
Co-translational Localization of an LTR-Retrotransposon RNA to the Endoplasmic Reticulum Nucleates Virus-Like Particle Assembly Sites
Retrotransposons are mobile elements that have invaded the genomes of organisms from bacteria to humans. Facilitated by host co-factors, retrotransposon proteins copy their RNA genomes into DNA that integrates into the host genome, causing mutations and genome instability. The yeast Ty1 element belongs to a family of retrotransposons that are related to infectious retroviruses. Ty1 RNA and its coat protein, Gag, assemble into virus-like particles, wherein the RNA is copied into DNA. It was not previously known how Ty1 RNA and Gag are concentrated in a specific cellular location to initiate the assembly of virus-like particles. In this study, we show that Ty1 RNA is brought to the presumptive assembly site during translation by the protein chaperone, signal recognition particle. As Ty1 RNA is translated, the nascent Gag polypeptide enters the lumen of the endoplasmic reticulum, where Gag adopts a stable conformation before returning to the cytoplasm to bind to translating Ty1 RNA. An interaction between Gag molecules bound to translating Ty1 RNA results in the nucleation of the virus-like particle assembly site. Our findings identify new host co-factors in retrotransposon mobility and suggest potential approaches to controlling retrotransposon-associated genome instability in aging and cancer.
Published in the journal:
. PLoS Genet 10(3): e32767. doi:10.1371/journal.pgen.1004219
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004219
Summary
Retrotransposons are mobile elements that have invaded the genomes of organisms from bacteria to humans. Facilitated by host co-factors, retrotransposon proteins copy their RNA genomes into DNA that integrates into the host genome, causing mutations and genome instability. The yeast Ty1 element belongs to a family of retrotransposons that are related to infectious retroviruses. Ty1 RNA and its coat protein, Gag, assemble into virus-like particles, wherein the RNA is copied into DNA. It was not previously known how Ty1 RNA and Gag are concentrated in a specific cellular location to initiate the assembly of virus-like particles. In this study, we show that Ty1 RNA is brought to the presumptive assembly site during translation by the protein chaperone, signal recognition particle. As Ty1 RNA is translated, the nascent Gag polypeptide enters the lumen of the endoplasmic reticulum, where Gag adopts a stable conformation before returning to the cytoplasm to bind to translating Ty1 RNA. An interaction between Gag molecules bound to translating Ty1 RNA results in the nucleation of the virus-like particle assembly site. Our findings identify new host co-factors in retrotransposon mobility and suggest potential approaches to controlling retrotransposon-associated genome instability in aging and cancer.
Introduction
Long terminal repeat (LTR)-retrotransposons are ubiquitous molecular symbionts of eukaryotic genomes whose mobility is responsive to environmental and developmental cues and can result in host cell genome remodeling. These retroelements are the evolutionary progenitors of retroviruses, which have acquired env genes, and with them, the ability of their nucleocapsids to undergo exocytosis and infection of a naïve cell [1]. In contrast, LTR-retrotransposons lack env genes and replicate intracellularly. Because of their streamlined genomes and complex life cycles, both retroviruses and LTR-retrotransposons rely extensively on host cell factors to proliferate, yet much remains to be learned about the role of host cell pathways in retroelement replication.
Our understanding of the mechanism of LTR-retrotransposon replication is derived largely from the study of Ty elements in Saccharomyces cerevisiae. Ty1 elements comprise the most abundant and most active family of LTR-retrotransposons in budding yeast. Ty1 contains a gag ORF that encodes a single structural protein with capsid and nucleocapsid functions, and a pol ORF, which encodes protease (PR), integrase (IN) and reverse transcriptase (RT) activities. A 5.7 kb sense-strand RNA expressed from genomic Ty1 elements functions both as an mRNA and as the genomic RNA of nucleocapsids, or VLPs. Ty1 RNA is reverse transcribed in cytoplasmic VLPs to form a DNA copy (cDNA). The Ty1 cDNA is transported to the nucleus and inserted into the host cell genome by integration or more rarely, homologous recombination [2].
Ty1 RNA is translated into two precursor proteins, p49-Gag and p199-Gag-Pol, the latter a result of programmed translational frameshifting from gag to pol. The p49-Gag and p199-Gag-Pol proteins multimerize to assemble into VLPs. Gag binds Ty1 RNA and encapsidates it as a dimer into the VLP during assembly [3], [4]. Initiated by autocatalytic processing of p20-PR from the p199-Gag-Pol precursor, proteolytic processing of p49-Gag and p199-Gag-Pol to mature p45-Gag, p20-PR, p71-IN and p63-RT is thought to occur within the assembled VLP [5], [6], [7], [8].
Ty1 RNA and Gag co-localize in microscopically distinct cytoplasmic RNA foci known as T bodies [9] or retrosomes [10]. Ty1 retrosomes partially co-localize with P bodies, and many factors involved in translational repression and P body formation are also activators of Ty1 retrosome formation and retrotransposition. In fact, virtually all 5′-3′ mRNA decay and nonsense-mediated decay factors that have been analyzed are required for post-translational steps in Ty1 retrotransposition [11], [12]. Nonetheless, Ty1 retrosomes are functionally distinct from P bodies, as retrosomes disassemble during glucose starvation, which stimulates P body formation, and retrosomes are stable when translational elongation is blocked by cycloheximide, which triggers P body disassembly [9], [11]. These findings led to the idea that P body functions promote the clustering of Ty1 RNA molecules into distinct cellular foci whose formation promotes VLP assembly [10], [13]. Retrosomes are thought to be nascent VLP assembly sites because they can form in the absence of proteolytic processing of Ty1 proteins [14]. Moreover, Gag visualized by immunoelectron microscopy is found in cytoplasmic clusters that are associated with VLPs when VLP formation is induced by overexpression of Ty1 RNA [11], [15]. In P body mutants xrn1Δ and lsm1Δ, a lack of distinct Ty1 retrosomes is correlated with decreased clustering of VLPs. While the appearance of dispersed VLPs is increased in these mutants, Ty1 cDNA does not accumulate, suggesting that assembly of VLPs within the retrosome is critical for Ty1 replication [11].
Studies with plasmid-borne, galactose-inducible Ty1 (pGAL1:Ty1) elements in strains lacking endogenous Ty1 expression have demonstrated that retrosomes do not form when the gag ORF is not translated or when Gag lacking its C-terminal RNA binding domain is expressed [14], [16]. Beyond a role for functional Gag, very little is known about the requirements for the nucleation of VLP assembly sites. For example, it is not known whether Ty1 RNA is partitioned into separate pools of mRNA and genomic RNA, or whether translating Ty1 RNA can be packaged into VLPs. Moreover, the mechanism by which Ty1 RNA and Gag are directed to the presumptive VLP assembly site has not been described. One scenario that has been proposed is that Gag binds Ty1 RNA during or shortly after translation, thereby triggering its sequestration from translation. Ty1 RNA-Gag complexes could then coalesce in foci in a manner mechanistically related to the sequestration of mRNA in P bodies [10]. However, Ty1 proteins do not display a cis-preference for mobilizing the RNA molecule by which they are encoded [17], indicating that Ty1 Gag may be separated spatially or temporally from its RNA template after translation.
The goal of the present study was to determine how Ty1 RNA and Gag are directed to the presumptive VLP assembly site. Specifically, we explored the hypothesis that Ty1 RNA is co-translationally localized to a specific subcellular domain, resulting in coordinated localization of Ty1 RNA and newly synthesized Gag to the presumptive VLP assembly site. Co-translational localization of mRNAs on RNC complexes is a major pathway for targeting mRNAs encoding secretory and membrane proteins to the ER so that the nascent peptides can be translocated across the ER membrane (reviewed in [18], [19]). Co-translational mRNA targeting to the ER is mediated by SRP, an evolutionarily conserved ribonucleoprotein complex that functions as a protein chaperone (reviewed in [20]). SRP has two major domains: the Alu domain, which binds the ribosome at the elongation-factor binding site and transiently pauses elongation, and the S domain, which binds a hydrophobic signal sequence in the nascent peptide. Dual binding of the ribosome and the signal domain of the nascent peptide is accompanied by a conformational change in SRP [21], and accounts for the high affinity of SRP for cognate RNC complexes [22]. SRP docks the RNC to the membrane-bound SRP receptor and aligns the nascent chain tunnel with the ER translocon, a channel through which the nascent peptide traverses the ER membrane as the mRNA template is translated (Figure 1). The ER chaperone, Kar2, interacting with the translocon-associated Sec63 complex, promotes translocation of nascent peptides to the ER lumen.

Here, we provide evidence that Ty1 RNA-nascent Gag translation complexes are associated with SRP, and nascent Gag is translocated to the lumen of the ER. Our data support a model in which Gag is folded into a stable conformation in the ER lumen and then retrotranslocated to the cytoplasm, where it binds Ty1 RNA on SRP-associated RNCs. Multimerization of Gag bound to Ty1 RNA-SRP-RNC complexes results in the coalescence of Ty1 RNA into retrosomes and likely promotes translational repression and packaging of Ty1 RNA in VLPs. These findings suggest that Ty1 RNA transitions between its role in translation and its role as the genomic RNA of VLPs. Moreover, the observation that nascent Gag is separated from its RNA template by transit into and out of the ER lumen uncovers a new mechanism by which Gag can associate with translating retroelement RNA without displaying a cis-preference for packaging its own RNA template.
Results
A defect in N-glycosylation results in Ty1 Gag instability
Deletion of DFG10, a gene encoding polyprenol reductase, was identified in a screen for Ty1 retrotransposition-defective mutants [23]. Dfg10 catalyzes the synthesis of dolichol, the precursor for N-linked protein glycosylation in the ER. Western blot analysis of Gag protein expressed from the ∼30 endogenous Ty1 elements demonstrated that the steady-state level of Gag is substantially decreased in the dfg10Δ mutant (Figure 2A). A C-terminal fusion of GFP to p45-Gag (Gag:GFP), expressed from the LTR promoter on a plasmid, was also present at a reduced level in the dfg10Δ mutant. Ty1 RNA foci were visualized by performing fluorescent in situ hybridization (FISH) with a Cy3-labeled antisense primer in the gag ORF and detecting the hybrid by fluorescent microscopy. Ty1 RNA failed to efficiently localize to foci, or retrosomes, in the dfg10Δ mutant (Figure 2B). Similarly, deletion of the ribosome biogenesis factor gene, BUD21, resulted in decreased Gag, Gag:GFP and Ty1 RNA foci. To determine whether the lack of Gag accumulation in these mutants results from a reduced level of Ty1 RNA, inefficient translation or protein instability, we performed northern blot analysis of Ty1 RNA and pulse-chase labeling followed by immunoprecipitation of Gag. The steady-state level of Ty1 RNA in a dfg10Δ mutant was very low relative to the congenic wild-type strain or the bud21Δ mutant (Figure 2C). Surprisingly though, the amount of labeled Gag that was immunoprecipitated immediately after pulse-labeling of proteins in a dfg10 mutant was 89% of that immunoprecipitated from the wild-type strain (Figure 2D, 0 min chase). In contrast, pulse-labeled Gag in the bud21Δ mutant was reduced to 26% of that in the wild-type strain, consistent with the proposed role for Bud21 in translation of Ty1 RNA [23]. These data indicate that synthesis of Gag is not significantly reduced by deletion of DFG10, despite the reduced steady-state level of Ty1 RNA. However, Gag is degraded rapidly after synthesis in the dfg10Δ mutant, as evidenced by the significant reduction in pulse-labeled Gag after a 30 or 60 min chase relative to the level in the wild-type strain at the same time points (Figure 2D). Thus, accumulation of Ty1 RNA and Gag is substantially reduced by a post-translational mechanism in the dfg10Δ mutant. The increased turnover of Gag in a mutant with a defect in N-linked glycosylation suggests that Ty1 Gag is degraded as a result of induction of the ER stress response. However, Ty1 Gag has not previously been shown to be associated with the ER.
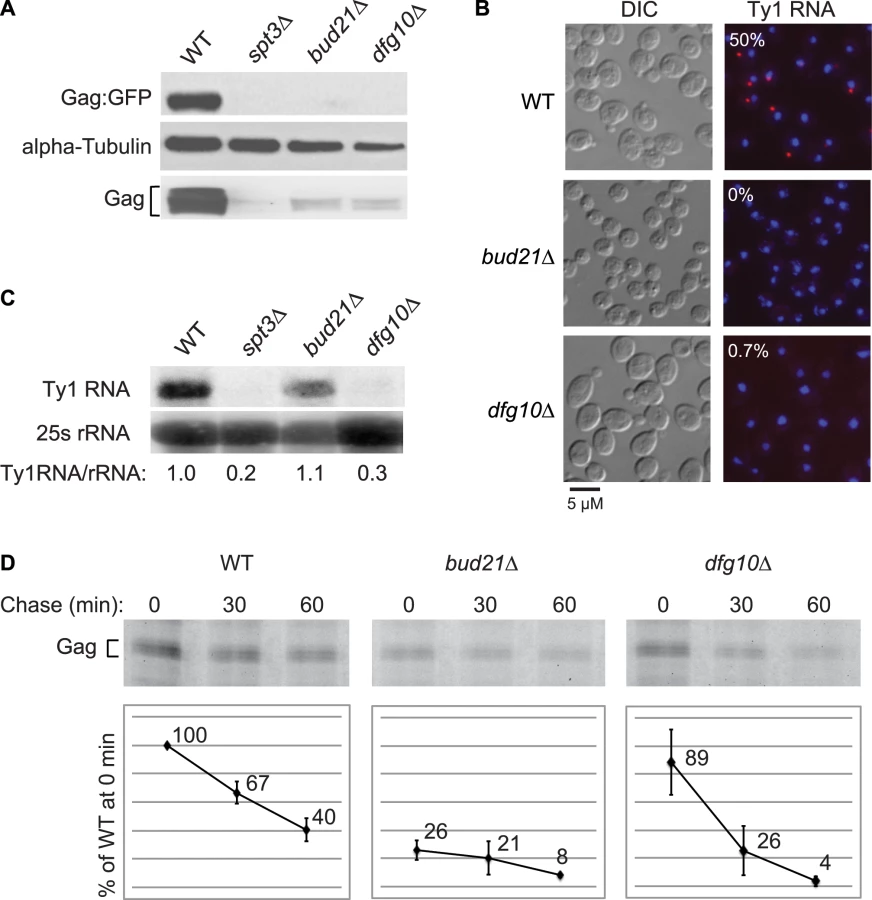
Gag is translocated to the ER lumen
Several experiments were performed to determine whether Ty1 Gag is associated with the ER membrane or is a soluble protein in the ER lumen. To investigate the possible association of Gag with the ER membrane, equilibrium density gradient centrifugation was performed to investigate the flotation behavior of Gag (Figure 3A). To provide a membrane-associated control protein for this analysis, we used a strain in which the KAR2 ORF, which encodes the ER chaperone, Kar2/BiP, was fused at the C-terminal end to the tandem affinity purification (TAP) tag. Kar2 is a soluble lumen protein; however, it is tethered to the membrane via interactions with the Sec63 complex and the Hrd1/Hrd3-Yos9 complex [18], [24], so Kar2 co-purifies primarily with membrane fractions [24], . Cell lysate adjusted to 2 M sucrose was loaded between higher and lower sucrose cushions in a step gradient (Figure 3A). After centrifugation of the gradient, nine fractions of equal volume were collected and analyzed by western blotting. Ty1 Gag was concentrated in high-density fractions 7 to 9. As expected, Kar2-TAP was found primarily in low-density fractions 3 to 5, but was also present in high-density fractions 7 to 9. In an identical analysis of a congenic ADH5-TAP strain, the cytosolic Adh5-TAP protein was found only in high-density fractions 7 to 9. The presence of Gag exclusively in high-density fractions suggests that little if any Gag associates with the ER membrane.

To determine whether Gag is present as a soluble protein in the lumen of the ER, we prepared ER microsomes from the KAR2-TAP strain. Microsomes are cell fractions enriched for closed vesicles of fragmented ER. Following disruption of microsomes with sodium carbonate, the membrane fraction was separated from the soluble lumen fraction by centrifugation. Western blot analysis was performed on total microsomes (T), as well as the membrane-enriched pellet (P) and lumen-enriched supernatant (S) fractions of sodium carbonate-extracted microsomes (Figure 3B). Ty1 Gag co-purified with total microsomes, as did Kar2-TAP. As expected, Kar2-TAP co-fractionated with both the pellet and the supernatant fractions of sodium carbonate-treated microsomes. Gag segregated primarily with the supernatant, which is indicative of its presence in the lumen of the ER. A low level of Gag was also present in the pellet fraction, which was unexpected because Gag was not apparent in low-density membrane fractions in the flotation assay (Figure 3A). The presence of Gag in the microsome pellet could result from aggregation of Gag during the incubation of microsomes with sodium carbonate. Alternatively, if a low level of Gag is transiently associated with the ER membrane, it might be detectable in these concentrated microsomal fractions as compared to the membrane fractions of the density gradient in the flotation assay.
As expected, Gag was present in microsomes prepared from the ADH5-TAP strain as well, but Adh5-TAP was not detected, suggesting that there was minimal contamination of microsome preparations with cytosolic proteins (unpublished result). To directly determine whether Gag association with ER microsomes is due to its presence in the ER lumen or to contamination of microsomes with cytosolic Gag, we treated the microsome preparation with trypsin (Figure 3C). If Gag is present on the outer surface of microsomes because of cytosolic contamination, it should be digested by trypsin; however, the level of Gag was not reduced by treatment with trypsin. Kar2-TAP protein was also resistant to trypsin digestion, which demonstrates that ER lumen proteins were protected in the microsome preparation. In contrast, trypsin efficiently digested small non-specific proteins, indicating that trypsin digested contaminating proteins under these conditions. When microsomes were treated with trypsin and Triton X-100, which disrupts the microsomal membrane, both Gag and Kar2-TAP were markedly reduced. Together, these findings demonstrate that some fraction of Gag is present in the lumen of the ER.
Ty1 RNA and Gag are associated with SRP-RNC complexes
A major pathway by which proteins enter the ER is co-translational translocation mediated by SRP (Figure 1). SRP binds a hydrophobic domain of nascent peptides, including N-terminal signal sequences and transmembrane domains; however, not all peptide domains that are recognized by SRP have been defined [26]. Therefore, it is conceivable that the Ty1 Gag polypeptide, although lacking a discernible N-terminal signal sequence or transmembrane domain, could be co-translationally targeted to the ER lumen by SRP. To explore the possibility, we examined the results of a genome-wide study of SRP targets by del Alamo et al. [26]. These authors identified a collection of yeast ORFs corresponding to mRNAs that associate with SRP-RNC complexes despite the absence of a signal sequence or transmembrane domain in the corresponding nascent polypeptide. Three ORFs that encode Ty1 Gag were present in this collection: YJR027W, YBL005W-A and YGR109W-A. These findings suggest that Ty1 RNA-ribosome-nascent Gag complexes are targets of the SRP chaperone.
As an independent test of the hypothesis that Ty1 RNA is translated in association with SRP, we treated cells of an SRP54-TAP strain with cycloheximide to stabilize SRP-RNC complexes, affinity-purified Srp54-TAP complexes and analyzed the co-purifying RNA by RT-PCR using gene-specific primers (Figure 4A). The enrichment of SRP subunit 7SL RNA and 18S rRNA in the Srp54-TAP purification confirmed that SRP-RNC complexes were purified with Srp54-TAP. As controls for non-specific binding, cells expressing TAP-tagged Lsm1, an activator of mRNA decapping, or no TAP tag were purified under identical conditions. The lack of enrichment of 7SL RNA and 18S rRNA in the control purifications indicates that SRP-RNC complexes were specifically enriched in the Srp54-TAP purification. Next, we assayed for the association of Ty1 RNA with SRP-RNC complexes. We found that Ty1 RNA was substantially enriched in the Srp54-TAP purification, whereas its enrichment was minimal or absent in the Lsm1-TAP and no-TAP controls, respectively. Furthermore, a Ty1 PCR product was not detected in a reaction lacking reverse transcriptase, demonstrating that amplification of Ty1 sequences was not due to the presence of contaminating DNA.
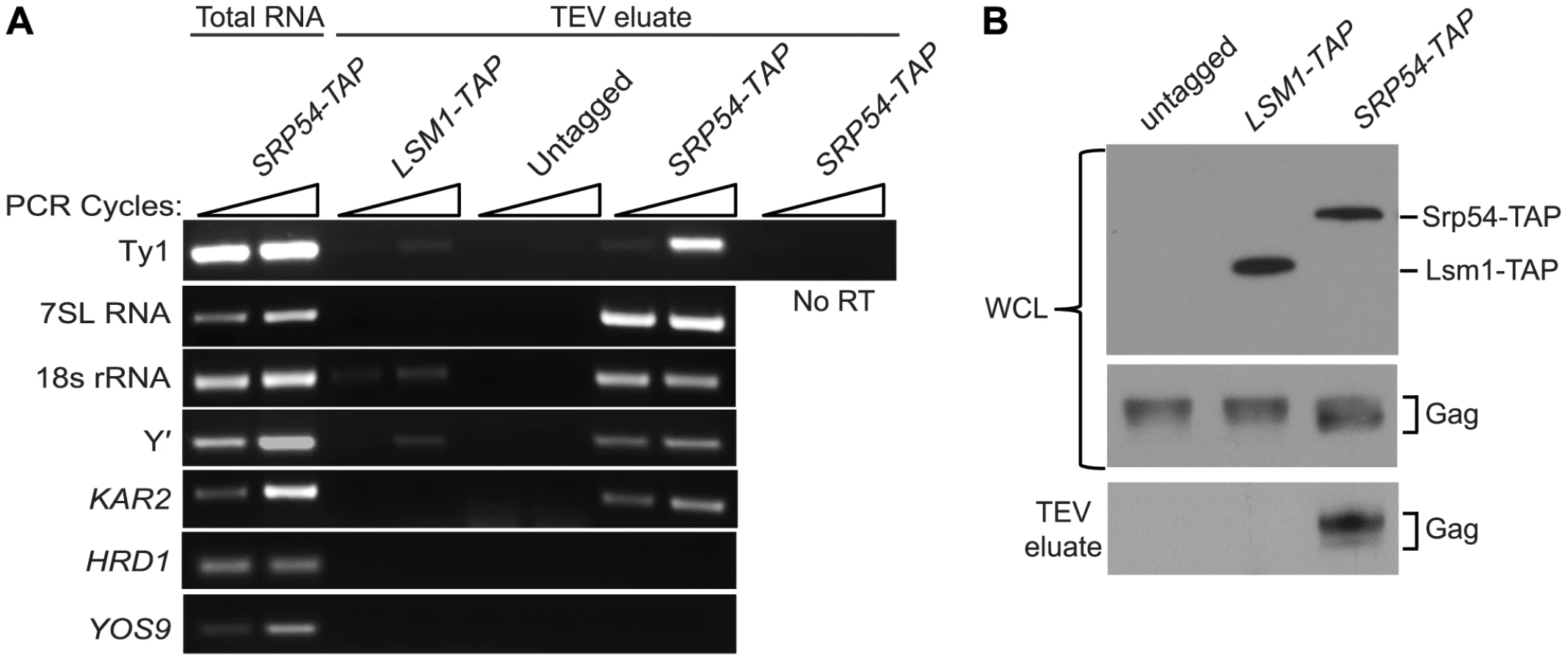
Three uncharacterized ORFs, YEL076C, YLR463C and YHL049C, corresponding to the mRNA of the subtelomeric repetitive element, Y′ were also identified as interacting with SRP-RNC complexes in the genome-wide study [26]. Notably, Y′ RNA is enriched in Ty1 VLPs [27]. We found that Y′ RNA was substantially enriched in the Srp54-TAP purification but not in the Lsm1-TAP or mock purification. Similar results were obtained with KAR2 mRNA, which is also known to interact with SRP during translation. On the other hand, mRNAs encoding the transmembrane protein Hrd1 and the ER-lumen protein Yos9, which undergo SRP-independent translation [26], were not enriched in Srp54-TAP complexes (Figure 3A). Together, these data confirm that SRP-associated RNC complexes were selectively purified in the Srp54-TAP affinity purification. Thus, we conclude that Ty1 RNA is likely translated in association with SRP and therefore, that Gag is translocated to the ER lumen during translation.
It has been proposed that binding of Ty1 Gag to translating Ty1 RNA represses its translation and promotes its packaging into VLPs [10]. Therefore, we assayed for the presence of Gag in affinity purified SRP-RNC complexes by western blotting. Gag co-purified with Srp54-TAP, but was not detected in the Lsm1-TAP or no TAP-tag purification (Figure 4B). The co-purification of Gag with SRP-associated translation complexes indicates that Gag is present not only in the ER lumen but also in the cytoplasm. Moreover, the results suggest that Gag binds Ty1 RNA that is being translated on SRP-RNC complexes. However, it is also possible that Gag bridges an interaction between non-translating Ty1 RNA and SRP-RNC complexes. To rule out this possibility and support the conclusion that Ty1 RNA is translated in association with SRP, we determined whether Ty1 RNA is associated with SRP-RNC complexes in the retrotransposition-defective rpl7aΔ mutant [23]. In this mutant, Ty1 RNA and Gag fail to co-localize in retrosomes, even though Ty1 RNA and Gag are present at wild type or modestly reduced levels, respectively (Figure S2 and unpublished data). These observations suggested to us that the interaction between Ty1 RNA and Gag is disrupted in the rpl7aΔ mutant. (The retrotransposition defect of the rpl7aΔ mutant will be described in detail elsewhere). We found that Ty1 RNA co-purified with 7SL RNA, 18S rRNA and Y′ RNA in Srp54-TAP complexes from the rpl7aΔ mutant (Figure S2A); however, no Gag was detected in the Srp54-TAP complexes (Figure S2B). In contrast, Gag co-purified with Srp54-TAP in the bud21Δ mutant, despite the low level of Gag in this mutant (Figure S2B). Thus, Ty1 RNA is present in SRP-RNC complexes even when Gag is not detectably associated, and therefore, Gag does not bridge the interaction between Ty1 RNA and SRP. Together, these results are consistent with a model in which Gag binds Ty1 RNA that is translated in association with SRP. Notably, the lack of Gag association with Ty1 RNA on SRP-RNC complexes in the rpl7aΔ mutant is correlated with an absence of Ty1 RNA and Gag localization in retrosomes (Figure S2C), raising the possibility that Gag binding to Ty1 RNA translation complexes is required for the nucleation of retrosomes.
Depletion of co-translational ER translocation factors reduces Gag stability and Ty1 retrotransposition
The association of Ty1 RNA with SRP-RNC complexes and the presence of Gag in the ER lumen suggest that Gag is co-translationally translocated to the ER lumen. SRP directs nascent polypeptides to the ER lumen by binding to the SRP receptor and delivering the nascent peptides to the ER translocon, (Figure 1). To further explore the role of the SRP-mediated translocation pathway in the synthesis of Gag, we examined the effect of mutations in genes encoding subunits of SRP, SRP receptor, ER translocon and translocon-associated complexes on Ty1 RNA and Gag levels. Because deletion of these genes results in severe growth defects, and because Ty1 retrotransposition is heat-sensitive, we used Decreased Abundance by mRNA Perturbation (DAmP) mutations. DAmP alleles contain a selectable marker, KanMX, inserted into the 3′ UTR region of the gene, which results in variable levels of mRNA destabilization and reduced gene expression [28]. To determine whether SRP-dependent translocation was compromised in the DAmP mutants, we used a phenotypic assay for translocation of a Pho8-Ura3 reporter protein to the ER [29] (Figure S3 and data not shown). The efficiency of Pho8-Ura3 translocation varied among strains carrying DAmP alleles of different co-translational translocation genes. Most mutants had partial (srp54-DAmP, srp72-DAmP and srp101-DAmP) or severe (srp68-DAmP, sec61-DAmP and sec63-DAmP) defects in Pho8-Ura3 translocation, and sec61-DAmP and srp68-DAmP mutants grew more slowly than the wild-type strain. Only the srp21-DAmP and srp102-DAmP mutants had no detectable deficiency in Pho8-Ura3 translocation relative to the wild-type strain. The relative steady-state level of Gag in each mutant was determined by western blot analysis (Figure 5A). Gag levels were reduced to varying degrees in eight mutants harboring DAmP alleles of genes encoding subunits of SRP (Srp21, Srp54, Srp68 and Srp72), the SRP receptor (Srp101 and Srp102), the ER translocon (Sec61) or the ER chaperone, Kar2. However, Gag levels were not decreased in the sec63-DAmP strain (Figure 5A), even though Pho8-Ura3 translocation was strongly compromised in this mutant (Figure S3). In contrast, Gag levels were markedly decreased in srp21-DAmP and srp102-DAmP mutants (Figure 5A), which lacked a detectable defect in Pho8-Ura3 translocation (Figure S3 and data not shown). Together, the data indicate that each of the nine DAmP mutants analyzed are hypomorphic; however, some of the mutations affect Gag accumulation differently than they do Pho8-Ura3 translocation. Overall, we find that Gag accumulation is reduced when co-translational ER translocation is compromised.

The results above suggest that the level of SRP could directly determine how much Ty1 Gag accumulates. To test this interpretation, we depleted functional SRP complexes by shutting off the expression of Srp54, which is required for SRP to translocate proteins to the ER [30], [31], and measuring the level of Ty1 Gag as a function of declining Srp54 levels (Figure 5B). An srp54Δ strain harboring plasmid pGAL1:SRP54-HA was shifted from galactose medium to glucose medium to halt transcription of the GAL1p:SRP54-HA cassette. Srp54-HA decreased from 0 to 28 hours after the carbon source shift, and Gag levels decreased concomitantly, whereas the level of the control protein, GAPDH was unaffected. These data indicate that the level of Gag is directly correlated with the level of functional SRP, suggesting that SRP-mediated ER translocation is necessary for the accumulation of Gag.
Depletion of Gag in mutants with hypomorphic alleles of SRP, SRP receptor and ER translocon subunit genes is correlated with a defect in retrotransposition of a chromosomal Ty1his3AI element (Figure 5C). The mobility of Ty1his3AI was measured quantitatively by determining the frequency of His+ prototroph formation in each strain [32]. Mutants with a small reduction in Gag accumulation, such as srp54-DAmP and srp72-DAmP, displayed an insignificant reduction in retrotransposition, while those with very low levels of Gag, such as srp68-DAmP and srp102-DAmP mutants, had a strong reduction in retrotransposition to ∼2% to 7% of that in a wild-type strain. The reduction in Gag levels and retrotransposition were not due to a decrease in Ty1 RNA levels, as the amount of Ty1 RNA in the hypomorphic srp54, srp68, srp72, sec61 or kar2 mutant was equivalent to that in the wild-type strain (Figure 5D). To determine whether these ER translocation mutants have defects in Gag synthesis or stability, we performed pulse-chase labeling and immunoprecipitation of Gag using two strains, kar2-DAmP and srp68-DAmP, which have low or undetectable steady-state levels of Gag (Figure 5E). After a 15-min pulse-label, the amount of labeled Gag detected in the kar2-DAmP and srp68-DAmP mutants was 96% or 85% of that in the wild-type strain, respectively. However, the level of pulse-labeled Gag was two or four-fold lower, respectively, after a 60 min chase in the kar2-DAmP or srp68-DAmP mutant relative to the wild-type strain (Figure 5E). The data demonstrate that Gag is synthesized efficiently but degrades more rapidly when ER translocation is compromised. We conclude that translocation to the ER is necessary for the stability of Gag. Possibly the oxidizing environment of the ER lumen enables Gag to adopt a stable conformation, or Gag may undergo post-translational modification in the ER. Since Gag also associates with SRP-RNC complexes in the cytoplasm (Figure 4B), the most plausible scenario to explain these findings is that Gag, once it folds into a stable conformation in the ER lumen, is retrotranslocated to the cytoplasm, where it associates with Ty1 RNA on SRP-RNC complexes.
Retrosomes accumulate when ER translocation is blocked by treatment with tunicamycin
A key question raised by the findings above is how Gag-associated Ty1 RNA-SRP-RNC complexes are temporally and spatially related to retrosomes, in which Ty1 RNA and Gag assemble into VLPs. We considered the possibility that co-translational localization of Ty1 RNA to the ER membrane and binding of Gag to Ty1 RNA-SRP-RNC complexes nucleate retrosomes. To test this hypothesis, we treated cells with tunicamycin and monitored the effect on retrosome abundance. Tunicamycin is an inhibitor of N-glycosylation that induces the unfolded protein response, which, in S. cerevisiae, does not attenuate translation initiation but impedes ER translocation [33], [34], [35]. Our expectation, based on the phenotype of the N-glycosylation defective mutant, dfg10Δ (Figure 2), is that degradation of Gag would be enhanced. In accordance with this expectation, steady-state levels of Gag decreased gradually with increasing time of exposure to tunicamycin from 1 to 18 hours (Figure 6A). To determine the effect of blocking ER translocation on the formation of retrosomes, Ty1 RNA foci were visualized in cells treated with tunicamycin for 8 hours, when Gag was decreased but not completely depleted, because Ty1 RNA foci do not form in the absence of Gag (Figure 2B). FISH and fluorescent microscopy were performed to detect Ty1 RNA. The number of Ty1 RNA foci increased substantially from 0.46 foci/cell in mock-treated cells to 1.28 foci/cell in tunicamycin-treated cells (Figure 6B). Elevated accumulation of Ty1 RNA foci comprise an increase in the percentage of cells that contained at least one retrosome from 34% to 90% and an increase in the percentage of cells with two or more retrosomes from 7% to 26%. Direct visualization of Gag-GFP expressed from plasmid pLTR:Gag1–401:GFP:ADH1TER showed that Gag-GFP and Ty1 RNA co-localize in virtually all cells with detectable Gag-GFP foci, consistent with the idea that the tunicamycin-induced foci are retrosomes. These findings are consistent with the hypothesis that coalescence of translating Ty1 RNA gives rise to retrosomes. However, we also considered two alternative models that are formally consistent with these results: First, Ty1 RNA bound by Gag could be recruited to stress granules during ER stress, resulting in the formation of abnormal Ty1 RNA-Gag foci; or second, treatment with tunicamycin could induce the coalescence of non-translating Ty1 RNA-Gag complexes, or VLPs, to form retrosomes.

To determine whether Ty1 RNA and Gag accumulate in stress granules when cells are treated with tunicamycin, we measured the induction of Ty1 RNA-Gag foci by tunicamycin in strains lacking stress-granule components Pub1 and Pbp1 (Figure 6B). Pub1, an ortholog of mammalian TIA-1, and Pbp1, an ortholog of mammalian ataxin-2, are both required for stress granule assembly in yeast and mammalian cells [36], [37], [38]. The absence of Pbp1 had no effect on the prevalence of Ty1 RNA foci or the co-localization of Gag in mock-treated cells or tunicamycin-treated cells. In the pub1Δ mutant, Ty1 RNA-Gag foci were absent from mock-treated cells, which is likely a consequence of the reduced Ty1 RNA stability and Gag levels in this mutant [39], [40]. However, treatment with tunicamycin for 8 hours completely suppressed the Ty1 RNA localization defect of the pub1Δ mutant, and remarkably, restored Ty1 RNA foci to levels approaching those in wild-type cells treated with tunicamycin. Moreover, Gag co-localized with Ty1 RNA foci in tunicamycin-treated pub1Δ cells. These data demonstrate that neither Pbp1 nor Pub1 is necessary for the accumulation of Ty1 RNA-Gag foci in tunicamycin-treated cells, and therefore the observed Ty1 RNA-Gag foci are not stress granules. Furthermore, the tunicamycin-induced formation of retrosomes in a pub1 mutant, which is normally depleted of Gag and therefore VLPs, strongly suggests that retrosomes are not nucleated by a coalescence of VLPs. Thus, stalled Ty1 RNA translation complexes that accumulate when ER translocation is blocked by inhibiting N-linked glycosylation likely nucleate retrosomes.
Retrosome abundance is a function of the rate of ER translocation and Gag accumulation
As an independent test of the model that retrosomes are nucleated by an accumulation of Ty1 RNA-RNC complexes, we determined whether retrosome formation increases when ER translocation is stalled by a limited depletion of SRP (Figure 7). To examine the effect of reducing the level of SRP, we used hypomorphic alleles of SRP54, SRP68 and SRP72, which encode components of the S domain of SRP. Depletion of these SRP subunits reduces the rate of SRP-dependent protein translocation [41]. In the srp54-DAmP and srp72-DAmP strains, which have a slight reduction in the steady-state level of Gag (Figure 7A) and a modest Pho8-Ura3 translocation defect (Figure S3 and data not shown), Ty1 RNA foci increased to 0.70 and 0.91 foci/cell, respectively, compared to 0.48 foci/cell in the wild-type strain (Figure 7B). Gag-GFP foci that were detected co-localized with Ty1 RNA foci, indicating that the foci are retrosomes. These data are consistent with the accumulation of Ty1 RNA-RNC complexes in foci when SRP is limiting, and thus translocation of Gag to the ER lumen is stalled. In contrast, the srp68-DAmP mutant, which has a very low level of Gag (Figure 7A) and a severe Pho8-Ura3 translocation defect (Figure S3), lacked Ty1 RNA foci. This phenotype is similar to that of the bud21Δ mutant, which also has a wild-type level of Ty1 RNA and a very low level of Gag (Figure 2). Together, the data suggest that a minimum level of Gag is needed for Ty1 RNA to coalesce in foci and support the idea that multimerization of Gag molecules bound to Ty1 RNA on SRP-RNC complexes nucleates the formation of retrosomes.

A further prediction of the model that retrosomes form by accumulation of Ty1 RNA-RNC complexes is that suppressing the translocation deficiency of the srp54-DAmP or srp72-DAmP mutant should reverse the elevated formation of retrosomes in each mutant. Treatment of srp hypomorphs with a very low concentration of cycloheximide suppresses their translocation deficiency by providing more time for SRP to sample RNC complexes for a cognate nascent peptide [31]. Therefore, we treated srp54-DAmP and srp72-DAmP mutants and the wild-type strain with 0.44 µg/ml cycloheximide for 30 min. Exposure to this low concentration of cycloheximide did not decrease the level of Gag in the srp54-DAmP or srp72-DAmP mutant (Figure 7A). In fact, Gag levels were slightly increased, consistent with the suppression of stalled translocation of Gag. In contrast, the number of Ty1 RNA foci decreased sharply, from 0.7 to 0.36 foci/cell in the srp54-DAmP strain and from 0.91 to 0.45 foci/cell in the srp72-DAmP strain. This effect of cycloheximide on the accumulation of retrosomes was specific for the srp hypomorphs, as there was no change in the number of Ty1 RNA foci/cell as a result of cycloheximide treatment of the wild-type strain. Thus, slowing down translational elongation to complement the limiting levels of SRP in the srp54 or srp72 hypomorph rapidly reverses the elevated formation of retrosomes. Together, the results demonstrate that retrosomes are dynamic foci formed by the coalescence of Ty1 RNA translation complexes at the ER.
Discussion
Translocation of Gag to the ER lumen by SRP-mediated translation of Ty1 RNA
This study revealed an unanticipated association of Ty1 RNA and Gag with the ER. Ty1 Gag co-purifies with the lumen fraction of ER microsomes and is protected from trypsin digestion by the microsomal membrane (Figure 3), providing direct biochemical evidence that Gag is translocated to the ER lumen. Although Gag is not a secreted protein and lacks a recognizable signal sequence, two lines of evidence suggest that SRP mediates the co-translational translocation of Gag across the ER membrane. First, Ty1 RNA is enriched in affinity-purified SRP-RNC complexes (Figure 4A) [26]. The enrichment of Ty1 RNA is observed even in a mutant that lacks a detectable association of Gag with SRP-RNC complexes, indicating that Gag does not bridge the association of Ty1 RNA with SRP (Figure S2). Second, disruption of ER translocation by depleting subunits of SRP, the SRP receptor, the ER translocon or the ER chaperone, Kar2/BiP, results in decreased accumulation of Gag (Figure 5A). In the translocation-deficient srp68-DAmP strain, which has a very low steady-state level of Gag, newly synthesized Gag is present at 85% of that in a wild-type strain but is rapidly degraded (Figure 5E). Therefore, most if not all of the nascent Gag is co-translationally localized to the ER lumen, or at least, nascent Gag that is synthesized in the cytoplasm does not contribute significantly to the cellular pool.
A model for the nucleation of presumptive VLP assembly sites
Our findings support a model in which the coalescence of Ty1 RNA-ribosome-nascent Gag complexes nucleates the formation of cytoplasmic Ty1 retrosomes, where the concentration of Ty1 RNA and proteins enables the assembly of VLPs (Figure 8). In this model, Ty1 RNA-ribosome-nascent Gag complexes are specifically recognized and bound by SRP, which docks the Ty1 RNA translation complex to the SRP receptor on the ER membrane. Nascent Gag is threaded through the ER translocon into the ER lumen. Gag adopts a stable conformation in the ER lumen and is subsequently retrotranslocated to the cytoplasm. When Gag enters the cytoplasm, it binds Ty1 RNA that is being translated on SRP-RNC complexes, perhaps aided by the proximity of Ty1 RNC complexes that are docked onto the ER membrane. Multimerization of Gag bound to Ty1 RNA-SRP-RNC complexes results in the coalescence of Ty1 RNA and Gag to form the presumptive VLP assembly site. When sufficient Gag is synthesized and bound to translating Ty1 RNA, Gag likely sequesters Ty1 RNA from translation and promotes its dimerization and packaging into assembling VLPs, although the details of this transition have not yet been elucidated.

In wild-type cells, VLP assembly is inefficient, as Ty1 VLPs are rarely detected, and only about 20% Ty1 RNA is protected from nuclease digestion by encapsidation in VLPs [11], [12], [15]. Nonetheless, retrosomes are visualized in 40% to 50% of wild-type cells, suggesting that there is sufficient Gag to drive the coalescence of Ty1 RNA-RNC complexes into foci in many cells, but insufficient Gag levels to enable the efficient transition of Ty1 RNA from translation to packaging in VLPs. Thus, retrosome formation might represent a bottleneck in the retrotransposition cycle in which Ty1 RNA-SRP-RNC complexes accumulate because a component of the ER translocation machinery is limiting. This model explains why retrosomes increase in number and size when Ty1 RNA is overexpressed [11], since increasing the level of Ty1 RNA would create a larger bottleneck of Ty1 RNA-SRP-RNC complexes awaiting recognition by the SRP receptor or ER translocon.
Several lines of evidence indicate that multimerization of Gag bound to Ty1 RNA on SRP-RNC complexes results in the nucleation of retrosomes. First, Gag co-purifies with SRP-RNC complexes (Figure 4B). In an rpl7aΔ mutant, in which SRP-RNC complexes are associated with Ty1 RNA but not Gag, Ty1 RNA-RNC complexes fail to coalesce in retrosomes (Figure S2). Second, both the bud21Δ mutant, which has inefficient Gag synthesis (Figure 2D), and the srp68-DAmP mutant, in which Gag is rapidly degraded (Figure 5E), lack retrosomes. Thus, the level of Gag in these mutants may be too low to enable coalescence of Ty1 RNA-SRP-RNC complexes into foci. Third, modestly reducing the rate of co-translational ER translocation increases the prevalence of retrosomes. Treatment of cells with tunicamycin dramatically increased Ty1 RNA foci that co-localize with Ty1 Gag. This tunicamycin-induced accumulation of Ty1 RNA-Gag foci occurs in the absence of Pbp1 and Pub1, which are required for stress granule formation. Ty1 VLP formation is likely to be severely impaired by the paucity of Ty1 RNA and Gag in the pub1Δ mutant [39], [40]; therefore, the tunicamycin-induced formation of retrosomes in the pub1Δ mutant further supports the idea that Ty1 RNA-RNC complexes, rather than VLPs, nucleate retrosomes. The accumulation of Ty1 retrosomes that results from limiting the amount of SRP in the srp54-DAmP and srp72-DAmP hypomorphs is completely reversed by exposure of cells to a very low concentration of cycloheximide. The fact that altering the efficiency of co-translational translocation rapidly alters the prevalence of retrosomes strongly suggests that the Ty1 RNA that coalesces in retrosomes is being translated. Overall, these findings support the idea that Ty1 RNA is localized to the retrosome during translation.
We previously showed that nearly all Ty1 RNA in the cell resides in very high molecular weight complexes whose migration in sucrose gradients is not significantly altered by treatment with EDTA, which causes the dissociation of ribosomes [12]. Thus, we concluded that Ty1 RNA was translationally repressed in complexes that appeared to be devoid of translating ribosomes. However, the data presented here clearly indicate that Ty1 RNA is translated in association with SRP. Moreover, translation of Ty1 RNA within these high molecular weight complexes is consistent with the broader molecular weight distribution of Ty1 ribonucleoprotein complexes in a mutant with a defect in 40S ribosomal subunit biogenesis [39]. The fact that very little Ty1 RNA is released into low molecular weight complexes when ribosomes are dissociated by EDTA suggests that nearly all Ty1 RNA, including that being translated, is in a complex with Gag multimers.
The co-purification of Gag with SRP-RNC complexes in which Ty1 RNA is translated suggests that Gag binding to translating Ty1 RNA promotes the transition of Ty1 RNA from translation to packaging in VLPs. Thus, our findings provide indirect evidence that Ty1 RNA is not partitioned into separate mRNA and genomic RNA pools. Binding of a retroelement RNA chaperone protein to its RNA during translation is thought to enable the preferential mobilization of the encoding RNA in cis, which results in mobilization of only those elements that encode functional proteins. For example, human L1 retrotransposon proteins show a strong cis-preference for mobilizing their own RNA template, presumably because they bind to L1 RNA co-translationally. As a consequence, only functional L1 elements transpose efficiently [42], [43]. However, Ty1 proteins efficiently package Ty1 RNA in trans, and defective elements are mobilized as efficiently as competent elements [17], [44], raising the question of how Gag associates with translating RNA without interacting preferentially with its own RNA template. A similar paradox is manifest by studies on the nucleation of HIV-1 assembly sites: while HIV-1 mRNA and genomic RNA reside in a single pool, and Gag binds HIV-1 RNA in the cytoplasm before transport to the plasma membrane assembly site, HIV-1 Gag does not display a cis-preference for packaging its encoding RNA [45], [46]. Our data reveal a novel mechanism by which Gag is temporarily separated from its RNA template by translocation to the ER and retrotranslocation to the cytoplasm before binding Ty1 RNA translation complexes. This model may have implications for other retrotransposons or retroviruses that display efficient trans-packaging of RNA.
The role of Gag in Ty1 RNA stability
The observation that Ty1 RNA levels are not reduced in mutants with very low levels of Gag, including the bud21Δ mutant (Figure 2C) and the srp68-DAmP mutant (Figure 5D) indicates that Gag binding to Ty1 RNA is probably not required for the exceptionally long half-life of Ty1 RNA [47]. Our conclusion that Gag is dispensable for Ty1 RNA stability differs from the conclusion reached in a recent study in which the authors analyzed a Ty1 transcript with a premature stop codon placed directly after the start codon [16]. The difference between our results may indicate that a minimal level of Ty1 RNA translation, rather than the presence of stable Gag, is required for Ty1 RNA stability. In the dfg10Δ mutant, Ty1 RNA is unstable (Figure 2B), although its degradation occurs subsequent to Gag synthesis (Figure 2D). Perhaps Ty1 RNA instability in the dfg10Δ strain is an indirect result of N-linked glycosylation, which is expected to induce unfolded protein accumulation in the ER, whereas Ty1 RNA is not destabilized in the srp68-DAmP strain because the cellular stress response to blocking ER translocation is distinct from that of the unfolded protein response.
SRP-mediated translocation of Gag to the ER and retrotranslocation to the cytoplasm
The essential role of SRP in the nucleation of presumptive Ty1 VLP assembly sites is particularly interesting in light of the association of 7SL RNA with nucleocapsids of several retroviruses [46], [48], [49]. It is not known whether the 7SL RNA has a function in retroviral replication, but it has been implicated in the incorporation of the human antiviral restriction factor APOBEC3G into HIV-1 particles [50]. In addition, SRP interacts co-translationally with the Gag polyprotein of the murine endogenous retrovirus, IAP, and brings Gag to the ER membrane, although Gag was not translocated to the ER lumen in an in vitro system [51]. Our data raise the possibility that the association of 7SL RNA with retroviral particles evolved from an ancient functional role of SRP in retrotransposition.
The SRP-mediated translocation of Ty1 Gag to the ER lumen and the presence of Gag in the cytoplasm raises many questions about the mechanism and purpose of Gag transit into and out of the ER lumen. For example, the target sequence in the nascent Gag peptide that is recognized by SRP is not known. A systematic identification of nascent peptides that interact with SRP demonstrated that approximately 20% of SRP targets lack a predicted N-terminal signal sequence or transmembrane domain [26]. Many of these nascent peptides, including Ty1 Gag, are encoded by mRNAs that are membrane-associated and therefore are validated targets of SRP. Binding of SRP to a hydrophobic domain of the nascent peptide is required for the high affinity association of SRP with RNC complexes and for translocation [22], so it is likely that Gag has a specific target sequence that is bound by SRP. Analysis of the Ty1-H3 Gag sequence with the Kyte-Doolittle algorithm reveals that the longest hydrophobic region of Gag resides in the C-terminal end of p45-Gag from amino acid 334 to 345 (NTVAELFLDIHA). This region is predicted to form an alpha-helical domain, which could promote recognition by SRP [52], [53]. Interestingly, amino acids 341 through 346 (LDIHAI) have been shown to be critical for the formation of VLPs [54], [55].
The findings reported here suggest that Gag that is not co-translationally translocated to the ER is retained in the cytoplasm and unstable, perhaps because it cannot fold properly. What aspect of the ER lumen environment promotes the stability of Gag? One possibility is that Gag is post-translationally modified in the lumen. Although the C-terminal domain of Gag harbors five to eight potential N-glycosylation sites revealed by NetNglyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and N-GlycoSite (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html) algorithms, digestion of Gag with Endo H did not alter its mobility on SDS-PAGE gel (unpublished result). Thus, there is no evidence that Gag is subject to N-glycosylation. The migration of mature Gag in two bands on SDS-PAGE gels indicates that processed Gag may be subject to post-translational modification (Figure S1). Indeed, phosphorylation of Ty1 Gag in cells treated with mating pheromone has been observed previously [56]; however, there is no evidence that these modifications are linked to transit through the ER. We suggest that a more likely possibility is that the oxidizing environment of the ER supports the folding of the Gag and Gag-Pol precursors into conformations that promote their stability, enabling them to return to the cytoplasm to bind Ty1 RNA and initiate VLP assembly. Notably, maturation of the Moloney murine leukemia virus (MMLV) Gag and Gag-Pol proteins is regulated by the redox environment. Proteolytic processing of MMLV Gag and Gag-Pol proteins is constrained in infected cells exposed to a mild oxidizing agent and induced by treatment of the immature viral particles with a reducing agent [57], [58]. Perhaps folding of Ty1 Gag and Gag-Pol in the ER lumen prevents premature maturation of Gag and Gag-Pol until they are retrotranslocated to the cytoplasm, where Gag can bind Ty1 RNA and multimerize to initiate the formation of Ty1 VLPs.
How is Gag retrotranslocated from the ER lumen to the cytoplasm? Misfolded ER proteins are retrotranslocated to the cytoplasm via the ER-associated degradation (ERAD) pathway, in which transport from the ER to the cytoplasm is coupled to polyubiquitination and proteosomal degradation (reviewed in [59]). Some proteins that are known to be retrotranslocated from the ER to the cytoplasm, such as A/B toxins that enter the ER following endocytosis [60], may utilize the ERAD pathway but escape proteasome degradation. One example of a cellular protein that is retrotranslocated from the ER is the mammalian and Trypanosoma cruzi calcium-binding chaperone, calreticulin [61], [62]. Calreticulin is targeted to the ER lumen via the canonical SRP-mediated pathway by recognition and cleavage of its N-terminal signal sequence. A fraction of lumenal calreticulin is subsequently retrotranslocated to the cytoplasm by a process that is regulated by the concentration of calcium in the ER. Several viruses are also known to hijack the retrotranslocation pathway to promote viral assembly. For example, the N-glycosylated ORF2 capsid protein of Hepatitis E virus is retrotranslocated to the cytoplasm. In this instance, retrotranslocation is dependent on glycosylation of the ORF2 protein in the ER [63]. Further studies aimed at understanding how Ty1 Gag is recognized as an ER substrate, what conformational changes or modifications are required for Gag stability and how Gag is retrotranslocated to the cytoplasm will likely provide significant insights into the host-retrotransposon relationship as well as illuminate poorly understood aspects of SRP target specificity and protein retrotranslocation from the ER to the cytoplasm.
Materials and Methods
Plasmids and yeast strains
The plasmid pLTRp:Gag1–401:GFP:ADH1TER is a LEU2-marked, CEN-based plasmid containing a Ty1 U3 promoter, 5′ UTR and GAG ORF from amino acid 1 to 401 (corresponding to the processed p45-Gag protein) fused to the GFP(S65T) ORF and followed by the ADH1 terminator. The GFP(S65T)-ADH1TER BamHI-EagI fragment was PCR-amplified from a DNA template derived from pFA6a-GFP(S65T)-His3MX [64]. Ty1-H3 sequence from nucleotide 238 to 1496 was PCR-amplified and fused to the GFP(S65T)-ADH1TER fragment by PCR splicing by overlap extension (SOEing). The Ty1 5′ UTR-Gag1–401:GFP(S65T)-ADH1TER was digested with XhoI and EagI and ligated into plasmid vector pRS415 digested with XhoI and EagI. The resulting plasmid was digested with ApaI and XhoI, and ligated to a ApaI-XhoI fragment containing the U3 region of the Ty1-H3 3′ LTR amplified by PCR with primers PJ762 and PJ763 to yield pLTRp:Gag1–401:GFP:ADH1TER. Primer PJ762 introduced an ApaI site upstream of the U3 sequence. PJ763 contained a single base pair mismatch that introduced an XhoI site at the 3′ end of the U3 sequence.
Plasmid pGAL1-SRP54-HA, carrying a GAL1P-SRP54-HA expression cassette on the URA3-based 2-micron vector, BG1805 [65], was obtained from Open Biosystems. Plasmid pMP234 [29], which harbors a PHO5P-PHO81–82:URA3 reporter cassette on LEU2-based CEN vector, pRS315, was obtained from Martin Pool (University of Manchester).
The Saccharomyces cerevisiae strains used in this study are derivatives of strain BY4741. The spt3Δ:kanMX, bud21Δ:kanMX, dfg10Δ:kanMX, pbp1Δ:kanMX and pub1Δ:kanMX mutant strains were obtained from Open Biosystems [66]. The srp21-DAmP, srp54-DAmP, srp68-DAmP, srp72-DAmP, srp101-DAmP, srp102-DAmP, sec61-DAmP, sec63-DAmP and kar2-DAmP strains were obtained from Thermo Scientific [28]. Strain JC6008 was constructed by introducing plasmid pLTRp:Gag1–401:GFP:ADH1TER into BY4741. Strains JC6009, JC6010, JC6011, JC6155, JC6157, JC6167, JC6169, and JC6172 were obtained by introducing plasmid pLTRp:Gag1–401:GFP:ADH1TER into spt3Δ:kanMX, bud21Δ:kanMX, dfg10Δ:kanMX, pbp1Δ:kanMX, pub1Δ:kanMX, srp54-DAmP, srp72-DAmP, and srp68-DAmP strains, respectively.
Strain BY4741 harboring a chromosomal allele of LSM1-TAP-HIS3MX, KAR2-TAP-HIS3MX or ADH5-TAP-HIS3MX strains were obtained from Open Biosystems [67]. Strain JC6177, a SRP54-TAP derivative of BY4741, was constructed by PCR-mediated gene disruption of BY4741 with a PCR product containing the TAP cassette flanked by sequences at the C-terminus of the SRP54 ORF. The PCR product was amplified from genomic DNA of the KAR2-TAP derivative of BY4741 using primers PJ1205 and PJ1206. The rpl7aΔ:KlURA3 SRP54-TAP strain JC6111, was constructed by PCR-mediated gene disruption of strain JC6177 with a PCR product containing the pGAL-I-SceI-HygB-KlURA3 cassette [68].
Strains JC3212 and the isogenic spt3Δ:kanMX derivative, JC5398 have been described previously [69]. Strains JC6183, JC6184, JC6185, JC6187, and JC6189 were constructed by PCR-mediated gene disruption of strain JC3212 with PCR products amplified with primers PJ1233 and PJ1234 and genomic DNA of the srp68-DAmP strain, primers PJ1231 and PJ1232 and genomic DNA of the srp54-DAmP strain, primers PJ1235 and PJ1236 and srp72-DAmP strain DNA, primers PJ1237 and PJ1238 and srp102-DAmP strain DNA, or primers PJ1293 and PJ1294 and kar2-DAmP strain DNA, respectively.
Strain JC6159 is a haploid srp54Δ:LEU2 ura3Δ0 leu2Δ0 his3Δ1 strain carrying plasmid pGAL1-SRP54-HA [65]. The strain was constructed by PCR-mediated gene disruption of a trp1:hisG/trp1:hisG derivative of strain BY4743 with a srp54Δ:LEU2 PCR product amplified with primers PJ1207 and PJ1208 and plasmid pRS405 as a template, thereby generating an srp54Δ:LEU2/SRP54 diploid strain. Plasmid pGal-SRP54-HA was transformed into the srp54Δ:LEU2/SRP54 diploid strain, and tetrads were dissected to obtain the segregant JC6159.
Primers used in plasmid and strain construction are provided in Table S1.
Western blot analyses
Strains were grown at 20°C, a temperature that is permissive for Ty1 retrosome formation and retrotransposition, to mid-log phase (OD600 of 0.4–0.6) unless otherwise noted. Total cell lysates were prepared as described by Yarrington et al. [70] and proteins were separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were incubated in phosphate-buffered saline (PBS) plus 0.05% Tween 20 and 1% nonfat milk containing a 1∶50,000 dilution of affinity-purified anti-VLP polyclonal antibody [12] to detect Ty1 Gag, a 1∶10,000 dilution of anti-GFP polyclonal antibody (Sigma) to detect Gag∶GFP, a 1∶7,500 dilution of peroxidase-Anti-Peroxidase (PAP) soluble complex (Sigma) to detect Kar2-TAP or Adh5-TAP, a 1∶5,000 dilution of anti-calmodulin binding protein (CBP) polyclonal antibody (Millipore) to detect Srp54-TAP, or a 1∶500 dilution of anti-HA (F-7) monoclonal antibody to detect Srp54-HA (Santa Cruz Biotechnology). Subsequently, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies and SuperSignal West Pico chemiluminescent substrate (Pierce), and then the blots were exposed to film. The PVDF membranes were stripped of antibody in 50 mM Tris-HCl (pH 7), 2% SDS, and 50 mM DTT at 70°C for 30 min and washed in 1× PBS several times. The membrane was incubated with a 1∶10,000 dilution of anti-alpha tubulin monoclonal antibody (Millipore) to detect alpha-tubulin, a 1∶7,500 dilution of anti-actin monoclonal antibody (Abcam) to detect actin, or a 1∶5,000 dilution of anti-GAPDH monoclonal antibody (Thermo Scientific) to detect GAPDH as a loading control, and bands were visualized as described above.
Northern blot analyses
Northern blot analysis of total RNA prepared from cells grown to mid-log phase in CSM-Leu 2% Glu or YPD broth at 20°C, was performed as described previously [27]. Plasmid pJC940 DNA was used as a template to synthesize a 32P-labeled riboprobe to detect Ty1 RNA [12]. Plasmid pDG513, a gift of David Garfinkel, was used as a template to synthesize a riboprobe to detect 25S rRNA. Bands were quantified by phosphorimaging.
Pulse-chase labeling and immunoprecipitation
Proteins were pulse-labeled by incubation of cells in culture with L-homopropargylglycine, an analog of L-methionine with an alkyne side chain. Strains were grown to mid-log phase in YPD broth at 20°C. 30 OD600 units of cells per strain were harvested and washed three times in 5 ml of CSM-Met 2% Glu media. Click-iT L-Homopropargylglycine (Life Technologies) was added to 5 ml cell resuspensions in CSM-Met broth to a final concentration of 80 µM. Cultures were incubated at 20°C with vigorous shaking, and equal OD600 units of cells were harvested at 0, 30 and 60 min time points. Cells were centrifuged at 1000× g for 1 min, and cell pellets were washed twice with 5 ml of chase media (CSM-Met 2% Glu+50 mM methionine).
Cell pellets were resuspended in 100 µl HB buffer [(25 mM Tris-Cl (pH7.5), 125 mM NaCl, 5 mM EDTA, 0.5% IPEGAL, Complete Mini, EDTA-free protease inhibitor cocktail (Roche)] and vortexed at 4°C for 3 min with 0.15 g of glass beads. Following addition of 5 µl of 10% SDS, the cell extract was boiled for 5 min, and 500 µl ice-cold HB buffer was added. The extract was centrifuged for 1 min at 13000× g, and 50 µl Protein A Sepharose (GE Healthcare) was added to the supernatant, which was incubated at 4°C for 4 hr. Anti-VLP polyclonal antibody was added to pre-cleared lysate and rotated at 4°C for 18 hr. 50 µl of Protein A Sepharose was added to each sample and rotated at 4°C for 30 min. Samples were washed 3 times with 500 µl IP wash buffer [50 mM Tris (pH7.5), 250 mM NaCl, 5 mM EDTA, 1% IPEGAL, 1× EDTA-free protease inhibitor cocktail (Roche)], and proteins were eluted from the Protein A Sepharose with 50 mM Glycine (pH 3.0) and equilibrated in 200 µl of 50 mM Tris-Cl (pH 8.0), 1% SDS.
Eluted proteins were precipitated by methanol/chloroform precipitation. Copper-catalyzed triazole formation click reactions were performed using Click-iT Cell Reaction Buffer Kit (Life Technologies) to conjugate TAMRA (Tetramethylrhodamine 5-Carboxamido-(6-Azidohexanyl), 5-isomer; Life Technologies) to HPG-labeled Gag. SDS loading buffer was added to each reaction, proteins were separated on 10% SDS-PAGE gels. The fluorescent signal of TAMRA-conjugated Gag was detected using a Typhoon Scanner with a 580 BP30 filter, and bands were quantified with ImageQuant TL Software (GE Healthcare).
Membrane flotation assay
Strain BY4741 was grown to mid-log phase in YPD broth at 20°C. The cells were harvested, washed, and then resuspended in Buffer F [50 mM HEPES-NaOH pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol supplemented with Complete Mini, EDTA-free protease inhibitor cocktail (Roche)] to an OD600 of 10. Glass beads were added and cells were agitated on a Vortex mixer for 3 min at 4°C. The extract was removed and centrifuged twice at 300× g for 2 min at 4°C. 50 µl of the supernatant was mixed with 300 µl of 2.3 M sucrose in Buffer F, to obtain lysate in 2.0 M sucrose. This lysate was layered onto a 300 µl cushion of 2.3 M sucrose in Buffer F in a centrifuge tube. 1.5 M sucrose in Buffer F (500 µl) and 0.25 M sucrose in Buffer F (350 µl) were successively layered onto the gradient and the tube was centrifuged in a Beckman SW55 rotor at 100,000× g for 4 hr at 4°C. A 150 µl fraction was removed from the top of the gradient and discarded. Nine 150 µl fractions were collected from the top of the gradient, and protein profiles were analyzed by western blot analysis as described above.
Microsome preparation, sodium carbonate extraction, and protease protection assay
Microsomes were prepared from a culture of strain BY4741 grown to mid-log phase in YPD broth at 20°C as described in Brodsky et al. [71]. The microsomes were resuspended in B88 buffer (20 mM HEPES, pH 6.8, 250 mM sorbitol, 150 mM KOAc, 5 mM MgOAc) to an OD280 of 10. Sodium carbonate extractions were performed by incubating 50 µl of microsomes with 1 ml extraction buffer [200 mM Na2Co3 (pH 11.5), 10 mM DTT, Complete Mini, EDTA-free protease inhibitor cocktail (Roche), 0.5 M sucrose, 2% glycerol] on ice for 30 min. Samples were centrifuged at 230,000× g at 4°C for 1 hr, and the pellet was dissolved in 1× SDS loading buffer. Proteins in the supernatant were precipitated by incubation on ice for 30 min with trichloroacetic acid (TCA) added to a final concentration of 10%, followed by a 10-min centrifugation at 16,060× g at 4°C. The pellet was solubilized in 1× SDS loading buffer, and proteins separated on 10% SDS-PAGE gels were analyzed by western blot analysis, as described above.
Protease protection assays were performed by incubating 50 µl of microsomes with and without 1% Triton X-100 with 0.2 mg/ml TPCK-treated trypsin from bovine pancreas (Sigma) for 15 min on ice. The reactions were stopped by the addition of 5× SDS loading buffer. Ty1 Gag and Kar2-TAP were detected by western blot analysis, as described above.
Affinity purification of TAP complexes, RNA and protein analysis
Srp54-TAP and control complexes were purified following the procedure of del Alamo et al [26]. Strain BY4741 and derivatives harboring a chromosomal SRP54-TAP or LSM1-TAP allele were grown to an OD600 of 0.6–0.7 at 20°C. Cycloheximide was added to a final concentration of 0.1 mg/ml and growth was continued for 1 min at 20°C with vigorous shaking. Cells were immediately harvested by filtration onto 0.45 µm pore size nitrocellulose filters (Whatman), and cells were resuspended in 0.5 ml of ice-cold buffer A [50 mM Hepes-KOH (pH 7.5), 140 mM KCl, 10 mM MgCl2, 0.1% NP-40, 0.1 mg/ml cycloheximide, 0.5 mM DTT, Complete Mini EDTA-free protease inhibitor cocktail (Roche), 0.2 mg/ml heparin, 50 U/ml Superasin (Ambion), and 50 U/ml RNAsin (Promega)]. The cell suspension was dripped into a 50-ml conical tube containing liquid nitrogen. Frozen cells were pulverized for six cycles of 3 min at 15 Hz, using a Retsch MM301 mixer mill. Sample chambers were chilled in liquid nitrogen before each pulverization cycle. Pulverized cells were thawed and resuspended in 5 ml of buffer A. Cell debris was removed by two sequential centrifugation steps at 8,000× g for 5 min at 4°C. A 100 µl aliquot of the supernatant was removed for total RNA isolation, and a 50 µl aliquot was removed for western blot analysis. The remaining supernatant was incubated with IgG-Sepharose 6 Fast Flow (GE Healthcare) at 4°C for 2 hr. Beads were washed once in 5 ml of buffer A for 2 min and 5 times in 1 ml buffer B [50 mM Hepes-KOH (pH 7.5), 140 mM KCl, 10 mM MgCl2, 0.01% NP-40, 10% glycerol, 0.5 mM DTT, 10 U/ml SUPERase-In (Life Technologies), 10 U/ml RNasin, 0.1 mg/ml cycloheximide] for 1 min. Beads were resuspended in 100 µl of buffer B, and 0.3 U/ml AcTEV protease (Invitrogen) was added. The samples were incubated for 2 hr at 16°C, and the eluate was recovered. A 50 µl aliquot of the eluate was mixed with 5× SDS loading buffer, and the proteins were separated on 10% SDS-PAGE gels. Srp54-TAP and Ty1 Gag proteins were analyzed by western blot analysis, as described above. Total RNA and RNA from the eluate were isolated by sequential extraction with Phenol/Chloroform [5∶1] (Amresco), Phenol/Chloroform/Isoamyl Alcohol [25∶24∶1] (Sigma), and chloroform followed by isopropanol precipitation with 15 µg of Glycoblue (Ambion) as carrier. Each RNA sample was treated with 0.04 U/µl Turbo DNase (NEB) for 30 min at 30°C and inactivated with DNase Inactivation Reagent for 5 min at room temperature, and cDNA was synthesized using the First-Strand cDNA Synthesis Kit (Affymetrix). PCR reactions were performed using 0.5 µg of cDNA as a template with gene-specific primers. Each reaction was subject to 29 or 32 cycles of amplification. PCR products were separated by electrophoresis on a 2% agarose gel stained with ethidium bromide. The sequence of gene-specific RT-PCR primers is provided in Table S2.
Srp54 depletion
Cells of strain JC6159 were diluted to an OD600 of 0.01–0.02 in CSM-Ura-Leu 2% Gal 2% Raf 2% Suc and grown at 20°C for 24 hours. Glucose was added to the culture to a final volume of 2%, and 4 OD600 units of cells were harvested, pelleted and frozen at different time points. Equal volumes of lysate prepared from each cell pellet were separated on 10% SDS-PAGE gels and analyzed by western blot analysis as described above.
Transposition frequency assays
To measure transposition of the chromosomal Ty1his3AI[Δ1]-3114 element [69], strain JC3212 and mutant derivatives were grown in YPD broth at 30°C to saturation. Cultures were diluted 1∶1000 in YPD broth and grown at 20°C for three days. 1–5 µl of a 1∶1000 dilution of each culture was plated on YPD agar to determine the colony forming units (CFU). Aliquots (1–2 ml) of each culture were plated on CSM-His 2% Glu agar, and all plates were incubated at 30°C for 3 days. The frequency of Ty1his3AI retrotransposition is the number of His+ colonies divided by the total number of CFU plated on CSM-His 2% Glu agar, which was determined from the colony count on YPD agar. The average frequency and standard error for each genotype tested were calculated from nine separate cultures.
Tunicamycin time course
A 250 ml culture of BY4741 harboring plasmid pLTRp:Gag1–401:GFP:ADH1TER was grown to an OD600 of 0.25 at 20°C in CSM-Leu 2% Glu broth, and 250 µl of 5 mg/ml tunicamycin in DMSO (Research Products International, Corp) or DMSO only was added. Four OD600 units of cells were harvested 0, 1, 4, 8, and 18 hr after addition of tunicamycin. Lysates of each cell pellet were separated on 10% SDS-PAGE gels and analyzed by western blot analysis, as described above.
FISH and fluorescence microscopy
Fluorescence in situ hybridization was performed essentially as described in Amberg et al. [72], with the following modifications. Strains were grown in YPD broth or, in the case of strains harboring plasmid pLTRp:Gag1–401:GFP:ADH1TER, CSM-Leu 2% Glu broth, to mid-log phase (OD600 of 0.4–0.6) at 20°C. For experiments with tunicamycin, a solution of tunicamycin in DMSO was added to a final concentration of 5 µg/ml, or an equivalent volume of DMSO was added, and cultures were grown for an additional 8 hr at 20°C. For experiments with cycloheximide, a solution of cycloheximide in DMSO was added to a final concentration of 0.44 µg/ml cycloheximide, or an equivalent volume of DMSO was added, and cultures were incubated with gentle turning at 20°C for 30 min. Cultures were treated with formaldehyde (4% final concentration) and incubated at room temperature for 15 min on a turning platform. Cells were collected by centrifugation, resuspended in 5 ml of 0.1 M KPO4 (pH 6.5)/4% formaldehyde, and rotated for 90 min at 23°C. Cells were washed twice with 0.1 M KPO4 (pH 6.5) and once in 1 ml wash buffer [0.1 M KPO4 (pH 6.5), 1.2 M sorbitol]. The cell pellet was resuspended in 1 ml wash buffer containing 500 µg of 100T Zymolyase (MP Biomedicals) and incubated for 30 min at 30°C. Spheroblasts were washed gently with wash buffer and resuspended in a volume of wash buffer approximately twice the volume of the pellet and transferred to 10-well slides (Electron Microscopy Sciences) pretreated with 0.1% poly-L-lysine (Sigma). After aspirating non-adhered cells, adhered cells were washed twice with 100 µl of 2× SSC (1× SSC = 0.15 NaCl, 0.015 M sodium citrate) per well. Cells in each well were incubated with 12 µl of prehybridization buffer (50% formamide, 10% dextran sulfate, 4× SSC, 0.02% polyvinyl pyrrolidone, 0.02% bovine serum albumin, 0.02% Ficoll-400, 125 µg/ml of tRNA, 500 µg/ml of denatured salmon sperm DNA) at 37°C for 1 hr in a humid chamber. Subsequently, 0.75 pmol of a Cy3-labeled gag anti-sense oligomer, PJ798 (5′ -/Cy3/TCT GTT TTG GAA GCT GAA ACG TGT AAC GGA TCT TGA TTT GTG TGG ACT TC - 3′), obtained from Integrated DNA Technologies, Inc., was added to each well, and slides were incubated for 12–18 hr at 37°C in a dark humid chamber. Slides were washed in 2× SSC for 3 min, 1× SSC for 5 min, 1× SSC containing 2 mg/ml DAPI for 3 min, 1× SSC for 5 min and 0.5× SSC for 5 min. After drying, coverslips were adhered to the slides with antifade solution [1.2% Mowiol-488 (Sigma), 3% glycerol, 50 mM Tris HCl (pH 8.5)]. Cells were visualized by fluorescence microscopy using a Zeiss Axioskop 200 M inverted microscope equipped with filter set: 31 (Cy3), 34 (DAPI) and 38 EX Band Pass 470/40 (GFP), at a magnification of 63 or 100×. A Q Imagining camera (or Hamamatsu ORCA ER) was used to obtain the images, which were then colored and merged in Openlab 4.0.4 software (Improvision) and modified with Photoshop CS software.
Pho8-Ura3 translocation assay
Strains harboring plasmid pMP234 [29] were grown overnight in CSM-Leu 2% Glu broth at 20°C to an OD600 of 0.6 to 1.0. The OD600 of each culture was adjusted to 0.5 by addition of CSM-Leu 2% Glu broth. A 6 µl aliquot of each culture, and 10-fold serial dilutions of each culture in water, were spotted onto selective media containing glucose and grown at 20°C for 4 to 7 days.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MalikHS, HenikoffS, EickbushTH (2000) Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res 10 : 1307–1318.
2. Voytas DF, Boeke JD (2002) Ty1 and Ty5 of Saccharomyces cerevisiae. In: Craig N, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Washington, DC: ASM Press. pp. 631–662.
3. PurzyckaKJ, LegiewiczM, MatsudaE, EizentstatLD, LusvarghiS, et al. (2013) Exploring Ty1 retrotransposon RNA structure within virus-like particles. Nucleic Acids Res 41 : 463–473.
4. FengYX, MooreSP, GarfinkelDJ, ReinA (2000) The genomic RNA in Ty1 virus-like particles is dimeric. J Virol 74 : 10819–10821.
5. AL-KhayatHAB, D., KenneyJM, RothJF, KingsmanAJ, Martin-RendonE, et al. (1999) Yeast Ty retrotransposons assemble into virus-like particles whose T-numbers depend on the C-terminal length of the capsid protein. J Mol Biol 292 : 65–73.
6. GarfinkelDJ, HedgeAM, YoungrenSD, CopelandTD (1991) Proteolytic processing of pol-TYB proteins from the yeast retrotransposon Ty1. J Virol 65 : 4573–4581.
7. MellorJ, FultonAM, DobsonMJ, RobertsNA, WilsonW, et al. (1985) The Ty transposon of Saccharomyces cerevisiae determines the synthesis of at least three proteins. Nucleic Acids Res 13 : 6249–6263.
8. MerkulovGV, LawlerJFJr, EbyY, BoekeJD (2001) Ty1 proteolytic cleavage sites are required for transposition: all sites are not created equal. J Virol 75 : 638–644.
9. MalagonF, JensenTH (2008) The T body, a new cytoplasmic RNA granule in Saccharomyces cerevisiae. Mol Cell Biol 28 : 6022–6032.
10. SandmeyerSB, ClemensKA (2010) Function of a retrotransposon nucleocapsid protein. RNA Biol 7 : 642–654.
11. CheckleyMA, NagashimaK, LockettSJ, NyswanerKM, GarfinkelDJ (2010) P-body components are required for Ty1 retrotransposition during assembly of retrotransposition-competent virus-like particles. Mol Cell Biol 30 : 382–398.
12. DutkoJA, KennyAE, GamacheER, CurcioMJ (2010) 5′ to 3′ mRNA decay factors colocalize with Ty1 gag and human APOBEC3G and promote Ty1 retrotransposition. J Virol 84 : 5052–5066.
13. BeckhamCJ, ParkerR (2008) P bodies, stress granules, and viral life cycles. Cell Host Microbe 3 : 206–212.
14. MalagonF, JensenTH (2011) T-body formation precedes virus-like particle maturation in S. cerevisiae. RNA Biol 8 : 184–189.
15. GarfinkelDJ, BoekeJD, FinkGR (1985) Ty element transposition: reverse transcriptase and virus-like particles. Cell 42 : 507–517.
16. CheckleyMA, MitchellJA, EizenstatLD, LockettSJ, GarfinkelDJ (2013) Ty1 gag enhances the stability and nuclear export of Ty1 mRNA. Traffic 14 : 57–69.
17. CurcioMJ, GarfinkelDJ (1994) Heterogeneous functional Ty1 elements are abundant in the Saccharomyces cerevisiae genome. Genetics 136 : 1245–1259.
18. GerstJE (2008) Message on the web: mRNA and ER co-trafficking. Trends Cell Biol 18 : 68–76.
19. HermeshO, JansenRP (2013) Take the (RN)A-train: localization of mRNA to the endoplasmic reticulum. Biochim Biophys Acta 1833 : 2519–2525.
20. NyathiY, WilkinsonBM, PoolMR (2013) Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim Biophys Acta 1833 : 2392–2402.
21. HalicM, BeckerT, PoolMR, SpahnCM, GrassucciRA, et al. (2004) Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427 : 808–814.
22. SaraogiI, ShanSO (2011) Molecular mechanism of co-translational protein targeting by the signal recognition particle. Traffic 12 : 535–542.
23. RislerJK, KennyAE, PalumboRJ, GamacheER, CurcioMJ (2012) Host co-factors of the retrovirus-like transposon Ty1. Mobile DNA 3 : 12.
24. BrodskyJL, SchekmanR (1993) A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J Cell Biol 123 : 1355–1363.
25. ZhangY, NijbroekG, SullivanML, McCrackenAA, WatkinsSC, et al. (2001) Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell 12 : 1303–1314.
26. del AlamoM, HoganDJ, PechmannS, AlbaneseV, BrownPO, et al. (2011) Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol 9: e1001100.
27. MaxwellPH, CoombesC, KennyAE, LawlerJF, BoekeJD, et al. (2004) Ty1 mobilizes subtelomeric Y′ elements in telomerase-negative Saccharomyces cerevisiae survivors. Mol Cell Biol 24 : 9887–9898.
28. BreslowDK, CameronDM, CollinsSR, SchuldinerM, Stewart-OrnsteinJ, et al. (2008) A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods 5 : 711–718.
29. DalleyJA, SelkirkA, PoolMR (2008) Access to ribosomal protein Rpl25p by the signal recognition particle is required for efficient cotranslational translocation. Mol Biol Cell 19 : 2876–2884.
30. HannBC, WalterP (1991) The signal recognition particle in S. cerevisiae. Cell 67 : 131–144.
31. OggSC, WalterP (1995) SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell 81 : 1075–1084.
32. CurcioMJ, GarfinkelDJ (1991) Single-step selection for Ty1 element retrotransposition. Proc Natl Acad Sci U S A 88 : 936–940.
33. MoriK (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146 : 743–750.
34. SchuckS, PrinzWA, ThornKS, VossC, WalterP (2009) Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol 187 : 525–536.
35. RubioC, PincusD, KorennykhA, SchuckS, El-SamadH, et al. (2011) Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol 193 : 171–184.
36. BuchanJR, MuhlradD, ParkerR (2008) P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol 183 : 441–455.
37. GilksN, KedershaN, AyodeleM, ShenL, StoecklinG, et al. (2004) Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 15 : 5383–5398.
38. NonhoffU, RalserM, WelzelF, PicciniI, BalzereitD, et al. (2007) Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18 : 1385–1396.
39. DakshinamurthyA, NyswanerKM, FarabaughPJ, GarfinkelDJ (2010) BUD22 affects Ty1 retrotransposition and ribosome biogenesis in Saccharomyces cerevisiae. Genetics 185 : 1193–1205.
40. DuttaguptaR, TianB, WiluszCJ, KhounhDT, SoteropoulosP, et al. (2005) Global analysis of Pub1p targets reveals a coordinate control of gene expression through modulation of binding and stability. Mol Cell Biol 25 : 5499–5513.
41. BrownJD, HannBC, MedzihradszkyKF, NiwaM, BurlingameAL, et al. (1994) Subunits of the Saccharomyces cerevisiae signal recognition particle required for its functional expression. EMBO J 13 : 4390–4400.
42. WeiW, GilbertN, OoiSL, LawlerJF, OstertagEM, et al. (2001) Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol 21 : 1429–1439.
43. DombroskiBA, MathiasSL, NanthakumarE, ScottAF, KazazianHHJr (1991) Isolation of an active human transposable element. Science 254 : 1805–1808.
44. XuH, BoekeJD (1990) Localization of sequences required in cis for yeast Ty1 element transposition near the long terminal repeats: analysis of mini-Ty1 elements. Mol Cell Biol 10 : 2695–2702.
45. JohnsonSF, TelesnitskyA (2010) Retroviral RNA dimerization and packaging: the what, how, when, where, and why. PLoS Pathog 6: e1001007.
46. JouvenetN, LaineS, Pessel-VivaresL, MougelM (2011) Cell biology of retroviral RNA packaging. RNA Biol 8 : 572–580.
47. MunchelSE, ShultzabergerRK, TakizawaN, WeisK (2011) Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell 22 : 2787–2795.
48. Onafuwa-NugaAA, KingSR, TelesnitskyA (2005) Nonrandom packaging of host RNAs in moloney murine leukemia virus. J Virol 79 : 13528–13537.
49. Onafuwa-NugaAA, TelesnitskyA, KingSR (2006) 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12 : 542–546.
50. WangT, TianC, ZhangW, LuoK, SarkisPT, et al. (2007) 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol 81 : 13112–13124.
51. FehrmannF, JungM, ZimmermannR, KrausslichHG (2003) Transport of the intracisternal A-type particle Gag polyprotein to the endoplasmic reticulum is mediated by the signal recognition particle. J Virol 77 : 6293–6304.
52. WangZ, JonesJD, RizoJ, GieraschLM (1993) Membrane-bound conformation of a signal peptide: a transferred nuclear Overhauser effect analysis. Biochemistry 32 : 13991–13999.
53. JonesJD, McKnightCJ, GieraschLM (1990) Biophysical studies of signal peptides: implications for signal sequence functions and the involvement of lipid in protein export. J Bioenerg Biomembr 22 : 213–232.
54. Martin-RendonE, MarfanyG, WilsonS, FergusonDJ, KingsmanSM, et al. (1996) Structural determinants within the subunit protein of Ty1 virus-like particles. Mol Microbiol 22 : 667–679.
55. MonokianGM, BraitermanLT, BoekeJD (1994) In-frame linker insertion mutagenesis of yeast transposon Ty1: mutations, transposition and dominance. Gene 139 : 9–18.
56. XuH, BoekeJD (1991) Inhibition of Ty1 transposition by mating pheromones in Saccharomyces cerevisiae. Mol Cell Biol 11 : 2736–2743.
57. CampbellS, OshimaM, MirroJ, NagashimaK, ReinA (2002) Reversal by dithiothreitol treatment of the block in murine leukemia virus maturation induced by disulfide cross-linking. J Virol 76 : 10050–10055.
58. ReinA, OttDE, MirroJ, ArthurLO, RiceW, et al. (1996) Inactivation of murine leukemia virus by compounds that react with the zinc finger in the viral nucleocapsid protein. J Virol 70 : 4966–4972.
59. BrodskyJL, SkachWR (2011) Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol 23 : 464–475.
60. SpoonerRA, LordJM (2012) How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr Top Microbiol Immunol 357 : 19–40.
61. AfsharN, BlackBE, PaschalBM (2005) Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol Cell Biol 25 : 8844–8853.
62. LabriolaCA, ConteIL, Lopez MedusM, ParodiAJ, CarameloJJ (2010) Endoplasmic reticulum calcium regulates the retrotranslocation of Trypanosoma cruzi calreticulin to the cytosol. PLoS One 5: e13141.
63. SurjitM, JameelS, LalSK (2007) Cytoplasmic localization of the ORF2 protein of hepatitis E virus is dependent on its ability to undergo retrotranslocation from the endoplasmic reticulum. J Virol 81 : 3339–3345.
64. WachA, BrachatA, Alberti-SeguiC, RebischungC, PhilippsenP (1997) Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast 13 : 1065–1075.
65. GelperinDM, WhiteMA, WilkinsonML, KonY, KungLA, et al. (2005) Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19 : 2816–2826.
66. WinzelerEA, ShoemakerDD, AstromoffA, LiangH, AndersonK, et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285 : 901–906.
67. GhaemmaghamiS, HuhWK, BowerK, HowsonRW, BelleA, et al. (2003) Global analysis of protein expression in yeast. Nature 425 : 737–741.
68. StoriciF, ResnickMA (2006) The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol 409 : 329–345.
69. MouZ, KennyAE, CurcioMJ (2006) Hos2 and Set3 promote integration of Ty1 retrotransposons at tRNA genes in Saccharomyces cerevisiae. Genetics 172 : 2157–2167.
70. YarringtonRM, RichardsonSM, Lisa HuangCR, BoekeJD (2012) Novel transcript truncating function of Rap1p revealed by synthetic codon-optimized Ty1 retrotransposon. Genetics 190 : 523–535.
71. BrodskyJL (2010) The use of in vitro assays to measure endoplasmic reticulum-associated degradation. Methods Enzymol 470 : 661–679.
72. AmbergDC, GoldsteinAL, ColeCN (1992) Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev 6 : 1173–1189.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle
- Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion
- Genetic Dissection of Photoreceptor Subtype Specification by the Zinc Finger Proteins Elbow and No ocelli
- GC-Rich DNA Elements Enable Replication Origin Activity in the Methylotrophic Yeast
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy