression differentiation is primarily a genic rather than a chromosomal region effect. Firmer conclusions on this matter await comprehensive analysis of population genomic and functional datasets.
Previous analysis of the D. melanogaster transcriptome revealed that roughly 30% of genes show a two-fold or greater difference in expression between the sexes [43,44], primarily as a result of male-biased expression. To determine whether male-biased genes are more likely than other genes to be differentially expressed between populations, we used a two-fold expression difference in modENCODE data from whole adult males vs. females [44] as the cut-off to categorize a gene as male-biased, and then compared this list to the genes expressed in our experiments. We found that a greater proportion of male-biased genes (6.1% at 21°C and 10.0% at 29°C) were differentially expressed between populations compared to other genes (5.2% at 21°C and 5.8% at 29°C; hypergeometric test, both P <0.001, Table 1). This is consistent with previous results that male-biased genes tend to exhibit greater expression variation than do other genes [36,45]. To investigate whether this enrichment is associated with gonadal expression, we estimated the proportion of differentially expressed genes between populations for testis-biased genes (as defined in [41]) and for other genes. This comparison revealed no evidence of increased likelihood of differential expression for testis-biased genes (hypergeometric test, both P >0.1, S6 Table), which suggests that the enrichment observed for male-biased genes is driven by somatically expressed, sexually dimorphic genes [43,46]. In support of this, we found no evidence that male-specific genes [44], which are typically testis-specific in expression, were enriched among the genes showing expression differences between populations (hypergeometric test, both P >0.1, S6 Table).
In D. simulans, 1206 (8.9%) and 821 (6.1%) genes were differentially expressed between populations at 21°C and 29°C, respectively (FDR <0.05, Fig 1A and 1B, S2 Fig, S2 Table). Thus, compared to D. melanogaster, D. simulans shows evidence of substantially more geographic expression differentiation at 21°C and slightly less geographic differentiation at 29°C. As was true for D. melanogaster, the proportion of genes differentially expressed between populations was significantly different for 21°C vs. 29°C (χ2 test, P <0.001), though unlike D. melanogaster, this proportion was higher at 21°C rather than 29°C (Table 1). Similar to D. melanogaster, magnitudes of expression differences for significantly differentially expressed genes were greater at 29°C (Table 1). Unlike D. melanogaster, in D. simulans we observed no difference between temperature treatments with respect to the mean expression difference between populations for all expressed genes (Wilcoxon rank sum test, P = 0.79). At 29°C, slightly more differentially expressed D. simulans genes showed higher expression in Panama than in Maine (Fig 1B, light blue line, 473 vs. 348, χ2 test, P = 0.002), while at 21°C, a similar number of genes showed higher vs. lower expression in Maine and Panama (Fig 1B, purple line, 623 vs. 583, χ2 test, P >0.1), similar to the pattern observed in D. melanogaster.
In D. simulans the only significantly over-represented GO terms among differentially expressed genes were structural constituent of cuticle (S3 Table, P = 0.002 at 29°C, P = 0.001 at 21°C) and structural molecule activity (P = 1.77E-04 at 29°C, P = 0.04 at 21°C). Thus, although D. simulans exhibits somewhat greater geographic differentiation for transcript abundance, the associated genes appear to be less obviously biased across gene functions compared to D. melanogaster. To investigate whether the relatively incomplete GO annotations in D. simulans contributed to the small number of enriched GO terms, we also used the GO terms associated with D. melanogaster orthologs to perform the enrichment analysis and found the same pattern (S3 Table), suggesting that species difference may reflect a real biological phenomenon. In D. simulans, 13 and 7 of 197 expressed TFs were differentially expressed at 21°C and 29°C, respectively. Two transcription factors, GD15807 (orthologous gene of D. melanogaster acj6) and GD11523 (orthologous gene of D. melanogaster lms), were differentially expressed between populations at both temperatures.
To investigate whether male-biased genes in D. simulans are more likely than other genes to be differentially expressed between populations, we used the same criteria as those used in D. melanogaster (Methods) to categorize genes as male-biased or not. We found that at 21°C a greater proportion of male-biased genes (10.0%) were differentially expressed between populations compared to other genes (8.6%; hypergeometric test, P = 0.006, Table 1) similar to the pattern observed in D. melanogaster. However, at 29°C we observed no difference in the proportion of differentially expressed genes for male-biased (6.2%) vs. other genes (6.1%) (hypergeometric test, P >0.1).
Chromosomal Distribution of Geographically Differentially Expressed Genes
In D. melanogaster, we observed no over or under-presentation of significantly geographically differentially expressed genes on the X chromosome vs. the autosomes (χ2 test, P >0.1, Table 2). However, considering each chromosome arm separately, 3R was enriched for geographically differentially expressed genes (χ2 test, P <0.001, Table 2) at both experimental temperatures. In D. melanogaster the frequencies of cosmopolitan chromosome inversions are often negatively correlated with latitude [14,47,48]. These inversions appear to have relatively small genomic effects on the magnitude of genomic differentiation between high and low latitude populations [15,49], with the exception of chromosome 3R, which is substantially more differentiated than the other arms [14,48]. This 3R-effect appears to be driven in large part by In(3R)Payne, which shows steep latitudinal clines [47,48]. To investigate the possible influence of cosmopolitan inversions on gene expression differentiation between populations, we compared the proportion of differentially expressed genes in regions spanned by In(3R)P, In(3R)Mo, In(3L)P, In(2L)t, In(2R)Ns, and In(3R)K relative to autosomal regions not spanned by inversions. For both temperatures, we observed a greater proportion of differentially expressed genes in regions spanned by In(3R)P and In(3R)K compared to other chromosomal regions (hypergeometric tests, P <0.001, S7 Table), supporting the idea that chromosome inversions contribute to expression differentiation on 3R. This concordance between genomic differentiation and expression differentiation for 3R supports the idea that cis-acting regulatory variants play an important role in gene expression differences between populations, which is expected given the importance of such variants for expression variation within populations [41,50]. Unlike D. melanogaster, there are no common chromosome inversions segregating in D. simulans [24]. D. simulans exhibited no heterogeneity across chromosome arms in the proportion of differentially expressed genes at either temperature. However, relative to autosomal genes, X-linked differentially expressed genes were slightly underrepresented at 29°C but not at 21°C (Table 2).

Parallel Expression Differences between High and Low Latitude Populations in D. melanogaster and D. simulans
To investigate whether these closely related species exhibit parallel patterns of geographic expression differentiation (possibly associated with the colonization of new habitats), we compared differentially expressed genes in Maine vs. Panama samples from both species, using only one-to-one orthologs that satisfied our minimum expression criteria in all samples (10,085 genes). We observed 106 and 76 genes that were geographically differentially expressed in both species for 21°C and 29°C, respectively (Table 3, S8 Table). This 12.1%-26.5% overlap corresponds to a 2-to-3 fold enrichment, (hypergeometric test P <8.92E-12 for 29°C, P <3.45E-31 for 21°C), which supports the idea that a significant component of geographic differentiation in gene expression is driven by local adaptation occurring in a similar manner in these two species, though it is worth keeping in mind that a majority of differentially expressed genes is not shared between species. The fact that most (82.5%) shared, differentially expressed genes exhibit the same direction of differential expression (both species exhibit higher expression in Panama or both species exhibit higher expression in Maine) (S8 Table, Fig 2), strongly supports the proposition that parallel geographic differentiation reflects parallel adaptation. There were no significantly enriched GO terms associated with these shared differentially expressed genes (FDR >0.1) for either temperature, though the small number of genes provides little power for detecting such enrichments. Five genes, tim (a circadian rhythms gene), Cyp6a19 (a cytochrome P450), Mur18b (a chitin binding gene), CG34461 (a chitin-based cuticle gene), and CG17752, showed significant geographic differentiation in gene expression in both species at both temperatures. The T-box transcription factor Doc1 was geographically differentially expressed in both species, showing higher expression in Maine at 21°C. Three likely transcription factors (based on ortholog gene function), GD10401 (ortholog of CG30431), GD20175 (ortholog of gl), and GD14098 (ortholog of CG6765) showed significant geographic differentiation in both species, all with higher expression level in Maine at 21°C. Genes that showed parallel expression differentiation exhibited no significant heterogeneity across chromosome arms (hypergeometric tests, all P >0.05). Among the genes showing parallel gene expression differences between populations there was no obvious trend for transcript abundance to be greater in either Panama or Maine (χ2 test, P >0.1). Finally, among all expressed one-to-one orthologs, we observed a highly significant excess of parallel expression differences (5851 parallel genes vs. 4617 opposite direction genes at 29°C, 6591 parallel vs. 3594 opposite genes at 21°C, χ2 test, both P <0.001), which supports the proposition that parallel selection responses may shape expression differences even for genes that are not significantly differentially expressed in our data.

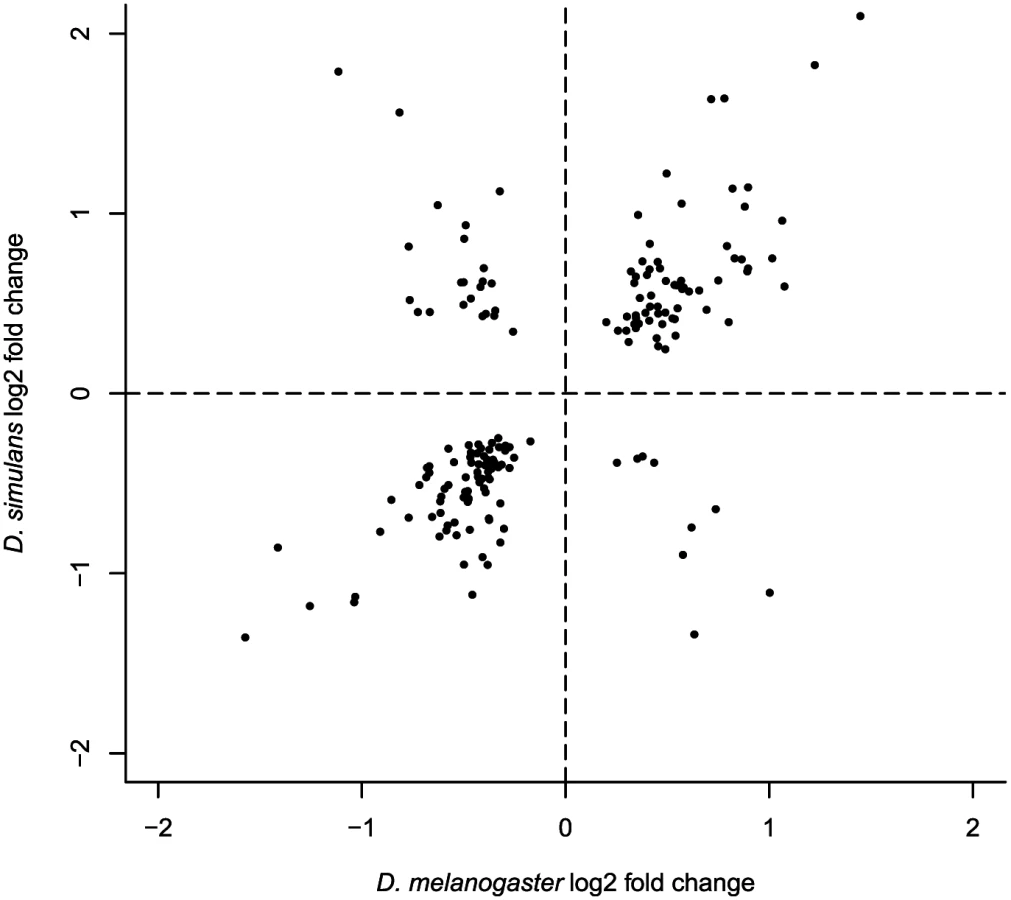
To investigate whether parallelism extends to the magnitude of population differences in transcript abundance, we estimated the interspecific correlation in fold change for genes exhibiting parallel significantly different expression. While the biased subset of data included makes it difficult to evaluate the correlation, the observed correlation (Pearson’s R2 of log2 fold changes = 0.80, Fig 2, S1 Text) appears to be remarkably high. This suggests (though by no means proves) that the relationship between expression variation and fitness variation may be quite similar for the genes influenced by spatially varying selection in the two species.
Parallel Differentiation in Gene Expression Plasticity
In D. melanogaster, 1329 (9.5%) and 2788 (19.9%) genes showed differential expression between 21°C and 29°C for Maine and Panama, respectively (Fig 1C and 1D, S3 Fig, Table 4, S9 Table), while 770 genes showed significant expression plasticity for both populations. The differences in plasticity between populations (9.5% vs. 19.9%) were highly significant (χ2 test, P <0.001), consistent with a previous microarray analysis of high and low latitude Australia populations [51]. In Maine, genes exhibiting significant temperature plasticity were enriched in structural constituent of chitin-based cuticle (P = 9.36E-4), signal peptides (P = 6.96E-04) and muscle protein (P = 2.86E-06). There was an underrepresentation of X-linked (relative to autosomal) genes showing expression plasticity for both populations (hypergeometric test, Panama P = 2.97E-05; Maine P = 6.92E-06; shared genes P = 3.06E-05), consistent with the trend reported for African and European populations [36]. Chromosome 3R was enriched for genes showing expression plasticity, but only in the Panama sample (hypergeometric test, P = 5.60E-07); this pattern is attributable to the enrichment of differentially expressed genes for inversions In(3R)K and In(3R)P. Focusing on the top 50 most differently expressed genes for temperature plasticity, 24 were shared between Panama and Maine populations, including the transcription factors ftz and twi.

In D. simulans of the 13,464 expressed genes, 1488 (11.1%) and 2147 (16.0%) showed differential expression between temperatures for Maine and Panama, respectively (Fig 1C and 1D, Table 4, S9 Table); 951 genes exhibited expression plasticity for both populations. Thus, as was the case for D. melanogaster, there appears to be greater expression plasticity for Panama than for Maine (χ2 test, P <0.001). Similar to the differences between species for geographic differentiation in expression at each experimental temperature, D. simulans exhibits greater expression plasticity than does D. melanogaster (χ2 test, P <0.001) for the Maine samples. However, compared to D. melanogaster, D. simulans exhibited a smaller proportion of genes with expression plasticity for Panama (χ2 test, P <0.001). For D. simulans, there was no significant difference in plasticity between X and autosome, however, chromosome 3R was enriched for differentially expressed genes in both populations (Panama, hypergeometric test, P = 2.96E-06; Maine, hypergeometric test, P = 0.01), in spite of the fact that unlike D. melanogaster, the D. simulans pattern cannot be associated with inversion polymorphism. Focusing on the top 50 most differently expressed genes for temperature plasticity, 6 and 9 genes in Panama and Maine, respectively, were involved in chitin-based cuticle development. Other highly differentially expressed genes in both Panama and Maine populations include the D. simulans orthologs of CG7214, Ance-3, tim, Rh5, retinin, Acyp, and boss, some of which have functions related with visual perception. Note that Ance-3 and Acyp are adjacent, suggesting the possibility of shared regulatory information associated with plasticity.
To compare expression plasticity in the two species, we restricted our analysis to one-to-one orthologs that satisfied our minimum expression criteria in all samples. Plasticity was highly conserved between species. For example, for Maine, 375 of 909 genes (41.25%) showed expression plasticity in both species (S10 Table), while for Panama, 861 of 1995 (42.16%) genes exhibited plasticity for both species (S10 Table). For Maine, all 375 genes that showed plasticity for both species exhibited the same direction of differential expression (i.e., both species showed higher expression at 21°C or both showed higher expression at 29°C). Of these 375 genes, 207 showed higher expression at 29°C while 168 showed higher expression at 21°C (χ2 test, P = 0.17). For Panama, among the 861 genes that showed plasticity for both species, 830 (96.4%) exhibited the same direction of differential expression; 282 and 548 of these genes showed greater at 29°C and 21°C, respectively. Thus, in Panama, there were significantly more shared genes that show higher expression at 21°C (χ2 test, P <0.0001). We observed 229 genes showing temperature plasticity in Maine and Panama for both species; 105 and 117 showed higher expression at 21°C and 29°C, respectively. Only 7 of these 229 genes showed different directions of expression plasticity across species, further supporting the idea that expression plasticity is generally conserved.
To compare geographic differences in expression plasticity in the two species, we identified for each species, the genes showing temperature related expression plasticity in either Maine or Panama (but not both) and then determined whether the same genes tend to show population-specific plasticity in the two species. Remarkably, the geographic differences in plasticity exhibited significant parallelism. There were 1804 (17.9%) and 1298 (12.9%) genes that showed geographic differences in plasticity in D. melanogaster and D. simulans, 380 (21.1% and 29.3%) of which were shared between the two species, which represents an at least 1.64 fold enrichment (hypergeometric test, P = 1.42E-27). Thus, there is a component of plasticity variation that is highly conserved and another component showing evidence of recent, parallel local adaptation.
Finally, we compared our D. melanogaster transcriptome data to previously published expression microarray data on expression plasticity for high and low latitude Australian D. melanogaster populations [51] reared at 18°C and 30°C. We compared the top 300 differentially expressed genes in Innisfail (Queensland, low latitude) to differentially expressed genes in Panama, and found that 129 genes were shared between the two populations. Similarly, the Cygnet (Tasmania, high latitude) and Maine populations shared 117 genes with temperature plasticity (S11 Table). 112 of 129 (86.8%) and 108 of 117 (92.3%) of the above genes showed the same pattern of increased vs. decreased expression in response to temperature for our data from Maine and Panama compared to Australia populations. This suggests, not surprisingly given the parallel temperature response differentiation observed between species, that high vs. low latitude D. melanogaster populations in the Americas and Australia exhibit similar patterns of gene expression temperature response.
Genetic Differentiation Associated with Geographic Variation in Transcript Abundance
Given previous work supporting the importance of cis-acting variants on gene expression variation in D. melanogaster [41,50,52,53], we investigated whether genes exhibiting geographic differences in transcript abundance also tend to harbor differentiated SNPs. To do so, we used our transcriptome data to estimate SNP frequencies in Maine and Panama [54] and then estimated FST (see methods). We found that genes differentially expressed at 29°C were enriched for highly differentiated SNPs (0.5% FST outlier) in the 3’UTRs, 5’UTR and CDS regions, while there was significant enrichment of outlier FST SNPs at 3’UTR and 5’UTR for genes differentially expressed at 21°C (Table 5, S12 Table, S13 Table), consistent with our observation that there is greater geographic differentiation at 29°C than at 21°C (Table 1). To determine whether the magnitude of genetic differentiation was associated with the magnitude of gene expression differentiation, we estimated for all expressed genes and for significantly differentially expressed genes the proportion of SNPs in the 0.5% and 0.25% tail FST and found no significant correlation between FST and expression differentiation for either gene set (Pearson’s r <0.1, ANOVA P >0.1). There are many possible explanations for the lack of such a correlation including various limitations of our data and the possible genetic complexity of regulatory variation and its interaction with fitness across genes.

Because estimates of SNP differentiation from RNA-seq data may be influenced by expression differentiation (depending on linkage disequilibrium between cis-eQTLs and nearby SNPs), we extended the analysis of genetic and expression differentiation using previously published genomic data from Maine and Florida populations of D. melanogaster [15]. We compared our significantly differentially expressed genes to the genes overlapping the 1% most extreme 1-kb FST windows in the genome [15]. We observed highly significant overlap between the two sets of genes at 29°C (P = 3.58E-07) and marginally significant overlap at 21°C (P = 0.001), which is consistent with our SNP-based analysis. These results are consistent with an important role for cis-acting variants in gene expression differences between populations [55,56].
The D. simulans genome annotation is of substantially lower quality than that of D. melanogaster, leading to reduced power to detect phenomena such as annotation enrichments. Nevertheless, we wanted to determine if this species also exhibits evidence of genetic differentiation in differentially expressed genes. We found that 3’ UTRs and CDSs of expressed genes differentially expressed at 21°C were significantly enriched with highly differentiated SNPs (Table 5) but only CDS regions were enriched with FST outliers at 29°C. Thus, similar to the pattern observed in D. melanogaster, the temperature associated with a greater number of differentially expressed genes tends to exhibit greater enrichment of SNP outliers.
Focusing on the outlier SNPs in the shared differentially expressed genes (106 at 21°C and 76 at 29°C, with a total of 177 genes), we observed that the corresponding genes were enriched for outlier SNPs to a greater extent than all differentially expressed genes (37 for D. melanogaster, 46 for D. simulans, 10 were shared, χ2 test, P = 0.018). Inspection of outlier SNPs in the shared differentially genes revealed no SNPs at homologous sites in the two species, consistent with relatively low levels of shared polymorphism in these species [57].
To investigate parallel geographic differentiation more broadly, we used data from all 10,085 expressed one-to-one orthologs to identify the SNPs in the top 0.5% and 0.25% of the FST distribution and their corresponding genes and gene annotation (UTRs or CDS). We first examined the 6867 one-to-one orthologs associated with UTR SNPs in both species. The 0.25% tail UTR SNP FST outliers were associated with 304 and 163 genes in D. melanogaster and D. simulans, respectively; the 12 genes shared between species represents a marginally significant (P = 0.057) enrichment. The comparable analysis using the 0.5% tail UTR outliers, yielded 555 and 337 genes in D. melanogaster and D. simulans, respectively, of which 42 were shared between species (P = 0.002). We then examined the 9479 orthologous genes with CDS SNPs in both species. We observed an excess of shared genes associated with the top 0.25% CDS SNPs (570 genes in D. melanogaster, 661 genes in D. simulans, 62 shared, P = 2.66E-05) and 0.5% (1032 genes in D. melanogaster, 1136 genes in D. simulans, 197 shared, P = 1.64E-12). These shared genes are associated with substantially longer CDS than other genes. To address the possibility that the excess of shared genes is simply an artifact of gene size, we created an empirical distribution by sampling gene sets of the same size as the observed data and for which the number of SNPs in each gene matched the number in the observed genes such that the total number of SNPs and distribution of SNPs across genes was exactly the same in the observed data and the sampled gene sets. We repeated this 1000 times in each species to generate an empirical distribution of the number of genes harboring at least one shared 0.25% CDS FST SNP. We found that the expected (median) number of shared CDS was 36 CDS, while the observed number of shared CDS was 62 (P <0.001). Of the 2032 and 1766 0.25% outlier SNPs identified in D. melanogaster and D. simulans, respectively, only 2 were outliers in both species.
Splice Junction and Isoform Differentiation
In D. melanogaster we observed 191 annotated splice junctions in 175 genes that showed significantly different expression in Maine vs. Panama at 21°C. At 29°C we observed differential usage of 732 splice junctions in 546 genes (S14 Table). Thus, as was the case for transcript abundance, it appears that there is more differentiation for alternative splice junction use at 29°C. In D. simulans, we observed 870 splice junctions (in 743 genes) that differed in abundance between Maine and Panama at 21°C and 432 (in 383 genes) at 29°C (S14 Table). This, too, is consistent with the transcript abundance data, and shows the two species have different degrees of expression differentiation at these two temperatures. We observed 20 and 21 splice junctions showing differential expression in both species at 21°C and 29°C, respectively (S15 Table); 37 of the 41 shared splicing junctions shared the same expression direction (for example both showed higher expression in Maine or Panama) in both species, which supports the idea that they have been influenced by parallel spatially varying selection.
We estimated relative isoform usage across D. melanogaster samples to formally identify possible instances of alternative transcripts exhibiting geographic differentiation. We found 373 and 414 isoforms differentially expressed at 21°C and 29°C, respectively. For most such genes, significant variation was observed only for one transcript. Furthermore, most of these differentially expressed alternative transcripts affected only the choice of UTR. This is consistent with version 5.55 of the D. melanogaster annotation, for which only 25.5% (3557 of 13937) genes were associated with multiple protein isoforms (the remaining isoforms differing only in the UTRs).
Population × Temperature Interaction
Gene-by-environment interactions (GEI) may play a role in the maintenance of genetic variation in the presence of spatially varying selection, even if the optimum phenotype does not vary with geography [58]. While we did not use defined individual genotypes in our experiments, we estimated population × temperature interactions as (Maine at 21°C-Maine at 29-C)-(Panama at 21°C-Panama at 29°C) following Levine et al. [51]; for convenience we refer to this as GEI. At an FDR of 0.05, 264 D. melanogaster genes showed significant GEI (S16 Table, Fig 3). GO analysis showed that these genes were highly associated with neurological system process, visual perception, sensory perception of light stimulus, and signal transduction (P <0.05, FDR<0.05, S17 Table), consistent with our finding that genes associated with vision show substantial geographic expression differences. Using the same approach in D. simulans, only 7 genes showed significant GEI (11 genes with FDR increased to 0.1, S16 Table). Thus, it appears that the species are quite different in the extent of GEI across the genome. To further investigate this potential species difference, we summarized gene expression data from both species to identify genes for which the rank order of transcript abundance in the two populations differs across temperatures (and further requiring the log2 of the sum of the absolute difference of the fold difference for the two temperatures to be greater than 0.5; genes satisfying these criteria can be thought of as showing the classic crossing of reaction norms). We observed 754 and 622 such genes in D. melanogaster and D. simulans, respectively. Thus, the proportion of genes showing this pattern is only slightly greater in D. melanogaster (χ2 test, P = 0.004), though the magnitude of the effect is substantially greater than it is in D. simulans. To further characterize the difference between species we determined the proportion of the 754 and 622 genes for which the slopes of the lines connecting the expression estimate for each temperature were of different sign (positive vs. negative) in the two populations. In D. melanogaster, 644 of the 754 genes showed such a pattern while only 268 of 622 genes showed such a pattern in D. simulans. Focusing on 200 genes exhibiting the greatest population × temperature interaction in each species, 196 D. melanogaster genes showed the classic crossover between populations at 21°C and 29°C while fewer than 150 D. simulans genes showed such a pattern. These patterns all support the idea that population × temperature interactions are of greater magnitude in D. melanogaster than in D. simulans.

Discussion
The most important conclusion from the data and analyses reported here is that these two species exhibit significant parallelism with respect to low vs. high latitude gene expression differences. The most parsimonious explanation for the quantitative parallelism is that for a subset of the genome the relationship between transcript abundance and fitness variation across heterogeneous environments is similar in the two species. Given previous population genomic analyses of these species [14,15] and the short timescales of colonization of the Americas for both species [11], selection on standing variation is likely to underlie geographic expression differences for both species. Whether or not the observed expression differentiation is associated with spatially varying selection in the ancestral ranges of the species is an open question. In both species, differentially expressed genes show enrichment for differentiated 3’UTR SNPs. Given the possible role of 3’ UTRs in regulating transcript abundance [59,60] and in light of previous findings of high levels of geographic differentiation in 3’UTRs in whole genome analyses [49], the hypothesis that such cis-acting variation is responsible for some of the parallel gene expression differentiation is worth investigating. While there is little evidence for shared differentiated 3’UTR SNPs in genes exhibiting parallel expression differences in the two species, which is consistent with previous analyses of these two species [57], the more general question of the possible role of parallel non-coding regulatory variation at the level of individual SNPs or regulatory elements in the two species awaits more comprehensive genetic and genomic analyses. It is also worth noting that we observed a substantial number of transcription factors that exhibited expression differences between populations; their role in generating the patterns observed here also remains to be determined.
In addition to the observed interspecific parallelism for transcript abundance, we found substantial parallelism for geographic differences in expression plasticity in the two species. This supports the hypothesis that spatially varying selection influences plasticity, in line with previous results suggesting that selection is the dominant process shaping variation in Drosophila [61]. We found for both species, greater expression plasticity for Panama than for Maine populations. Though the classic view is that selection may favor genotypes associated with greater phenotypic plasticity in temperate environments [62], our results do not support that view. If the regulation of expression plasticity often involves trans-acting elements [63,64] then the geographic patterns of expression plasticity observed here may be attributable to trans-acting elements such as transcription factors. Such hypotheses are certainly amenable to testing by further population genomic and functional analysis.
In spite of the evidence for parallelism, there are many differences between species with respect to differentially expressed genes. Moreover, the degree of functional annotation enrichment associated with expression differences is much greater in D. melanogaster than in D. simulans. One interpretation of this difference is that selection is distributed across a much greater range of biological functions in D. simulans than in D. melanogaster, which would be remarkable given their very recent ancestry and sympatric distribution in recently colonized habitats. Unfortunately, an alternative explanation for this difference is simply that the quality of functional annotation in the two species is very different, though our attempt to circumvent this limitation by using the GO terms associated with D. melanogaster orthologs for D. simulans did not show greater enrichment either. A high quality annotation of D. simulans will be indispensable for incisive analysis of similarities and differences between these species. Nevertheless, we can state with some confidence that the visual system of D. melanogaster appears to be influenced by strong spatially varying selection, while D. simulans reveals little evidence of a comparable phenomenon. We do not have any particularly attractive hypotheses to explain this difference. However, we note that one apparently major difference in the visual ecology of these species is that D. melanogaster requires no light for successful courtship while D. simulans does [65]. Alternatively, because the visual system has multiple functions including detection of visible and non-visible (UV) light, temperature response (such as TRP-dependent cold/hot response, see review [66]), locomotor [67,68], and circadian behaviors [68] or photoperiodism [69], it is difficult to formulate clearly articulated hypotheses on the possible interspecific differences in ecology that may impinge upon the local adaptation via the visual system in the two species. Cross-talk between pathways for perception of temperature and light [70,71] further complicates the situation. Finally, we cannot rule out a contribution of geographic differences in relative eye size to the observed GO enrichment in D. melanogaster. In D. simulans, we found cuticle genes are enriched among the genes showing expression differences between temperatures and between populations, while no such enrichment was observed in D. melanogaster. A recent genomic analysis [72] showed that cuticle genes in D. simulans are overrepresented among recently duplicated genes which may have undergone rapid adaptive evolution in D. simulans. Geographic variation in gene copy number could certainly be related to some of the observations reported here.
Another obvious genomic differences between species was that in D. simulans a greater proportion of genes showed geographic expression differences at 21°C, while in D. melanogaster a greater proportion of genes showed expression differences at 29°C. It is difficult to interpret this difference given only the available data, but it would certainly be of great interest to carry out comparable experiments on populations from the ancestral ranges of both species to investigate how the geography of ancestral and recently established populations might illuminate this species difference.
Another noteworthy species difference is the substantially greater population temperature interactions observed in D. melanogaster than in D. simulans. A possible explanation for these differences is that optimal D. melanogaster expression phenotypes are similar in high and low latitude populations experiencing different environments; under such circumstance alleles exhibiting GEI might be favored and exhibit different allele frequencies but the associated genotypes would have similar phenotypes. In this way of thinking, the weaker GEI in D. simulans is a consequence of the fact that compared to D. melanogaster, high and low latitude populations more often have different optimal expression phenotypes. Of course, such speculation offers no explanation for why these species might have such different expression/fitness functions in high vs. low latitude environments.
Finally, and in contrast to most previous studies of D. simulans, our results reveal as much (or more) geographic differentiation in D. simulans as in D. melanogaster. Perhaps the conventional wisdom that D. simulans exhibits less phenotypic clinality than does D. melanogaster (which is based on relatively few data) needs to be further evaluated [73] ideally in multiple environments. Additional studies, perhaps guided by genomic inferences such as those presented here, might provide evidence of parallel latitudinal differentiation for a great many intermediate phenotypes.
Materials and Methods
Sample Preparation
D. melanogaster and D. simulans females were collected from Fairfield, Maine (September 2011, Latitude: 44°37’N) and Panama City, Panama (January 2012, Latitude: 8°58’N), placed individually on vials and then shipped to the laboratory where they were maintained as isofemale lines. Isofemale lines were maintained at 25°C on a standard yeast-cornmeal-agar food medium. Experimental animals were generated from these lines in Spring 2012. To generate experimental animals for each line, 40 freshly laid eggs were picked and placed onto a vial, which was then incubated at either of two temperature treatments, 21°C or 29°C, at synchronized 12 : 12 hour Light:Dark. Virgin males were collected within 3 hours after eclosion. After 48 hours an individual male was randomly picked from each isofemale line without anesthetics to create a biological replicate for each population and temperature; a total of three biological replicates were created for each population/temperature combination. Flies were frozen in Trizol and stored at -80°C before RNA was extracted following a standard Trizol-chloroform extraction protocol. The numbers of isofemale lines contributing to each biological replicate were as follows. For the 21°C treatment, we used 29 D. melanogaster Panama strains and 30 Maine strains; we used 17 D. simulans Panama strains and 29 Maine strains. For the 29°C treatment, we used 30 D. melanogaster Panama strains and 30 Maine strains; we used 15 D. simulans Panama strains and 30 Maine strains. We used a relatively large number of strains from each population with three replicates. Thus, although we have no method of partitioning expression variance across individuals within and between populations, we make the reasonable assumption that for the majority of genes we obtain useful estimates of mean population expression phenotypes.
Sequencing, Assembly and Data Filtering
Poly(A)+ RNA was prepared from an aliquot of each total RNA sample. Individual libraries were constructed using the Illumina Truseq RNA kit with insert size 170–200 bp and sequenced on an Illumina HiSeq machine using paired-ends chemistry and 90 cycles. Clean reads were deposited to NCBI under BioProject number PRJNA260940 and SRA number SRP047141. Filtered, clean reads for each sample/replicate were aligned independently to a reference genome using the Bowtie-based TopHat program [74]. D. melanogaster reads were mapped to reference genome (FlyBase r5.55). D. simulans reads were mapped to dsimv2 [75] and reference genome (FlyBase r1.4). Only uniquely mapped reads (Q >20 for bases and Q >30 for reads) were kept for further analysis. For each species, there were more than 300 million paired-end reads (60 gigabases) aligned to the each genome.
Gene Differential Expression Analysis
We measured differential expression with DESeq2 [76] (version 1.4.5), edgeR [77] (version 3.0.8) and voom-limma [78] (version 3.20.8). We first adopted Bedtools to estimate a read count for each gene. Genes with a minimum 10 counts average were kept for further analysis. We then used the Bioconductor package (version 2.14) in R to analyze read counts. We used DESeq2, edgeR, and voom-limma with empirical Bayes estimation and exact tests based on a negative binomial model [77] or linear modeling [78]. The Benjamini—Hochberg procedure was used to control the false discovery rate [79] for all methods. We observed that the different methods returned highly consistent count estimates. Here were present results from DESeq2 differentially expressed genes because these results showed the greatest consistency compared to the other two methods. For gene plasticity results, only genes showing significant differences between temperatures by at least two methods were kept for further analysis. All major conclusions related to gene lists, GO enrichment, and parallel expression differences between species were supported by either method of normalization. We also used Cufflinks [80] to calculate the expression value (FPKM) with upper quantile normalization. To examine differential expression of genes/transcript between different samples, we used Cuffdiff2 [80] with all three biological replicates, and then obtained genes/transcripts with significant differences (P <0.05 and the FDR <0.05 after Benjamini-Hochberg correction for multiple-testing). Using this program, we also estimated isoform usage for each gene.
Orthologous Genes Analysis
To infer orthologous genes in D. melanogaster and D. simulans, we used MCscan [81] to determine synteny relationships for all genes in both reference genomes. We then identified reciprocal best hits for all genes in each pairwise species comparison using BLASTP (at alignment length >50%, similarity >70%). Genes having reciprocal best hits and shared synteny were defined as orthologous. Single-copy genes that were reciprocal best hits but were not syntenic were also defined as orthologs because such cases are easily explained by genomic rearrangements. Using this approach we identified about 11,000 genes as orthologous in the two species, of which, 10,085 genes were expressed in the dataset, and thus used to investigate parallel gene expression differentiation. We used modENCODE developmental stage RNAseq data from whole male and whole female adults [44] to identify male-biased genes in D. melanogaster. We used modENCODE D. simulans male (NCBI SRA SRR166817 and SRR166818) and female (NCBI SRA SRR166815 and SRR166816) to identify D. simulans male-biased genes. Only high quality unique reads were mapped to each reference sequence using TopHat, which were then used to estimate expression with Cufflinks/Cuffdiff2. For genes that showed FPKM >2 in the male sample, if expression was more than two fold greater than that observed for the female sample, we defined the gene as male-biased. Using this method, we identified 3920 and 3546 such genes for D. melanogaster and D. simulans, respectively. We calculated the tissue specificity index (tau) of each gene using FlyAtlas data [42]. For each gene, we obtain a tau value and recorded the tissue in which expression was highest. To identify testis-biased genes we used FlyAtlas data with tau >0.9 [41]. We also downloaded male-specific genes from Graveley et al. [44].
Data Comparison and Analysis
We used a FDR adjusted P-value <0.05 as the cutoff for differential expression. For geographic comparisons we also calculated absolute fold changes for all genes to estimate genome-wide expression variation. The fold changes were transformed to absolute fold change values (Panama population expression as 1). Differential gene expression enrichment was tested using hypergeometric or χ2 tests. All gene location, UTR, CDS information used in the analysis were downloaded and extracted from FlyBase r5.55. GEI interaction was estimated following Levine et al. [51] using the limma [78] package. Inversion breakpoints were from Corbett-Detig and Hartl [82]. We generated empirical distributions to determine whether differentially expressed genes are physically clustered. We randomly picked a number genes on each chromosome arm corresponding to the observed differentially expressed genes, calculated the distance between genes, and repeated this 1000 times. We compared the median distances separating genes in the observed differentially expressed genes and the randomly sampled genes.
Splicing Isoforms
We extracted the reads that spanned the junctions from TopHat-mapped bam files. For each such read we identified the junction site (including intron location, chromosome location and left and right of the junction sites) and calculated the read count (coverage) for each junction (as well as introns). We then extracted annotated intron information from FlyBase (data extracted from D. melanoagaster FlyBase r5.55 and D. simulans r1.4) and compared the introns/junctions between identified ones with annotated introns. We then fed the junction counts into R and calculated if splice junction abundance was heterogeneous with FDR <0.05. For alternative splicing, we used Cufflinks and Cuffdiff2 to detect different splicing isoforms and their expression with FDR <0.05. To identify shared differential junction use between species, we converted D. simulans junction/intron coordinates to the D. melanogaster assembly using UCSC liftOver.
Functional Annotation
We used DAVID [33] to compare enrichment for functional terms among groups of genes. DAVID’s tools use a modified Fisher’s exact test (the EASE score) to determine the extent of enrichment for a subset of genes compared to a specified background. GO categories that were significantly enriched at a false discovery rate <0.05 and Bonferroni corrected P value <0.05 were used. We also used GOTermFinder [34] to confirm the DAVID results for D. melanogaster genes.
Transcriptomic FST Analysis
We took advantage of the high coverage RNA-seq data to calculate transcriptomic FST in both D. melanogaster and D. simulans. For reads that aligned to D. melanogaster and D. simulans genome, we removed sites that had coverage <10 and those for which a SNP was supported by only a single read. SNPs and coverage were then calculated and extracted by SAMtools mpileup [83]. SNP frequencies for each population (3 biological replicates pooled together; median coverage for the SNPs were 494 for D. melanogaster and 220 for D. simulans) were then calculated. In order to obtain the first dataset of population SNP differentiation, we calculated a non-conventional FST using RNA-seq data. SNP FST were estimated by Popoolation2 [84]; we removed SNPs with frequency <0.01. We then identified SNPs in top 1%, 0.5% and 0.25% FST tail on each chromosome arm. Each SNP was assigned to one of three categories, 5’ UTR, 3’ UTR or CDS. We then determined the overlap between genes associated with tail SNPs and those showing geographic differential expression at 29°C or 21°C. For D. simulans, we used FlyBase r1.4 CDS coordinates. For 3’ UTR and 5’ UTR, we used the UCSC liftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver) coordinates from D. melanogaster UTRs. We determined whether differentiated expressed genes were enriched in locations with high FST using 1kb FST data generated by Reinhardt et al. [15]. We found the conclusions using transcriptomic FST were consistent with conclusions using genomic FST from Reinhardt et al. [15]. Hypergeometric tests were used to calculate the P-values and expressed UTRs, CDSs or mRNAs were used as the population size for each test.
To determine whether the number of shared genes/CDS with FST outliers in D. simulans and D. melanogaster is influenced by gene/CDS size, we carried out 1000 independent bootstraps to obtain an empirical distribution of shared outlier genes considering the number of SNPs in each gene. We first counted the number of correlated outlier SNPs in the genes that have 0.25% FST tail, for example in D. melanogaster there were 386 genes having one SNP outlier, 196 genes having two SNP outliers, 2 to 86 genes that has 3 to 9 SNP outliers and 6 genes with more than 10 SNP outliers. In D. simulans we calculated the same SNP outlier properties for each gene. We then randomly picked genes/CDS that have number SNPs equal to or greater than the observed SNPs in the shared genes in each species, and then calculated the number of shared orthologous genes/CDS in D. melanogaster and D. simulans. After repeating 1000 times, we obtained the empirical distribution and P-values.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Futuyma D. Evolution. Third Edit. Sinauer Associates, Inc. 2013; p. 656.
2. Carson HL. Three flies and three islands: parallel evolution in Drosophila. Proc Natl Acad Sci U S A. 1974;71 : 3517–3521. 4530320
3. Anderson PR, Oakeshott JG. Parallel geographical patterns of allozyme variation in two sibling Drosophila species. Nature. 1984;308 : 729–731
4. Sucena E, Delon I, Jones I, Payre F, Stern DL. Regulatory evolution of shavenbaby/ovo underlies multiple cases of morphological parallelism. Nature. 2003;424 : 935–938. 12931187
5. Wood TE, Burke JM, Rieseberg LH. Parallel genotypic adaptation: When evolution repeats itself. Genetica. 2005;123 : 157–170. 15881688
6. Steiner CC, Weber JN, Hoekstra HE. Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biol. 2007;5 : 1880–1889.
7. Linnen CR, Kingsley EP, Jensen JD, Hoekstra HE. On the origin and spread of an adaptive allele in deer mice. Science. 2009;325 : 1095–8. doi: 10.1126/science.1175826 19713521
8. Rosenblum EB, Römpler H, Schöneberg T, Hoekstra HE. Molecular and functional basis of phenotypic convergence in white lizards at White Sands. Proc Natl Acad Sci U S A. 2010;107 : 2113–2117. doi: 10.1073/pnas.0911042107 20080544
9. Grant BS, Owen DF, Clarke CA. Parallel rise and fall of melanic peppered moths in America and Britain. J Hered. 1996;87 : 351–357.
10. Jeffery WR. Regressive evolution in Astyanax cavefish. Annu Rev Genet. 2009;43 : 25–47. doi: 10.1146/annurev-genet-102108-134216 19640230
11. David J, Capy P. Genetic variation of Drosophila melanogaster natural populations. Trends Genet. 1988;4 : 106–111. 3149056
12. Singh RS, Long AD. Geographic variation in Drosophila: From molecules to morphology and back. Trends Ecol Evol. 1992;7 : 340–345. doi: 10.1016/0169-5347(92)90127-W 21236059
13. Hoffmann AA, Shirriffs J, Scott M. Relative importance of plastic vs genetic factors in adaptive differentiation: Geographical variation for stress resistance in Drosophila melanogaster from eastern Australia. Funct Ecol. 2005;19 : 222–227.
14. Fabian DK, Kapun M, Nolte V, Kofler R, Schmidt PS, Schlötterer C, et al. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Mol Ecol. 2012;21 : 4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x 22913798
15. Reinhardt JA, Kolaczkowski B, Jones CD, Begun DJ, Kern AD. Parallel geographic variation in Drosophila melanogaster. 2014;197 : 361–373. doi: 10.1534/genetics.114.161463 24610860
16. Keller A. Drosophila melanogaster’s history as a human commensal. Curr Biol. 2007; 17: R77–81. 17276902
17. Stephan W, Li H. The recent demographic and adaptive history of Drosophila melanogaster. Heredity. 2007;98 : 65–68. 17006533
18. Duchen P, Zivkovic D, Hutter S, Stephan W, Laurent S. Demographic inference reveals African and European admixture in the North American Drosophila melanogaster population. Genetics. 2013;193 : 291–301. doi: 10.1534/genetics.112.145912 23150605
19. Dean MD, Ballard JWO. Linking phylogenetics with population genetics to reconstruct the geographic origin of a species. Mol Phylogenet Evol. 2004;32 : 998–1009. 15288072
20. Aquadro CF, Lado KM, Noon WA. The rosy region of Drosophila melanogaster and Drosophila simulans. I. Contrasting levels of naturally occurring DNA restriction map variation and divergence. Genetics. 1988;119 : 875–88. 2900794
21. Begun DJ, Lindfors HA, Kern AD, Jones CD. Evidence for de novo evolution of testis-expressed genes in the Drosophila yakuba/Drosophila erecta clade. Genetics. 2007;176 : 1131–1137. 17435230
22. Knibb WR. Chromosome inversion polymorphisms in Drosophila melanogaster II. Geographic clines and climatic associations in Australasia, North America and Asia. Genetica. 1982;58 : 213–221.
23. Turner TL, Levine MT, Eckert ML, Begun DJ. Genomic analysis of adaptive differentiation in Drosophila melanogaster. Genetics. 2008;179 : 455–473. doi: 10.1534/genetics.107.083659 18493064
24. Ashburner M, Lemeunier F. Relationships within the melanogaster species subgroup of the genus Drosophila (Sophophora). I. Inversion polymorphisms in Drosophila melanogaster and Drosophila simulans. Proc R Soc B Biol Sci. 1976;193 : 137–157. 5729
25. Singh RS, Choudhary M, David JR. Contrasting patterns of geographic variation in the cosmopolitan sibling species Drosophila melanogaster and Drosophila simulans. Biochem Genet. 1987;25 : 27–40. 3107542
26. Singh RS. Population genetics and evolution of species related to Drosophila melanogaster. Annu Rev Genet. 1989;23 : 425–53. 2515792
27. Arthur AL, Weeks AR, Sgrò CM. Investigating latitudinal clines for life history and stress resistance traits in Drosophila simulans from eastern Australia. J Evol Biol. 2008;21 : 1470–1479. doi: 10.1111/j.1420-9101.2008.01617.x 18811666
28. Gibert P, Moreteau B, Pétavy G, Karan D, David JR. Chill-coma tolerance, a major climatic adaptation among Drosophila species. Evolution. 2001;55 : 1063–1068. 11430643
29. Imasheva AG, Bubli OA, Lazebny OE. Variation in wing length in Eurasian natural populations of Drosophila melanogaster. Heredity. 1994;72 : 508–514. 8014061
30. James AC, Azevedo RB, Partridge L. Cellular basis and developmental timing in a size cline of Drosophila melanogaster. Genetics. 1995;140 : 659–666. 7498744
31. van’t Land J, van Putten P, Zwaan B, Kamping A, van Delden W. Latitudinal variation in wild populations of Drosophila melanogaster: heritabilities and reaction norms. J Evol Biol. 1999;12 : 222–232.
32. Calboli FCF, Kennington WJ, Partridge L. QTL mapping reveals a striking coincidence in the positions of genomic regions associated with adaptive variation in body size in parallel clines of Drosophila melanogaster on different continents. Evolution. 2003;57 : 2653. 14686541
33. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4 : 44–57. doi: 10.1038/nprot.2008.211 19131956
34. Boyle EI, Weng S, Gollub J, Jin H, Botstein D. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20 : 3710–5. 15297299
35. Telonis-Scott M, Hallas R, McKechnie SW, Wee CW, Hoffmann AA. Selection for cold resistance alters gene transcript levels in Drosophila melanogaster. J Insect Physiol. 2009;55 : 549–555. doi: 10.1016/j.jinsphys.2009.01.010 19232407
36. Hutter S, Saminadin-Peter SS, Stephan W, Parsch J. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biol. 2008;9: R12. doi: 10.1186/gb-2008-9-1-r12 18208589
37. Huylmans AK, Parsch J. Population - and Sex-Biased Gene Expression in the excretion organs of Drosophila melanogaster. G3 (Bethesda). 2014;4 : 2307–15. doi: 10.1534/g3.114.013417 25246242
38. Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005: re3. 15728426
39. Chen Y, Lee SF, Blanc E, Reuter C, Wertheim B, Martinez-Diaz P, et al. Genome-wide transcription analysis of clinal genetic variation in Drosophila. PLoS One. 2012;7: e34620. doi: 10.1371/journal.pone.0034620 22514645
40. Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21 : 650–659. 15388519
41. Zhao L, Saelao P, Jones CD, Begun DJ. Origin and spread of de novo genes in Drosophila melanogaster populations. Science. 2014;343 : 769–772. doi: 10.1126/science.1248286 24457212
42. Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39 : 715–720. 17534367
43. Parisi M, Nuttall R, Edwards P, Minor J, Naiman D, Lü J, et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5: R40. 15186491
44. Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471 : 473–479. doi: 10.1038/nature09715 21179090
45. Meiklejohn CD, Parsch J, Ranz JM, Hartl DL. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci U S A. 2003;100 : 9894–9899. 12907700
46. Chang PL, Dunham JP, Nuzhdin S V, Arbeitman MN. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics. 2011;12 : 364. doi: 10.1186/1471-2164-12-364 21756339
47. Voelker RA, Cockerham CC, Johnson FM. Inversions fail to account for allozyme clines. Genetics. 1978;88 : 515–527. 17248810
48. Kapun M, Van Schalkwyk H, McAllister B, Flatt T, Schlötterer C. Inference of chromosomal inversion dynamics from Pool-Seq data in natural and laboratory populations of Drosophila melanogaster. Mol Ecol. 2014;23 : 1813–1827. doi: 10.1111/mec.12594 24372777
49. Kolaczkowski B, Kern AD, Holloway AK, Begun DJ. Genomic differentiation between temperate and tropical Australian populations of Drosophila melanogaster. 2011;187 : 245–260. doi: 10.1534/genetics.110.123059 21059887
50. Massouras A, Waszak SM, Albarca-Aguilera M, Hens K, Holcombe W, Ayroles JF, et al. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 2012;8: e1003055. doi: 10.1371/journal.pgen.1003055 23189034
51. Levine MT, Eckert ML, Begun DJ. Whole-genome expression plasticity across tropical and temperate Drosophila melanogaster populations from Eastern Australia. Mol Biol Evol. 2011;28 : 249–56. doi: 10.1093/molbev/msq197 20671040
52. Wittkopp PJ, Haerum BK, Clark AG. Evolutionary changes in cis and trans gene regulation. Nature. 2004;430 : 85–8. 15229602
53. Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 2012;13 : 59–69. doi: 10.1038/nrg3095 22143240
54. Gayral P, Melo-Ferreira J, Glémin S, Bierne N, Carneiro M, Nabholz B, et al. Reference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap. PLoS Genet. 2013;9: e1003457. doi: 10.1371/journal.pgen.1003457 23593039
55. Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8 : 206–216. 17304246
56. Saminadin-Peter SS, Kemkemer C, Pavlidis P, Parsch J. Selective sweep of a cis-regulatory sequence in a non-African population of Drosophila melanogaster. Mol Biol Evol. 2012;29 : 1167–1174. doi: 10.1093/molbev/msr284 22101416
57. Langley CH, Stevens K, Cardeno C, Lee YCG, Schrider DR, Pool JE, et al. Genomic Variation in Natural Populations of Drosophila melanogaster. Genetics. 2012;192 : 533–598. doi: 10.1534/genetics.112.142018 22673804
58. DeWitt TJ, Scheiner SM. Phenotypic plasticity: functional and conceptual approaches. New York Oxford Univ Press. 2004; 247.
59. Kuersten S, Goodwin EB. The power of the 3’ UTR: translational control and development. Nat Rev Genet. 2003;4 : 626–637. 12897774
60. Merritt C, Rasoloson D, Ko D, Seydoux G. 3′ UTRs Are the primary regulators of gene expression in the C. elegans germline. Curr Biol. 2008;18 : 1476–1482. doi: 10.1016/j.cub.2008.08.013 18818082
61. Mitchell-Olds T, Willis JH, Goldstein DB. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat Rev Genet. 2007;8 : 845–856. 17943192
62. Gibert P, Capy P, Imasheva A, Moreteau B, Morin JP, Petavy G, et al. Comparative analysis of morphological traits among Drosophila melanogaster and D. simulans: genetic variability, clines and phenotypic plasticity. Genetica. 2004;120 : 165–179. 15088656
63. Li Y, Álvarez OA, Gutteling EW, Tijsterman M, Fu J, Riksen JAG, et al. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2006;2 : 2155–2161.
64. Grishkevich V, Yanai I. The genomic determinants of genotype X environment interactions in gene expression. Trends in Genetics. 2013;8 : 479–487. doi: 10.1016/j.tig.2013.05.006 23769209
65. Spieth HT, Hsu TC. The influence of light on the matsping behavior of seven species of the Drosophila melanogaster species group. Evolution. 1950;4 : 316–325.
66. Barbagallo B, Garrity PA. Temperature sensation in Drosophila. Curr Opin Neurobiol. 2015;34C: 8–13. doi: 10.1016/j.conb.2015.01.002 25616212
67. Busto M, Iyengar B, Campos AR. Genetic dissection of behavior: modulation of locomotion by light in the Drosophila melanogaster larva requires genetically distinct visual system functions. J Neurosci. 1999;19 : 3337–44. 10212293
68. Kaneko H, Head LM, Ling J, Tang X, Liu Y, Hardin PE, et al. Circadian rhythm of temperature preference and its neural control in Drosophila. Curr Biol. 2012;22 : 1851–7. doi: 10.1016/j.cub.2012.08.006 22981774
69. Claridge-Chang A, Wijnen H, Naef F, Boothroyd C, Rajewsky N, Young MW. Circadian Regulation of Gene Expression Systems in the Drosophila Head. Neuron. 2001;32 : 657–671. 11719206
70. Aho AC, Donner K, Hydén C, Larsen LO, Reuter T. Low retinal noise in animals with low body temperature allows high visual sensitivity. Nature. 1988;334 : 348–50. 3134619
71. Juusola M, Hardie RC. Light adaptation in Drosophila photoreceptors: II. Rising temperature increases the bandwidth of reliable signaling. J Gen Physiol. 2001;117 : 27–42. 11134229
72. Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, Thornton KR. Landscape of standing variation for tandem duplications in Drosophila yakuba and Drosophila simulans. Mol Biol Evol. 2014;31 : 1750–1766. doi: 10.1093/molbev/msu124 24710518
73. David JR, Allemand R, Capy P, Chakir M, Gibert P, Pétavy G, et al. Comparative life histories and ecophysiology of Drosophila melanogaster and D. simulans. Genetica. 2004;120 : 151–163. 15088655
74. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25 : 1105–1111. doi: 10.1093/bioinformatics/btp120 19289445
75. Hu TT, Eisen MB, Thornton KR, Andolfatto P. A second generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res. 2013;23 : 89–98. doi: 10.1101/gr.141689.112 22936249
76. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15 : 550. 25516281
77. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26 : 139–140. doi: 10.1093/bioinformatics/btp616 19910308
78. Smyth GK, Ritchie M, Thorne N. Linear Models for Microarray Data User’ s Guide. Bioinformatics. 2011;20 : 3705–3706.
79. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57 : 289–300.
80. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28 : 516–520. doi: 10.1038/nbt.1626 20436463
81. Tang H, Wang XX, Bowers JE, Ming R, Alam M, Paterson AH, et al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18 : 1944–1954. doi: 10.1101/gr.080978.108 18832442
82. Corbett-Detig RB, Hartl DL. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 2012;8: e1003056. doi: 10.1371/journal.pgen.1003056 23284285
83. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25 : 1754–1760. doi: 10.1093/bioinformatics/btp324 19451168
84. Kofler R, Pandey RV, Schlötterer C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics. 2011;27 : 3435–3436. doi: 10.1093/bioinformatics/btr589 22025480
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Drosophila Spaghetti and Doubletime Link the Circadian Clock and Light to Caspases, Apoptosis and Tauopathy
- Autoselection of Cytoplasmic Yeast Virus Like Elements Encoding Toxin/Antitoxin Systems Involves a Nuclear Barrier for Immunity Gene Expression
- Parp3 Negatively Regulates Immunoglobulin Class Switch Recombination
- PERK Limits Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy