
Cleavage Factor I Links Transcription Termination to DNA Damage Response and Genome Integrity Maintenance in
DNA damage occurs constantly in living cells and needs to be recognized and repaired to avoid mutations. DNA repair is particularly relevant for lesions occurring in actively transcribed DNA strands because the RNA polymerase cannot proceed through a damaged site. Stalled RNA polymerases and persisting DNA lesions can lead to genome instability or cell death. Specific mechanisms to repair obstructing DNA lesions are found from bacteria to higher eukaryotes, their malfunction leading to severe genetic syndromes in humans. Termination of transcription comprises cleavage and polyadenylation of the nascent transcript and displacement of the RNA polymerase from its DNA template. These processes, which are crucial for cell viability and growth in eukaryotes, require two major multi-subunit complexes in budding yeast. Here, we found that one of these complexes, Cleavage Factor I (CFI), participates in the cellular response to DNA damage. In addition, we found that CFI dysfunction leads to replication defects, conceivably mediated by stalled RNA polymerases, rendering cell cycle checkpoints mandatory to prevent genomic instability. Our findings emphasize the importance of coordinating transcription termination, DNA damage response and replication in the maintenance of genomic stability suggesting that CFI plays a fundamental function in the coupling of these processes.
Published in the journal:
. PLoS Genet 10(3): e32767. doi:10.1371/journal.pgen.1004203
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004203
Summary
DNA damage occurs constantly in living cells and needs to be recognized and repaired to avoid mutations. DNA repair is particularly relevant for lesions occurring in actively transcribed DNA strands because the RNA polymerase cannot proceed through a damaged site. Stalled RNA polymerases and persisting DNA lesions can lead to genome instability or cell death. Specific mechanisms to repair obstructing DNA lesions are found from bacteria to higher eukaryotes, their malfunction leading to severe genetic syndromes in humans. Termination of transcription comprises cleavage and polyadenylation of the nascent transcript and displacement of the RNA polymerase from its DNA template. These processes, which are crucial for cell viability and growth in eukaryotes, require two major multi-subunit complexes in budding yeast. Here, we found that one of these complexes, Cleavage Factor I (CFI), participates in the cellular response to DNA damage. In addition, we found that CFI dysfunction leads to replication defects, conceivably mediated by stalled RNA polymerases, rendering cell cycle checkpoints mandatory to prevent genomic instability. Our findings emphasize the importance of coordinating transcription termination, DNA damage response and replication in the maintenance of genomic stability suggesting that CFI plays a fundamental function in the coupling of these processes.
Introduction
All cells are continuously exposed to DNA damaging agents, which can arise from exogenous sources or from endogenous metabolic processes. The DNA damage response (DDR) includes the activation of checkpoints and induction of DNA repair pathways. DNA lesions can generate structural distortions that interfere with basic cellular functions like transcription and replication. Such helix-distorting DNA lesions are generally handled by nucleotide excision repair (NER), which can be divided into global genome repair (GG-NER) and transcription-coupled repair (TC-NER) sub-pathways, depending on whether the DNA lesion is located anywhere in the genome or on the transcribed strand (TS) of an active gene, respectively. At transcribed genes, TC-NER acts when elongating RNA polymerase (RNAP) stalls at bulky DNA lesions such as UV-induced cyclobutane pyrimidine dimers (CPDs) (reviewed in [1], [2]). Transcription down-regulation and proteasome-mediated degradation of engaged RNAPII take place as part of the DDR to UV-induced damages [3], [4]. In humans, defects in TC-NER are responsible for two severe genetic disorders called Cockayne Syndrome (CS) and UV Sensitivity Syndrome (reviewed in [5], [6]). In S. cerevisiae, the major TC-NER factor is Rad26, the yeast homologue of CS protein B (CSB) [7]. However, residual TC-NER activity remains in the absence of Rad26, indicating that other factors are also involved in the process [7], [8]. Mutations in several transcription and messenger ribonucleoprotein (mRNP) biogenesis factors including the RNAPII subunit Rpb9, THO, THSC/TREX-2, Paf1, and Ccr4-NOT are partially defective in TC-NER in yeast [9]–[12].
During the past few years it has become clear that the different mRNA processing steps (including 5′-end capping, splicing, and 3′-end cleavage), mRNP export, and transcription are connected to each other (reviewed in [13]) and that surveillance mechanisms ensure that these processes occur in a coordinated manner (reviewed in [14]). THO and THSC/TREX-2 both work at the interface between transcription elongation, mRNP biogenesis and export and defects are characterized by a strong transcription-dependent hyperrecombination phenotype (reviewed in [15], [16]). THO might also act in the process of transcription termination, as in vitro assays suggest that THO mutants lead to polyadenylation defects [17]. Interestingly, other factors required for proficient TC-NER also function during transcription termination. The Paf1 transcription elongation factor contributes to the recruitment of 3′-end processing factors necessary for accurate transcription termination (reviewed in [18]). The Ccr4-NOT complex acts, among other gene expression functions, during transcription elongation and interacts with mRNP export factors (reviewed in [19]).
In the yeast Saccharomyces cerevisiae, the transcription termination machinery can be divided into three different sub-complexes: cleavage factor IA (CFIA), cleavage factor IB (CFIB), and cleavage and polyadenylation factor (CPF). CFIA is comprised of the Rna14, Rna15, Pcf11, and Clp1 proteins. CFIB consists of the RNA-binding protein Hrp1, which is tightly associated with CFIA. The CPF complex is a large complex that can be further classified into the cleavage factor II (CFII) made out of the Cft1, Yhh1, Pta1, Brr5, Ysh1, Cft2, and Ydh1 proteins; the polyadenylation factor I made of Fip1, Yth1, and Psf1; and other proteins including the Pap1 polymerase. In vitro reconstitution of the cleavage reaction demonstrated that it requires the joint action of CFIA, CFIB, and CFII [20], [21], while additional proteins such as the 5′-3′-exoribonuclease Rat1 are required for termination downstream of poly(A) sites in vivo and dismantling of RNAPII complexes in vitro [22]–[24]. In addition to their role in cleavage, many of the components of the cleavage machinery are required for transcription termination downstream of the poly(A) site and polyadenylation of the transcript (reviewed in [25], [26]). Notably, the CFIA rna14-1 and rna15-1 mutants suffer from transcription elongation defects and increase in transcription-dependent hyper-recombination [27], suggesting that the CFIA complex serves important functions in transcription beyond termination and 3′-end processing.
To assess the possible function of RNA 3′-processing and transcription termination on TC-NER, we analysed the impact of a number of mutations on the DDR and the repair of UV-induced lesions. We found that CFI mutants become sensitive to UV in the absence of GG-NER, but surprisingly are proficient for CPD repair. By contrast, DDR is compromised in those cells, as seen by RNAPII degradation and checkpoint activation analyses upon UV irradiation. In addition, we show that rna14-1 cells are impaired in cell cycle progression and rely on a functional G1/S checkpoint to prevent genomic instability and cell death. Our study reveals that CFI functions in DDR and is required for genomic integrity maintenance in yeast.
Results
CFI mutants are UV-sensitive in the absence of global genome repair
We first analysed the sensitivity of several transcription termination mutants to DNA damage in the absence of Rad7, a protein required for GG-NER in yeast. Growth of each double mutant was compared to the growth of rad7Δ after irradiation with UV light and in the presence of the UV-mimetic agent 4-nitroquinoline 1-oxide (4-NQO) (Figure 1A). The rna14-1 rad7Δ, rna15-1 rad7Δ, and hrp1-5 rad7Δ double mutants were significantly more affected by UV irradiation or 4-NQO than the respective single mutants, while the remaining assayed alleles (pcf11-2, rat1-1, and yhh1-3) were not. Notably, deletion of the RAD26 gene, which encodes the main TC-NER factor, further increased the sensitivity of rna14-1 rad7Δ and hrp1-5 rad7Δ mutants, indicating that the rna14-1 and hrp1-5 alleles are not epistatic to rad26Δ (Figure 1B).

Because UV sensitivity in the absence of GG-NER is a phenotype mostly associated with TC-NER deficiencies, we tested whether functional CFI was required for proficient TC-NER by monitoring the repair rates of the transcribed (TS) and non-transcribed (NTS) strands of the constitutively expressed RPB2 gene in rna14-1, rna15-1, and hrp1-5 cells (Figure 2, A and B). With the exception of the 60 min. time-point in rna14-1, which is seemingly lower than the wild type on the TS, no significant differences were observed between the repair rates of the mutants and the wild type in either RPB2 strand. Repair experiments were thus performed in rad7Δ and rna14-1 rad7Δ cells. As can be seen in Figure 2 (A and B), both strains show a similar low repair on the NTS and are repair-proficient on the TS. Together, our results indicate that the rna14-1, rna15-1, and hrp1-5 mutants are repair-proficient for CPDs. Because deficiencies in NER may cause an increase in recombinational repair and rna14-1 cells show moderate hyper-recombination [27], we assessed whether recombination increased upon UV irradiation in rna14-1, rad7Δ, and rna14-1 rad7Δ cells. For this, we used a direct-repeat (LY) and an inverted-repeat (SU) plasmid-based system [28]. As expected, rad7Δ cells show an increase in recombination upon UV-damage in both systems (13 - and 35-fold, Figure S1). However, recombination frequencies did not increase upon UV irradiation in rna14-1 cells, suggesting that UV damage is efficiently repaired by NER. Notably, the rna14-1 rad7Δ double mutant shows UV-dependent increase in recombination frequency as compared to the rad7Δ mutants in the direct-repeat system -but not in the inverted-repeat system - suggesting that these cells suffer from increased genomic instability that is not linked to increased CPD repair deficiencies.
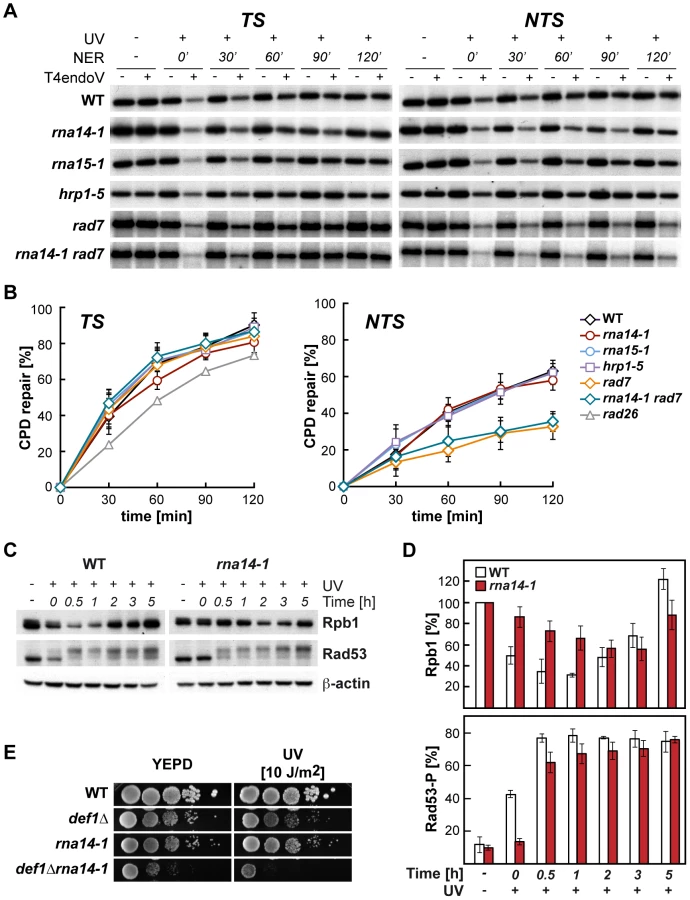
The DNA damage response is delayed in rna14-1 mutants
The cellular response to UV-induced damage involves, in addition to checkpoint activation, proteosomal degradation of RNAPII [3]. To check the functionality of the DDR in rna14-1 cells, we analysed the stability of Rpb1, the largest subunit of RNAPII, and activation of the Rad53 checkpoint protein upon UV irradiation by Western analysis (Figure 2, C and D). Interestingly, UV-induced Rpb1 degradation was less pronounced and severely delayed in rna14-1 cells as compared to the wild type. Activation of the DNA-damage checkpoint, monitored by the appearance of hyper-phosphorylated Rad53 upon UV irradiation was delayed in rna14-1 cells as compared to the wild type, in which Rad53 phosphorylation occurs immediately upon UV irradiation. In addition, the rna14-1 mutation did not increase the sensitivity to UV or 4-NQO of cells lacking either one of the DNA-damage checkpoint proteins Rad9 and Mec1 (Figure S2), suggesting that CFI might act within the canonical checkpoint pathways. To gain more insights into the function of CFI in the cellular response to UV-induced damage, Rpb1 stability and Rad53 phosphorylation were also analysed in cells bearing the rna15-1, hrp1-5 and pcf11-2 mutations (Figure S3). Both rna15-1 and pcf11-2 cells were partially impaired in UV-induced Rpb1 degradation while hrp1-5 cells behaved similarly to the wild type. However, Rad53 phosphorylation was delayed in the rna15-1 and hrp1-5 mutants but not in pcf11-2 cells. These interesting results suggest that UV-induced Rpb1 degradation might not depend on Rad53 activation. Previously, deletion of the DEF1 gene was shown to increase the sensitivity to UV in the absence of GG-NER without affecting DNA repair at the molecular level and to impair UV-dependent Rpb1 degradation [29]. Thus, we assayed viability and sensitivity of rna14-1 def1Δ, rna15-1 def1Δ, hrp1-5 def1Δ, and rat1-1 def1Δ double mutants to assess possible genetic interactions and observed strong synthetic sickness even in the absence of exogenous damage in all strains except hrp1-5 def1Δ (Figures 2E and S4). These interesting genetic interactions suggest that Def1 and CFI might have complementary functions for cell growth, which eventually rely on alternative ways to regulate RNAPII turnover. Although the penetrance of the different alleles differs depending on the analysed phenotype, our data indicate that CFI is required for the cellular response to UV-induced damage.
The ability to withstand DNA damage is reduced in CFI mutants
Sensitivity analysis of different termination mutants to distinct DNA damaging agents revealed that the rna14-1, rna15-1, and hrp1-5 mutants were sensitive to Phleomycin and to methyl methansulfonate (MMS) in contrast to the pcf11-2, rat1-1, and yhh1-3 cells, which were either slightly or not sensitive to those genotoxic agents (Figure 3A). Interestingly, the three alleles conferring significant sensitivity were those that increase the UV-sensitivity of rad7Δ mutants. To assess whether this phenotype was general rather than specific to GG-NER, we generated double mutants of rna14-1 with mutations in representative genes with known functions in the different DNA repair pathways, including homologous recombination (HR), non-homologous end joining (NHEJ), post-replicative repair (PRR), mismatch repair (MMR), base excision repair (BER) and NER (Figure 3B). Interestingly, the rna14-1 mutant showed synthetic growth defects even in the absence of exogenous damage with several repair mutants, including rad52Δ, ku70Δ, lig4Δ, and rad1Δ. These growth defects are further sustained by DNA content profiling FACS analysis (Figure S5). In addition, synthetic UV/4-NQO sensitivity was observed in all double mutants but rna14-1 ogg1Δ ntg1Δ ntg2Δ. Thus, our results indicate that Rna14 dysfunction makes cells unable to cope with high levels of DNA damage and rely on functional repair pathways even in the absence of exogenous damage.

To check whether these genetic interactions might arise from expression defects of DNA repair genes, mRNA expression was analysed by microarrays in rna14-1 and rna15-1 cells (Table S1). The results obtained with the two mutants were highly similar (Figure S6). Analysis of gene ontology terms of genes with higher (> 2-fold) and lower (< 2-fold) expression as compared to wild-type levels revealed that many genes involved in the DNA damage and/or stress response are induced in these mutants (Table S2), including genes such as OGG2, PRX1, DNL4, LIF1, RAD2 or MAG1. In addition, we found out that in rna14-1 or rna15-1 cells, the down-regulated genes were on the average longer than those of the entire genome, while the up-regulated genes were shorter (Figure S6), but DNA repair genes were not specifically down regulated. Thus the results rule out that the reduced capability of CFI mutants to withstand DNA damage is due to reduced transcription of repair protein encoding genes. On the contrary, the elevated expression of DNA damage and/or stress response transcripts suggests that CFI mutants may accumulate DNA damage or structures that impose a steric hindrance to DNA metabolic processes.
CFI mutants show severe replication defects
Transcription and replication need to occur in a coordinated manner in order to avoid conflicts that can result in genetic instability (reviewed in [30], [31]). To assess whether the CFI dysfunction affects replication, we first analysed sensitivity of several mutants to hydroxyurea (HU), a drug that slows replication down by reducing the pool of available deoxyribonucleotides (Figure 4A). Notably, the alleles that conferred sensitivity to HU were rna14-1, rna15-1, and hrp1-5, while the others did not at concentrations assayed. Since the expression of genes encoding ribonucleotide reductase components were not affected in rna14-1 and rna15-1 (Table S1), the observed HU sensitivity might reflect DNA replication impairment. Next we analysed plasmid loss in rna14-1 cells as a way to measure replication efficiency genetically (Figure 4B). Our results show that less than 5% rna14-1 cells maintained the pRS315 centromeric plasmid after about 10 divisions in non-selective medium as compared to the 50% value of wild-type cells. FACS analysis of cell cycle progression upon release from α-factor-mediated G1-arrest revealed that rna14-1 mutants remain trapped in G1 and suffer from a delay in S-phase entry as compared to the wild type (Figure 4C). For a specific analysis of initiation and progression of replication, we monitored BrdU incorporation upon release from α-factor-mediated G1-arrest at three different early origins (Figure 4D). DNA was immunoprecipitated with anti-BrdU antibody and BrdU enrichment at each locus was analysed by real-time qPCR with specific primers. Importantly, strong defects in replication were observed in rna14-1 mutants, as ARS activation was significantly reduced and occurred at later time points than in wild-type cells. Thus, cell-cycle progression is severely compromised in rna14-1 cells.

rna14-1 relies on a functional G1/S checkpoint to avoid genomic instability
Because G1 to S-phase progression was markedly delayed in rna14-1 cells, we asked whether persistent G1/S checkpoint activation might be responsible for the observed cell-cycle delay. Deprivation of Sic1, a protein that is required for the G1/S checkpoint, suppressed the S-phase entry defects in the rna14-1 mutant upon release from α-factor-mediated G1-arrest as seen by FACS analysis (Figure S7). To evaluate the consequences of forcing S-phase entry in rna14-1 mutants by SIC1 deletion, we analysed phosphorylated H2A (H2A-P) levels by Western analysis (Figure 5A). Our results indicate that the rna14-1 sic1Δ mutant accumulates DNA damage, as seen by the large amount of H2A-P. We then analysed recombination and Rad52-foci accumulation to gain insight into the impact of G1/S-checkpoint bypass in rna14-1 cells. As rna14-1 sic1Δ shows severe growth defects at 30°C (Figure S8), recombination was scored at 26°C, a semi-permissive temperature for the rna14-1 mutant, in a direct-repeat (LYΔNS) as well as an inverted-repeat (TINV) plasmid-based system [28] (Figure 5B). A significant increase in recombination frequency was observed in the double rna14-1 sic1Δ mutants with respect to the frequencies of either single mutant in both systems. Rad52-foci accumulation was monitored in cells transformed with plasmid pWJ1344 expressing a Rad52-YFP fusion protein using fluorescence microscopy. As can be seen in Figure 5C, the percentage of S/G2 cells with Rad52-foci was significantly higher in the rna14-1 sic1Δ double mutant (≈35%) than in the single mutants (<20%). Altogether, these results indicate that a functional G1/S checkpoint is essential to avoid genomic instability and/or cell death in rna14-1 cells.

Discussion
In this study, we asked whether transcription termination might contribute to DNA repair by TC-NER in S. cerevisiae. We found that the rna14-1, rna15-1, and hrp1-5 alleles of CFI confer increased UV and 4-NQO sensitivities in the absence of GG-NER, but surprisingly do not affect CPD repair in a transcribed gene. Importantly, we show that both checkpoint activation and RNAPII degradation are delayed in UV-irradiated CFI-deficient cells and that the rna14-1 mutation interacts genetically with mutations affecting several DNA repair pathway, including HR, NHEJ, MMR, PPR, and NER, in some cases even in the absence of exogenous DNA damage. Our data indicate that CFI participates in DDR in yeast and that this function is needed to cope with high amount of DNA damage. Additionally, we demonstrate that the rna14-1 mutation leads to severe cell cycle progression hindrance and that a functional G1/S checkpoint becomes essential in restraining genomic instability when CFI function is impaired.
Although the precise mechanisms underlying termination downstream of poly(A) sites and 3′-end processing of RNAPII-transcribed genes remains unresolved, it certainly requires cooperation among several factors, including CFI, CPF, Pap1, Rat1 and even the RNAPII holoenzyme (reviewed in [32], [33]). CFIA is progressively recruited to RNAPII during elongation and peaks at poly(A) sites [34], [35]. Its role in transcription termination and 3′-end processing is recapitulated by ongoing transcription past poly(A) sites and in vitro cleavage and polyadenylation defects in CFI mutants [36]–[38]. The CFIB factor Hrp1 binds throughout transcribed genes [39] and displays in vitro cleavage and polyadenylation defects when mutated [40], [41]. We found that CFIA rna14-1 and rna15-1 as well as the CFIB hrp1-5 alleles increased the UV and 4-NQO sensitivities of cells deficient in GG-NER and led to Phlemomycin and MMS sensitivities while the CFIA pcf11-2, CPF yhh1-3, and the rat1-1 alleles did not (see Figures 1 and 3A). On the other hand, UV-induced Rpb1 degradation is impaired in rna14-1, rna15-1 and pcf11-2 but not in hrp1-5 while Rad53-phosphorylation upon UV irradiation is delayed in rna14-1, rna15-1 and hrp1-5 but not in pcf11-2 cells (Figures 2C, 2D and S3). Thus it appears that the penetrance of each particular mutation depends on the assayed phenotype. Indeed, different pcf11 alleles differ in phenotype strength as seen by RNAPII chromatin immunoprecipitation (ChIP) on the ADH1 and PMA1 genes [42]. However, transcriptional read-through or 3′-end processing defects alone might not be sufficient to impair the DDR as ongoing transcription past poly(A) sites are also observed in yhh1-3 and rat1-1 mutants, and yhh1-3 is deficient in 3′-end cleavage and polyadenylation as well [22], [36], [43]. One possibility could be that the requirement of CFI function for the DDR could rely on intrinsic sensing activity or specific interaction with DDR factors, thus enabling CFI to coordinate transcription termination and DDR.
UV irradiation was shown to lead to 3′-end processing inhibition along with targeted RNAPII degradation in human cells, these responses seemingly being mediated by direct interaction between CstF, the functional homologue of yeast CFI, and BRCA1/BARD1 [44], [45]. The link between DDR and 3′-end processing is further supported by the observations that partial depletion of the CstF-50 subunit leads to increased UV sensitivity, reduced ability to ubiquitinate RNAPII in response to UV and defects in CPD repair in human cells [46]. Our results show a notable divergence with respect to the human system though, as no CPD repair defects were observed in yeast CFI mutants (see Figure 2A and 2B). Another difference between yeast and human is the observation that poly-adenylated mRNAs get stabilized upon UV irradiation in yeast [47], while transcript deadenylation takes place under damaging conditions in humans, mediated by DNA damage-dependent physical interaction between CstF and the PARN deadenylase [48]. In addition, it has recently been shown that targeted variation of poly(A) site usage occurs in response to 4-NQO treatment in yeast, possibly as a consequence of transient depletion of CPF subunits [49]. Altogether, these findings suggest that transcription termination factors participate in DDR, a multiple-sided system fundamental for cell survival under genotoxic stress conditions.
The cellular response to UV damage involves global down-regulation of transcriptional activity concomitantly with high expression of a subset of stress-induced genes and proteosomal-mediated degradation of RNAPII major subunit Rpb1. Notably, UV-induced Rpb1 degradation is delayed in CFI-deficient cells (see Figures 2C, 2D and S3), RNAPII turnover being thus impaired. Interestingly, transcription termination factors - including CFI - interact with the transcription initiation factor TFIIB and this interaction is required for the formation of gene loops both in yeast and humans [50]–[53]. Gene looping has been proposed to enable the efficient recycling of RNAPII and to contribute to transcription regulation by acting on promoter directionality and transcriptional memory (reviewed in [54], [55]). It is thus conceivable that gene looping may also function to control transcription and RNAPII turnover under DNA damaging conditions. This idea is supported by recent work showing that TFIIB may function as a general transcriptional switch in humans, as it is dephosphorylated during genotoxic stress thus losing its interaction with CstF, while direct interaction between CstF and the p53 tumor suppressor ensures the recruitment of termination factors to the promoter of stress-induced genes [56].
The persistence of stalled RNAPII on transcribed genes is known to impede the progression of the replication machinery and to be one of the causes underlying transcription-associated recombination (TAR) (reviewed in [30], [31]). Recently, inhibition of Rho-dependent transcription termination in bacteria has been shown to induce double-strand breaks depending on replication, suggesting that Rho might function in the release of obstructing RNAP during replication [57]. It is possible that CFI might act on paused RNAP, whether or not stalled at a DNA damage, contributing to its displacement and thus allowing progression of an oncoming replication fork. Over the last few years, growing evidence supports a role for co-transcriptionally formed RNA-DNA hybrids (R-loops) as a source of TAR (reviewed in [58]). Noteworthy, several transcription termination and 3′-end processing mutants have been shown to accumulate R-loops in yeast (including pcf11-2 and rna15-58) [59]. It is thus possible that stalled RNAPIIs accumulate at DNA damages or other structures such as R-loop in CFI mutants, leading to steric hindrances to the replication machinery that would account for the observed cell cycle progression defects (see Figure 4). The mechanisms by which stalled RNAPIIs or structures presenting steric hindrance to replication are sensed to activate the G1/S cell cycle checkpoint, which is required to restrain genetic instability in rna14-1 cells (see Figure 5), are currently unknown. Interestingly, the Sen1/SETX helicase - a component of the NRD transcription termination complex - prevents R-loop accumulation at transcription termination sites both in yeast and humans [60], [61]. In addition to its association with transcribed units, yeast Sen1 is also found at replication forks, contributing to prevent deleterious outcomes of the putative collisions between the transcription and replication machineries [62]. Noteworthy, Sen1 interacts physically with the NER repair protein Rad2 and the sen1-1 mutation increases the UV sensitivity of cells lacking RAD2 [63], suggesting further connections between transcription termination, replication, and DNA repair.
Altogether, our results support a model in which CFI dysfunction impairs DDR, probably leading to the accumulation of endogenous DNA lesions, and hinders DNA replication possibly due to the accumulation of RNAPs, whether or not stalled at DNA damages, thus rendering the G1/S checkpoint mandatory to prevent genomic instability (see Figure 6). Our findings emphasize the importance of coordinating transcription termination, DDR and replication in the maintenance of genomic stability and suggest that CFI plays a fundamental function in the coupling of these processes.

Materials and Methods
Yeast strains and plasmids
All strains used were isogenic to W303, and are listed in Table S3. Newly generated strains were obtained either by direct transformation or by genetic crosses. Plasmids used for recombination tests were pRS314-LYΔNS, pRS316-TINV, pRS314-LY and pRS314-SU [28].
UV survival curves and assays
For cell survival, yeast cells were grown in YEPD rich medium to an OD600 of 0.6. 10-fold serial dilutions were dropped on YEPD plates, irradiated with the indicated dose of UV-C light, and incubated in the dark at 30°C for 3 days. For the 4-NQO, Phleomycin, MMS, CPT and HU sensitivity assays, the serial dilutions were dropped on YEPD plates containing the indicated amounts of genotoxic agents and incubated in the dark at 30°C for 3 days. UV survival curves were performed as described [9]. UV-C irradiation was performed using a BS03 UV irradiation chamber and UV-Mat dosimeter (Dr. Gröbel UV-Elektronik GmbH).
Gene - and strand-specific repair assays
CPD repair at the RPB2 gene was analysed as described [64]. Briefly, cells were grown at 30°C in YEPD rich medium, irradiated in SD medium w/o amino acids with 200 J/m2 UV-C light (BS03 UV irradiation chamber), the medium supplemented to YEPD rich and the cells incubated at 30°C in the dark for recovery. DNA from the different time-points was extracted, cut with NsiI and PvuII restriction enzymes (Roche) and aliquots were either treated with T4-endonuclease V (Epicentre) or left untreated. DNA was electrophoresed in 1.3% alkaline agarose gels, blotted to Nylon membranes and hybridized with radioactively labelled strand-specific DNA probes, which were obtained by primer extension. Sequences of the primers are listed in Table S4. Membranes were analysed and quantified with a PhosphorImager (Fujifilm FLA5100). The average of the initial damage generated was 0.025 CPD/kb. To allow direct comparison between different strains, repair curves were calculated as the fraction of CPDs removed versus time. The initial damage was set to 0% repair.
Expression microarray analysis
Cells were grown at 30°C in YEPD medium to an OD660 of 0.6. Total RNAs were purified (RNeasy Midi kit, Qiagen) and expression profiling performed using the Affymetrix platform (see Table S1). The relative RNA levels for all yeast genes were determined using an Affymetrix microarray scanner and processed using the robust multiarray average method. Statistical data analyses were performed using the limma package (affylmGUI interface) of the R Bioconductor project (http://www.bioconductor.org). For each strain, microarray analysis was conducted in triplicate. All values presented represent the average of these three determinations. Genes were considered significantly up - or down-regulated when their expression values were > or < 2-fold, respectively (parameters: false discovery rate-adjusted p-value<0.01, B-statistic value>2, and average log2intensity A>7). The expression data for each mutant has been deposited in NCBI's Gene Expression Omnibus (accession number GSE50947).
Recombination and plasmid-loss assays
Plasmid loss was monitored as the percentage of cells that lost centromeric plasmid pRS315 upon growth in non-selective media. Individual transformants were inoculated in 5 ml YEPD and grown at 30°C to OD660 0.6. Cells were plated on YEPD or SC-leu to determine the percentage of plasmid loss. Six individual transformants were analysed for each genotype.
Recombination frequencies were determined as the average value of the median frequencies obtained from at least three independent fluctuation tests performed at 26°C each from six independent colonies according to standard procedures [28].
Replication analysis
Isogenic wild-type and rna14-1 strains deleted for the BAR1 gene and carrying several copies of the Herpes simplex thymidine kinase (TK) under the control of the strong constitutive GPD promoter were obtained by genetic crosses with strain SY2201 (E. Schwob). Cells were grown in YEPD, incubated for 2.5 h with 0.125 µg/ml α-factor, washed twice with pre-warmed YEPD and released into S phase by addition of 1 µg/ml pronase. BrdU (200 µg/ml) was added to the cultures prior to release. Cell cycle progression was monitored by flow cytometry on a FACSCalibur (BD Bioscience) using CellQuest software. Chromatin immunoprecipitation was carried out as described [65] with minor modifications. Briefly, Sodium Azide (0.1%) was added to each sample and cells were broken in a multi-beads Shocker (MB400U, Yasui Kikai, Japan) at 4° in lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% triton X-100, 0.1% sodium deoxicholate) and sonicated. Immunoprecipitation was performed using anti-BrdU antibody (MBL) attached to magnetic beads coated with Protein A (Invitrogen). Input and precipitated DNA were analysed by RT qPCR (7500FAST Applied Biosystems). Relative BrdU incorporation at a given region was calculated relative to the signal at a late replicating region (Chr. V, position 242210–242280, [66]) in the same sample. Primer sequences are listed in Table S4.
Detection of Rad52-YFP
Rad52-YFP foci from log-phase cells transformed with plasmid pWJ1344 were visualized with a DM600B microscope (Leica) as previously described [67] with minor modifications. Individual transformants were grown to early-log-phase at 26°C, incubated at 30°C for 4 hours, fixed for 10 minutes in 0.1 M KiPO4 pH 6.4 containing 2.5% formaldehyde, washed twice in 0.1 M KiPO4 pH 6.6, and resuspended in 0.1 M KiPO4 pH 7.4. A total of 617 wild type, 947 rna14-1, 733 sic1Δ, and 820 rna14-1 sic1Δ cells derived from at least three different transformants were analysed.
Cell extracts and western analysis
Detection of Rpb1, Rad53, H2A-P, and β-actin was accomplished by Western analysis of TCA-precipitated proteins separated in 4–20% Cristerion TGX gradient PAGE (Biorad). Antibodies 8WG16 (Rpb1, Covance), sc-20169 (Rad53, Santa Cruz Biotechnology), ab15083 (H2A-P, Abcam) and ab8224 (β-actin, Abcam) were used. For quantification, secondary antibodies conjugated to IRDye 680CW or 800CW (LI-COR) were used, the blot scanned in an Odyssey IR scanner and analysed with Image Studio 2.0 software (LI-COR). For Western analysis after UV irradiation, cells were grown in YEPD rich medium to mid-log-phase, resuspended in SD media lacking amino acids to an OD660 of 0.6 and irradiated with UV-C light in a BS03 UV irradiation chamber (Dr. Gröbel UV-Elektronik GmbH) at 100 J/m2. Medium was supplemented to YEPD rich and cells incubated in the dark at 30°C for recovery.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GaillardH, AguileraA (2013) Transcription coupled repair at the interface between transcription elongation and mRNP biogenesis. Biochim Biophys Acta 1829 : 141–150.
2. HanawaltPC, SpivakG (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9 : 958–970.
3. BeaudenonSL, HuacaniMR, WangG, McDonnellDP, HuibregtseJM (1999) Rsp5 ubiquitin-protein ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II in Saccharomyces cerevisiae. Mol Cell Biol 19 : 6972–6979.
4. RockxDA, MasonR, van HoffenA, BartonMC, CitterioE, et al. (2000) UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci U S A 97 : 10503–10508.
5. CleaverJE, LamET, RevetI (2009) Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 10 : 756–768.
6. SpivakG (2005) UV-sensitive syndrome. Mutat Res 577 : 162–169.
7. van GoolAJ, VerhageR, SwagemakersSM, van de PutteP, BrouwerJ, et al. (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J 13 : 5361–5369.
8. VerhageRA, van GoolAJ, de GrootN, HoeijmakersJH, van de PutteP, et al. (1996) Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol Cell Biol 16 : 496–502.
9. GaillardH, WellingerRE, AguileraA (2007) A new connection of mRNP biogenesis and export with transcription-coupled repair. Nucleic Acids Res 35 : 3893–3906.
10. GaillardH, TousC, BotetJ, Gonzalez-AguileraC, QuinteroMJ, et al. (2009) Genome-wide analysis of factors affecting transcription elongation and DNA repair: a new role for PAF and Ccr4-not in transcription-coupled repair. PLoS Genet 5: e1000364.
11. LiS, SmerdonMJ (2002) Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J 21 : 5921–5929.
12. TatumD, LiW, PlacerM, LiS (2011) Diverse roles of RNA polymerase II-associated factor 1 complex in different subpathways of nucleotide excision repair. J Biol Chem 286 : 30304–30313.
13. LunaR, GaillardH, Gonzalez-AguileraC, AguileraA (2008) Biogenesis of mRNPs: integrating different processes in the eukaryotic nucleus. Chromosoma 117 : 319–331.
14. SchmidM, JensenTH (2010) Nuclear quality control of RNA polymerase II transcripts. Wiley Interdiscip Rev RNA 1 : 474–485.
15. LunaR, RondonAG, AguileraA (2012) New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim Biophys Acta 1819 : 514–520.
16. RondonAG, JimenoS, AguileraA (2010) The interface between transcription and mRNP export: from THO to THSC/TREX-2. Biochim Biophys Acta 1799 : 533–538.
17. SaguezC, SchmidM, OlesenJR, GhazyMA, QuX, et al. (2008) Nuclear mRNA surveillance in THO/sub2 mutants is triggered by inefficient polyadenylation. Mol Cell 31 : 91–103.
18. JaehningJA (2010) The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta 1799 : 379–388.
19. CollartMA, PanasenkoOO (2012) The Ccr4—not complex. Gene 492 : 42–53.
20. ChenJ, MooreC (1992) Separation of factors required for cleavage and polyadenylation of yeast pre-mRNA. Mol Cell Biol 12 : 3470–3481.
21. GrossS, MooreC (2001) Five subunits are required for reconstitution of the cleavage and polyadenylation activities of Saccharomyces cerevisiae cleavage factor I. Proc Natl Acad Sci U S A. 98 : 6080–6085.
22. KimM, KroganNJ, VasiljevaL, RandoOJ, NedeaE, et al. (2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432 : 517–522.
23. LuoW, JohnsonAW, BentleyDL (2006) The role of Rat1 in coupling mRNA 3′-end processing to transcription termination: implications for a unified allosteric-torpedo model. Genes Dev 20 : 954–965.
24. PearsonEL, MooreCL (2013) Dismantling Promoter-driven RNA Polymerase II Transcription Complexes in Vitro by the Termination Factor Rat1. J Biol Chem 288 : 19750–19759.
25. RichardP, ManleyJL (2009) Transcription termination by nuclear RNA polymerases. Genes Dev 23 : 1247–1269.
26. ProudfootNJ (2011) Ending the message: poly(A) signals then and now. Genes Dev 25 : 1770–1782.
27. LunaR, JimenoS, MarinM, HuertasP, Garcia-RubioM, et al. (2005) Interdependence between transcription and mRNP processing and export, and its impact on genetic stability. Mol Cell 18 : 711–722.
28. Gomez-GonzalezB, RuizJF, AguileraA (2011) Genetic and molecular analysis of mitotic recombination in Saccharomyces cerevisiae. Methods Mol Biol 745 : 151–172.
29. WoudstraEC, GilbertC, FellowsJ, JansenL, BrouwerJ, et al. (2002) A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature 415 : 929–933.
30. GaillardH, Herrera-MoyanoE, AguileraA (2013) Transcription-Associated Genome Instability. Chem Rev 113 : 8638–8661.
31. KimN, Jinks-RobertsonS (2012) Transcription as a source of genome instability. Nat Rev Genet 13 : 204–214.
32. KuehnerJN, PearsonEL, MooreC (2011) Unravelling the means to an end: RNA polymerase II transcription termination. Nat Rev Mol Cell Biol 12 : 283–294.
33. MischoHE, ProudfootNJ (2013) Disengaging polymerase: terminating RNA polymerase II transcription in budding yeast. Biochim Biophys Acta 1829 : 174–185.
34. AhnSH, KimM, BuratowskiS (2004) Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell 13 : 67–76.
35. KimM, AhnSH, KroganNJ, GreenblattJF, BuratowskiS (2004) Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J 23 : 354–364.
36. AmraniN, MinetM, WyersF, DufourME, AggerbeckLP, et al. (1997) PCF11 encodes a third protein component of yeast cleavage and polyadenylation factor I. Mol Cell Biol. 17 : 1102–1109.
37. BirseCE, Minvielle-SebastiaL, LeeBA, KellerW, ProudfootNJ (1998) Coupling termination of transcription to messenger RNA maturation in yeast. Science 280 : 298–301.
38. Minvielle-SebastiaL, PrekerPJ, KellerW (1994) RNA14 and RNA15 proteins as components of a yeast pre-mRNA 3′-end processing factor. Science 266 : 1702–1705.
39. KomarnitskyP, ChoEJ, BuratowskiS (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev 14 : 2452–2460.
40. KesslerMM, HenryMF, ShenE, ZhaoJ, GrossS, et al. (1997) Hrp1, a sequence-specific RNA-binding protein that shuttles between the nucleus and the cytoplasm, is required for mRNA 3′-end formation in yeast. Genes Dev 11 : 2545–2556.
41. BarnwalRP, LeeSD, MooreC, VaraniG (2012) Structural and biochemical analysis of the assembly and function of the yeast pre-mRNA 3′ end processing complex CF I. Proc Natl Acad Sci U S A. 109 : 21342–21347.
42. KimM, VasiljevaL, RandoOJ, ZhelkovskyA, MooreC, et al. (2006) Distinct pathways for snoRNA and mRNA termination. Mol Cell 24 : 723–734.
43. DichtlB, BlankD, SadowskiM, HubnerW, WeiserS, et al. (2002) Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J 21 : 4125–4135.
44. KimHS, LiH, CevherM, ParmeleeA, FonsecaD, et al. (2006) DNA damage-induced BARD1 phosphorylation is critical for the inhibition of messenger RNA processing by BRCA1/BARD1 complex. Cancer Res 66 : 4561–4565.
45. KleimanFE, Wu-BaerF, FonsecaD, KanekoS, BaerR, et al. (2005) BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev 19 : 1227–1237.
46. MirkinN, FonsecaD, MohammedS, CevherMA, ManleyJL, et al. (2008) The 3′ processing factor CstF functions in the DNA repair response. Nucleic Acids Res 36 : 1792–1804.
47. GaillardH, AguileraA (2008) A novel class of mRNA-containing cytoplasmic granules are produced in response to UV-irradiation. Mol Biol Cell 19 : 4980–4992.
48. CevherMA, ZhangX, FernandezS, KimS, BaqueroJ, et al. (2010) Nuclear deadenylation/polyadenylation factors regulate 3′ processing in response to DNA damage. EMBO J 29 : 1674–1687.
49. GraberJH, NazeerFI, YehPC, KuehnerJN, BorikarS, et al. (2013) DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res 23 : 1690–1703.
50. SinghBN, HampseyM (2007) A transcription-independent role for TFIIB in gene looping. Mol Cell 27 : 806–816.
51. WangY, FairleyJA, RobertsSG (2010) Phosphorylation of TFIIB links transcription initiation and termination. Curr Biol 20 : 548–553.
52. Al HusiniN, KudlaP, AnsariA (2013) A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription. PLoS Genet 9: e1003722.
53. MedlerS, Al HusiniN, RaghunayakulaS, MukundanB, AldeaA, et al. (2011) Evidence for a complex of transcription factor IIB with poly(A) polymerase and cleavage factor 1 subunits required for gene looping. J Biol Chem 286 : 33709–33718.
54. ShandilyaJ, RobertsSG (2012) The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochim Biophys Acta 1819 : 391–400.
55. HampseyM, SinghBN, AnsariA, LaineJP, KrishnamurthyS (2011) Control of eukaryotic gene expression: gene loops and transcriptional memory. Adv Enzyme Regul 51 : 118–125.
56. ShandilyaJ, WangY, RobertsSG (2012) TFIIB dephosphorylation links transcription inhibition with the p53-dependent DNA damage response. Proc Natl Acad Sci U S A 109 : 18797–18802.
57. WashburnRS, GottesmanME (2011) Transcription termination maintains chromosome integrity. Proc Natl Acad Sci U S A 108 : 792–797.
58. AguileraA, Garcia-MuseT (2012) R loops: from transcription byproducts to threats to genome stability. Mol Cell 46 : 115–124.
59. StirlingPC, ChanYA, MinakerSW, AristizabalMJ, BarrettI, et al. (2012) R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26 : 163–175.
60. MischoHE, Gomez-GonzalezB, GrzechnikP, RondonAG, WeiW, et al. (2011) Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 41 : 21–32.
61. Skourti-StathakiK, ProudfootNJ, GromakN (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42 : 794–805.
62. AlzuA, BermejoR, BegnisM, LuccaC, PicciniD, et al. (2012) Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151 : 835–846.
63. UrsicD, ChinchillaK, FinkelJS, CulbertsonMR (2004) Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res 32 : 2441–2452.
64. GaillardH, WellingerRE, AguileraA (2009) Methods to study transcription-coupled repair in chromatin. Methods Mol Biol 523 : 141–159.
65. HechtA, GrunsteinM (1999) Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol 304 : 399–414.
66. Gomez-GonzalezB, Garcia-RubioM, BermejoR, GaillardH, ShirahigeK, et al. (2011) Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J 30 : 3106–3119.
67. LisbyM, RothsteinR, MortensenUH (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci U S A 98 : 8276–8282.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle
- Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion
- Genetic Dissection of Photoreceptor Subtype Specification by the Zinc Finger Proteins Elbow and No ocelli
- GC-Rich DNA Elements Enable Replication Origin Activity in the Methylotrophic Yeast
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy