
The Kil Peptide of Bacteriophage λ Blocks Cytokinesis via ZipA-Dependent Inhibition of FtsZ Assembly
Bacterial antibiotic resistance is a serious concern, particularly its role in hospital-acquired infection. Viruses that infect bacteria (bacteriophage) can kill their host, and some prevent the bacterial cell from reproducing during that process. Since their discovery, phage have been considered a potential tool against bacterial infection, but little is known regarding how phage-encoded factors may inhibit bacterial cell division. Understanding the interaction between phage factors and the targeted host systems is therefore a critical research goal. Our report focuses on E. coli and λ, a well-studied phage that infects it. λ contains a gene, kil, whose expression prevents E. coli from dividing, causing cells to grow into long filaments that die. Here we report that Kil protein prevents an essential bacterial protein, FtsZ, from properly assembling into the structure needed for cell division. Our data show that Kil can inhibit FtsZ assembly directly in vitro, but that ZipA, another essential cell division protein, enhances its activity on FtsZ in vivo. The results of our study elucidate one way that a phage naturally inhibits bacterial reproduction, which could serve as a target for rational antibiotic design.
Published in the journal:
. PLoS Genet 10(3): e32767. doi:10.1371/journal.pgen.1004217
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004217
Summary
Bacterial antibiotic resistance is a serious concern, particularly its role in hospital-acquired infection. Viruses that infect bacteria (bacteriophage) can kill their host, and some prevent the bacterial cell from reproducing during that process. Since their discovery, phage have been considered a potential tool against bacterial infection, but little is known regarding how phage-encoded factors may inhibit bacterial cell division. Understanding the interaction between phage factors and the targeted host systems is therefore a critical research goal. Our report focuses on E. coli and λ, a well-studied phage that infects it. λ contains a gene, kil, whose expression prevents E. coli from dividing, causing cells to grow into long filaments that die. Here we report that Kil protein prevents an essential bacterial protein, FtsZ, from properly assembling into the structure needed for cell division. Our data show that Kil can inhibit FtsZ assembly directly in vitro, but that ZipA, another essential cell division protein, enhances its activity on FtsZ in vivo. The results of our study elucidate one way that a phage naturally inhibits bacterial reproduction, which could serve as a target for rational antibiotic design.
Introduction
The replication and lytic functions of bacteriophage λ rapidly diminish Escherichia coli viability and lead to ultimate host death by lysis [1]. Decades-old research uncovered a secondary mode of λ-induced host cell death using a defective prophage containing only the immunity region and the PL operon. Under derepressed conditions, E. coli cells containing this defective prophage filament and eventually die.
The PL operon (Figure 1A, top) contains no genes essential for lytic growth except N, but consists of accessory genes that may be essential in certain circumstances, such as the red recombination genes (exo, bet, and gam) [2]. A series of nested deletions beginning at attL and int, and removing successive prophage genes toward PL, initially defined the region of this secondary, lysis-independent killing function [3]. These deletions identified a putative gene located in this region named kil (host killing by an induced λ prophage) responsible for host cell filamentation, loss of viability, and ultimate death. However, because kil overlaps the gam and cIII genes [4], some questions remained concerning the exact identity of kil and the possible influence of gam and cIII on host killing [2], [5]. Later experiments further mapped Kil activity to the annotated kil open reading frame [6]. Additionally, separate experiments inducing expression of the annotated kil open reading frame from a plasmid verified that this region was responsible for causing cell filamentation and a loss of viability [7].

Although assumed to encode a protein, the product of the annotated kil gene has not been identified. Likewise, the host cell target of the kil gene product (Kil) is unknown. Given the strong cell filamentation and loss-of-viability phenotypes associated with kil expression, we reasoned that Kil likely targets a component of the E. coli cell division apparatus.
Cytokinesis in most bacteria studied to date involves assembly of the highly conserved prokaryotic tubulin-homolog FtsZ into a ring-shaped structure at the nascent site of divison [8]. FtsZ undergoes GTP-dependent polymerization into single stranded polymers in vitro, which in turn are able to bundle together through lateral interactions that are enhanced by certain buffer conditions or bundling agents [9].
In E. coli, the assembly of the FtsZ ring at mid-cell prior to division is stabilized and linked to the membrane through the formation of a ‘proto-ring’ that includes the essential transmembrane-anchored ZipA and membrane-associated FtsA proteins [10]. Once the proto-ring is formed, a coterie of both essential and non-essential proteins is recruited in a partially step-wise fashion to form a mature complex termed the divisome. The divisome contains all the components necessary to divide the cell through a combination of constriction, a switch from lateral cell wall growth to septum (crosswall) formation, and cell separation [11].
Cell survival requires proper coordination of division with other cell cycle events, such as growth, DNA replication, and chromosome segregation. This coordination is largely controlled by precisely regulating the timing and position of FtsZ ring formation by altering FtsZ assembly dynamics [12]. In E. coli, this regulation is chiefly controlled through the combined activities of the Min system (MinCDE) and the nucleoid-occlusion factor SlmA [8], [13]. SlmA is a protein that binds specific regions of the chromosome and prevents FtsZ from assembling over unsegregated nucleoids [14], [15], [16], [17], [18]. The Min system functions to prevent FtsZ from assembling in DNA-free regions of cell poles through the inhibitory activity of MinC, whose localization is controlled by MinD and MinE [13], [19], [20],[21]. Another well-described inhibitor of FtsZ assembly is SulA, which is activated following DNA damage as part of the SOS response to inhibit cell division until genetic errors are corrected [22], [23], [24].
Whereas numerous additional host factors that regulate cell division have been described in several species [11], [25], little work has been done characterizing potential regulation by phage factors. Rac prophage and bacteriophage Mu each contain a gene also named kil [26], [27]. Despite their identical names, however, these kil genes and their predicted peptide products have no significant similarity to one another or to λ kil. Rac kil expression prevents FtsZ ring formation, which blocks cell division resulting in filamentation; no details on the direct target or the mechanism of its activity have been reported [26]. In contrast, expression of the Mu kil gene results in spherical E. coli cells [27], indicative of inhibiting cell elongation by this temperate member of the Myoviridae. The cryptic lambdoid prophage Qin (Kim) and its widespread relatives also contain two factors known to affect cell division [28]: DicB, which acts in place of MinD to bring MinC into position to inhibit FtsZ ring formation [29], [30], and dicF, which encodes an antisense RNA that inhibits ftsZ translation [31]. Finally, the cryptic e14 phage-like element of E. coli contains the sfiC gene, which inhibits FtsZ assembly following SOS induction similarly to SulA (SfiA) [32], [33], [34].
Here, we report that λ kil expression from a defective prophage, from a plasmid, or during induction of a complete, lytic-competent λ lysogen inhibits cell division due to a block in FtsZ ring formation. We verify that Kil encodes a peptide, and show that it acts independently of well-characterized host systems of FtsZ assembly regulation. We further identify and characterize Kil-resistant ftsZ and zipA mutant alleles, demonstrate inhibition of FtsZ assembly in vitro by an affinity-tagged Kil derivative, and discuss potential models for Kil activity on FtsZ assembly. Finally we address the relevance of kil to λ biology, demonstrating that Kil acts during normal lytic growth of λ phage and suggesting that this activity can delay cell lysis.
Results
Expression of kil interferes with E. coli cell division
To investigate the role of kil expression on E. coli cell division, we first used a bacterial strain (CC4506) harboring a defective λ prophage in which a modified operon is under control of a temperature-sensitive (ts) allele (cI857) of the phage CI repressor. In this kil+ strain, a point mutation destroys the start codon of cIII, a cat cassette replaces sieB to ea10, and a tet cassette replaces gam through int (Figure 1A). Consistent with previous findings [6], CC4506 cells formed long, non-dividing filaments after derepression of the PL operon by thermo-inactivation of the CI857 repressor at 42°C (Figure 1B), and lost viability (Figure 1C). In contrast, an isogenic strain with the start codon of kil destroyed by a point mutation (CC4512), continued to divide normally following induction of the prophage and retained viability, similar to the W3110 background strain (Figure 1B & C).
The loss of cell viability, presumably caused by the division blockage elicited by kil expression, could be rescued in the short term by returning E. coli filaments to low temperature conditions that restored CI857 repressor activity (Figure 1C, bottom left). However, prolonged kil expression at 42°C (∼6 hours) prevented restoration of growth even after returning cells to conditions that repress kil expression (Figure 1D).
To identify the potential target of kil expression, we screened a pBR322 library of E. coli chromosomal fragments for multi-copy suppression of kil-induced toxicity at 42°C. Consistent with kil expression targeting cell division, multi-copy expression of an ∼5.4-kb fragment including the ftsQAZ operon completely suppressed toxicity. Similarly, low-copy (pSC101) expression of the ftsQAZ operon alone suppressed kil-induced toxicity, but less efficiently (Figure 1E). Analysis of the kil-expressing strain containing pSC101 with ftsQZ, ftsQA, or ftsZ alone confirmed that this suppression derives specifically from ftsZ (but not ftsQA) over-expression. Further supporting the link between the toxicity of kil expression and cell division, an independent multi-copy suppressor screen identified sdiA (Figure 1E), whose product increases ftsQAZ operon transcription [35].
Kil inhibits FtsZ ring formation
Filamentation of E. coli cells could arise either from a failure to form FtsZ rings at the nascent division site, or from a defect in divisome maturation and septal synthesis after FtsZ ring assembly. Over-expression of ftsZ could conceivably help suppress either of these mechanisms to permit Kil-resistance. We therefore determined by immunofluorescence microscopy whether FtsZ rings continued to form in E. coli cells following derepression of kil under PL control.
At 30°C, when PL is repressed, both CC4506 (kil+) and CC4512 (kil−) cells were normal-sized on average (3.1±0.1 and 3.1±0.2 µm, respectively) compared to W3110 wild-type control cells (3.1±0.2 µm) (Figure 2A, top). Immunofluorescence microscopy (IFM) with antibody against E. coli FtsZ demonstrated that cells of each strain contained a single band of FtsZ signal localized at midcell in the majority (92.4±1.3% to 94.0±1.5%) of the populations, on average (Figure 2B, top).

However, upon a shift to growth at 42°C and the resulting kil expression, CC4506 cells continued to grow at a normal rate into nondividing filaments (>10 µm), before reaching stationary phase (Figure 2A, bottom). In contrast, W3110 and CC4512 control strains still displayed normal average cell lengths at 42°C (3.3±0.55 and 3.2±0.48 µm, respectively). Kil induction also led to a rapid (<5 minutes) loss in FtsZ rings observable by IFM, leaving only a few cells with normal FtsZ localization on average (8.3±2.2%). FtsZ localization in W3110 cells was unaffected on average (92.1±1.7%) until 90 minutes post-temperature-shift when cells entered stationary phase and FtsZ ring frequency decreased sharply (Figure 2B, bottom).
Notably, although CC4512 (kil−) cells displayed normal cell lengths during growth at 42°C (Figure 2A, bottom), this strain background still showed a small, but significant, effect on FtsZ ring formation in the first minutes following temperature shift, with ring frequency dropping to an average of 73.0±4.6%. However, by 20 minutes, these cells had recovered normal FtsZ ring frequencies (92.7±0.07%), unlike the CC4506 kil+ counterpart strain where FtsZ ring frequency remained below 10% (Figure 2B, bottom). This suggests that some other factor in CC4512 has a minor and transient effect on FtsZ assembly upon shift to high temperature.
IFM micrographs corresponding to the data in Figure 2A & B showed normal medial FtsZ localization in both W3110 and CC4512 kil− strains after five minutes post temperature shift to 42°C (Figure 2C) and all time points observed thereafter (data not shown). CC4506 kil+ cells showed normal FtsZ localization at 30°C, but FtsZ immunostaining became patchy and diffuse after only five minutes of kil induction at 42°C. This mislocalization continued throughout growth at 42°C, sometimes forming into broad, unproductive FtsZ foci (possibly helices) along the cell filament (Figure 2C). This pattern, typical upon FtsZ ring formation inhibition in vivo, suggests that FtsZ is unable to form a coherent ring-shaped structure in the presence of Kil, thus preventing septum formation and cell division.
Due to the observed kil-independent, transient effect on FtsZ ring formation (Figure 2B, bottom), we chose to study the effects of kil outside of any phage context by expressing the gene from a plasmid. This enabled us to be certain that any observed effects were caused by Kil alone, and eliminated the need to expose E. coli cells to temperature shock. We cloned the kil gene downstream of the arabinose-inducible promoter in pBAD33 and transformed the resulting plasmid (pDH104) into W3110. W3110 pDH104 cells grown in non-inducing conditions divided normally, comparable to the W3110 background containing empty vector, indicating that uninduced levels of Kil are low. Consistent with the prophage results, expression of kil from the plasmid proved sufficient to induce cell filamentation through a rapid loss of FtsZ ring formation (Figure 2D). Similar results were also obtained by cloning kil under Plac (IPTG-inducible) control in pRR48 (data not shown).
Kil does not alter FtsZ levels or act through well-characterized endogenous systems of FtsZ assembly inhibition
Proper FtsZ assembly and subsequent cell division in E. coli is dependent on levels of FtsZ relative to other division components [36]. Though alterations in FtsZ levels are not a normal part of division regulation in E. coli [37], conditions that artificially elevate or reduce available FtsZ result in FtsZ mislocalization and cell filamentation [38]. A trivial explanation for the loss of FtsZ ring formation upon the induction of λ kil is that its product, the presumed Kil peptide (see below), somehow alters FtsZ protein levels. A known example of this occurs with the cryptic prophage-derived dicF, which encodes a small RNA that blocks ftsZ mRNA translation by an antisense mechanism [31]. However, quantitative immunoblotting of FtsZ in CC4506 cells induced for kil expression showed levels of FtsZ approximately equivalent to those in uninduced CC4506 counterparts or in W3110 and CC4512 controls (Figure 3A). Furthermore, induction of kil from pBAD33 or pRR48 in W3110 similarly had no effect on FtsZ compared to levels seen in uninduced cells or cells with empty plasmid (data not shown). These results suggest that the kil product does not act by altering FtsZ levels.

Another possibility is that Kil acts indirectly through an endogenous system of FtsZ assembly inhibition, such as the SOS-response activated SulA [22], [23], [39], [40], MinC [41], [42], [43], [44], or the nucleoid-occlusion factor SlmA [14], [15], [16], [17]. This was important to rule out because another phage-derived dic gene, dicB, encodes a small protein that inhibits FtsZ ring assembly by mimicking MinD to recruit MinC to midcell [29], [30].
To investigate a possible contribution of the SOS-inducible protein, SulA, in Kil activity, we examined the effects of kil induction from pBAD33 in a WM1074 ΔsulA::kan background. WM1074 is a wild-type derivative of MG1655, whereas the characterizations of Kil presented above utilized the K12 derivative W3110. Expression of kil from pBAD33 in WM1074 caused filamentation and inhibition of FtsZ ring formation (Figure 3B) comparable to that seen in W3110 (Figure 2D). Addition of 0.2% arabinose to sulA− cells resulted in cell filamentation (Figure 3C) indistinguishable from that seen for the sulA+ background, verifying that Kil does not act through SulA and that it confers a similar phenotype in a different strain background.
As with the ΔsulA::kan background, induction of kil from pBAD33 in ΔminCDE::kan or ΔslmA::kan backgrounds also increased cell filamentation (Figure 3C), arguing that neither MinC nor SlmA are involved in Kil activity. As shown previously, minCDE mutant cells already have division defects due to the presence of extra FtsZ rings, and are elongated. Upon kil induction all FtsZ rings disappear into irregular patchy localization, and the already-long minCDE mutant cells grow into even longer filaments. Together these data demonstrate that unlike dicF or DicB, the kil product does not act through any of the well-characterized endogenous host systems that regulate FtsZ assembly.
Isolation and characterization of ftsZ alleles resistant to kil expression
The preceding results suggest that Kil inhibits FtsZ ring formation, potentially through direct interaction with FtsZ. To further investigate this interaction, we generated ftsZ mutant alleles by recombineering with randomly mutagenized ftsZ PCR fragments, selecting for those that allowed survival at 42°C in a thermo-inducible kil+ and thermo-sensitive ftsZ (ftsZ84) strain (see Materials and Methods). This strategy identified two mutant alleles of ftsZ with increased resistance to kil expression, ftsZV208A and ftsZL169R (Figure 4A). According to a recently reported Mycobacterium tuberculosis FtsZ crystal structure [45], both of these residues are located in the subunit interface of FtsZ protofilaments. Specifically, FtsZV208 lies in the conserved T7-loop region, which during polymerization inserts into the nucleotide-binding region near the FtsZL169 residue in the N-terminal domain of an adjacent subunit (Figure 4B).

To verify that these alleles confer Kil resistance, we replaced the wild-type ftsZ gene of a fresh kil+ strain with each mutant allele. Spot dilutions of the resulting strains verified that the ftsZV208A or ftsZL169R alleles conferred resistance to Kil compared to the wild-type ftsZ allele control, although the appearance of the spots suggests that the strains acquired suppressors fairly readily at 42°C (Figure 4C).
IFM on the kil+ strains harboring ftsZV208A or ftsZL169R in place of the native wild type allele showed normal-sized cells with FtsZ rings present at 30°C when kil expression is repressed (Figure 4D). However, in addition to normal medial ring localization, FtsZV208A appeared to also form frequent rings or puncta at cell poles (marked by arrowheads) and occasional ring doublets (marked by an asterisk). Similarly, FtsZL169R sometimes formed aberrant FtsZ structures (marked by asterisks), along the long axis of the cell. These observations suggest that although these FtsZ mutants are competent for assembly and cell division, they seem to be overly stable and insensitive to endogenous regulators of FtsZ assembly.
Unlike in the wild-type ftsZ allele background, short-term exposure to kil expression at 42°C did not cause cell filamentation in ftsZV208A or ftsZL169R backgrounds (Figure 4D), consistent with their partial resistance to Kil as seen in spot dilutions (Figure 4C). The apparent insensitivity of the mutant FtsZV208A and FtsZL169R proteins to endogenous regulation seen at 30°C is even more obvious at 42°C upon PL operon derepression. For example, FtsZV208A localizes predominantly to one cell pole at higher temperature (only ∼14% midcell localization after 40 min of kil induction). FtsZL169R, in contrast, forms multiple rings throughout the length of cells at higher temperatures, likely contributing to their slightly longer cell lengths. These cells also contain apparent inclusions visible by DIC microscopy (Figure 4D), which may be from high levels of non-interactive Kil. Importantly, the inclusion bodies and mutant FtsZ localization abnormalities did not arise from inappropriate FtsZ levels in the cell, as both mutant FtsZs were present at levels comparable to the wild-type control (Figure 4E).
The above spot dilution and IFM data demonstrate that the isolated ftsZV208A and ftsZL169R mutant alleles confer partial resistance to kil as expressed from PL upon de-repression at 42°C, permitting FtsZ assembly, albeit aberrant, and preventing cell filamentation. To further address the Kil-resistance of these mutant ftsZ alleles and avoid the mutant FtsZ mislocalization seen at higher temperatures, the above strains were transformed with pRR48-kil and investigated by expressing kil by IPTG induction at 30°C.
In contrast to the resistance of the ftsZ mutant alleles to kil expressed at 42°C from PL, they were unable to confer resistance to short-term induction of kil from plasmid pRR48 (1 mM IPTG). Rings formed by FtsZWT, FtsZV208A, or FtsZL169R were all rapidly lost upon pRR48-kil induction (Figure 4F, top and center panels), leading to cell filamentation (Figure 4F, bottom panels). It is possible that the levels of Kil obtained from plasmid expression were higher than those obtained from its native PL context, but we noticed that longer inductions of kil from PL also ultimately overcame the initial resistance of FtsZV208A or FtsZL169R (data not shown). These results suggest that although these ftsZ alleles confer partial resistance to Kil from their abnormal assembly characteristics, Kil is still able to overcome these mutant FtsZ proteins under some conditions.
FtsZ alleles resistant to inhibition by Kil are also resistant to inhibition by MinC or SulA
The abnormal localization of FtsZV208 and FtsZL169R, such as to cell poles, suggested that these mutants may have increased filament stability and a certain general resistance to endogenous FtsZ assembly inhibitors, not just the action of λ Kil. Additionally, WM1074 cells transduced from ftsZWT to ftsZV208A or ftsZL169R had a minicell-producing phenotype (data not shown), further implicating resistance to FtsZ assembly inhibition by MinC.
To determine whether the ftsZV208A and ftsZL169R alleles make the strains generally resistant to FtsZ assembly inhibitors, we monitored resistance to over-expression of sulA or to a his-minCD translational fusion (minCD). We transformed the kil+ (under chromosomal PL control) strains harboring ftsZV208A or ftsZL169R with either pDSW210-his-minCD or pBAD33-sulA and grew them at 30°C, where kil is not expressed whether or not the plasmids are induced. Induction of either minCD or sulA caused FtsZWT cell filamentation and a loss of FtsZ ring formation after 40 min. However, in the presence of FtsZV208 or FtsZL169R, high levels of SulA or MinCD had no apparent effect on cell length in the same time frame (Figure 5A & B). Consistent with prior results, FtsZV208 and FtsZL169R localized normally at 30°C in non-inducing conditions for either plasmid. Similar to observations for the FtsZ mutants upon kil expression at 42°C (Figure 4C), induction of sulA, and particularly minCD, at 30°C caused both FtsZV208 and FtsZL169R to mislocalize, often at cell poles (Figure 5A & B). This suggests that, in contrast to FtsZWT, both FtsZ mutants are able to assemble in the presence of high SulA or MinCD levels, but that this assembly is abnormal and persists at new cell poles following division, similar to what was seen for these FtsZ mutant proteins in the presence of Kil.
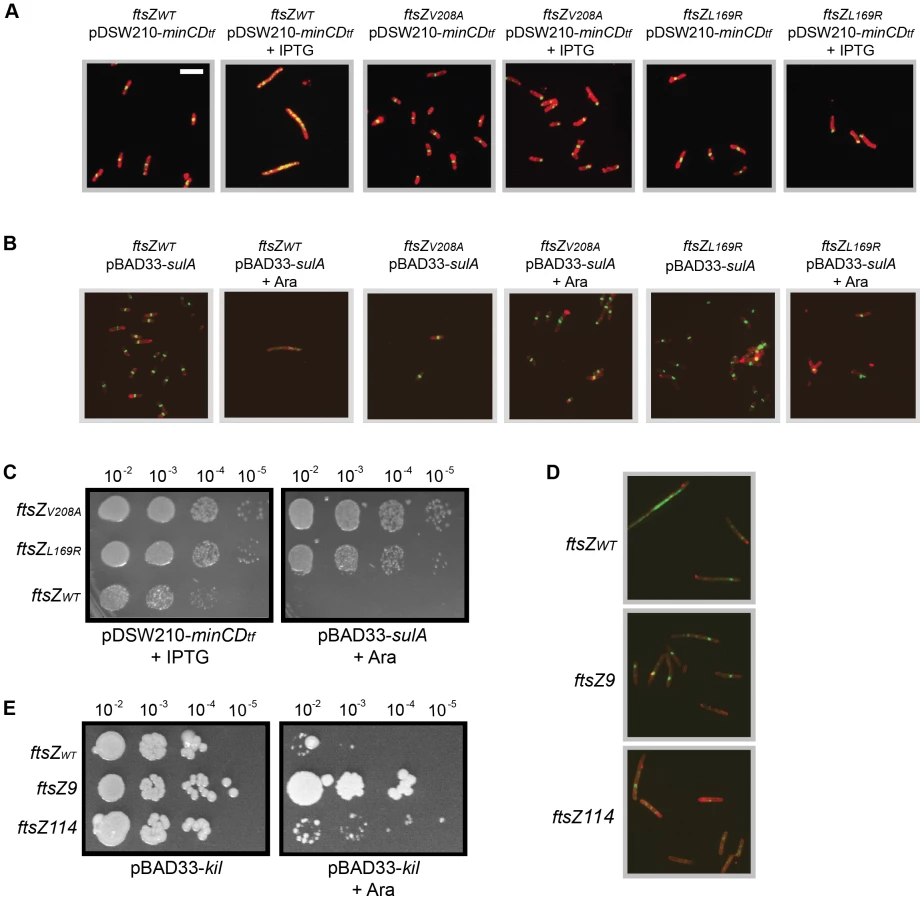
The continued growth and division of ftsZV208A or ftsZL169R mutant cells upon short-term induction of either minCD or sulA suggests that these cells remain more viable than wild-type cells, despite many mislocalized FtsZ rings. Spot dilutions of ftsZWT, ftsZV208A, or ftsZL169R cells carrying pDSW210-his-minCD or pBAD33-sulA onto plates with relevant inducers at 30°C (to ensure no kil expression) verified that both ftsZV208A and ftsZL169R increased cell viability upon sulA over-expression (Figure 5C). Although minCD over-expression blocked wild-type FtsZ ring formation and led to cell filamentation (Figure 5A), these cells remained partially viable in spot dilutions upon full induction with IPTG (1 mM). However, ftsZV208A and ftsZL169R conferred an approximately 10-fold improvement in that viability (Figure 5C). As expected, cell growth was normal in the absence of inducer (data not shown). As with kil resistance (Figure 4D), the ftsZV208A allele showed somewhat stronger resistance to minCD and sulA over-expression compared to the ftsZL169R allele (Figure 5C).
Previously characterized ftsZ mutant alleles, ftsZ9 (ftsZ18ΩV-G – a two residue insertion) and ftsZ114 (ftsZF268C), show general resistance to both MinC and SulA [46], [47], [48]. We therefore tested whether these alleles were also resistant to λ kil expression. For these experiments we utilized the original PB143 ftsZ− background strains that contain ftsZWT, ftsZ9, or ftsZ114 on a low-copy number plasmid under constitutive expression (pBEF0, pBEF9, and pBEF114) and transformed each with pBAD33-kil. FtsZWT and FtsZ114, which displays ∼50% GTPase activity of the wild-type FtsZ, were unable to resist inhibition by Kil and failed to form detectable FtsZ rings in the presence of Kil. However, FtsZ9, which is nearly devoid of GTPase activity (10% of normal) more strongly resisted Kil inhibition and was able to form some detectable FtsZ rings (Figure 5D). This result in the presence of Kil is comparable to the partial, weak resistance of FtsZ114 (and the relatively strong resistance of FtsZ9) to inhibition by SulA or MinC [46], [47], [48]. The ability of these strains to form colonies with or without kil induction correlates well with their Kil resistance by IFM (Figure 5E).
Kil activity requires ZipA in vivo
The ftsAR286W (ftsA*) gain of function mutation can bypass the loss of several normally essential cell division proteins, including ZipA or FtsK, as well as partially suppress an ftsQ ts mutant [49], [50], [51], [52]. This ability to stabilize the divisome prompted us to ask whether ftsAR286W cells might be more resistant to Kil. However, we found that ftsAR286W conferred no resistance to Kil produced from pRR48, as cells formed extensive filaments (data not shown). As a control, we also tested a ftsAR286W ΔzipA::aph double mutant for resistance to Kil, expecting similar results. Strikingly, these double mutant cells were entirely resistant to kil expression from pRR48, manifesting neither filamentation nor a loss of FtsZ ring localization following induction (Figure 6A). This strain background was also resistant to induction of kil from pBAD33 (data not shown). These results suggested that the absence of zipA, not the presence of ftsAR286W, imparts resistance to Kil. Note that zipA+ ftsAR286W cells are slightly shorter, while ΔzipA ftsAR286W cells are slightly longer than their wild-type counterparts [49].

To verify that the observed kil-resistance is specifically caused by an absence of zipA, we added the gene back in trans by introducing the compatible plasmid pKG110-zipA into the ΔzipA ftsAR286W strain containing pRR48-kil. Uninduced levels of zipA expressed from pKG110 in these cells did not perturb cell division on their own but were sufficient to restore kil-sensitivity (Figure 6B). Cells with the empty vector or expressing another early division protein, zapA [53], [54], [55], did not restore Kil-sensitivity in this ΔzipA ftsAR286W strain (Figure 6B). Unlike the isolated Kil-resistant ftsZ alleles, ΔzipA ftsAR286W cells remained sensitive to both SulA and MinC inhibition (Figure 6C), arguing that the Kil resistance of the ΔzipA ftsAR286W strain is not a general effect on FtsZ. Taken together, these results strongly suggest that ZipA is specifically required for the effect of Kil on FtsZ assembly and subsequently cell division.
Additional Kil-resistant isolates map to zipA
If ZipA were required for Kil-mediated inhibition of FtsZ as inferred from the above genetic experiments, we reasoned that it should be possible to isolate mutations in the essential zipA gene that confer Kil-resistance in a wild-type ftsA background without abrogating ZipA's normal function in the divisome. The powerful phenotype of Kil-induced cell filamentation and death allowed us to isolate spontaneous Kil-resistant mutants. To discourage the isolation of mutations that simply reduce kil expression or inactivate the kil product itself, we constructed a ‘double kil’ strain harboring both pBAD33-kil and pRR48-kil in WM1074. We isolated spontaneous Kil-resistant colonies by plating cultures of this strain on LB agar with both IPTG and arabinose (Figure 7A). Several independent isolates were saved and their zipA loci amplified for DNA sequencing.
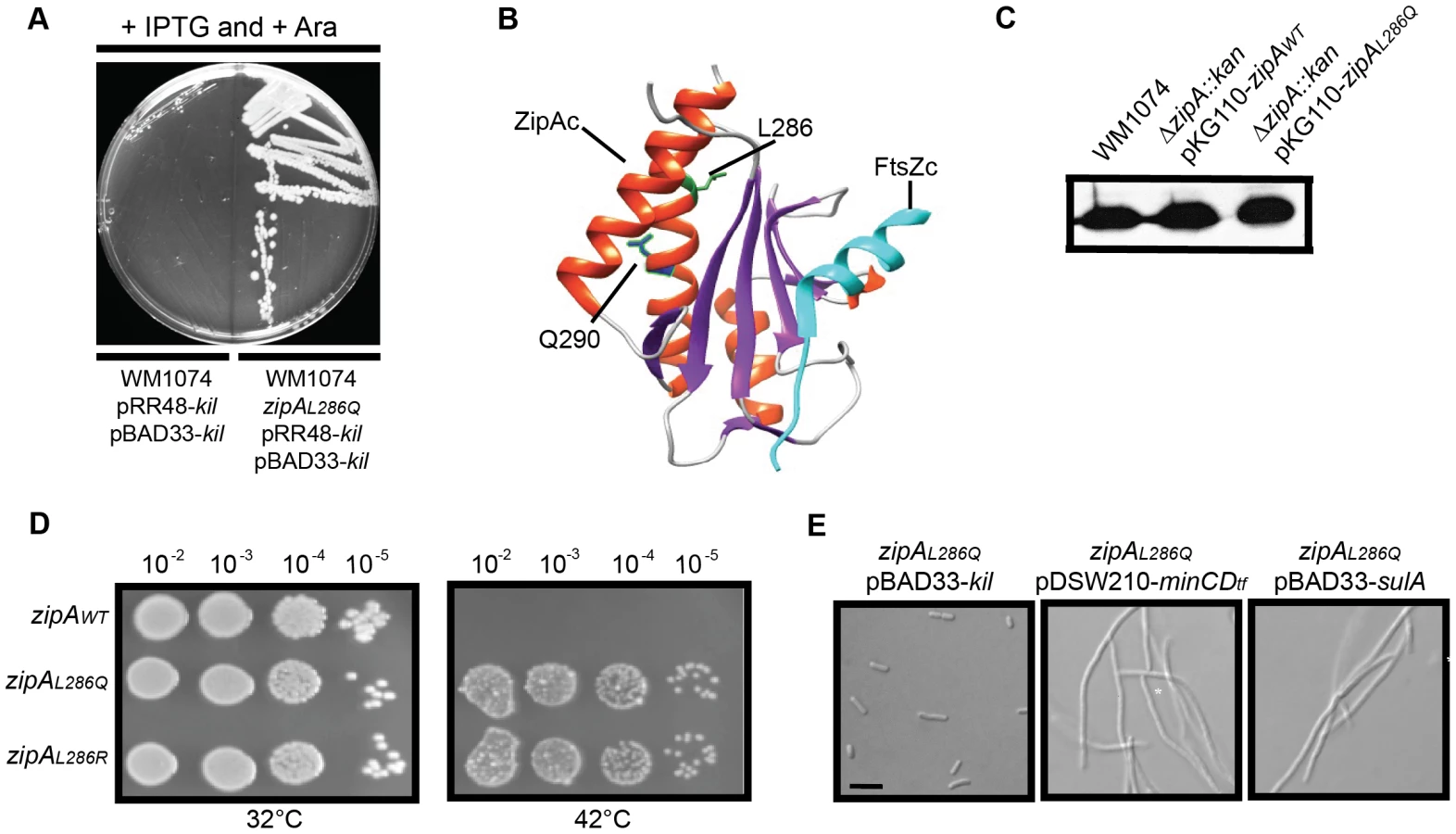
All isolated kil resistant strains contained one of two mutations in zipA, encoding ZipAL286Q and ZipAL286R, respectively. The C-terminal domain of ZipA containing this residue (ZipAC) directly interacts with the C terminus of FtsZ [56], [57], [58] (Figure 7B). As both alleles seemed to have similar properties, we chose ZipAL286Q for further investigation.
To assess the general activity of ZipAL286Q in cell division, we first cloned it into pKG110 and transformed the resulting plasmid into a WM1074 wild-type background. We then introduced the ΔzipA::aph allele by P1 transduction. The transduction efficiencies into cells harboring pKG110-zipAL286Q or pKG110-zipAWT were similar, verifying that ZipAL286Q functions normally in cell division in place of the essential ZipAWT. Low-level induction of either zipAWT or zipAL286Q from pKG110 (with 0.5 µM sodium salicylate) in the WM1074 ΔzipA::aph background resulted in cellular levels of ZipA comparable to those in wild-type cells (Figure 7C) and caused no discernable differences in growth or division phenotypes. Additionally, overproduction of ZipAWT [59] or ZipAL286Q caused cell filamentation with multiple FtsZ rings present (data not shown).
To verify the Kil-resistance of zipAL286Q, we replaced the zipAWT gene of the W3110 strain harboring kil under PL control with zipAL286Q (or zipAL286R) by recombineering. The resulting strains displayed normal cell division phenotypes and resistance to kil upon derepression of promoter at 42°C (Figure 7D and data not shown). Finally, a fresh WM1074 wild-type background transduced with zipAL286Q linked to a ptsH<>tet marker was resistant to kil expression from pRR48, but remained sensitive to both SulA and MinCD inhibition (Figure 7E), demonstrating that this zipA allele is specific for resistance to Kil. Finally, random mutagenesis of zipA by PCR generated a mutation in a nearby residue, Q290R, (Figure 7B) which conferred the same level of Kil resistance to cells as the L286 mutations (data not shown), further arguing for the importance of this region of ZipA for Kil activity in vivo.
The kil gene encodes a protein that interacts with ZipA and FtsZ
Early research on the kil gene identified a putative and abundantly produced protein product [60]. However, subsequent research established that this protein was actually λ Ea10, and that kil is likely expressed at only low levels from the PL promoter of λ phage ([2] and see below). Initial attempts to identify, purify, or chemically synthesize the protein product of the kil gene failed, prompting us to investigate whether the kil gene truly encodes a protein. Previous work established that insertion of a stop codon within the 5′ portion of kil or disruption of its stop codon abolishes Kil activity ([6] and Figure 1), but did not definitely show whether the loss of activity was caused by changes at the protein level or the nucleotide alterations themselves.
To test this definitively, we inserted an amber stop mutation into the thirty-first codon (normally encoding tyrosine) of kil (kiltyr31UAG) to terminate translation prematurely. Cells harboring the kiltyr31UAG allele under PL control survived at 42°C. To determine if cell survival was due to interrupted translation or due to a mutation in the RNA, we introduced the supF gene, which encodes a suppressor tRNA that would supply the originally encoded tyrosine amino acid at the amber mutation. The presence of supF restored Kil activity, confirming the product of kil functions as a protein (Figure 8A).

To obtain purified Kil protein, we cloned the kil gene into a pET vector, adding an N-terminally encoded FLAG-tag to create a his6-flag-kil fusion for expression and protein purification by cobalt-affinity chromatography. Purification of His6-FLAG-Kil yielded a single prominent band on Coomassie-stained SDS polyacrylamide gels that migrates near its predicted ∼10.5 kDa molecular weight with an estimated purity of >95% (Figure 8B). Induction of his6-flag-kil in BL21(DE3) cells inhibited cell division, indicating that the tagged fusion is active (data not shown). We found that optimal induction occurred in the absence of zipA and in the presence of extra FtsZ; we therefore preferentially expressed his6-flag-kil in a BL21(DE3) ftsAR286W ΔzipA::aph pBS58 background.
Surprisingly, his6-flag-kil expression seemed to augment Kil activity in vivo, blocking cell division even in the absence of zipA (data not shown). Notably, this phenotype occurs with all tested Kil fusion derivatives, and no instances of complete suppression of any fusions have been isolated to date. With no antibody against Kil, we cannot know if this effect is merely because Kil fusions permit higher protein levels compared to wild-type untagged Kil, or if this effect arises from the increased length/size of a tagged Kil fusion.
Nonetheless, because ZipA is required for the activity of normal, untagged Kil on FtsZ assembly in vivo, we investigated whether Kil interacts directly with ZipA or FtsZ. We incubated purified, renatured His6-FLAG-Kil (see Materials and Methods) in the presence of α-FLAG M2 affinity resin with or without purified FtsZ and/or purified, His-tagged ZipA C-terminal domains (denoted ZipAC for wild type zipA and ZipA*C for zipAL286Q). Following binding of the resin by His6-FLAG-Kil, samples were washed extensively and retained proteins bound to the resin and/or His6-FLAG-Kil were visualized on Coomassie-stained SDS-polyacrylamide gels (Figure 8C). As expected, His6-FLAG-Kil remained bound to the resin following the washes, whereas FtsZ, ZipAC, or ZipA*C were not resin-associated when incubated on their own without any His6-FLAG-Kil. However, FtsZ and each ZipAC remained in samples following washes in the presence of His6-FLAG-Kil, arguing in favor of direct specific interactions between His6-FLAG-Kil and FtsZ, and between His6-FLAG-Kil and either ZipAC. In support of this, bovine serum albumin added to all the input mixtures at concentrations similar to those of FtsZ and ZipAc showed no specificity for the resin. Notably, His6-FLAG-Kil – ZipAC interacts similarly with wild-type ZipAC and the mutant ZipA*C, and the His6-FLAG-Kil – FtsZ interaction does not seem to be significantly augmented by either ZipAC under the conditions of this experiment. Together these data suggest that Kil is capable of interacting independently with either FtsZ or the C-terminal domain of ZipA, and the L286Q mutation in ZipA does not prevent it from interacting with His6-FLAG-Kil.
Purified Kil inhibits FtsZ bundling in vitro
Our in vivo evidence suggests that Kil inhibits FtsZ assembly, and that this activity normally requires the presence of ZipA. To test whether Kil could directly act to inhibit FtsZ assembly, we used sedimentation assays with purified FtsZ and His6-FLAG-Kil purified from the BL21(DE3) ftsAR286W ΔzipA::aph pBS58 background to ensure that little ZipA would be present in reactions. Not surprisingly, purified FtsZ does contain a small amount of copurified ZipA (data not shown), barely detectable by immunoblotting and not visible at the FtsZ concentrations used for sedimentation reactions.
We initiated FtsZ assembly with the addition of 1 mM GTP and then sedimented polymer bundles of FtsZ by centrifugation. Sedimentation primarily detects polymer bundles of FtsZ, and in our buffer conditions; little FtsZ sedimented in the presence of GTP alone, consistent with the relatively poor bundling capability of E. coli FtsZ [61]. Addition of calcium increased FtsZ polymer bundling and thus sedimentation, as previously reported [62], [63]. Strikingly, addition of His6-FLAG-Kil to FtsZ assembly reactions greatly reduced the amount of polymer sedimentation in the presence of calcium (Figure 8D), suggesting that the purified tagged Kil can directly inhibit FtsZ assembly in vitro.
To address the potential contribution of ZipA to Kil activity on FtsZ, we added purified ZipAC or ZipA*C to additional sedimentation reactions. As previously reported [56], ZipAC increased polymer bundling, measured as sedimentation, similar to the effect of calcium. ZipA*C also enhanced FtsZ sedimentation to similar levels. As with calcium, addition of His6-FLAG-Kil to the assembly reactions greatly reduced polymer sedimentation in the presence of either ZipAC or ZipA*C (Figure 8E). His6-FLAG-Kil co-sedimented with the small fraction of FtsZ polymer bundles that assembled in its presence, either with calcium (Figure 8D) or the ZipAC domains, but did not sediment on its own (Figure 8E). This suggests that Kil may multimerize in the presence of FtsZ and/or form stable Kil-FtsZ complexes. Together, these data demonstrate that His6-FLAG-Kil interacts with FtsZ and inhibits its assembly in vitro, and suggest that the normal importance of ZipA in vivo for this activity is bypassed by the in vitro conditions.
To test the effect of Kil on FtsZ GTP hydrolysis rates, we performed GTPase activity assays under conditions identical to the sedimentation assays. GTP hydrolysis by FtsZ is induced by monomer-monomer interactions within a newly formed protofilament. If Kil inhibits FtsZ assembly by sequestering monomers, similar to SulA, it should strongly inhibit FtsZ GTP hydrolysis activity. If instead Kil severs FtsZ protofilaments or decreases FtsZ polymer bundling, this would increase the pools of monomers available to make new protofilaments, which should increase GTP hydrolysis.
The background GTPase activity signal from His6-FLAG-Kil, ZipAC, or ZipA*C was negligible (∼0.5 GTP/min/FtsZ), and FtsZ displayed a rate of ∼3.2–4.1 GTP/min/FtsZ independent of bundling agent (Figure 8F). The presence of Kil in assembly reactions caused a small but significant increase in FtsZ GTP hydrolysis to 4.3–5.3 GTP/min/FtsZ. The effect of Kil was most pronounced in the presence of calcium (a 1.4-fold increase) and least pronounced in the presence of ZipA*C (Figure 8F). Though modest, the across-the-board increase in GTP hydrolysis in the presence of Kil is consistent with either inhibition of FtsZ protofilament extension, severing protofilaments, or inhibition of FtsZ protofilament bundling.
Kil-mediated inhibition of host cell division during phage infection
Our data inspire a model in which Kil interacts with FtsZ in a ZipA-dependent manner in vivo to inhibit FtsZ ring assembly and cause cell filamentation. The location of kil in the λ genome (Figure 1A) suggests that it would be expressed transiently during initial establishment of lysogeny and then upon exit of lysogeny when entering the lytic cycle. Normally, induction of a λ lysogen leads to rapid host cell lysis, but the potential role and importance of kil in this process is unknown. We therefore chose to investigate kil in its native context in intact E. coli λ lysogens, with the cI857 allele to facilitate universal λ lysogen activation.
Induction of a λ lysogen by a shift to 42°C led to an increase in cell length up to 40 minutes post-induction (Figure 9A), at which time cells began to lyse. This increase in cell length is similar to that observed upon similar PL derepression in a defective prophage (Figure 2A), but does not proceed as rapidly or reach the point of extreme filamentation seen during kil expression. Induction of the λ lysogen also led to a rapid loss of FtsZ rings (Figure 9B).

The observed increase in cell length and rapid, complete loss of FtsZ ring assembly following induction of the λ lysogen did not occur in an isogenic kil− mutant. Although kil− cells still entered the lytic cycle with no observable delay, cell length did not increase significantly in the absence of kil (Figure 9A). Although FtsZ ring formation was partially inhibited under these conditions, perhaps by other λ components or through indirect effects on host cell physiology, the majority of kil− cells still contained FtsZ rings sufficient to maintain division (Figure 9B). These data indicate that kil expression has a dramatic effect on FtsZ ring formation, and subsequently on cell length, during the induction of a λ lysogen into the lytic cycle.
Finally, to ask whether kil− phage are compromised for reproduction, we monitored a single cycle of phage growth of cI857 and a nonpolar deletion of kil, cI857kilΔ7, in MG1655 at 39°C (Figure 9C). Overall phage yields were not significantly affected by the absence of kil, although we observed a reproducibly shorter lysis time for the cI857 kilΔ7 phage relative to kil. Kil effects on host cell lysis have been previously reported [7], [64].
Discussion
The results reported here establish that the Kil protein of bacteriophage λ blocks host cell division by rapidly ablating FtsZ rings. In reactions with FtsZ purified from ZipA-deficient cells, purified tagged Kil prevents sedimentation of FtsZ bundled either by calcium or purified ZipAC. In addition, purified tagged Kil interacts with FtsZ in the presence or absence of ZipA. Together, these results suggest that Kil can interact directly with FtsZ in vitro to disrupt FtsZ protofilament bundling. However, ZipA is required for wild-type Kil to disrupt FtsZ rings normally in vivo, and point mutations in either FtsZ or ZipA can effectively resist FtsZ ring disruption by Kil. Excess FtsZ can also protect against inhibition by Kil in vivo, presumably by enhancing protofilament assembly and/or titrating out the inhibitor.
A model for Kil-mediated inhibition of FtsZ ring assembly
The normal involvement of ZipA in vivo suggests that Kil may interact with both FtsZ and ZipA in the cell, a possibility supported by our in vitro interaction data. One explanation for the in vivo ZipA requirement is that ZipA may act specifically to recruit Kil to FtsZ protofilaments (Fig. 9D). The location of the mutations in ZipA that confer resistance to Kil is consistent with this idea. Although these mutations are in the FtsZ-binding domain of ZipA, the mutated L286 residue is not predicted to interact directly with the C-terminus of FtsZ, facing away from it in the co-crystal structure [58]. The L286 mutations could have an allosteric effect on ZipA-FtsZ interactions, but cells carrying zipAL286Q divide normally, so these interactions cannot be grossly altered. Moreover, over-expression of zipAL286Q causes cell division defects similar to that seen with zipAWT (data not shown), further arguing that the mutant is not dysfunctional in its effects on FtsZ assembly.
According to this model, ZipAL286Q would be less able to recruit Kil to midcell than ZipAWT, presumably because of decreased ZipA-Kil interactions. However, our in vitro interaction data suggest no difference between His6-FLAG-Kil interaction with ZipAC or ZipA*C. This disparity could stem from the additional length/size of the His6-FLAG-Kil fusion (that seems to act independently of ZipA in vivo unlike untagged Kil), or from the absence of full-length ZipA in the in vitro experiments. Additionally, results from the sedimentation experiment suggest that the high concentrations of Kil present in reactions might cause it to multimerize in the presence of FtsZ. Further work is required to clarify the precise mechanism of Kil activity and the role of ZipA in this activity.
The proposed ZipA-mediated recruitment of Kil to FtsZ in vivo is reminiscent of mechanisms proposed for other FtsZ inhibitors. For example, the cryptic prophage-derived DicB protein uses ZipA to recruit the DicB/MinC complex to the membrane-associated FtsZ ring [65]. However, unlike DicB, Kil inhibits FtsZ directly and does not require MinC for its inhibitory activity (Figure 3). The B. subtilis EzrA protein is related to ZipA in that it binds FtsZ and shares ZipA's unusual topology of N-terminal integral membrane domain and C-terminal cytoplasmic domain. A mutant of EzrA lacking this N-terminal transmembrane domain is able to inhibit FtsZ assembly directly in vitro, but in vivo this mutant does not localize to the membrane, thus lowering the local concentration of EzrA to a point where its interaction with FtsZ is no longer inhibitory [66]. By analogy, ZipA might serve to concentrate Kil near the membrane where FtsZ is located, enhancing its inhibition of FtsZ assembly (a function no longer needed in vitro). Kil is predominantly hydrophobic, and although it is not predicted to contain any transmembrane segment, it is predicted to have peripheral membrane association [67]. If this is the case, interactions with ZipA could still enhance localization of Kil specifically to midcell and/or position Kil optimally to interfere with FtsZ assembly within the in vivo environment that is absent in our in vitro FtsZ assembly assays.
Normal FtsZ assembly dynamics ensure that FtsZ rings properly form both temporally and spatially to link cell division with other cell cycle events [12]. Kil acts independently of the well-characterized host-derived systems of regulation, but may act on FtsZ assembly in a generally similar fashion. In support of this notion, our two Kil-resistant ftsZ alleles, ftsZV208A and ftsZL169R, display a general resistance to multiple forms of FtsZ assembly inhibition, and ftsZ alleles previously reported for SulA and MinC-resistance [46], [47], [48] also show similar degrees of resistance to Kil. Despite having lesions in the conserved T7-loop and near the nucleotide-binding region, these two mutant FtsZs are capable of FtsZ ring assembly in vivo, although many rings form at potential division sites other than midcell, including cell poles. This is consistent with a bias away from disassembly and towards stabilization. Such a bias would lead to resistance to disassembly factors such as MinC, even if Kil inhibits by a different mechanism. This resistance is incomplete, because Kil produced from a plasmid rapidly ablates FtsZV208A and FtsZL169R rings, and even Kil produced from the PL operon eventually overwhelms their ability to form rings and encourages acquisition of suppressors.
Recent publication of a crystal structure for a GDP-bound protofilament of FtsZ from Mycobacterium tuberculosis implicates residues adjacent to those corresponding to V208 and L169 of E. coli FtsZ as being involved in conformational changes at the subunit interface between straight, GTP-bound filament and curved, GDP-bound filament forms [45]. The general resistance of the FtsZV208A and FtsZL169R mutants to disassembly factors suggests that they may favor the GTP-bound form and have reduced GTP hydrolysis. The properties of other inhibitor-resistant FtsZ mutants fit this idea. The FtsZ9 mutant has extremely low GTPase activity and is highly resistant to SulA, MinC or Kil. In contrast, the FtsZ114 mutant, which has ∼50% of normal GTPase activity, confers only partial resistance to Kil or MinC [48]. The inhibitory activity of His6-FLAG-Kil on FtsZ filament bundling would be predicted to induce greater FtsZ subunit turnover and thus increase GTP hydrolysis, which is what we observe, and is similar to the effects of some other factors that inhibit FtsZ bundling such as EzrA [66], [68]. Consistent with Kil blocking FtsZ bundling, FtsZ localization in the presence of Kil becomes patchy and diffuse, possibly into broad helices, suggesting that FtsZ filaments still assemble, but fail to condense into a mature, bundled ring. An alternative explanation for the increased GTPase activity is that Kil may also prevent FtsZ protofilament elongation, which would increase the pool of free FtsZ monomers available to form dimers and oligomeric filaments capable of hydrolyzing GTP.
Role of bacteriophage-encoded cell division inhibitors
A handful of other phage-encoded factors have been identified that block bacterial cell division, including a newly-described T7 gene product that also acts on FtsZ [69]. Other factors act through host-cell regulation of FtsZ assembly (DicB and dicF [28], [29]) or through an unknown mechanism (Kil of prophage Rac [26]). The lytic SPO1 phage of Bacillus subtilis encodes a peptide that inhibits host division prior to lysis [70], but acts at a stage following FtsZ ring formation (Haeusser and Margolin, unpublished data). Therefore, it seems that a variety of phage are able to target host cell division through unique peptides that act by diverse mechanisms. Suppression of host cell division by cryptic prophage-encoded factors has previously been implicated in increasing host adaptation to stress and resistance to antibiotics [71], and it is possible that kil benefits the host in some way during the lysogenic state.
In contrast to inhibiting FtsZ ring formation, a recent report described the role of B. subtilis Φ29 p1 in promoting phage replication through association with assembled FtsZ [72]. This strategy allows Φ29 to utilize an existing host cell scaffold to organize and optimize viral DNA production, but how a phage would benefit from perturbing FtsZ ring formation and inhibiting cell division is less clear. The original studies on kil− λ phage suggested that they still replicate, assemble, and lyse the host comparably to kil+ counterparts, with similar burst sizes [3], [6], [73]. However, these studies used cIII67 mutant phage, which may be more prone to lysogeny and mask a kil− growth phenotype. Here, we show that Kil activity is evident upon lytic induction of λ, and demonstrate that, in a single cycle of growth, the kil− mutant phage has a shorter latent period and causes earlier cell lysis. Although we did not observe a decrease in yield for the kil mutant phage, in some circumstances a shorter latent period may result in lower phage yieldl. The larger average volume of kil+ cells relative to kil− cells might translate into a longer time to induce lysis. Cell size changes may also affect the lysis-lysogeny decision [74].
ln addition to affecting the timing of lysis, kil could benefit phage by creating a non-compartmentalized host that permits easier and more accurate phage reproduction. In this scenario, activation of cell division and construction of a septum could interfere with excision of the λ lysogen, its replication, or its packaging. Previous studies with kil from the lambdoid P22 phage of Salmonella found that kil expression was required for efficient lytic growth of abc− mutants lacking anti-RecBCD activity, increasing burst size by eight fold [75]. This suggests Kil activity becomes important during conditions of high recombination frequency. By analogy, B. subtilis Maf induces a temporary block in cell division during natural competence development that permits efficient, uninterrupted DNA recombination [76].
The lack of an obvious kil− phenotype for λ phage in original studies [3] and the subtle defects reported here could arise from biased, unnatural laboratory growth conditions, or an overlapping function with other phage components, such as gam. The abrupt but temporary loss of FtsZ rings upon PL derepression in the absence of kil and the relatively large percentage of cells that lose FtsZ ring formation even in an otherwise complete kil− λ lysogen suggest that Kil may not act completely independently. Future experiments will focus on clarifying the molecular mechanism of Kil-mediated FtsZ ring disruption and uncovering its role in λ phage biology.
Materials and Methods
Strains and growth conditions
All E. coli strains used are listed in Table 1. Standard genetic methods including transformation and P1 vir transduction were used for strain construction. Recombineering methods for strain construction [77], [78] are described in a section below.

Cells were grown in Luria-Bertani (LB) medium at 30°C or 32°C, as indicated, for temperature-sensitive (ts) strains under permissive conditions and 42°C under non-permissive conditions, or at 37°C for all non-ts strains. Optical density readings at 600 nm (OD600) were measured using a UV-1601 or UV-1800 spectrophotometer (Shimadzu). LB medium was supplemented with ampicillin (50 µg/ml; Fisher Scientific), kanamycin (50 µg/ml; Sigma-Aldrich), chloramphenicol (20 µg/ml; Acros Organics), tetracycline (10 µg/ml; Sigma-Aldrich), and spectinomycin (100 µg/ml; Sigma-Aldrich), as needed. Gene expression from vectors derived from pET28-, pET15 - (Novagen – EMD Millipore), and pRR48 [79] was induced with 1 mM isopropyl-β-D-galactopyranoside (IPTG) (Fisher Scientific). Gene expression from pBAD33-derived vectors [80] was induced at a final concentration of 0.2% L-(+)-arabinose (Ara) (Sigma-Aldrich). Gene expression from pKG110-derived vectors (J.S. Parkinson, University of Utah) was either uninduced or induced with 0.5 µM sodium salicylate (Mallinkrodt), as indicated. A PCR test [81] confirmed that LT447 and LT1566 were monolysogens.
DNA and protein manipulation and analysis
Standard protocols or manufacturers' instructions were used to isolate plasmid DNA, as well as for restriction endonuclease, DNA ligase, PCR, and other enzymatic treatments of plasmids and DNA fragments. Enzymes were purchased from New England BioLabs, Inc. (NEB) or Invitrogen. Plasmid DNA was prepared using the Wizard Plus SV Minipreps DNA Purification Kit, PCR and digest reactions were cleaned up using the Wizard SV Gel and PCR Clean-up System, and chromosomal DNA was prepared using the Wizard Genomic DNA Purification Kit (Promega). KAPA HiFi HotStart DNA polymerase (Kapa Biosystems) or Phusion High-Fidelity DNA polymerase (Thermo Scientific – NEB) or Platinum Taq (Invitrogen) were used for PCR reactions in a MyCycler Thermal Cycler (Bio-Rad). Oligonucleotides were purchased from Sigma-Aldrich or IDT and their sequences are listed in Table S1. The final versions of all cloning products were sequenced to verify their construction. DNA sequencing was performed by GeneWiz (South Plainfield, NJ), SeqWright (Houston, TX) or SAIC-Frederick, Inc. (Frederick, MD). DNA bands were visualized using an AlphaImager Mini System (ProteinSimple) and DNA concentrations were estimated with a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific). Protein concentrations were determined using the Coomassie Plus Assay (Themo Scientific – Pierce). Molecular graphics and analyses were performed with the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Plasmid construction
All plasmids are listed in Table 2.

Kil expression plasmids: For pRR48-kil, the kil coding sequence was amplified from CC4506 chromosomal DNA with primers DPH199 and 200; the PCR product was digested with PstI and SpeI, then ligated into a pRR48-derivative where the original multiple cloning site had been replaced (NdeI to SalI) with that of pKG110. For pDH104 (pBAD33-kil), the kil coding sequence with its native ribosome-binding site was amplified from CC4506 chromosomal DNA with primers DPH217 and 202; the PCR product was digested with XmaI and PstI, then ligated into pBAD33.
Toxic overexpression of minCD: For pWM2737 (pDSW210-his-minCD) the minD coding sequence was amplified with primers WM960 & 356, the PCR product was digested with SalI and HindIII, then ligated into pWM2735 (pDSW210-his6-minC) [44] to create sequence encoding an uninterrupted His-tagged, MinCD translational fusion (minCDtf).
Complementation of ΔzipA cells with the zipAL286Q allele: pDH145 (pKG110-zipAL286Q) was constructed as previously published for pKG110-zipA [82], but using DPH615 (zipAL286Q) chromosomal DNA as template. Plasmid pKG110-zapA was constructed by Daisuke Shiomi in the same manner as pKG116-zapA [82].
Plasmids for protein purifications: For pET28-zipAC the C-terminal ZipA domain coding sequence was amplified by Tushar Beuria with primers WM269 & 268; the PCR product was digested with BamHI and XhoI, then ligated into pET28a. As primer WM268 did not include a stop codon, this construct produces His6-ZipAC-His6. Construction of pDH146 (pET28-zipA*C) was therefore performed identically, but with DPH615 chromosomal DNA as template, to produce a double His-tagged version of ZipAC that harbors the L286Q mutation. For pDH139 (pET28-flag-kil) the kil coding sequence with its stop codon was amplified from CC4506 chromosomal DNA in two steps (primers DPH309 & 170, followed by DPH310 & 170) to add sequence encoding a FLAG-tag to the 5′ end of kil, in-frame with the His-tag-encoding codons of the plasmid. The resulting product was digested with BamHI and HindIII, then ligated into pET28a. Plasmid pDH149 (pET15-flag-kil) was constructed by digesting pDH139 with NcoI and XhoI, then ligating the his6-flag-kil insert into identically-digested pET15b.
General recombineering methods
Recombineering was done using published methods [77], [78]. Briefly, the Red functions were expressed either from the defective prophage or from a recombineering plasmid such as pSIM18, a hygromycin resistant plasmid encoding the temperature-sensitive CI857 repressor and a portion of the PL operon containing gam, exo, and bet. Log phase cells propagated at 32°C were subjected to a 15 minute heat pulse in a 42°C shaking water bath to induce the Red functions, quickly chilled in an ice-water slurry, and prepared for electroporation by washing. DNA, either double - or single-stranded, was introduced into the cells by electroporation. When a drug marker was selected, the cells were allowed to recover for several hours in 1 mL broth before plating. When point mutations were introduced with ∼70 base oligonucleotides, the cells were plated non-selectively on LB agar plates after a 30 minute recovery time, either at 42°C for direct selection of Kil-resistant alleles or at 32°C for screening by PCR, as described below. In some cases oligonucleotides with additional sequence changes in third positions (wobble positions) were used to create mutations. As described [83], these additional wobble changes were placed near the change of interest, creating a configuration of mispairs that is not recognized by the E. coli mismatch repair system, without changing the amino acid sequence. This allows both high-efficiency recombineering and detection of recombinant chromosomes with PCR. When wobble changes were introduced, between 12–20 colonies were analyzed, using one oligonucleotide that hybridized specifically to the recombinant sequence, paired with a second nearby oligonucleotide; this primer pair should not yield product with the parental sequence. Positive candidates were purified to single colonies and about a dozen of these single colonies were again subjected to the same PCR analysis. The region of interest from positive candidates identified in the second round of PCR screening was sequenced. Sequences for oligonucleotides used are listed in Table S1.
Construction of a non-polar deletion of kil in λ cI857 phage
A seven-codon in-frame deletion within kil, removing codons 20–26, was initially constructed in XTL241 (HME6 gam<>cat-sacB) and then moved to a phage. The deletion was built into a hybrid oligonucleotide, LT793, which along with an oligonucleotide to the downstream gam gene, LT795, were used to amplify a PCR product containing the deletion and spanning the region containing the counter-selectable marker. Recombineering was used to replace the cat-sacB. After introduction of the PCR product into electrocompetent XTL241 induced for the Red functions, cultures were grown overnight and recombinants were selected on sucrose plates at 32°C. Candidate colonies were purified on sucrose and confirmed to be chloramphenicol-sensitive. Primers AW24 and LT795 were used to amplify the region containing the deletion, which was confirmed by sequencing. In contrast to the kil+ control, cells containing the deletion did not filament after several hours of growth at 42°C, and formed colonies after overnight growth at 42°C. A lysate of λ cI857red3 was grown on cells containing the seven-codon deletion to allow marker rescue from the defective prophage onto the phage. The lysate was plated on LT352. Plaques were picked into 0.5 mL TMG (10 mM Tris base, 10 mM MgS04 0.01% gelatin) and 2 µl of the pickate was used for PCR with oligonucleotides AW24 and LT795: the difference in size between the deletion and the wild-type alleles was resolvable on a 1.2% agarose gel. The deletion phage was purified by another round of plating on LT352 and a high titer plate lysate was grown from a purified plaque on C600.
Isolation of multicopy suppressors of kil
CC4506 was transformed with a pBR322-based library containing 1.5–5 kb Sau3A fragments of LE392 genomic DNA cloned into pHDB3 [84] and transformants were selected on LB ampicillin agar at 42°C. Ampicillin-resistant colonies that survived Kil induction were pooled, plasmid DNA was isolated and CC4506 was transformed with this enriched plasmid population to confirm suppression. The Sau3A library was kindly provided by Nadim Magdalani (NIH). Plasmid pJP10 was isolated by this procedure. Plasmid pNB15 was isolated by a similar procedure except the pBR322-based library was constructed by cloning 2–4 Kb Sau3A fragments of JS549 genomic DNA into pBR322 cut with BamHI.
Isolation of Kil-resistant ftsZ alleles
The ftsZ gene was amplified from W3110 with oligonucleotides XMZ325 and XMZ326, using standard PCR conditions. This ftsZ PCR product was used as a template for mutagenic PCR [85] with the same oligonucleotides. This randomly mutagenized pool of ftsZ PCR fragments was used for recombineering into strain AW34, which carries the thermo-sensitive ftsZ84 allele (G105S) linked to leuD::Tn10, a defective λ prophage with thermo-inducible kil expression, and a mutS<>amp allele to prevent host mismatch repair during recombination. After a 30 minute recovery in broth at 30°C, aliquots were plated on nitrocellulose filters on LB plates and incubated at 32°C for 3 hours. Filters were then transferred to 42°C pre-warmed LB plates and these plates were subsequently incubated overnight at 42°C to select for growth. This procedure simultaneously selects for recombinants that replace ftsZ84 with the wild type allele and for resistance to λ Kil expression. A lysate of P1 vir was grown on a pool of the temperature-resistant isolates. This lysate was used to transduce the Kil-expressing strain AW41, selecting tetracycline resistance at 42°C. Thermo-resistant colonies were purified and their ftsZ gene was amplified with colony PCR; the resulting PCR products were sequenced. Several isolates of the ftsZV208A mutation were obtained, and only one isolate of ftsZL169R. One isolate of each of the mutant types was chosen for further characterization. The mutations were introduced into a fresh background by P1 transduction at 32°C, selecting for the linked leuD<>Tn10 and screening for temperature resistance.
Isolation of Kil-resistant zipA alleles
Competent WM1074 cells were sequentially transformed with pBAD33-kil and pRR48-kil and the resulting strain was verified to have a kil+ phenotype upon induction of kil from either plasmid. A culture of this ‘double kil’ strain was grown to mid-logarithmic phase and induced with IPTG and arabinose simultaneously. 100 µL of stationary phase culture was plated on LB agar supplemented with appropriate antibiotics, IPTG, and arabinose and grown overnight at 37°C. All resulting colonies were purified, then grown in liquid media to freeze samples. Chromosome purification and PCR amplification of the zipA locus in the saved isolates showed that all contained a zipAL286Q mutation, while the original WM1074 pBAD33-kil pRR48-kil strain contained zipAWT sequence.
A second isolation of spontaneous Kil-resistant mutants was carried out using a similar protocol, with the only difference being that cells were grown in liquid culture under non-inducing conditions into stationary phase, then plated on medium containing IPTG and arabinose. All isolated colonies from this second experiment contained a zipAL286R mutation.
Using ssDNA recombination to isolate, map and confirm suppressor mutations
Recombineering using ssDNA can be very efficient with up to 75% of the cells being recombinants [83]. Therefore, recombineering with ssDNA can be used to isolate and map mutations, and these techniques were used throughout this work. For example, when mutagenic PCR was used to isolate zipA mutations, in one case sequencing revealed a double mutation, zipAA245T,Q290R. We designed and ordered oligonucleotides to make each mutation separately via recombineering. Recombineering-proficient cells prepared on CC4506 were transformed with each oligonucleotide separately or both together, outgrown for 30 minutes, then diluted and spot-titered on LB plates at 30°C and 42°C. We found that only one of the oligonucleotides, MH82, was necessary and sufficient to create a mutation that suppressed Kil-dependent killing.
Spot titers
Cells used for spot titers were taken from the same cultures used for fixation unless as noted below. A ten-fold dilution of these cultures was taken approximately forty minutes after induction (or control conditions), unless otherwise noted, and then serially diluted into fresh LB media in a 96-well plate using a multichannel pipette. A flame-sterilized and cooled, metal-pronged tool was then used to replica-plate spots of serially diluted culture onto LB plates with added components and incubation conditions as indicated in the text and figures. Photos of plates were taken in a FluoroChem 8800 system with its accompanying camera and software (Alpha Innotech).
Spot titers in Figures 4C and 7D were done as follows: An overnight culture was diluted 100-fold and cells were grown at 30° for 2 hours in LB. Ten-fold serial dilutions of these cultures were made in TMG (see above) and 10 µl was spotted on pre-warmed LB plates and incubated at the indicated temperatures.
Determining loss of viability after exposure to Kil
To generate the data in Figure 1D, an overnight culture of CC4506 was diluted 70-fold into 15 mL of LB broth and grown to an OD600∼0.25 at 32°. The culture was diluted in 10-fold increments from 100 to 10−5. At time “0”, 0.1 mL samples of appropriate dilutions were spread on prewarmed 42° LB plates on which we had placed a sterile 82 mm (diameter) nitrocellulose filter. After the indicated time, sterile forceps were used to move the filter to a 32° plate. These LB plates were incubated overnight at 32° and colonies counted.
Cell fixation, microscopy, and analysis
Overnight cultures were started from −80°C strain stocks and grown under appropriate antibiotic selection and permissive conditions. Overnight cultures were diluted into fresh medium, grown to mid-logarithmic phase, then the OD600 of individual cultures was adjusted to a uniform OD600 = 0.025 with fresh medium. Cultures were grown for approximately 2 doublings at permissive conditions and then shifted to non-permissive conditions (or kept as permissive controls) at time point zero. Unless otherwise indicated, samples of culture for microscopy were taken approximately 40 minutes after shifting to non-permissive conditions.
Cells were fixed with methanol and processed for immunofluorescence microscopy (IFM) as previously published [86] using lysozyme (Sigma) treatment for 5 minutes and antibodies diluted in bovine serum albumin (Fisher Scientific). Primary polyclonal rabbit α-FtsZ [87] was used at 1/2500, secondary goat α-rabbit-AlexaFluor 488 (Molecular Probes) and wheat germ agglutinin conjugated to rhodamine (Molecular Probes), to visualize cell wall, were used at 1/200. For DNA staining, 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes) was used at 0.5 µg/mL. Micrograph images were captured on an Olympus BX60 microsope with a Hamamatsu C8484 camera using HC Image software (Hamamatsu). Cell length and ring frequency measurements were taken with the ObjectJ extension [88] (http://simon.bio.uva.nl/objectj/) of ImageJ [89] (National Institutes of Health) using a minimum number of 100 cells and images were minimally adjusted for brightness/contrast using Adobe Photoshop CS4. Microsoft Excel 2008 for Mac (v. 12.3.6) was used for data tabulation and calculatio
Immunoblot analysis
Samples were prepared, subjected to SDS-PAGE on 20% acrylamide gels, and transferred as published [90]. Transfer of His6-FLAG-Kil samples was done in 10 mM CAPS pH 10.0, 10% methanol transfer buffer with a two-hour transfer time. Mouse monoclonal α-His primary antibody (Sigma-Aldrich) and affinity-purified rabbit polyclonal α-FtsZ [87] were used at 1/5000. Affinity-purified rabbit polyclonal α-ZipA [49] was used at 1/1000. Goat α-mouse and α-rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) (Sigma-Aldrich) were used at 1/10,000. A SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo Scientific – Pierce) was used for HRP detection; blots were exposed to film and developed using a Konica SRX101A Film Processor (Konica Minolta).
Protein purification
Proteins were induced for purification from pET vectors (Novagen – EMD Millipore) in BL21(DE3) [91] backgrounds as 2 L cultures that were grown to OD600∼0.7 at 30°C, at which point 1 mM IPTG was added. (Volumes in this and subsequent steps were scaled down by a factor of 10 for copurification assays). Cultures were left overnight at 30°C to induce protein and were harvested in the morning by spinning in a Beckman J2-21 centrifuge with a JA-17 rotor at 7,000 rpm. Cell pellets were then washed in buffer (50 mM sodium phosphate pH 8.0; 300 mM NaCl), re-centrifuged, and stored as cell pellets at −80°C.
For purifications, cell pellets were thawed on ice, resuspended in 30 mL of the sodium phosphate wash buffer (except for His-tagged Kil, see below) with 1 mg/mL lysozyme and an EDTA-Free c0mplete Protease Inhibitor Cocktail Tablet (Roche), and incubated on ice for 30 minutes. Cells were disrupted by sonication on ice (Branson Sonifier 250; 50% level, output 4) in a series of six alternating 30-second periods of sonication and 30-second rest periods. Cell lysates were clarified of debris by spinning at 40,000 rpm at 4°C for 45 minutes in an 80 Ti rotor with an Optima XL-100K Ultracentrifuge (Beckman).
E. coli FtsZ was purified from WM971 cell lysates by successive 20% and 30% ammonium sulfate cuts. Following the second cut, protein was resuspended in polymerization buffer (50 mM MES pH 6.5; 50 mM KCl; 2.5 mM MgCl2; 1 mM EGTA; 10% sucrose), flash-frozen in liquid nitrogen and stored at −80°C.
His-tagged ZipAC proteins were purified by gravity flow over water-washed and buffer-equilibrated cobalt-conjugated resin with a 5 mL bed volume at 4°C. Imidazole at pH 8.0 was added to 10 mM in cell lysates for loading onto the column. Following loading, columns were washed successively in ten-column volumes of purification buffer (50 mM HEPES pH 7.5; 300 mM NaCl) with 10 mM, 25 mM, and 50 mM imidazole. His-tagged ZipACs were eluted in 10 mL purification buffer+250 mM imidazole. Eluted proteins were concentrated and subjected to buffer exchange into polymerization buffer using Amicon Ultra-10K Centrifugal Filter Devices (Millipore), flash-frozen in liquid nitrogen, and stored at −80°C.
His6-FLAG-Kil was primarily induced in, and purified from, BL21(DE3) ftsAR286W ΔzipA::aph pBS58 cells. The exception was for copurification experiments, where the fusion was also prepared from zipAWT+ or zipAL286Q+ backgrounds, as noted. Following induction, His6-FLAG-Kil was found to be predominantly associated in insoluble inclusion bodies, leading to very low yields under native conditions, even in an ftsAR286W ΔzipA::aph background. We therefore purified His6-FLAG-Kil under denaturing conditions according to the QIAexpressionist Handbook (QIAGEN, 2003) protocol, followed by renaturation by dialysis. Purity was estimated at >95% by Coomassie staining.
Lysates from His6-FLAG-Kil-expressing cells were prepared similarly as described above, except cell pellets were resuspended in denaturing lysis buffer (100 mM sodium phosphate, pH 8.0, 10 mM Tris-Cl, 6M guanidine hydrochloride) with an EDTA-Free c0mplete Protease Inhibitor Cocktail Tablet (Roche), but without lysozyme. After incubation for 30 minutes at room temperature, cells were sonicated as described above.
These His6-FLAG-Kil-containing lysates were then loaded by gravity flow onto water-washed and denaturation lysis buffer-equilibrated cobalt-conjugated resin with a 5 mL bed volume at room temperature. Columns were washed in ten-column volumes of freshly prepared denaturing wash buffer (100 mM sodium phosphate, pH 6.3, 10 mM Tris-Cl, 8 M urea). Denatured His6-FLAG-Kil was eluted in 10 mL freshly prepared denaturing elution buffer (100 mM sodium phosphate, pH 4.5, 10 mM Tris-Cl, 8 M urea). The eluted protein was then renatured by dialysis into polymerization buffer overnight and through the following day (three 2 L changes of buffer in total). Renatured His6-FLAG-Kil was concentrated using Amicon Ultra-3K Centrifugal Filter Devices (Millipore), flash-frozen in liquid nitrogen, and stored at −80°C.
Interaction and sedimentation assays
To assay interaction between purified proteins, 200 mL samples were prepared in polymerization buffer containing 6 µM bovine serum albumin. FtsZ at 5 µM, His6-FLAG-Kil at 10 µM, and/or His6-tagged ZipAC domains (WT and L286Q) at 5 µM were included as indicated. 50 µL of 50% buffer-equilibrated α-FLAG M2 affinity resin (Sigma-Aldrich) were added and samples were incubated at room temperature, mixing, for one to three hours for binding. Samples were then loaded onto gravity flow columns and the resin was washed with 25 mL polymerization buffer. Following washes, resin was recovered in 250 µL buffer, SDS-PAGE sample buffer was added and samples were boiled and separated by 20% SDS-PAGE followed by Coomassie blue staining.
Sedimentation assays were performed essentially as previously described [66] in a Beckman TL-100 Ultracentrifuge, but using a TLA 100.3 rotor at 70,000 rpm with appropriate adaptors and speed-resistant tubes (Beckman). 100 µL samples were prepared in polymerization buffer with FtsZ at 5 µM, His6-FLAG-Kil at 10 µM, His6-tagged ZipAC domains (WT and L286Q) at 5 µM, and GTP at 1 mM. Components were added in the following order: polymerization buffer, Kil buffer or Kil, FtsZ, ZipAC, and GTP. For reactions with calcium-induced bundling, CaCl2 was added to 1 mM after GTP addition.
GTPase activity assay
GTPase activities were determined using the EnzChek Phosphate Assay Kit (Molecular Probes) in reactions set up with the same concentrations and buffers as for sedimentation assays, but in a 96 well plate and with the required purine nucleoside phosphorylase enzyme and 7-Methyl-6-thio-D-guanosine (MESG) substrate components. Reactions were initiated by adding one half the reaction volume as buffer with FtsZ alone, simultaneously via multi-channel pipette, into the other half of the reaction volume that contained all other components. (For reactions without FtsZ, buffer with GTP alone was used to simultaneously initate reactions). OD360 readings were taken every 30 seconds using a Synergy Mx Microplate Reader (BioTek). GTP hydrolysis rates were calculated based on a phosphate standard curve.
Phage one-step growth assay
One-step growth was done according to Frank Stahl (unpublished). Briefly, 10 mL MG1655 was growth at 39°C in tryptone broth to OD600 = 0.4 (∼1.5×108 per mL). The cells were pelleted and suspended in TMG (see above) and incubated at 37°C for 30 minutes to starve the cells. NaCN was added at 2×10−3 M and 1.8 mL was dispensed to two small glass-plating tubes. To initiate the growth curve, 0.2 mL phage stock at 1.5×108 per mL was added to the cell suspension. Phage adsorption was monitored over a 30 minute period by mixing 0.1 mL infected cells into 4.9 mL tryptone broth containing 0.25 mL chloroform and plating appropriate dilutions on C600 host cells. After a 30 minute adsorption the infected cells were diluted 100-fold into tryptone broth and two further dilutions were made into 39°C tryptone broth, one 100-fold and one 5×103-fold. At various times, 0.1 mL samples were taken from these dilutions and plated immediately on C600.
Supporting Information
Zdroje
1. WeisbergRA, GallantJA (1967) Dual function of the lambda prophage repressor. J Mol Biol 25 : 537–544.
2. Court D, Oppenheim AB (1983) Phage lambda's accessory genes. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory. 251–278.
3. GreerH (1975) The kil gene of bacteriophage lambda. Virology 66 : 589–604.
4. Daniels D, Schroeder J, Szybalski W, Sanger F, Coulson A, et al.. (1983) Complete annotated lambda sequence. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory. 519–676.
5. BahlH, EcholsH, StrausDB, CourtD, CrowlR, et al. (1987) Induction of the heat shock response of E. coli through stabilization of sigma 32 by the phage lambda CIII protein. Genes Dev 1 : 57–64.
6. SergueevK, YuD, AustinS, CourtD (2001) Cell toxicity caused by products of the PL operon of bacteriophage lambda. Gene 272 : 227–235.
7. ReisingerGR, RietschA, LubitzW, BlasiU (1993) Lambda kil-mediated lysis requires the phage context. Virology 193 : 1033–1036.
8. de BoerPAJ (2010) Advances in understanding E. coli cell fission. Curr Opin Microbiol 13 : 730–737 doi: 10.1016/j.mib.2010.09.015
9. EricksonHP, AndersonDE, OsawaM (2010) FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74 : 504–528 doi: 10.1128/MMBR.00021-10
10. HuangKH, Durand-HerediaJ, JanakiramanA (2013) FtsZ ring stability: of bundles, tubules, crosslinks, and curves. J Bacteriol 195 : 1859–1868 doi: 10.1128/JB.02157-12
11. EganAJF, VollmerW (2013) The physiology of bacterial cell division. Ann N Y Acad Sci 1277 : 8–28 doi: 10.1111/j.1749-6632.2012.06818.x
12. RombergL, LevinPA (2003) Assembly dynamics of the bacterial cell division protein FtsZ: poised at the edge of stability. Annu Rev Microbiol 57 : 125–154.
13. LutkenhausJ (2007) Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu Rev Biochem 76 : 539–562.
14. ChoH, BernhardtTG (2013) Identification of the SlmA active site responsible for blocking bacterial cytokinetic ring assembly over the chromosome. PLoS Genet 9: e1003304 doi: 10.1371/journal.pgen.1003304
15. ChoH, McManusHR, DoveSL, BernhardtTG (2011) Nucleoid occlusion factor SlmA is a DNA-activated FtsZ polymerization antagonist. Proc Natl Acad Sci U S A 108 : 3773–3778 doi: 10.1073/pnas.1018674108
16. TonthatNK, AroldST, PickeringBF, Van DykeMW, LiangS, et al. (2011) Molecular mechanism by which the nucleoid occlusion factor, SlmA, keeps cytokinesis in check. EMBO J 30 : 154–164 doi: 10.1038/emboj.2010.288
17. TonthatNK, MilamSL, ChinnamN, WhitfillT, MargolinW, et al. (2013) SlmA forms a higher-order structure on DNA that inhibits cytokinetic Z-ring formation over the nucleoid. Proc Natl Acad Sci U S A 110 : 10586–10591 doi: 10.1073/pnas.1221036110
18. BernhardtTG, de BoerPAJ (2005) SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol Cell 18 : 555–564.
19. LooseM, Fischer-FriedrichE, HeroldC, KruseK, SchwilleP (2011) Min protein patterns emerge from rapid rebinding and membrane interaction of MinE. Nat Struct Mol Biol 18 : 577–583 doi: 10.1038/nsmb.2037
20. ParkK-T, WuW, BattaileKP, LovellS, HolyoakT, et al. (2011) The Min oscillator uses MinD-dependent conformational changes in MinE to spatially regulate cytokinesis. Cell 146 : 396–407 doi: 10.1016/j.cell.2011.06.042
21. ParkK-T, WuW, LovellS, LutkenhausJ (2012) Mechanism of the asymmetric activation of the MinD ATPase by MinE. Mol Microbiol 85 : 271–281 doi: 10.1111/j.1365-2958.2012.08110.x
22. BiE, LutkenhausJ (1993) Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J Bacteriol 175 : 1118–1125.
23. ChenY, MilamSL, EricksonHP (2012) SulA inhibits assembly of FtsZ by a simple sequestration mechanism. Biochem 51 : 3100–3109 doi: 10.1021/bi201669d
24. HuismanO, D'AriR, GottesmanS (1984) Cell-division control in Escherichia coli: specific induction of the SOS function SfiA protein is sufficient to block septation. Proc Natl Acad Sci U S A 81 : 4490–4494.
25. AdamsDW, ErringtonJ (2009) Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol 7 : 642–653 doi: 10.1038/nrmicro2198
26. ConterA, BouchéJP, DassainM (1996) Identification of a new inhibitor of essential division gene ftsZ as the kil gene of defective prophage Rac. J Bacteriol 178 : 5100–5104.
27. WaggonerBT, SultanaK, SymondsN, KarlokMA, PatoML (1989) Identification of the bacteriophage Mu kil gene. Virology 173 : 378–389.
28. FaubladierM, BouchéJP (1994) Division inhibition gene dicF of Escherichia coli reveals a widespread group of prophage sequences in bacterial genomes. J Bacteriol 176 : 1150–1156.
29. de BoerPA, CrossleyRE, RothfieldLI (1990) Central role for the Escherichia coli minC gene product in two different cell division-inhibition systems. Proc Natl Acad Sci U S A 87 : 1129–1133.
30. JohnsonJE, LacknerLL, de BoerPAJ (2002) Targeting of (D)MinC/MinD and (D)MinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ rings. J Bacteriol 184 : 2951–2962.
31. TétartF, BouchéJP (1992) Regulation of the expression of the cell-cycle gene ftsZ by DicF antisense RNA. Division does not require a fixed number of FtsZ molecules. Mol Microbiol 6 : 615–620.
32. JafféA, D'AriR, NorrisV (1986) SOS-independent coupling between DNA replication and cell division in Escherichia coli. J Bacteriol 165 : 66–71.
33. MaguinE, LutkenhausJ, D'AriR (1986) Reversibility of SOS-associated division inhibition in Escherichia coli. J Bacteriol 166 : 733–738.
34. D'AriR, HuismanO (1983) Novel mechanism of cell division inhibition associated with the SOS response in Escherichia coli. J Bacteriol 156 : 243–250.
35. WangXD, de BoerPA, RothfieldLI (1991) A factor that positively regulates cell division by activating transcription of the major cluster of essential cell division genes of Escherichia coli. EMBO J 10 : 3363–3372.
36. DaiK, LutkenhausJ (1992) The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J Bacteriol 174 : 6145–6151.
37. WeartRB, LevinPA (2003) Growth rate-dependent regulation of medial FtsZ ring formation. J Bacteriol 185 : 2826–2834.
38. LutkenhausJ, AddinallSG (1997) Bacterial cell division and the Z ring. Annu Rev Biochem 66 : 93–116.
39. DajkovicA, MukherjeeA, LutkenhausJ (2008) Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J Bacteriol 190 : 2513–2526 doi: 10.1128/JB.01612-07
40. TruscaD, ScottS, ThompsonC, BramhillD (1998) Bacterial SOS checkpoint protein SulA inhibits polymerization of purified FtsZ cell division protein. J Bacteriol 180 : 3946–3953.
41. DajkovicA, LanG, SunSX, WirtzD, LutkenhausJ (2008) MinC spatially controls bacterial cytokinesis by antagonizing the scaffolding function of FtsZ. Curr Biol 18 : 235–244 doi: 10.1016/j.cub.2008.01.042
42. HuZ, LutkenhausJ (2000) Analysis of MinC reveals two independent domains involved in interaction with MinD and FtsZ. J Bacteriol 182 : 3965–3971.
43. ShenB, LutkenhausJ (2010) Examination of the interaction between FtsZ and MinCN in E. coli suggests how MinC disrupts Z rings. Mol Microbiol 75 : 1285–1298 doi: 10.1111/j.1365-2958.2010.07055.x
44. ShiomiD, MargolinW (2007) The C-terminal domain of MinC inhibits assembly of the Z ring in Escherichia coli. J Bacteriol 189 : 236–243.
45. LiY, HsinJ, ZhaoL, ChengY, ShangW, et al. (2013) FtsZ protofilaments use a hinge-opening mechanism for constrictive force generation. Science 341 : 392–395 doi: 10.1126/science.1239248
46. BiE, LutkenhausJ (1990) Analysis of ftsZ mutations that confer resistance to the cell division inhibitor SulA (SfiA). J Bacteriol 172 : 5602–5609.
47. BiE, LutkenhausJ (1990) Interaction between the min locus and ftsZ. J Bacteriol 172 : 5610–5616.
48. PichoffS, LutkenhausJ (2001) Escherichia coli division inhibitor MinCD blocks septation by preventing Z-ring formation. J Bacteriol 183 : 6630–6635.
49. GeisslerB, ElrahebD, MargolinW (2003) A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A 100 : 4197–4202.
50. GeisslerB, ShiomiD, MargolinW (2007) The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology 153 : 814–825.
51. PichoffS, ShenB, SullivanB, LutkenhausJ (2012) FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA's self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol 83 : 151–67 doi: 10.1111/j.1365-2958.2011.07923.x
52. GeisslerB, MargolinW (2005) Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol Microbiol 58 : 596–612.
53. DajkovicA, PichoffS, LutkenhausJ, WirtzD (2010) Cross-linking FtsZ polymers into coherent Z rings. Mol Microbiol 78 : 651–668 doi: 10.1111/j.1365-2958.2010.07352.x
54. Gueiros-FilhoFJ, LosickR (2002) A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev 16 : 2544–2556.
55. SmallE, MarringtonR, RodgerA, ScottDJ, SloanK, et al. (2007) FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. J Mol Biol 369 : 210–221.
56. HaleCA, RheeAC, de BoerPA (2000) ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J Bacteriol 182 : 5153–5166.
57. KuchibhatlaA, BhattacharyaA, PandaD (2011) ZipA Binds to FtsZ with High Affinity and Enhances the Stability of FtsZ Protofilaments. PLoS ONE 6: e28262 doi: 10.1371/journal.pone.0028262
58. MosyakL, ZhangY, GlasfeldE, HaneyS, StahlM, et al. (2000) The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J 19 : 3179–3191.
59. HaleCA, de BoerPA (1997) Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88 : 175–185.
60. GreerH, AusubelFM (1979) Radiochemical identification of the kil gene product of bacteriophage lambda. Virology 95 : 577–580.
61. BuskePJ, LevinPA (2012) Extreme C-terminus of the bacterial cytoskeletal protein FtsZ plays a fundamental role in assembly independent of modulatory proteins. J Biol Chem 287 (14) 10945–10957 doi: 10.1074/jbc.M111.330324
62. MukherjeeA, LutkenhausJ (1999) Analysis of FtsZ assembly by light scattering and determination of the role of divalent metal cations. J Bacteriol 181 : 823–832.
63. YuXC, MargolinW (1997) Ca2+-mediated GTP-dependent dynamic assembly of bacterial cell division protein FtsZ into asters and polymer networks in vitro. EMBO J 16 : 5455–5463.
64. VolpiL, GhisottiD, SironiG (1983) Evidence of cell fragility caused by gene kil following lambda induction. Virology 128 : 166–175.
65. JohnsonJE, LacknerLL, HaleCA, de BoerPAJ (2004) ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J Bacteriol 186 : 2418–2429.
66. HaeusserDP, SchwartzRL, SmithAM, OatesME, LevinPA (2004) EzrA prevents aberrant cell division by modulating assembly of the cytoskeletal protein FtsZ. Mol Microbiol 52 : 801–814.
67. KleinP, KanehisaM, DeLisiC (1985) The detection and classification of membrane-spanning proteins. Biochim Biophys Acta 815 : 468–476.
68. ChungK-M, HsuH-H, YehH-Y, ChangB-Y (2007) Mechanism of regulation of prokaryotic tubulin-like GTPase FtsZ by membrane protein EzrA. J Biol Chem 282 : 14891–14897.
69. KiroR, Molshanski-MorS, YosefI, MilamSL, EricksonHP, et al. (2013) Gene product 0.4 increases bacteriophage T7 competitiveness by inhibiting host cell division. Proc Natl Acad Sci U S A 110 : 19549–19554.
70. StewartCR, DeeryWJ, EganESK, MylesB, PettiAA (2013) The product of SPO1 gene 56 inhibits host cell division during infection of Bacillus subtilis by bacteriophage SPO1. Virology 447 : 249–253 doi: 10.1016/j.virol.2013.09.005
71. WangX, KimY, MaQ, HongSH, PokusaevaK, et al. (2010) Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1 : 147 doi: 10.1038/ncomms1146
72. Ballesteros-PlazaD, HolgueraI, ScheffersD-J, SalasM, Muñoz-EspínD (2013) Phage 29 protein p1 promotes replication by associating with the FtsZ ring of the divisome in Bacillus subtilis. Proc Natl Acad Sci U S A 110 : 12313–12318 doi: 10.1073/pnas.1311524110
73. SergueevK, CourtD, ReavesL, AustinS (2002) E. coli cell-cycle regulation by bacteriophage lambda. J Molec Biol 324 : 297–307.
74. St-PierreF, EndyD (2008) Determination of cell fate selection during phage lambda infection. Proc Natl Acad Sci U S A 105 : 20705–20710 doi: 10.1073/pnas.0808831105
75. SemerjianAV, MalloyDC, PoteeteAR (1989) Genetic structure of the bacteriophage P22 PL operon. J Mol Biol 207 : 1–13.
76. BrileyKJr, PrepiakP, DiasMJ, HahnJ, DubnauD (2011) Maf acts downstream of ComGA to arrest cell division in competent cells of B. subtilis. Mol Microbiol 81 : 23–39 doi: 10.1111/j.1365-2958.2011.07695.x
77. SawitzkeJA, ThomasonLC, CostantinoN, BubunenkoM, DattaS, et al. (2007) Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol 421 : 171–199.
78. ThomasonL, CourtDL, BubunenkoM, CostantinoN, WilsonH, et al. (2007) Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 1.16.1–1.16.24.
79. StuddertCA, ParkinsonJS (2005) Insights into the organization and dynamics of bacterial chemoreceptor clusters through in vivo crosslinking studies. Proc Natl Acad Sci U S A 102 : 15623–15628.
80. GuzmanLM, BelinD, CarsonMJ, BeckwithJ (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 : 4121–4130.
81. PowellBS, RivasMP, CourtDL, NakamuraY, TurnboughCLJr (1994) Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res 22 : 5765–5766.
82. ShiomiD, MargolinW (2007) Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Mol Microbiol 66 : 1396–1415.
83. SawitzkeJA, CostantinoN, LiXT, ThomasonLC, BubunenkoM, et al. (2011) Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407 : 45–59 doi: 10.1016/j.jmb.2011.01.030
84. UlbrandtND, NewittJA, BernsteinHD (1997) The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88 : 187–196.
85. WilsonDS, KeefeAD (2000) Random mutagenesis by PCR. Curr Protoc Mol Biol Unit 8.3. doi: 10.1002/0471142727.mb0803s51
86. Levin PA (2001) Light microscopy techniques for bacterial cell biology. In: Sansonetti P, Zychlinsky A, editors. Methods in Microbiology 31: Molecular Cellular Microbiology. Academic Press Ltd., London. 115–132.
87. YuXC, TranAH, SunQ, MargolinW (1998) Localization of cell division protein FtsK to the Escherichia coli septum and identification of a potential N-terminal targeting domain. J Bacteriol 180 : 1296–1304.
88. VischerN, WoldringhCL (1994) Object-Image: an interactive image analysis program using structured point collection. Binary 6 : 160–166.
89. SchneiderCA, RasbandWS, EliceiriKW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9 : 671–675.
90. BusiekKK, ErasoJM, WangY, MargolinW (2012) The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. J Bacteriol 194 : 1989–2000 doi: 10.1128/JB.06683-11
91. StudierFW, MoffattBA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189 : 113–130.
92. OlivaMA, TrambaioloD, LöweJ (2007) Structural insights into the conformational variability of FtsZ. J Mol Biol 373 : 1229–1242.
93. YuD, EllisHM, LeeEC, JenkinsNA, CopelandNG, et al. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97 : 5978–5983.
94. BabaT, AraT, HasegawaM, TakaiY, OkumuraY, et al. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2 : 2006.0008.
95. SunQ, MargolinW (2001) Influence of the nucleoid on placement of FtsZ and MinE rings in Escherichia coli. J Bacteriol 183 : 1413–1422.
96. ChanW, CostantinoN, LiR, LeeSC, SuQ, et al. (2007) A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res 35: e64.
97. BeallB, LoweM, LutkenhausJ (1988) Cloning and characterization of Bacillus subtilis homologs of Escherichia coli cell division genes ftsZ and ftsA. J Bacteriol 170 : 4855–4864.
98. ThanedarS, MargolinW (2004) FtsZ exhibits rapid movement and oscillation waves in helix-like patterns in Escherichia coli. Curr Biol 14 : 1167–1173.
99. WeissDS, ChenJC, GhigoJM, BoydD, BeckwithJ (1999) Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol 181 : 508–520.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle
- Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion
- Genetic Dissection of Photoreceptor Subtype Specification by the Zinc Finger Proteins Elbow and No ocelli
- GC-Rich DNA Elements Enable Replication Origin Activity in the Methylotrophic Yeast
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy