
The and Hybrid Incompatibility Genes Suppress a Broad Range of Heterochromatic Repeats
Sister species capable of mating often produce hybrids that are sterile or die during development. This reproductive isolation is caused by incompatibilities between the two sister species' genomes. Some hybrid incompatibilities involve genes that encode rapidly evolving proteins that localize to heterochromatin. Heterochromatin is largely made up of highly repetitive transposable elements and satellite DNAs. It has been hypothesized that rapid changes in heterochromatic DNA drives the changes in these HI genes and thus the evolution of reproductive isolation. In support of this model, we show that two rapidly evolving HI proteins, Lhr and Hmr, which reproductively isolate the fruit fly sister species D. melanogaster and D. simulans, repress transposable elements and satellite DNAs. These proteins also help regulate the length of the atypical Drosophila telomeres, which are themselves made of domesticated transposable elements. Our data suggest that these proteins are part of the adaptive machinery that allows the host to respond to changes and increases in heterochromatin and to maintain the activity of genes located within or adjacent to heterochromatin.
Published in the journal:
. PLoS Genet 10(3): e32767. doi:10.1371/journal.pgen.1004240
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004240
Summary
Sister species capable of mating often produce hybrids that are sterile or die during development. This reproductive isolation is caused by incompatibilities between the two sister species' genomes. Some hybrid incompatibilities involve genes that encode rapidly evolving proteins that localize to heterochromatin. Heterochromatin is largely made up of highly repetitive transposable elements and satellite DNAs. It has been hypothesized that rapid changes in heterochromatic DNA drives the changes in these HI genes and thus the evolution of reproductive isolation. In support of this model, we show that two rapidly evolving HI proteins, Lhr and Hmr, which reproductively isolate the fruit fly sister species D. melanogaster and D. simulans, repress transposable elements and satellite DNAs. These proteins also help regulate the length of the atypical Drosophila telomeres, which are themselves made of domesticated transposable elements. Our data suggest that these proteins are part of the adaptive machinery that allows the host to respond to changes and increases in heterochromatin and to maintain the activity of genes located within or adjacent to heterochromatin.
Introduction
As populations diverge, their ability to reproduce with each other diminishes. Hybrid incompatibility (HI), the reduced viability and fertility of interspecific hybrids, is a major cause of reproductive isolation between nascent species and thus an important contributor to speciation. Many of the genes causing HI show evidence of adaptive evolution, typically manifest as excessive numbers of amino-acid-changing mutations compared to neutral expectations [1], [2]. These data do not, however, imply that natural selection acts directly on HI phenotypes. Rather, the prevailing model of HI formulated by Dobzhansky and Muller (D-M) emphasizes that incompatibilities evolve in two distinct steps. First, two or more loci diverge independently in two nascent species. Then, if these species later interbreed, these diverged genes may interact to cause deleterious HI phenotypes. The key insight of the D-M model is that hybrid lethality and sterility evolve as byproducts of intraspecific divergence [1].
Adaptive evolution therefore does ultimately lead to HI, but if we wish to identify the evolutionary forces that drive the divergence of HI genes, then we need to understand the function of these genes within species. The mechanisms by which HI genes cause sterility or lethality are important but separate issues. In fact, it remains uncertain whether the wild type functions of HI genes are generally predictive of the deleterious phenotypes that they cause within hybrids.
Pinpointing the function of HI genes and the causes of their adaptive evolution is a challenging goal. For example, the Hybrid male rescue (Hmr) gene causes large reductions in hybrid fitness [3]. Loss-of-function mutations in D. melanogaster, however, have only moderate effects on fertility and provide few insights into mechanistic underpinnings [4]. The nucleoporins provide an intriguing counterexample. Several have been implicated in hybrid lethality and found to evolve under adaptive evolution [5]. Mutations in nucleoporin subunits are lethal in D. melanogaster, but the genes have many pleiotropic functions and the challenge is to pinpoint which one(s) are driving evolutionary divergence.
Here we investigate two hybrid lethality genes, Lethal hybrid rescue (Lhr) and Hmr, which interact to cause F1 hybrid male lethality between D. melanogaster and D. simulans [6]. Both genes show extensive divergence in their coding sequences that is consistent with positive selection [6], [7]. For Hmr this sequence divergence appears to be required for hybrid lethality because the D. melanogaster ortholog of Hmr causes hybrid lethality but the D. simulans ortholog does not [7]. For Lhr, however, both orthologs have hybrid lethal activity, with D. simulans Lhr having greater activity due to its higher expression level in hybrids [8]. That study left open the possibility that Lhr coding sequence divergence makes some contribution to hybrid lethality. Furthermore we found that Lhr from the more diverged species D. virilis has no hybrid lethal activity, suggesting that more extensive coding sequence divergence does have substantial functional consequences [9].
These previous studies leave unanswered the fundamental question of what evolutionary force is driving adaptive sequence change, and necessitate a detailed understanding of Hmr and Lhr function within each of the hybridizing species. Loss of function alleles of Hmr and Lhr are strong suppressors of hybrid lethality, but are largely viable within D. melanogaster and D. simulans, respectively [10], [11].
Lhr (also known as HP3) protein localizes to heterochromatin [6], [12]. Several other Drosophila HIs also involve heterochromatin or heterochromatin proteins, which is intriguing because genome size varies widely among Drosophila, largely as a consequence of variation in repetitive DNAs that make up the heterochromatin [13], [14]. Heterochromatin may have a much wider role in incompatibility because repetitive DNA variation is the major cause of the ∼1000-fold variation in genome size among multi-cellular eukaryotes [15]. These DNAs can increase in copy number by general host processes such as unequal crossing over and duplication [16]. Alternatively, they may increase copy number by selfish properties such as transposition for TEs [17] and meiotic drive for satellite DNAs [18]. In either case, over-proliferation can be deleterious to their host species by causing genome instability, leading to the evolution of host defense mechanisms [19]. For example, one major mechanism is the piRNA pathway, where small (23–30 nt) RNAs derived from TE sequences are used to silence TE activity [20]. There are also hints that the piRNA pathway may regulate satellite DNAs [21]. Interestingly, piRNA regulatory genes often show signatures of adaptive evolution among Drosophila species [22].
Genetic conflicts with selfish DNAs have been proposed as an important driver of HI [1], [2], [23], but little is known about what specific sequences are interacting with HI genes. D. simulans and D. melanogaster have great potential for addressing this question because they differ substantially from each other in genome size [14], satellite DNA content [13], [14], and in both the types and number of TEs that they harbor [24]. Here we report that Hmr and Lhr are required to repress transcription from both TEs and satellite DNAs. Hmr and Lhr also regulate telomeres, a third specialized type of heterochromatic sequence that serves to protect the ends of linear chromosomes [25] and is composed of rapidly evolving DNA and proteins [26]–[28]. Telomere variation can affect host fitness and genome stability, and has been proposed as another potential source of meiotic drive [27], [29]. We used a D. simulans mutation in Lhr, comparative cytology, and interspecific complementation with Hmr transgenes to identify classes of TEs and satellites that are regulated differentially between the species. We conclude that Hmr and Lhr provide an adaptive defense against multiple classes of repetitive DNA sequences that change rapidly in evolutionary time, can reduce host fitness, and have high potential to provoke genetic conflict.
Results
Lhr and Hmr form a complex with HP1a
Lhr protein localizes to a subdomain of pericentric heterochromatin in early embryos [8]. To explore possible similarities with Hmr, we examined the localization of Hmr with a 3X-HA epitope-tagged Hmr transgene (see Materials and Methods). mel-Hmr-HA colocalizes with HP1a and H3K9me2 at heterochromatin in nuclear cycle 14 embryos (Figure 1A). We then used Immuno-FISH to determine its localization relative to specific heterochromatic satellite DNA sequences. mel-Hmr-HA does not overlap with the X-linked 359-bp satellite but colocalizes with dodeca, a GC-rich pericentromeric satellite on chromosome 3. This pattern mimics that seen previously with Lhr [8]. Additionally, mel-Hmr-HA colocalizes with GA-rich repeats and the 2L3L satellite in embryos (Figure 1B). Colocalization between mel-Hmr-HA with both dodeca and GA-rich repeats is also observed in ovarian nurse cells from Hmr3; mel-Hmr-HA females, indicating that localization is not a consequence of overexpression (Figures S1B, C). Unlike Lhr [8], mel-Hmr-HA localizes to the nucleolus in early embryos (Figure 1C), suggesting that Hmr may have some functions distinct from Lhr.

The largely similar localization patterns of Hmr and Lhr raise the possibility that they physically interact. We performed co-immunoprecipitation (co-IP) studies from embryo extracts and found that mel-Lhr-HA and mel-Hmr-FLAG co-IP (Figure 1D). mel-Lhr-HA was previously shown to express at wild type levels [8], and mel-Hmr-FLAG is expressed significantly lower than wild type levels (Figure S2), demonstrating that these results are not due to overexpression. Lhr was previously shown to bind to, co-localize with, and be dependent on HP1a for correct heterochromatic localization [6], [9], [12], [30]. We therefore tested if HP1a also associates with Hmr. IPs with HP1a pulled down mel-Lhr-HA and mel-Hmr-FLAG, but the reciprocal IPs failed to pull down detectable HP1a (Figure 1E).
Yeast two-hybrid assays show that Hmr and Lhr from D. melanogaster interact, suggesting that the co-IP reflects a direct interaction between the proteins (Figure 1F). This interaction is likely mediated via the BESS domains within Lhr and Hmr [6], a 40 amino-acid motif found in 19 proteins in D. melanogaster that has been implicated in protein-protein interactions and homo-oligomerization [31]. We also found that the D. simulans orthologs interact, as do the heterospecific combinations; the strength of interactions varied widely but exploring the potential significance of this result will require a more quantitative assay.
We next examined protein localization in mutant backgrounds to test the potential mutual dependence of Lhr and Hmr for their localization to heterochromatin. We made a D. melanogaster Lhr mutation by recombining a mini-white gene into the Lhr locus to create the LhrKO allele (Figure S3A). In LhrKO, transcription from Lhr but not flanking genes is greatly reduced, and no Lhr protein is detectable (Figure S3B, C). These results demonstrate that LhrKO is a strong loss of function allele, which we confirmed in hybrid rescue crosses (see Materials and Methods).
Lhr-HA levels are greatly reduced in Hmr3 mutant embryos but when examined at high gain a small amount of Lhr-HA is detectable in heterochromatin (Figure 1G). This result suggests that Hmr is not absolutely required to localize Lhr to heterochromatin, though it remains possible that some Hmr protein is made in the Hmr3 mutant. In a reciprocal experiment, Hmr-HA localization appears normal in LhrKO (Figure 1H). In combination with previous results, our data suggest that Lhr localization to heterochromatin depends on HP1a, and that Hmr stabilizes Lhr.
Lhr is required for female fertility
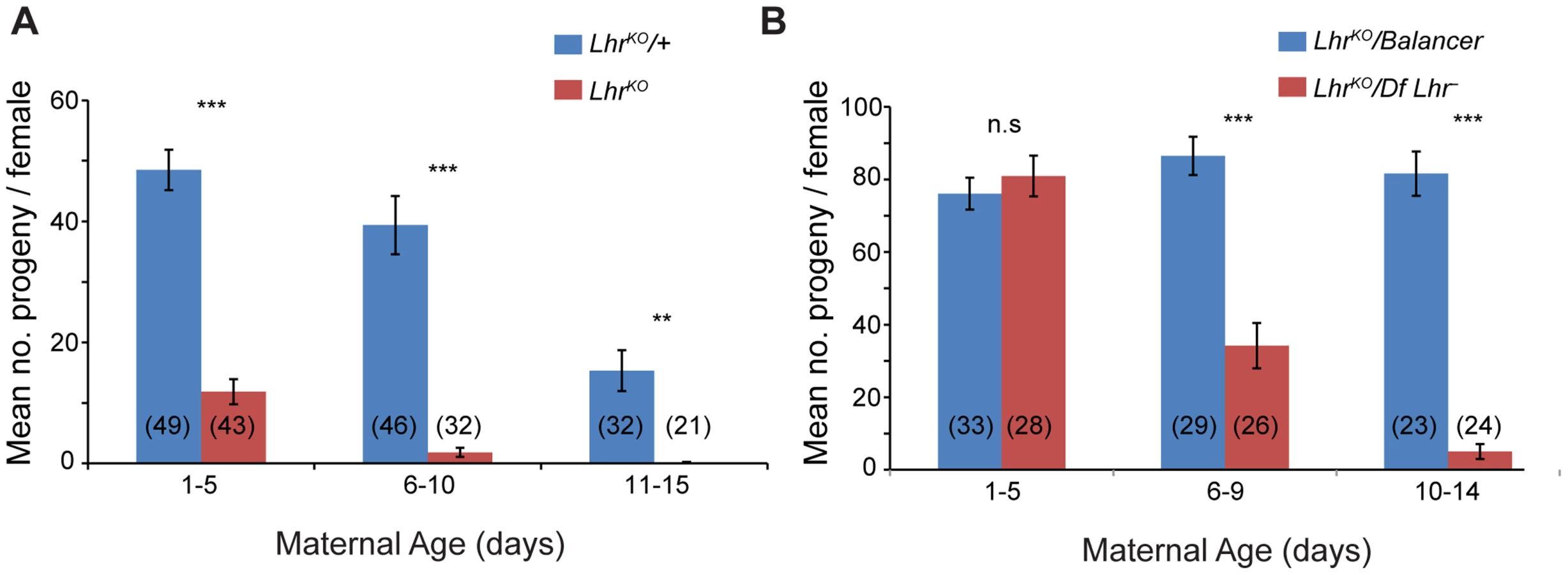
LhrKO flies are almost fully viable (22.25% compared to the expected 25% in crosses between heterozygotes at 27°; p<0.05 by Chi-squared; N = 2813 total flies scored). However, comparison of LhrKO with a background-matched Lhr+ control (see Materials and Methods) showed that LhrKO females have substantially lower fertility, particularly at higher temperatures. One to five day old LhrKO females display only a fraction of the fertility of LhrKO/+ and later become sterile (Figure 2A). We confirmed this in a different Lhr− background where a similar reduction in fertility occurs at later ages (Figure 2B). In a separate experiment we found that the hatch rate of the eggs laid by LhrKO/LhrKO mothers is low and declines with increasing maternal age (Table S1). This LhrKO female fertility phenotype is strikingly similar to that of Hmr mutants [4], suggesting that Hmr and Lhr may function in a common regulatory pathway.
Lhr and Hmr are required to repress transposable elements
We performed an RNA-Seq comparison of ovaries from LhrKO and Lhr+ to investigate the cause of this fertility reduction and discovered a widespread increase in transposable element (TE) transcripts. Using two different TE mapping methods (see Materials and Methods) we found that transcripts from 99 families were at least 2-fold upregulated, with 38 elements being at least 10 fold upregulated (Figure 3A; Table S2). Mis-regulated TEs include elements with germline expression such as the telomeric non-LTR retrotransposons HeT-A (350.7 fold) and TART (51.76 fold), the LTR retrotransposon copia (19.8 fold), and the DNA transposon bari-1 (44.7 fold). TEs expressed only in the somatic follicle cells, such as Gypsy (3.8 fold) and Zam (7 fold) were also upregulated. In addition, qRT-PCR in two different genetic backgrounds confirmed the massive increase in HeT-A transcript levels (185–846-fold; Figure S4). These results demonstrate that the telomeric TEs are especially sensitive to Lhr regulation.

We also performed RNA-Seq analysis of an Hmr mutant (Df(1)Hmr−/Hmr3, abbreviated below as Hmr−). We compared it to a heterozygous control (Df(1)Hmr−/y w Hmr+, abbreviated below as Hmr−/Hmr+) because it closely matches the genetic background of the mutant genotype, and also serves as a control for Hmr transgenic genotypes that are described below. We found that 55 different TE families are upregulated at least 2 fold in Hmr mutants, with 14 being upregulated at least 10 fold (Figure 3B; Table S3). Notably, the telomeric retrotransposons HeT-A and TART are again among the most highly upregulated. Strikingly, the TEs affected by Hmr are largely a subset of Lhr-regulated TEs, suggesting that they act together to regulate multiple TE families (Figure 3C). The smaller number of mis-regulated families in Hmr− likely reflects the fact that we are comparing Hmr− mutants to heterozygotes, but Lhr mutants to wild type.
Since some germline TE repressor genes also regulate somatic TE expression [32], we performed RNA-Seq to compare TE expression between 72–76 hour-old Df(1)Hmr−/Y and Hmr+/Y D. melanogaster male larvae. This also served as a control for experiments described below to address whether TE mis-expression may be contributing to hybrid lethality. We found that 31 TEs exhibit a statistically significant ≥2 fold upregulation (Figure 3D; Table S4), but there are two striking differences compared to Hmr mutant ovaries. First, different TEs are affected, with the telomeric retrotransposons in particular not upregulated in the larvae. Second, the magnitude of TE derepression is lower in larvae.
Lhr and Hmr affect expression of heterochromatic genes
We next examined potential effects on protein-coding genes. Remarkably few genes (11 in Hmr−; 0 in LhrKO) show a statistically significant misregulation in either Lhr or Hmr mutants (FDR 0.05; Tables S5, S6). However, a comparison of fold change in the expression of all heterochromatic versus all euchromatic genes found that heterochromatic genes are downregulated to a greater extent for both mutants, although the effect is stronger in LhrKO (Figure 4). Lhr preferentially associates with heterochromatic genes in an embryonic cell culture line [12]; our results suggest that Lhr and Hmr have a small positive effect on expression of some heterochromatic genes.

Lhr and Hmr mutants have long telomeres
Drosophilidae have lost the telomerase-based mechanism of telomere elongation and instead use the regulated transposition of the HeT-A, TART and TAHRE retrotransposons [33]. Strikingly, these were among the 3 most strongly affected TEs in LhrKO and Hmr− ovaries (Figure 3). We therefore investigated in more detail the localization of Lhr and Hmr proteins to the telomere [6]. Cytological markers on polytene chromosomes have been used to describe three distinct regions in the telomere, with HP1a localizing exclusively to the “cap”, a proteinacous structure at the most distal end of telomeres [25], [28].
mel-Lhr-HA and mel-Hmr-HA overlap with HP1a, showing that Lhr and Hmr localize to the cap but not to more proximal regions (Figure 5A, B). Localization is not due to the doubling of the dosage of these proteins in the transgenic lines because it also occurs in the Hmr3; Hmr-HA/Hmr-HA and LhrKO/+; Lhr-HA/+ genotypes (Figure S5). The localization of Lhr and Hmr to the cap, the primacy of the cap in the regulation of telomeric length, and the increase in the transcript levels of telomeric retro-transposons in Lhr and Hmr mutants led us to ask if these mutations cause long telomeres. We quantitated HeT-A DNA copy number by qPCR in LhrKO flies maintained at 27°C separately from its matched wild-type control strain for ∼40 generations. We found that HeT-A copy number increased approximately 6 fold in LhrKO (Figure 5C). We also examined HeT-A DNA copy number in an Hmr3 mutant stock, and found ∼4–16 fold higher abundance than in the Hmr+ stocks y w and Canton-S (Figure 5D).

Satellite DNA transcripts are upregulated in Lhr and Hmr mutants
Hmr and Lhr both localize to pericentric heterochromatin, which is largely composed of TEs and satellite DNAs. The potential effects of heterochromatin proteins on the levels of transcripts from satellites have not been widely explored. We therefore used our RNA-Seq data to examine transcript levels from 143 repeats in a repeat-sequence database (see Materials and Methods). Transcripts from most repeats are found at low abundance in Lhr+ with only 17 producing more than 10 reads (Table S7). Four different satellite classes are significantly higher in LhrKO versus Lhr+ ovaries, including three that collectively make up more than 8% of the D. melanogaster genome [13]: AAGAC, AACAC, and the GA-rich satellites (Figure 6a). The GAGAA satellite showed the strongest effect, with an approximately 30-fold increase.

These results raise the question of whether transcriptional regulation of specific satellite DNAs reflects a direct association with Lhr. Lhr was not previously tested for association with either GA-rich satellites, which are found on all chromosomes in D. melanogaster [34], or with the AACAC satellite found on chromosomes 2 and Y [35]. We found that Lhr-HA colocalizes extensively with the GA-rich and AACAC satellites in the nurse cell nuclei of early stage egg chambers (Figure 6B, S1A).
In our Hmr RNA-Seq data the number of reads mapping to each repeat family was generally very small, but 3 satellite families are significantly derepressed by at least 4 fold in Hmr− (Figure 6C; Table S8), including GAGAA, which has a 19 fold increase in expression. This finding is consistent with the localization of mel-Hmr-HA to GA-rich satellites above (Figure 1B). Additionally, the satellite Z37541, which binds nuclear lamins, is upregulated 5 fold in Hmr− [36].
Although Lhr-HA localizes to the dodeca satellite [8]; we detected very few reads in either our Lhr+ or LhrKO samples; likewise we did not find upregulation of dodeca in our Hmr RNA-Seq data. We conclude that Hmr and Lhr proteins are required to regulate transcript levels of a subset of satellites to which they localize.
siRNA and piRNA patterns are largely normal in LhrKO
The wide spectrum of TEs derepressed in Lhr and Hmr mutants is similar to mutations in piRNA regulatory genes such as Ago3 and aub that post-transcriptionally regulate TEs via small-RNA-mediated silencing [37], [38]. We therefore investigated a range of phenotypes that are associated with defects in the piRNA pathway. Ago3 and aub mutants disrupt Vasa localization to the peri-nuclear small-RNA processing center, the nuage, and exhibit drastic reductions in the piRNA fraction (23–30 nt) [38], [39]. We found, however, that Vasa localizes normally in LhrKO (Figure 7A). We then sequenced the small RNA pool in LhrKO and found that the piRNA level is broadly comparable to Lhr+ with only a minor reduction in longer piRNAs (Figure 7B). This pattern contrasts with mutants such as aub and spn-E that show a severe loss of piRNAs [39]. We looked more closely for TE-specific defects and found that piRNAs mapping to most individual TE families are comparable between Lhr+ and LhrKO (Figure 7C; Table S9). We also examined “ping-pong” processing, which produces piRNAs from opposing strands with a characteristic 10 nucleotide overlap [38], [39]. Ping-pong scores are generally higher in Lhr+ (Figure 7D; Table S10) but several points argue against there being a significant defect in ping-pong or piRNA processing in LhrKO. First, the magnitude of the difference between genotypes is low, with the ping-pong score being > = 2-fold higher in Lhr+ for only 26/140 TEs. Furthermore, half of these 26 have ping-pong scores <0.10 in Lhr+ (Table S10), suggesting that those TE families are not significantly processed by ping-pong in wild type flies. Second, these differences in ping-pong scores between Lhr+ and LhrKO are much milder compared to mutations in genes such as spn-E [39]. Third, many of the TEs showing differences in ping-pong scores are not strongly depressed in LhrKO. Conversely, many TEs that are strongly derepressed in LhrKO, including HeT-A, have ping-pong scores that are comparable to wild-type. Fourth, some TEs with elevated mRNA levels also show increased ping-pong signatures, probably because of increased processing through a functional ping-pong pathway. We suggest therefore that the moderate trend towards reduced ping-pong scores in LhrKO does not reflect a failure in the ping-pong cycle. Instead, it may result from a skew in the ratio of sense∶antisense piRNAs, because LhrKO flies have high levels of TE transcripts that can be processed into sense piRNAs. An analogous argument has been made for mutations in the Drosophila Gtsf1/asterix gene, which derepress TEs and give an altered ratio of sense and antisense piRNAs but appear to do so downstream of piRNA biogenesis [40].

We searched further for possible defects in piRNA production by examining piRNAs that map to 122 primary-piRNA-generating heterochromatic clusters [41]. piRNAs originating from most of the major clusters are not significantly affected in LhrKO but 16 and 11 of the 122 clusters are at least two-fold higher or lower, respectively, in LhrKO (Figure 7E; Table S11). Some of the most strongly affected clusters are associated with telomeres. Cluster 3 consists entirely of telomeric retrotransposons and is upregulated 4.3 fold in LhrKO. Sub-telomeric cluster 11 shows a complete loss of unique piRNAs, while clusters 33 and 4 are 2.6 and 2.9 fold downregulated, respectively. These 3 clusters consist mainly of HETRP telomere-associated (TAS) repeats and are therefore not expected to contribute to TE repression; their misregulation instead suggests that Lhr is required for regulating chromatin states at telomeres.
The siRNA pathway has also been implicated in repressing TEs in the ovary [42]–[44]. We found that siRNAs mapping to the vast majority of TE families, including those mapping to HeT-A, are not significantly different between LhrKO and Lhr+, suggesting that Lhr is not generally required for siRNA biogenesis (Figure 7F; Table S12). Taken together, our results indicate that defects in small RNA synthesis are not the cause of TE derepression in LhrKO. An intriguing possibility is that Lhr is a piRNA-dependent effector of TE silencing.
Comparing Lhr function in D. simulans and D. melanogaster
We propose that the dynamic sequence turnover of repetitive DNAs is the selective pressure driving the adaptive sequence divergence of Lhr and Hmr. This hypothesis implies that the localization and/or function of the Lhr protein have changed between species, due to co-evolution with species-specific repetitive DNAs. The Lhr1 allele in D. simulans [10] presents a rare opportunity to compare the function of a rapidly evolving heterochromatin protein between sibling species. We performed RNA-Seq from ovaries of Lhr1 females and a matched Lhr+ control (see Materials and Methods). We found essentially no Lhr transcript reads in the Lhr1 mutant strain (Table S13), strongly suggesting that this allele is null.
D. simulans has many of the same satellites as D. melanogaster but they are generally of lower abundance [13]. We therefore first examined satellite DNA expression in the Lhr1 and Lhr+ (control) RNA-Seq data. Unlike in D. melanogaster LhrKO, we found few satellite reads in either genotype and no significant differences between them. We conclude that Lhr has a unique role in D. melanogaster to repress satellite DNA transcription. The AACAC satellite that Lhr co-localizes with in D. melanogaster (Figure 6B) is absent in D. simulans [35]. The GAGAA satellite is also drastically different in D. simulans, being eight-fold less abundant and found only on the sex chromosomes [13], [35]. To determine if this interspecific difference in satellite content reflects divergent localization of Lhr orthologs, we examined D. simulans ovaries expressing a previously characterized sim-Lhr-HA transgene [8]. While Lhr-HA is juxtaposed to dodeca in both species, as previously described [8], the strongest foci in D. simulans do not overlap with GAGAA (Figure 8A). These results demonstrate that Lhr has evolved distinct localization patterns to at least two satellites between D. melanogaster and D. simulans.
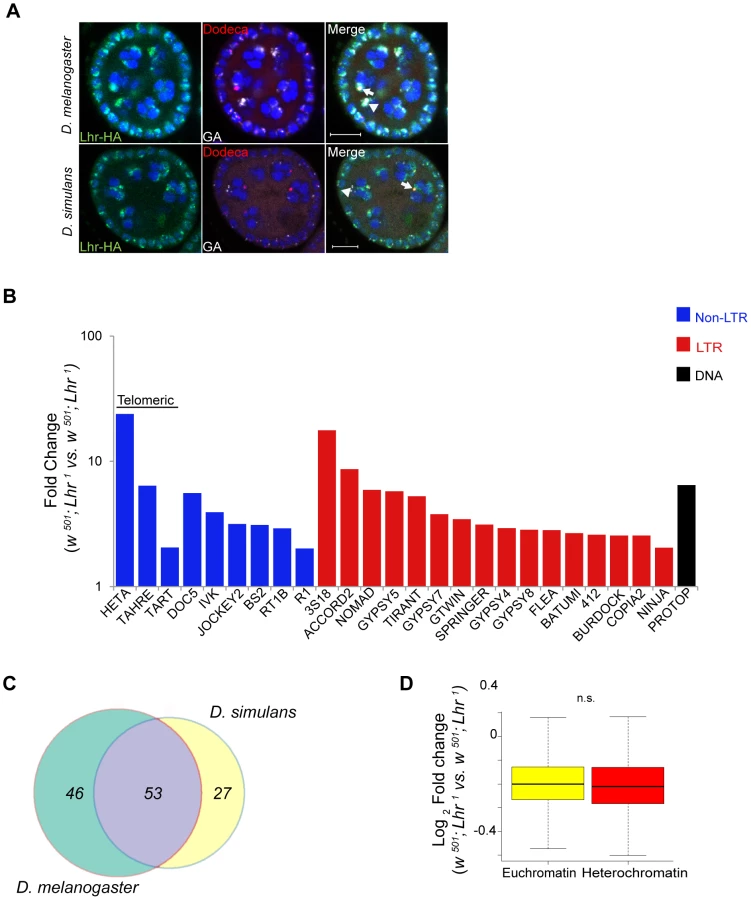
We next examined TE expression and discovered a broad spectrum of TEs derepressed in D. simulans Lhr1, with 80 TEs showing a greater than two-fold up-regulation (Figure 8B; Table S14). Upregulated TEs again include the telomeric transposable elements HeT-A, TART, and TAHRE, other germline elements such as Nomad, and somatic TEs such as Zam and Gypsy 5. 53 transposable elements were commonly mis-regulated in both D. melanogaster and D. simulans, showing that the function of Lhr in repressing TEs is broadly conserved between species (Figure 8C). However, the fold increases of most individual TE families are lower than seen in D. melanogaster LhrKO. For example, HeT-A is 352 fold upregulated in LhrKO but only 23.8 fold upregulated in Lhr1.
We further discovered that Lhr loss in D. simulans does not significantly affect the expression of heterochromatic genes (Figure 8D, Table S13), in contrast with our similar analysis of LhrKO in D. melanogaster (Figure 4A). This result suggests that pericentric genes in D. melanogaster are more sensitive to changes in heterochromatin state than in D. simulans. Overall, our results demonstrate that Lhr function correlates with the increased repeat content and larger amount of heterochromatin found in D. melanogaster.
Comparison of Hmr ortholog function
To examine the functional consequences of Hmr divergence, we took an alternative approach of transforming sim-Hmr transgenes into D. melanogaster. We found that sim-Hmr-HA, like mel-Hmr-HA, localizes to heterochromatin in D. melanogaster (Figure 9A).

To examine potential differences in TE and satellite regulation, we used parallel mel-Hmr-FLAG and sim-Hmr-FLAG transgenes, crossed them into an Hmr− background (Df(1)Hmr−/Hmr3), and performed RNA-Seq on ovarian mRNA. Our expectation was that divergence of Hmr between the orthologs might manifest as the failure of sim-Hmr-FLAG to complement the derepression of TEs in Hmr−.
As a control for the function of the transgenes, we compared the heterozygous wild type Hmr−/Hmr+ to Hmr−; ø{mel-Hmr-FLAG}/+, as each genotype has one wild type copy of Hmr+. The majority of the upregulated TEs in Hmr− (Figure 3B) are suppressed by the mel-Hmr-FLAG transgene; however, 9 out of 182 families ranged from 2 to 9 times more highly expressed in Hmr−; ø{mel-Hmr-FLAG}/+ than Hmr−/Hmr+ (Figure 9B). This result suggests that mel-Hmr-FLAG does not fully complement the Hmr mutant phenotype, which may reflect its decreased expression compared to a wild type allele (Figure S2), though it is also possible that some differences may result from TE polymorphisms that remain between the strains. qRT-PCR also demonstrated that sim-Hmr-FLAG expresses in D. melanogaster at ∼3× the level of mel-Hmr-FLAG (Figure S2), a difference previously seen with Lhr transgenes [8]. Because Hmr is a negative regulator of TE expression, we suggest that this expression difference will not bias against our goal of identifying TEs that are not fully repressed by sim-Hmr-FLAG.
We did not find any difference in satellite DNA expression; however, we found 11 TE families that are differentially expressed between the transgenic genotypes (Figure 9C). Five are more highly expressed in Hmr−; ø{mel-Hmr-FLAG}/+ with fold changes ranging from 2–3, of which 3 are incompletely repressed by mel-Hmr-FLAG in the control cross described above (Transpac, Tirant, and Batumi). The differential expression of these 5 families likely reflects the inability of mel-Hmr-FLAG to fully complement Hmr− and the higher expression level of sim-Hmr-FLAG.
More intriguing are 6 TE families that are 2–6× more highly expressed in Hmr−; ø{sim-Hmr-FLAG}/+ than in Hmr−; ø{mel-Hmr-FLAG}/+, implying that sim-Hmr-FLAG is unable to fully complement the derepression of these elements. BS and Doc6 (also known as Juan) elements are present at a mean frequency of about 0.1 in a population of Portuguese D. melanogaster [45] and have low pairwise identity in the reference genome [46], suggesting that they are likely active. The mean population frequencies of 4 of the other families (BS3, Circe, Helena, and FW2) are near 1, suggesting that these TEs are fixed and therefore currently inactive in D. melanogaster. Helena, though, appears to have been active more recently within D. simulans [47]. We suggest that BS, Doc6 and Helena are candidates for future investigation of co-evolution with Hmr in either D. melanogaster or D. simulans.
Transposable elements are upregulated in hybrids
In light of our discovery that Lhr and Hmr are required for TE repression within D. melanogaster and D. simulans, we investigated TE activity in lethal (Hmr+) hybrid male larvae. Because most TEs have different expression levels between D. melanogaster and D. simulans, we defined mis-regulated TEs as being at least two-fold higher than both parental species, as done in a previous analysis [48]. We found that 42 LTR and non-LTR elements are significantly upregulated in lethal (Hmr+) hybrid male larvae with 2 others being downregulated (Figure 10A; Table S15).

We next examined whether TE misregulation correlates with hybrid lethality by comparing the lethal Hmr+ hybrid males to viable Hmr− hybrid males (Figure 10B, Table S16). The expression of 29 TEs is significantly lower in Hmr− hybrids. Because Hmr functions as a repressor of TEs in D. melanogaster male larvae (Figure 3C), these differences may reflect a general difference between lethal and viable hybrids rather than the presence or absence of Hmr activity. In fact, only 4 of the 29 TEs downregulated in Hmr− hybrid male larvae are upregulated in Hmr− D. melanogaster male larvae (Table S4).
In addition, we found modest increases (2–4 fold) in the activity of 5 TE families in living hybrids. None of these are significantly upregulated in Hmr− D. melanogaster male larvae (Table S4). They include TAHRE and may reflect higher levels of cell proliferation in viable hybrids. Taken together our results suggest that TE overexpression is unlikely to be causing hybrid lethality.
Discussion
Lhr and Hmr interact with HP1a
We and others previously reported that Lhr (also known as HP3) interacts with HP1a [6], [9], [12], [30]. Here we report that Hmr also interacts with Lhr, and both are present in a complex together with HP1a. Consistent with this interaction, many of the roles we report here for Lhr and Hmr have been described for HP1a, including localizing to heterochromatin, regulating TE and pericentric gene expression, and controlling telomere length [49]–[51]. However, unlike mutations in Su(var)205 which enodes HP1a [52], mutations in Hmr and Lhr are viable. Furthermore, Hmr and Lhr do not localize to the 359 bp satellite which forms a substantial fraction of X-linked pericentric heterochromatin Figure 1; [ref. 8]. These findings suggest that Hmr and Lhr are not ubiquitous heterochromatin proteins, leaving open the intriguing question of what guides their localization specificity.
The interaction of Hmr and Lhr with HP1a has recently been independently reported [53; AA Alekseyenko and M. Kuroda, personal communication]. Thomae et al. [53] also report other findings similar to ours here including repressive effects of Hmr and Lhr on TEs in somatic tissues and their localization to telomeres. Several conclusions are similar between the two studies and with previously published conclusions. Thomae et al. [53] observe upregulation of TEs in hybrids but conclude that they are unlikely to be the direct cause of hybrid lethality, a conclusion we reach below using different methods. Their conclusion that hybrids are highly sensitive to Hmr dosage is in concordance with previous studies, such as the previous observation that a ∼9.7 kb Hmr+ transgene causes dosage-dependent lethality to hybrid females [3]. This conclusion also fits well with the discovery that hybrids are highly sensitive to Lhr dosage [8].
One area of possible discrepancy is the viability effects and cellular phenotypes associated with Hmr and Lhr mutants versus RNAi knockdown. Thomae et al report a high rate of mitotic defects in Lhr RNAi knockdown tissue culture cells, yet we found that LhrKO flies are almost fully viable (see Results), as are Lhr RNAi knockdown animals [53]. We also have not observed the lethality or morphological defects in Hmr mutants that are reported for Hmr RNAi cells and animals [53]. For example, Aruna et al. [4] found reduced longevity but no effect on viability up to eclosion of flies carrying the Df(1)Hmr− allele, a deletion of the 5′ end of Hmr. Further work is necessary to determine if these discrepancies reflect phenotypes associated with the use of RNA interference or differences between assaying whole animals versus tissue-culture cells, such as the aneuploid state of cultured cell lines [54].
Rapidly evolving heterochromatin proteins and repetitive DNA variation
Several HIs involve heterochromatin proteins or heterochromatic sequences, leading to the suggestion that genetic conflicts between selfish DNAs and host fitness are an important force that is driving the evolution of HI [1], [2], [23], [55].
TE and satellite abundance varies widely among species and is a major contributor to genome-size variation. The evolutionary causes of this variation have been widely debated for many years [56]. When considering genetic conflict theories, it is important to first exclude alternative evolutionary causes of repetitive DNA variation. One explanation is neutrality, with repeat variation governed by mutational processes, in particular the balance between insertions and deletions [57]. Insertion/deletion models are particularly appropriate for inactive and degenerate TEs, and perhaps also for certain classes of satellites that are no longer homogenized by concerted evolution [58].
Selectionist models fit better for active repeats, and must be invoked if the adaptive evolution of heterochromatin proteins is proposed to reflect co-evolution with repetitive DNA. One model is that some repeats are co-opted for host functions. Drosophila's telomeric retrotransposons are a relevant example that is discussed below. We also consider three, non-mutually exclusive selective costs associated with repetitive DNA when discussing the evolution of Hmr and Lhr
One potential cost arises from the overall load of repetitive DNAs, including increased genome size and instability. A second is direct genetic conflict. We define genetic conflict here to refer to fitness costs imposed by selfish DNAs that have evolved specific mechanisms to increase their transmission [59]. Such conflicts could be caused by highly active individual repeats, for example during hybrid dysgenesis caused by introduction of a TE family into naive strains [60]. Finally, genetic conflicts can have indirect costs, such as pleiotropic fertility defects caused by repeat expansions involved in meiotic drive [61].
Hmr and Lhr repress transposable elements
TEs define selfish DNA [56]. They infect most genomes, can self-mobilize and increase their copy number, and destabilize genomes via spontaneous mutations, ectopic recombination, and deleterious increases in genome size [62], [63]. Adaptive evolution of TE-defense genes can therefore be readily interpreted as the host species responding to the fitness cost of TEs [19].
Like Hmr and Lhr, many piRNA pathway genes are also evolving under positive selection [22]. This raises the possibility that Lhr and Hmr are co-evolving with the piRNA pathway proteins. However, the lack of major perturbations in the piRNA pool in LhrKO suggests that Lhr and Hmr function downstream or independently of piRNA biogenesis. Piwi, guided by piRNA, has been proposed to recruit repressive heterochromatin components including HP1a and histone methyl transferases to transposable elements [51], [64]. One possibility is that Lhr and Hmr function downstream of HP1a to repress TEs via RNA degradation machinery such as the nuclear exosome [65].
We note that Ago3 is moderately down-regulated in both LhrKO (3.4 fold) and Hmr− (∼2 fold) (Tables S5, S6), likely because the gene is peri-centromeric. Two results demonstrate that this modest reduction in Ago3 cannot explain the broad effects on TEs in Hmr and Lhr mutants. First, Ago3 expression is unaffected in D. simulans Lhr1, which also shows widespread TE derepression. Second, Ago3 mutants have major disturbances to their piRNA pool [38], which we did not observe in LhrKO (Figure 7).
Hmr and Lhr regulate telomeres
While TE repression is typically viewed in terms of genetic conflicts, the relationship between Lhr, Hmr and the telomeric TEs resembles symbiosis. These TEs have been domesticated by Drosophila species for tens of millions of years to serve a vital host function, and thus are not considered selfish DNA [33], [66]. The telomeric TEs were among the most strongly derepressed in Hmr and Lhr mutants, in some cases more than 100 fold. We also observed increases in HeT-A DNA copy number in Hmr and Lhr stocks. Increased telomeric TE expression does not necessarily increase HeT-A DNA copy number and cause longer telomeres, suggesting that multiple factors control telomere length [67]. If so, then Lhr and Hmr must control multiple processes at the telomere. This is supported by the localization of both proteins to the telomere cap, a protective structure that prevents telomere fusions [28]. The strong reduction in LhrKO of piRNAs from three TAS-repeat containing sub-telomeric piRNA clusters is particularly intriguing. piRNA production from clusters is dependent on them maintaining a heterochromatic state [68], which could explain why Lhr is required for TAS piRNA expression while it acts as a repressor in most other circumstances.
Hmr and Lhr regulate species-specific satellite DNAs
We discovered several striking examples that suggest species-specific co-evolution of Hmr and Lhr with satellite DNAs. We found that D. melanogaster Hmr and Lhr proteins localize to and repress transcripts from GA-rich satellites. GA-rich satellites are ∼8 fold less abundant in D. simulans [13] but are cytologically detectable; nevertheless we find that sim-Lhr does not localize to them. GA-rich satellites also have low abundance in the outgroup species D. erecta [13], implying that the differential abundance with D. simulans reflects an increase in D. melanogaster. Similarly we discovered that mel-Lhr-HA localizes to AACAC in D. melanogaster, a repeat that is absent in D. simulans [69]. Furthermore, we detected moderate up-regulation of several other satellite transcripts only in D. melanogaster. Our results suggest that Lhr and Hmr may have evolved in D. melanogaster to mitigate the deleterious consequences of satellite expansion, which can include ectopic recombination, increased genome size, and destabilized chromosome segregation [16], [70].
Satellite transcripts have been reported from various tissues in wild type D. melanogaster [71], [72] but little is known about their production. They could be products of either non-specific transcription or read-through from adjacent TEs. Increased levels of satellite transcripts are observed in D. melanogaster spn-E mutants, suggesting that RNA interference or piRNA pathways control satellite transcript levels [21].
Is the adaptive evolution of Hmr and Lhr driven by diverging heterochromatic repeats?
We find that at a broad scale, Lhr and Hmr from both D. melanogaster and D. simulans regulate heterochromatic repetitive DNAs but very few genes. This finding is consistent with previous analyses demonstrating that some functions of these genes are conserved between species [4], [7]–[9]. But many of the repeats regulated by Lhr and Hmr are rapidly evolving, raising the question of whether specific repetitive DNAs are directly driving the adaptive evolution of the Lhr and Hmr coding sequences between species. A simple prediction is that D. simulans orthologs should fail to fully repress such repeats when placed into D. melanogaster, a prediction that we tested for Hmr.
The BS non-LTR retrotransposon is significantly derepressed in D. melanogaster Hmr− and LhrKO, and in D. simulans Lhr1 mutants. Interestingly, BS appears to be transpositionally active in D. melanogaster but inactive in D. simulans [73]. One interpretation is that BS was active in the common ancestor and regulated by Hmr and Lhr. The genes would continue to co-evolve with BS in D. melanogaster, making the sim-Hmr ortholog less effective at repressing BS elements in D. melanogaster. In this scenario Hmr and Lhr are engaged in a recurrent genetic conflict with BS elements that leads to their sequence divergence. Consistent with this prediction we found significantly higher expression in Hmr−; ø{sim-Hmr-FLAG}/+ compared to Hmr−; ø{mel-Hmr-FLAG}/+.
Copia shows a different pattern, with ∼20-fold up-regulation in LhrKO but only ∼2-fold in Lhr1 (and only when mapping to the consensus-sequence database), as well as significant derepression in Hmr−. Copia expression level can be high in D. melanogaster but is variable among populations. In contrast, copia elements in D. simulans typically contain deletions in regulatory elements required for expression, and transcripts are undetectable by Northern blot analysis [74]. These results suggest that Hmr and Lhr could be D. melanogaster host factors that defend against a TE that is currently active within the species. However, copia was fully repressed in Hmr−; ø{sim-Hmr-FLAG}/+, demonstrating that adaptive divergence of Hmr by itself does not affect copia regulation.
Overall, we found surprisingly few cases of overexpression associated with Hmr divergence, including no effects on satellite DNAs (Figure 9). We also note that most of the TEs identified other than BS and Doc6 are likely transpositionally inactive in D. melanogaster [45], which makes it more challenging to fit a scenario of direct and recurrent evolution between Hmr and specific TEs.
We suggest several possible interpretations of these results. One is that Hmr and Lhr adaptive divergence is in fact driven largely or solely by BS and/or Doc6, a hypothesis that will require understanding the mechanism by which Hmr and Lhr affect expression of these TEs. Second is that Hmr and Lhr may be co-evolving with other genes, and that multiple diverged genes need to be replaced simultaneously in order to detect their effects on other TEs and satellite DNAs. Third is that more sensitive assays are needed, for example monitoring TE transposition rates over multiple generations. A fourth possibility is an alternative to genetic conflict scenarios that arises from population-genetic models. These models suggest that the fitness costs of individual TE families are likely extremely weak under most circumstances. The adaptive evolution of repressor proteins may therefore reflect the cumulative load of repeats within a genome [22]. This alternative view could be applicable to Hmr and Lhr since they repress a large number of TEs and satellites. Finally, Hmr and Lhr may have additional unidentified phenotypes that are also the targets of adaptive evolution.
Repeat load, adaptation and hybrid incompatibilities
D. simulans has a smaller genome with ∼4-fold less satellite DNA [13], [14] and significantly fewer TEs [24], [75] compared to D. melanogaster. This large difference in repeat content between D. melanogaster and D. simulans may have wider consequences. We found reduced expression from pericentric heterochromatin genes in Hmr and Lhr mutants in D. melanogaster. This reduction may reflect the fact that pericentric genes have evolved to use heterochromatin proteins such as Lhr and Hmr to maintain gene expression in a repeat-rich environment [76]. Pericentric genes in species with fewer repeats would presumably not require these proteins. Consistent with this model, we found that Lhr loss in D. simulans has a negligible impact on pericentric gene expression. This finding suggests that Lhr and Hmr have an adaptive role in blocking effects on gene expression arising from increasing repetitive DNA copy number.
If each genome is uniquely adapted to its repetitive DNA content, then the shock of hybridization may lead to misregulation of TEs and satellites. TEs are activated in various animal and plant hybrids but the consequences, if any, for hybrid fitness are largely unclear [77]. We found substantial TE misregulation in hybrid male larvae (Figure 10A). Since these hybrids are agametic [78], this TE expression comes from somatic tissues. The fitness cost of this upregulation is unclear as somatic TE overexpression is not necessarily lethal within D. melanogaster [79], [80]. Comparison of lethal Hmr+ and viable Hmr− hybrid males demonstrates that lethal hybrids have more TE expression (Figure 10B) than the viable hybrids, which in turn have more TE expression than either of its parents. However, this TE misregulation seems unconnected with Hmr as the TEs differentially expressed between Hmr+ and Hmr− hybrid male larvae are largely distinct from those between Hmr+ and Hmr− D. melanogaster male larvae. Further, while Hmr− causes rampant TE over-expression within D. melanogaster, it is associated with reduced TE levels in hybrids. These observations argue that the TE derepression in hybrids is unrelated to the pure species function of Hmr. This finding is consistent with previous genetic studies that demonstrate that the wild type Hmr+ allele causes hybrid lethality and thus behaves as a gain-of-function allele in hybrids [81], [82]. More generally it underscores the unique nature of the hybrid genetic background [1]. Somatic TE overexpression may result from breakdown in the siRNA or piRNA pathways due to incompatibilities among multiple rapidly evolving TE regulators.
One clear example is known where a species-specific difference in a satellite DNA causes incompatibility between Drosophila species [83]. But the toll caused by heterochromatic differences may more commonly be indirect, as heterochromatin proteins diverge in response to changes in heterochromatic DNA repeats. Recent work suggests that hybrid female sterility may be caused by incompatibilities among rapidly evolving piRNA proteins rather than by species-specific differences in TEs [48]. We suggest that the role of Hmr and Lhr in regulating the activity of three highly dynamic classes of heterochromatin has led to their recurrent adaptive evolution, and secondarily, to their involvement in interspecific hybrid lethality.
Materials and Methods
Construction of the LhrKO mutant
We used the pW25 donor vector and ends-out homologous recombination method to make an Lhr mutant allele [84]. The donor vector was designed to recombine a w+ marker into Lhr and simultaneously remove 26 bp of the coding region. iProof (Biorad) was used to PCR amplify two genomic fragments from y; cn bw sp (D. melanogaster) genomic DNA. The 3768 bp Lhr upstream fragment, including 128 bp of the coding region of Lhr, was amplified with primers LUF-Fwd: 5′ - ttggcgcgccAACAGGGTCGGCTGTCACATTT and LUF-Rev: 5′-ttggcgcgccGCGAGCATCTCCATGAGCAG (Tm = 63°C) and cloned into the AscI site of pW25 using the underlined sequences. The 3935 bp Lhr downstream fragment that includes 806 bp of the Lhr coding region was amplified with primers LDF-Fwd: 5′-AAGCGGCCGCAGGTGGAGCCCAAAATGGACG and LDF-Rev: 5′ - AAGCGGCCGCCACACATTGCGAATGCA G AAA (Tm = 65°C) and cloned into the NotI site using the underlined sequences. Restriction digestion was used to pick a clone in which the 2 inserts and the mini-white gene were in the same orientation.
The construct was injected into a strain of w1118 (Genetic Services) and a transgenic line, P{w+, Lhr-KO}5-1, with a lethal insertion on the X chromosome was obtained. P{w+, Lhr-KO}5-1/FM6 females were crossed to y w; P{ry+, hs-flpase}, P{v+ hs-I-Sce}/TM6, Ubx males. Two to three day-old larvae were heat shocked and P{w+, Lhr-KO}5-1/y w P{ry+, hs-flpase}, P{v+ hs-I-Sce}/+ female progeny were crossed to w1118 males. Rare w+ sons were screened for homologous recombination events by PCR. Primer pairs Lhr-f1 5′ - TTCGCACGTTGTGTTCAAGTAA-3′, /Lhr-r1 5′-GTAGCTTTCTCTTGGCGCTCTT-3′ and Lhr-f2 5′ - AACGTGCTCGTAGCTTTGGT-3′/, Lhr-r2 5′-TCGCGAAAATACTTCCGTCT-3′ (Tm = 58°C) produce no amplicons in the presence of the white insertion. Attempts to remove the w+ marker by Cre recombination were unsuccessful and the w+-disrupted Lhr locus was designated as LhrKO.
To test the genetic effects of this mutation, we took advantage of a recent observation that a deficiency chromosome which deletes D. melanogaster Lhr can weakly rescue D. melanogaster-D. mauritiana hybrid males to the pharate adult stage [8]. When we crossed LhrKO homozygous females to D. mauritiana males at 18°, we obtained 10.6% rescue of live males (17 males and 161 females). The stronger rescue observed here may be due to the fact that the mothers of the cross were homozygous for the LhrKO allele, since Lhr likely has strong maternal expression based on its high protein abundance in early embryos [8].
Hmr transgenes
A D. melanogaster Hmr-FLAG transgene was made by inserting a 3× FLAG tag sequence [85] immediately upstream of the stop codon of Hmr using fusion PCR into plasmid p72, which is a pCaSpeR2 vector containing a ∼9.7 kb fragment of the Hmr region [3]. Two Hmr fragments (L-arm and R-arm) were amplified from p72 with iProof polymerase by using primer pairs 739/738 and 736/740, respectively. The primers 738 and 736 contain sequence encoding the FLAG tag and partially overlap to allow fusion in the subsequent stage. The primers 739 and 740 were combined with L-arm and R-arm products to produce a fused partial fragment of Hmr containing the 3× FLAG sequence. This fragment was cloned into the pCR-BluntII-Topo vector (Invitrogen) and sequenced completely between the AvrII and KpnI restriction sites. The AvrII/KpnI fragment was then cloned into the corresponding sites of the p72 plasmid. A 300 bp fragment containing the attB site was then PCR amplified from plasmid pTA-attB (gift from Dr. Michele Calos) using primers 502 and 503 and cloned into the NotI site. This fragment was digested with NotI (on the ends of 502 and 503), gel purified, and inserted into the NotI site of the plasmid containing Hmr-FLAG. We refer to this transgene as mel-Hmr-FLAG.
A D. melanogaster Hmr-HA transgene was made by inserting a 3XHA epitope tag between codons 466 and 467 of Hmr. Primers 215/1246 and 1247/495 were used to amplify 573 and 316 bp fragments, respectively. Primers 1246 and 1247 overlap and encode the HA tag. Fusion PCR containing these 2 products and primers 215/495 was performed. The PCR product was cloned into pCR-Blunt II-TOPO, and the insert was checked by sequencing. The insert was then cloned using SpeI and BsiWI back into a modified p72 containing an attB site inserted into the NotI site. The orientation and presence of the HA tag were checked by double digests and PCR. We refer to this transgene as mel-Hmr-HA.
A D. simulans Hmr-FLAG transgene was made by inserting the 3× FLAG tag sequence upstream of the stop codon in p89, a pBluescript II KS(+) plasmid containing the D. simulans Hmr insert that was used for the p92 transformation construct in [7]. Primers 751/753 and 750/752 were used to amplify 1.3 kb and 1.8 kb fragments of the insert, respectively, which were then joined by fusion PCR using primers 750/751. The fusion PCR product was cloned into pCR-Blunt II-TOPO and confirmed by sequencing. The insert was designed to have an HpaI site near one end and a NotI site near the other. The NotI site was destroyed during cloning; however, the pCR-Blunt II-TOPO vector contains a NotI site within 40 bp of the destroyed sequence. The insert was then cloned back into p89 using HpaI and NotI. The orientation of the insert, as well as the addition of the FLAG tag, was checked by double digest with ClaI and HpaI. The D. simulans Hmr-FLAG insert was then removed as a SacII fragment. Klenow enzyme was used to fill-in the ends to allow cloning into the StuI site of pCaSpeR2 containing an attB site inserted at its NotI site. We refer to this transgene as sim-Hmr-FLAG.
The D. simulans Hmr-HA transgene was made from plasmid p89 by inserting the HA tag at the region orthologous to mel-Hmr-HA [7]. Primers 135/1365 and 1247/1364 were used to amplify 861 bp and 827 bp fragments, respectively, from the p89 template, and were fused together using primers 1364/135. The fusion PCR product was then cloned into pCR-Blunt II-TOPO and the entire insert was checked by sequencing. The insert was then cloned back into p89 using SpeI and BlpI. Blunt end ligation, used for sim-Hmr-FLAG above, proved inefficient for transferring the insert into the transformation vector. Therefore an XbaI site was added to the 3′ end of Hmr-HA by amplifying the entire insert using primers 1402/1403. The PCR product was then gel purified and cloned back into pCR-Blunt II-TOPO. The polylinker contains an XbaI site 5′ to the insert, allowing us to clone the entire insert into the XbaI site of pCaSpeR2 containing an attB site inserted at its NotI site. We refer to this transgene as sim-Hmr-HA.
Oligonucleotides for Hmr transgenes (all written 5′-3′). 739: AGCCAAATTGCCGACAGTAGCCAAG; 738: ATCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCAGGCGGTGGCGGATTGACCTTG; 736: GACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGTAGCTCTCGAAACTTTTGGCACACGTAG; 740: TTGTACTGCCATTAGGTATAGCTAACCATCC; 502: AAACCCGCGGCCGCATGCCCGCCGTGACCGTC; 503: AAACCCGCGGCCGCGATGTAGGTCACGGTCTCG; 152: TCTTCTTAGACTGCGGGTTG; 215: CAGCGCATGCGCGGCACCGTAT; 1246: ATAGTCCGGGACGTCATAGGGATAGCCCGCATAGTCAGGAACATCGTATGGGTACATTGCACTGTTGGTCATGCTCGT; 1247: TCCCTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTAC;GCTAGCACTGCCACAAGCATTGG; 495: GACACGCCCGTTCCCATAGT; 751: ACAGCGATTTGCGCAAGCCG; 753: TCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCAGGCGGTGGCGGATTTGCCTTCTTGGCGTATTTAGA; 750: GTGAATTGTAATACGACTCACTATAGGGCG; 752: GACGGTGATTATAAAGATCATGACATCGATTACAAGGATGACGATGACAAGTAGCTCTCGAATCATTGGCACACG; 135: GAGGAGGACCCCACCTATAACTAC; 1365: ATAGTCCGGGACGTCATAGGGATAGCCCGCATAGTCAGGAACATCGTATGGGTATGCACTGTTAGAAATGCTTGTGCTG; 1364: GCTGGCAATTTGGACTTTGT; 1402: GCGGGCGGTCATTATTAA; 1403: TATCTAGAGCGGCCGCGAGCTCTAATA.
Transgenic fly lines
φC31-mediated transgenesis was performed by Genetic Services using the P{CaryP}attP2 integration site at cytological position 68A4 [86]. Site specificity of integration was checked by PCR assays described in references [8], [87]. D. melanogaster transformants were recognized by their w+-eye color and were crossed to a y w strain. Wild type activity of the Hmr-HA and Hmr-FLAG transgenes was tested for complementation of an Hmr rescue mutation in hybrids as done previously for Hmr+ transgenes [3], [7]. Here we crossed Df(1)Hmr−, y w v/FM6; ø{mel-Hmr-HA}/+ females to D. simulans w501 males. We recovered 193 w501/Y; +/+ hybrid males but only 1 w501/Y; ø{mel-Hmr-HA}/+ hybrid male, demonstrating that the transgene is Hmr+. Likewise, we crossed Df(1)Hmr−, y w v; ø{mel-Hmr-FLAG}/+ females to D. simulans v males, and recovered 451 v females, 258 w males and only 3 w+ males.
Drosophila strains
LhrKO was outcrossed to w1118 for six generations. Sibling crosses were then used to generate a homozygous w1118; LhrKO/LhrKO (abbreviated as LhrKO), a heterozygous LhrKO/+, and a wildtype w1118; Lhr+/Lhr+ line (abbreviated as Lhr+). All experiments with Lhr in this paper use these matched mutant and sibling controls unless otherwise specified. The D. simulans Lhr1 allele is caused by an insertion in the 5′ UTR and appears to make no transcript by RT-PCR [6]. Lhr1 was outcrossed to the inbred wild-type line w501 for 3 generations to generate the stock w501; Lhr1 (abbreviated as Lhr1) and w501, Lhr+ (abbreviated as Lhr+). Lhr-HA transgenes were described previously [8]. y w F10 was created by single-pair matings between siblings for 10 generations.
We refer to the P{EPgy2}Hmr3 allele that is marked with y+ and w+ described in [4] as Hmr3. Df(1)Hmr−, y w v, abbreviated as Df(1)Hmr−, is described in [88]. In order to match backgrounds for the Hmr RNA-Seq experiments, the Hmr3 stock and the transgenic lines (mel-Hmr-FLAG and sim-Hmr-FLAG) were outcrossed to y w F10 for 6 generations and then made homozygous.
Fertility assays
Individual 1–2 day old virgin LhrKO and LhrKO/+ sibling females, obtained from crosses of LhrKO/+ at 27°C, were crossed to two w1118 males. Flies were transferred to a fresh vial every 5 days for 15 days. Vials in which either the female or both males were missing or dead were not scored or transferred. To create the heteroallelic siblings LhrKO/Df(2R)BSC44 and LhrKO/SM6a, LhrKO/LhrKO were crossed to the Lhr− deletion stock Df(2R)BSC44/SM6a [6]. The fertility assay was carried out as above except vials were flipped every 4–5 days.
Hatch rate assays
LhrKO/+ or LhrKO/LhrKO females were crossed to w1118 males at 27°C. Egg lays were carried out on grape juice/agar plates for 3 hour periods at either 2–3 days, 5–6 days or 10–11 days after eclosion of the female parents. The plates were maintained at 27°C and monitored over the next 24–36 hours for hatched eggs.
Crosses for generating Hmr genotypes for RNA-Seq of ovarian mRNA
y w Hmr3; +/+ females were crossed to y w; ø{mel-Hmr-FLAG}/ø{mel-Hmr-FLAG} males. F1 males were crossed to Df(1)Hmr−/FM6; +/+ females to generate both y w Hmr3/Df(1)Hmr−; ø{mel-Hmr-FLAG}/+ and y w Hmr3/Df(1)Hmr−; +/+. Similarly, y w Hmr3; +/+ females were crossed to y w; ø{sim-Hmr-FLAG}/ø{sim-Hmr-FLAG} males. F1 males were crossed to Df(1)Hmr−/FM6; +/+ females to generate y w Hmr3/Df(1)Hmr−; ø{sim-Hmr-FLAG}/+. Lastly, y w; +/+ females were crossed to y w; ø{mel-Hmr-FLAG}/ø{mel-Hmr-FLAG} males. F1 males were crossed to Df(1)Hmr−/FM6; +/+ females to generate the heterozygous wildtype control, y w/Df(1)Hmr−; +/+. These crosses were done at 27°C and in triplicate to generate 3 biological replicates.
Crosses for generating pure-species and hybrid samples for RNA-Seq of larvae
The Df(1)Hmr−, y w v/FM7i, P{w+mC = ActGFP}JMR stock (abbreviated as Df(1)Hmr−/FM7i, GFP) was described previously [88]. A stock with the matching Hmr+ genotype, y w v/FM7i, P{w+mC = ActGFP}JMR (abbreviated as Hmr+/FM7i, GFP) was created by crossing y w v/Y males with Df(1)Hmr−/FM7i, GFP females. FM7i, GFP/Y males from this Hmr+ stock were then crossed to Df(1)Hmr−/FM7i, GFP females for 10 generations in order to make the autosomal backgrounds comparable between the two stocks.
To generate hybrids, Df(1)Hmr−/FM7i, GFP or Hmr+/FM7i, GFP were crossed to v/Y D. simulans males. For each cross, 6 replicates were made each containing 25 0–12 hour-old virgin females and 50 4–6 day-old virgin males. Hybrid larval sons not carrying the balancer were selected by their y− mouth hook and GFP− body phenotypes. Additionally, some crosses were allowed to develop to ensure that only Df(1)Hmr− crosses produced hybrid sons. To generate D. melanogaster samples, 3 replicates of 10 Df(1)Hmr−/FM7i, GFP or Hmr+/FM7i, GFP virgin females were crossed to 15 FM7i, GFP/Y males. Larval sons not carrying the balancer were selected by y− and GFP− phenotypes. To generate D. simulans samples, 3 replicates of 10 y w D. simulans virgin females were crossed to 15 v/Y D. simulans males. Larval sons were selected by y−.
Preparation of protein lysates for semi-quantitative western blots
50 mg of 1–17 hr embryo collections were dounced 30 times with a tight pestle in 500 ul buffer A1 (15 mM HEPES, pH = 7.5; 15 mM NaCl; 60 mM 1M KCl; 4 mM MgCl2; 0.5% TritonX-100; 0.5 mM DDT) and then centrifuged for 5 minutes at 4°C. The pellet was washed with 500 µl buffer A1 and centrifuged. This process was repeated another two times. The pellet was lysed by douncing in 200 µl SDS lysis buffer (500 µl 10% SDS, 200 µl 1M Tris, pH = 8.0, 40 µl 0.5M EDTA, 100 µl 100× protease inhibitor, 10 µl 0.5M EGTA, 50 µl 100 mM PMSF, 9.1 ml water). The lysate was allowed to rotate at 4°C for 20 minutes and then centrifuged. The supernatant was removed, quantitated using the Bradford assay and was run on an SDS-PAGE gel.
Anti-Lhr antibodies and western blots
An Lhr cDNA was cloned into pDEST17 (Invitrogen). The expressed protein from E. coli was purified using Ni-Ag beads under denaturing conditions (8M urea), dialyzed down to 2M urea and injected into rabbits (Cocalico). The antisera was then purified by coupling purified His-Lhr to CnBr-activated Sepharose beads in the presence of 1% Triton-X and removing urea by dialysis. Antisera was eluted in 0.2 M glycine, pH 2.8 and then neutralized with 1M Tris, pH 8.5. The antibody failed to detect Lhr in immunofluorescent experiments but was used for Western blots in Figure S3 at 1∶4000 in 5% milk-TBST and HRP conjugated anti-rabbit secondary antibody at 1∶2000 dilution. HA-tagged Lhr was detected with 1∶1000 dilution of rat anti-HA (Roche, 3F10) and HP1a was detected with a 1∶700 dilution of mouse monoclonal supernatant (C1A9, DSHB).
Co-immunoprecipitation
0∼16 hour-old embryos were collected, dechorionated and snap frozen in liquid nitrogen. Embryos were then resuspended to 10× embryo volume of Buffer A (10 mM Tris-Cl pH 8.0, 300 mM sucrose, 3 mM CaCl2, 2 mM Mg acetate2, 0.1% Triton X-100, 0.5 mM DTT, 0.5 mM PMSF) and homogenized with a dounce homogenizer. The homogenized lysate was centrifuged at 700 g for 10 minutes at 4° to pellet the nuclei. The supernatant was removed, the pelleted washed once in Buffer A, the nuclei centrifuged again and then resuspended in 1× embryo volume of Buffer MN (15 mM Tris-Cl pH 7.4, 250 mM sucrose, 60 mM KCl, 1.0 mM CaCl2, 0.5 mM DTT, 1× protease inhibitor cocktail). The nuclear lysate was sonicated briefly, micrococcal nuclease added to a concentration of 500 units/ml, and the chromatin digested for 1 hour at 4° with gentle agitation. EDTA and Triton X-100 were then added to a concentration of 5 mM and 0.1% respectively, to inactivate nuclease activity and solubilize the proteins, followed by incubation at 4° for 1 hour. After a second brief sonication, the digest was centrifuged at 12,000 g for 10 min at 4° and the supernatant was collected. 50 µl of the chromatin digest was diluted in IP Wash Buffer (50 mM Tris-Cl pH 7.4, 100 mM NaCl, 0.1% Triton X-100) with 1× protease inhibitor cocktail to a final volume of 125 µl per co-immunoprecipitation mixture. 15 µl of protein G-conjugated magnetic beads and 2–5 µl of antibody were added followed by incubation for 4 hours at 4° with gentle agitation. The beads were washed 3 times in IP Wash Buffer. The immunoprecipitated proteins were then eluted by boiling the beads in 1× Laemmli sample buffer for 5 minutes and analyzed by immunoblotting.
RT-PCR and qRT-PCR assays
RNA extraction, cDNA synthesis and qRT-PCR assays were performed as in reference [8], using 2–5 µg of RNA. qRT-PCR experiments included three technical replicates of three separate biological replicates. Primers included: Lhr-f1 5′caccATGAGTACCGACAGCGCCGAGGAA, Lhr-r1 5′ ACACTTGGTTTTCGGCACATC CGC, Lhr-f2 5′ GTAGCTTTCTCTTGGCGCTCTT, Lhr-r2 5′ GTAAGTGAACTGAAGCTGC GTTGG, EDTP-F 5′GCTGGCAGGTGG TTACCGACA, EDTP-R 5′CGTGGCCAGGTTCA TGGATGA, Bap55-F 5′ CCGAGAGTC TCTTTGACAATGCA, and Bap55-R 5′GCCTCTT CGTACTCCTGCGA. Hmr-f1 5′ TAAGTTCGCCTTCCGCACATACC and Hmr-r1 5′ GACCAGAAACCTGAGTTGCTCCA. HeT-A and RpL32 (also known as Rp49) transcript levels were measured with primers from reference [89].
qPCR of HeT-A DNA copy number
The Invitrogen DNEasy kit was used to make genomic DNA from LhrKO and Lhr+ female carcasses that were free of ovarian tissue. Primers Het-s2 and Het-as2 amplify from the coding sequence of HeT-A [90]. HeT-A copy number was normalized to RpL32 (also known as Rp49) copy number using primers from reference [89].
RNA-Seq samples
For samples from ovaries, flies were kept at 27°C for several generations prior to and during the experiment. Freshly eclosed females were collected and aged 2–3 days and then transferred to fresh food with yeast paste for another 2–3 days. RNA was extracted, from ovaries dissected in chilled 1× PBS, using Trizol. Ovarian mRNA-Seq libraries were constructed at the Epigenomics Core Facility at Weill Cornell Medical College using the poly(A) enrichment method. Libraries were sequenced using the Illumina HiSeq2000 platform to produce 50 bp single reads which were then trimmed for quality and filtered to remove rRNA reads. One biological sample each from LhrKO and Lhr+ was duplexed and run in a single lane. 51,193,832 filtered reads were obtained for Lhr+ and 41,688,028 reads for LhrKO. Three biological replicates each of D. simulans w501 and Lhr1 ovarian mRNA libraries were run on a single lane and the number of filtered reads ranged from 36,472,726 to 43,449,879. For experiments with Hmr, two biological replicates were included for each genotype and all 8 samples were multiplexed in a single lane. The number of filtered reads for each sample ranged from 23,863,381 to 27,490,644. For larval samples, around 30 larvae were collected for each genotype and flash frozen in liquid N2. RNA was extracted from 2 biological replicates of each genotype using Trizol. Larval RNAseq libraries were generated and bar-coded using the TruSeq kit, and run in one lane of an Illumina HiSeq 2000 100 bp yielding 13,707,247 to 20,373,267 filtered reads per sample, except for one library which produced only 7,840,004 reads.
RNA-Seq analysis
Reads mapping to either rRNA or repetitive DNA were filtered out using Bowtie [91] and the filtered reads were mapped to the unmasked D. melanogaster genome using Tophat [92]. The BAM file outputs were used by Cuffdiff with the -b option [93]. All *.fasta and *.gtf files were based on the release 5.68 of the D. melanogaster genome from ENSEMBL. To find differentially expressed genes in D. simulans, we aligned reads to the D. melanogaster genome with Tophat, allowing two mis-matches. While this approach could potentially reduce mapping ability for diverged genes, it allowed us to take advantage of the better assembly and annotation of the D. melanogaster genome.
To maximize the TEs considered in our analyses, we mapped reads to two different databases using Bowtie. First, reads were uniquely mapped to a database consisting of all the annotated TE insertions in the D. melanogaster and D. simulans genomes [48]; we refer to this as the individual-insertion database. While this database likely represents most TE families present in our stocks, some TEs may either be absent from the assembled genome or be represented by copies that are sufficiently diverged such that they impact our ability to correctly assess transcript levels. These elements include the telomeric element TAHRE, which has only a few insertions in the genome and is known to be absent from the reference genome since only two telomeres are included in the assembly [94]. Therefore we also mapped reads, allowing for either 0 mismatches when aligning reads from D. melanogaster or 3 mismatches when aligning reads from D. simulans or hybrids, to a database consisting of the consensus sequences of the annotated TEs and repeats found in Repbase as well as de novo predicted TEs generated by piler-DF using the 12 Drosophila genomes [48]; we refer to this as the consensus-sequence database. Only reads that mapped uniquely within the same family were included in the subsequent analyses of differential expression. Mismatches allowed for each alignment are mentioned in figure legends. Statistical significance of differential expression among TEs was calculated with F.E.T. in the DEG-seq package [95].
To analyze reads mapping to satellite DNAs, we built a database using a curated file from the Berkeley Drosophila Genome Project (http://www.fruitfly.org/sequence/sequence_db/na_re.dros) which itself was constructed from GenBank sequences. This file includes some mis-annotated TEs and non-satellite sequences. We counted reads that mapped to these repeats without any mismatches and calculated statistical significance of differential expression among satellites with F.E.T. in the DEG-seq package.
Small RNA sequencing and analysis
Libraries were prepared as described but no oxidation was carried out [38]. Briefly, total RNA was extracted from 5–6 day old LhrKO and Lhr+ ovaries using the mirVANA kit (Invitrogen). Total RNA was size fractionated on a 15% Urea-PAGE gel to enrich for 18–29 nt small RNA, excised and eluted and then subjected to 2S rRNA depletion. This small RNA was ligated to a 3′ RNA adapter, gel purified, and then ligated to a 5′ DNA adapter. The adapter-ligated small RNAs were reverse transcribed and PCR amplified. The amplified PCR products were gel purified, quantified and sequenced in two lanes of a HISeq 2000 machine.
Only reads with a 3′ adapter were kept, which was then removed using a custom script [48]. These reads were binned by size as either miRNA/siRNA (17–22 nt) or piRNA (23–30 nt). rRNA, tRNA and snoRNA sequences were filtered from these reads and the remaining reads were further filtered to keep only those reads that mapped to either the unmasked genome,or the satellite DNA database described above, or Repbase consensus sequences [96]. These filtered reads included 89,953,149 piRNA reads and 40,859,119 siRNA reads in LhrKO, and 120,143,855 piRNA reads and 36,388,192 siRNA reads in Lhr+.
piRNA reads were mapped uniquely to all D. melanogaster sequences from Repbase using Bowtie, allowing for one mismatch. Ping-Pong scores were calculated using reads mapped with up to 1 mismatch, as described in reference [48]. For mapping to piRNA clusters, we built an index using sequences extracted from the Release 5 DM3 genome on the UCSC genome database and GenBank with coordinates of individual piRNA clusters obtained from reference [41]. piRNA reads were uniquely mapped to piRNA clusters with zero mismatches and significance for differential expression was calculated using F.E.T implemented in DEG-seq. siRNA reads were mapped uniquely to all D. melanogaster sequences from Repbase with Bowtie, without allowing for any mismatches.
Immuno-fluorescence and Immuno-FISH
Immunofluorescence and FISH were performed on embryos and ovaries as described in references [4], [83]. Polytene chromosomes were dissected in 0.7% NaCl, squashed, and fixed in 1.8% PFA, 45% acetic acid for 17 minutes. They were then washed in 1% Triton X in PBS for 10 minutes, then washed in 5% milk in PBS for 1 hour, incubated with primary antibody overnight at 4°C, washed in 5% milk in PBS for 10 minutes, incubated with secondary antibody for 1 hour at room temperature, and then washed for 10 minutes in buffer A (0.15M NaCl, 0.2% NP40 substitute, 0.2%Tween 20) followed by 10 minutes in buffer B (0.20M NaCl, 0.2% NP40 substitute, 0.2%Tween 20).
Rat anti-HA antibody (Roche, 3F10) was used at 1∶100, rat anti-Vasa (DSHB) was used at 1∶25, Fibrillarin (Abcam, Ab5281) was used at 1∶100, anti-HP1a antibody (C1A9, DSHB) was used at 1∶100. Alexa fluorophore-conjugated secondary antibodies were used to detect the primary antibody. Fluorescently labeled probes against GA-rich satellites, AACAC, 2L3L, 359 bp and dodeca were obtained from Sigma with sequences described in references [8], [83], [97]. Imaging was carried out using a Zeiss 710 confocal microscope at Cornell University's Microscopy and Imaging Facility.
Yeast two-hybrid assays
A full-length coding-sequence plasmid of D. melanogaster Hmr was made by correcting 3 frame-shift errors in the RE54143 cDNA [3]. Two errors in exon 5 were replaced by ligating in a ∼1.6 kb XbaI-HindIII fragment from the LD22117 cDNA, followed by replacement of a 2172 bp NdeI-ZraI fragment from the p83 genomic clone [3]. The coding sequence was then PCRd out and cloned into pENTR/D-TOPO. The D. simulans Hmr CDS was PCRd out of cDNA and cloned into pENTR/D-TOPO. The Lhr plasmids and yeast two-hybrid destination vectors and assays are described in reference [6].
Data access
Illumina sequence data from this study are available from the NCBI website under BioProject number PRJNA236022.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MaheshwariS, BarbashDA (2011) The genetics of hybrid incompatibilities. Annu Rev Genet 45 : 331–355.
2. PresgravesDC (2010) The molecular evolutionary basis of species formation. Nat Rev Genet 11 : 175–180.
3. BarbashDA, SiinoDF, TaroneAM, RooteJ (2003) A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc Natl Acad Sci U S A 100 : 5302–5307.
4. ArunaS, FloresHA, BarbashDA (2009) Reduced fertility of Drosophila melanogaster Hybrid male rescue (Hmr) mutant females is partially complemented by Hmr orthologs from sibling species. Genetics 181 : 1437–1450.
5. TangS, PresgravesDC (2009) Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science 323 : 779–782.
6. BrideauNJ, FloresHA, WangJ, MaheshwariS, WangX, BarbashDA (2006) Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science 314 : 1292–1295.
7. BarbashDA, AwadallaP, TaroneAM (2004) Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol 2: e142.
8. MaheshwariS, BarbashDA (2012) Cis-by-Trans regulatory divergence causes the asymmetric lethal effects of an ancestral hybrid incompatibility gene. PLoS Genet 8: e1002597.
9. BrideauNJ, BarbashDA (2011) Functional conservation of the Drosophila hybrid incompatibility gene Lhr. BMC Evol Biol 11 : 57.
10. WatanabeTK (1979) A gene that rescues the lethal hybrids between Drosophila melanogaster and D. simulans. Jpn J Genet 54 : 325–331.
11. HutterP, AshburnerM (1987) Genetic rescue of inviable hybrids between Drosophila melanogaster and its sibling species. Nature 327 : 331–333.
12. GreilF, de WitE, BussemakerHJ, van SteenselB (2007) HP1 controls genomic targeting of four novel heterochromatin proteins in Drosophila. EMBO J 26 : 741–751.
13. Lohe A, Roberts P (1988) Evolution of satellite DNA sequences in Drosophila. In: Verma RS, editors. Heterochromatin, Molecular and Structural Aspects. Cambridge: Cambridge Univ. Press. pp. 148–186.
14. BoscoG, CampbellP, Leiva-NetoJT, MarkowTA (2007) Analysis of Drosophila species genome size and satellite DNA content reveals significant differences among strains as well as between species. Genetics 177 : 1277–1290.
15. GregoryTR (2005) Synergy between sequence and size in large-scale genomics. Nat Rev Genet 6 : 699–708.
16. CharlesworthB, SniegowskiP, StephanW (1994) The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371 : 215–220.
17. HickeyDA (1982) Selfish DNA: a sexually-transmitted nuclear parasite. Genetics 101 : 519–531.
18. WalkerPM (1971) Origin of satellite DNA. Nature 229 : 306–308.
19. BlumenstielJP (2011) Evolutionary dynamics of transposable elements in a small RNA world. Trends Genet 27 : 23–31.
20. KhuranaJS, TheurkaufW (2010) piRNAs, transposon silencing, and Drosophila germline development. J Cell Biol 191 : 905–913.
21. UsakinL, AbadJ, VaginVV, de PablosB, VillasanteA, GvozdevVA (2007) Transcription of the 1.688 satellite DNA family is under the control of RNA interference machinery in Drosophila melanogaster ovaries. Genetics 176 : 1343–1349.
22. LeeYCG, LangleyCH (2012) Long-term and short-term evolutionary impacts of transposable elements on Drosophila. Genetics 192 : 1411–1432.
23. JohnsonNA (2010) Hybrid incompatibility genes: remnants of a genomic battlefield? Trends Genet 26 : 317–325.
24. LeratE, BurletN, BiémontC, VieiraC (2011) Comparative analysis of transposable elements in the melanogaster subgroup sequenced genomes. Gene 473 : 100–109.
25. AndreyevaEN, BelyaevaES, SemeshinVF, PokholkovaGV, ZhimulevIF (2005) Three distinct chromatin domains in telomere ends of polytene chromosomes in Drosophila melanogaster Tel mutants. J Cell Sci 118 : 5465–5477.
26. MeffordHC, TraskBJ (2002) The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet 3 : 91–102.
27. AndersonJA, GillilandWD, LangleyCH (2009) Molecular population genetics and evolution of Drosophila meiosis genes. Genetics 181 : 177–185.
28. RaffaGD, CiapponiL, CenciG, GattiM (2011) Terminin: a protein complex that mediates epigenetic maintenance of Drosophila telomeres. Nucleus 2 : 383–391.
29. ZwickME, SalstromJL, LangleyCH (1999) Genetic variation in rates of nondisjunction: association of two naturally occurring polymorphisms in the chromokinesin nod with increased rates of nondisjunction in Drosophila melanogaster. Genetics 152 : 1605–1614.
30. GiotL, BaderJS, BrouwerC, ChaudhuriA, KuangB, et al. (2003) A protein interaction map of Drosophila melanogaster. Science 302 : 1727–1736.
31. BhaskarV, CoureyAJ (2002) The MADF-BESS domain factor Dip3 potentiates synergistic activation by Dorsal and Twist. Gene 299 : 173–184.
32. PerratPN, DasGuptaS, WangJ, TheurkaufW, WengZ, et al. (2013) Transposition-driven genomic heterogeneity in the Drosophila brain. Science 340 : 91–95.
33. PardueM-L, DebarysheP (2011) Adapting to life at the end of the line: How Drosophila telomeric retrotransposons cope with their job. Mob Genet Elements 1 : 128–134.
34. LoheAR, HillikerAJ, RobertsPA (1993) Mapping simple repeated DNA sequences in heterochromatin of Drosophila melanogaster. Genetics 134 : 1149–1174.
35. PlateroJS, CsinkAK, QuintanillaA, HenikoffS (1998) Changes in chromosomal localization of heterochromatin-binding proteins during the cell cycle in Drosophila. J Cell Biol 140 : 1297–1306.
36. BarichevaEA, BerriosM, BogachevSS, BorisevichIV, LapikER, et al. (1996) DNA from Drosophila melanogaster beta-heterochromatin binds specifically to nuclear lamins in vitro and the nuclear envelope in situ. Gene 171 : 171–176.
37. VaginVV, SigovaA, LiC, SeitzH, GvozdevV, ZamorePD (2006) A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313 : 320–324.
38. LiC, VaginVV, LeeS, XuJ, MaS, et al. (2009) Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137 : 509–521.
39. MaloneCD, BrenneckeJ, DusM, StarkA, McCombieWR, et al. (2009) Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137 : 522–535.
40. DönertasD, SienskiG, BrenneckeJ (2013) Drosophila Gtsf1 is an essential component of the Piwi-mediated transcriptional silencing complex. Genes Dev 27 : 1693–1705.
41. BrenneckeJ, AravinAA, StarkA, DusM, KellisM, et al. (2007) Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128 : 1089–1103.
42. BlumenstielJP, HartlDL (2005) Evidence for maternally transmitted small interfering RNA in the repression of transposition in Drosophila virilis. Proc Natl Acad Sci U S A 102 : 15965–15970.
43. CzechB, MaloneCD, ZhouR, StarkA, SchlingeheydeC, et al. (2008) An endogenous small interfering RNA pathway in Drosophila. Nature 453 : 798–802.
44. RozhkovNV, AravinAA, ZelentsovaES, SchostakNG, SachidanandamR, et al. (2010) Small RNA-based silencing strategies for transposons in the process of invading Drosophila species. RNA 16 : 1634–1645.
45. KoflerR, BetancourtAJ, SchlöttererC (2012) Sequencing of pooled DNA samples (Pool-Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster. PLoS Genet 8: e1002487.
46. BergmanCM, BensassonD (2007) Recent LTR retrotransposon insertion contrasts with waves of non-LTR insertion since speciation in Drosophila melanogaster. Proc Natl Acad Sci U S A 104 : 11340–11345.
47. RebolloR, LeratE, KleineLL, BiémontC, VieiraC (2008) Losing helena: the extinction of a drosophila line-like element. BMC Genomics 9 : 149.
48. KelleherES, EdelmanNB, BarbashDA (2012) Drosophila interspecific hybrids phenocopy piRNA-pathway mutants. PLoS Biol 10: e1001428.
49. EissenbergJC, ElginSC (2000) The HP1 protein family: getting a grip on chromatin. Curr Opin Genet Dev 10 : 204–210.
50. SavitskyM, KravchukO, MelnikovaL, GeorgievP (2002) Heterochromatin protein 1 is involved in control of telomere elongation in Drosophila melanogaster. Mol Cell Biol 22 : 3204–3218.
51. WangSH, ElginSCR (2011) Drosophila Piwi functions downstream of piRNA production mediating a chromatin-based transposon silencing mechanism in female germ line. Proc Natl Acad Sci U S A 108 : 21164–21169.
52. EissenbergJC, JamesTC, Foster-HartnettDM, HartnettT, NganV, et al. (1990) Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc Natl Acad Sci U S A 87 : 9923–9927.
53. ThomaeAW, SchadeGOM, PadekenJ, BorathM, VetterI, et al. (2013) A Pair of Centromeric Proteins Mediates Reproductive Isolation in Drosophila Species. Dev Cell 27 (4) 412–24.
54. ZhangY, MaloneJH, PowellSK, PeriwalV, SpanaE, et al. (2010) Expression in aneuploid Drosophila S2 cells. PLoS Biol 8: e1000320.
55. SawamuraK (2012) Chromatin evolution and molecular drive in speciation. Int J Evol Biol 2012 : 301894.
56. DoolittleWF (2013) Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A 110 : 5294–5300.
57. PetrovDA (2002) Mutational equilibrium model of genome size evolution. Theor Popul Biol 61 : 531–544.
58. ShepelevVA, AlexandrovAA, YurovYB, AlexandrovIA (2009) The evolutionary origin of man can be traced in the layers of defunct ancestral alpha satellites flanking the active centromeres of human chromosomes. PLoS Genet 5: e1000641.
59. WerrenJH (2011) Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A 108 Suppl 2 : 10863–10870.
60. Bregliano J-C, Kidwell MG (1983) Hybrid dysgenesis determinants. In: Mob Genet Elements. Academic Press, New York.
61. FishmanL, SaundersA (2008) Centromere-associated female meiotic drive entails male fitness costs in monkeyflowers. Science 322 : 1559–1562.
62. GonzálezJ, PetrovDA (2012) Evolution of genome content: population dynamics of transposable elements in flies and humans. Methods Mol Biol 855 : 361–383.
63. LeeYCG, LangleyCH (2010) Transposable elements in natural populations of Drosophila melanogaster. Philos Trans R Soc Lond B Biol Sci 365 : 1219–1228.
64. KlenovMS, SokolovaOA, YakushevEY, StolyarenkoAD, MikhalevaEA, et al. (2011) Separation of stem cell maintenance and transposon silencing functions of Piwi protein. Proc Natl Acad Sci U S A 108 : 18760–18765.
65. YamanakaS, MehtaS, Reyes-TurcuFE, ZhuangF, FuchsRT, et al. (2013) RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature 493 : 557–560.
66. VillasanteA, AbadJP, PlanellóR, Méndez-LagoM, CelnikerSE, de PablosB (2007) Drosophila telomeric retrotransposons derived from an ancestral element that was recruited to replace telomerase. Genome Res 17 : 1909–1918.
67. Shpiz S, Kalmykova A (2012) Control of Telomere Length in Drosophila. In: Li, B, editor. Review of Selected Topic of Telomere Biology. Rijeka: InTech. pp. 33–56.
68. RanganP, MaloneCD, NavarroC, NewboldSP, HayesPS, et al. (2011) piRNA Production Requires Heterochromatin Formation in Drosophila. Curr Biol 21 (16) 1373–9.
69. SageBT, CsinkAK (2003) Heterochromatic self-association, a determinant of nuclear organization, does not require sequence homology in Drosophila. Genetics 165 : 1183–1193.
70. FerreePM, PrasadS (2012) How can satellite DNA divergence cause reproductive isolation? Let us count the chromosomal ways. Genet Res Int 2012 : 430136.
71. BonaccorsiS, GattiM, PisanoC, LoheA (1990) Transcription of a satellite DNA on two Y chromosome loops of Drosophila melanogaster. Chromosoma 99 : 260–266.
72. HeB, CaudyA, ParsonsL, RosebrockA, PaneA, et al. (2012) Mapping the pericentric heterochromatin by comparative genomic hybridization analysis and chromosome deletions in Drosophila melanogaster. Genome Res
73. GranzottoA, LopesFR, VieiraC, CararetoCMA (2011) Vertical inheritance and bursts of transposition have shaped the evolution of the BS non-LTR retrotransposon in Drosophila. Mol Genet Genomics 286 : 57–66.
74. CsinkAK, McDonaldJF (1995) Analysis of copia sequence variation within and between Drosophila species. Mol Biol Evol 12 : 83–93.
75. DowsettAP, YoungMW (1982) Differing levels of dispersed repetitive DNA among closely related species of Drosophila. Proc Natl Acad Sci U S A 79 : 4570–4574.
76. YasuharaJC, WakimotoBT (2006) Oxymoron no more: the expanding world of heterochromatic genes. Trends Genet 22 : 330–338.
77. CastilloDM, MoyleLC (2012) Evolutionary Implications of Mechanistic Models of TE-Mediated Hybrid Incompatibility. Int J Evol Biol 2012 : 698198.
78. KerkisJ (1933) Development of gonads in hybrids between Drosophila melanogaster and Drosophila simulans. J Exper Zool 66 : 477–509.
79. KawamuraY, SaitoK, KinT, OnoY, AsaiK, et al. (2008) Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 453 : 793–797.
80. GhildiyalM, SeitzH, HorwichMD, LiC, DuT, et al. (2008) Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 320 : 1077–1081.
81. BarbashDA, RooteJ, AshburnerM (2000) The Drosophila melanogaster Hybrid male rescue gene causes inviability in male and female species hybrids. Genetics 154 : 1747–1771.
82. OrrHA, IrvingS (2000) Genetic analysis of the Hybrid male rescue locus of Drosophila. Genetics 155 : 225–231.
83. FerreePM, BarbashDA (2009) Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol 7: e1000234.
84. GongWJ, GolicKG (2004) Genomic deletions of the Drosophila melanogaster Hsp70 genes. Genetics 168 : 1467–1476.
85. HernanR, HeuermannK, BrizzardB (2000) Multiple epitope tagging of expressed proteins for enhanced detection. Biotechniques 28 : 789–793.
86. GrothAC, FishM, NusseR, CalosMP (2004) Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166 : 1775–1782.
87. VenkenKJT, HeY, HoskinsRA, BellenHJ (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 : 1747–1751.
88. BarbashDA, LoriganJG (2007) Lethality in Drosophila melanogaster/Drosophila simulans species hybrids is not associated with substantial transcriptional misregulation. J Exp Zool B Mol Dev Evol 308 : 74–84.
89. PaneA, WehrK, SchüpbachT (2007) zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Dev Cell 12 : 851–862.
90. KlenovMS, LavrovSA, StolyarenkoAD, RyazanskySS, AravinAA, et al. (2007) Repeat-associated siRNAs cause chromatin silencing of retrotransposons in the Drosophila melanogaster germline. Nucleic Acids Res 35 : 5430–5438.
91. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
92. TrapnellC, PachterL, SalzbergSL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25 : 1105–1111.
93. TrapnellC, WilliamsBA, PerteaG, MortazaviA, KwanG, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28 : 511–515.
94. GeorgeJA, DeBaryshePG, TraverseKL, CelnikerSE, PardueM-L (2006) Genomic organization of the Drosophila telomere retrotransposable elements. Genome Res 16 : 1231–1240.
95. WangL, FengZ, WangX, WangX, ZhangX (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26 : 136–138.
96. JurkaJ, KapitonovVV, PavlicekA, KlonowskiP, KohanyO, WalichiewiczJ (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110 : 462–467.
97. Dernburg AF (2000) In situ hybridization to somatic chromosomes. In: Drosophila protocols. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. pp. 22–55.
98. SmithCD, EdgarRC, YandellMD, SmithDR, CelnikerSE, et al. (2007) Improved repeat identification and masking in Dipterans. Gene 389 : 1–9.
99. KaminkerJS, BergmanCM, KronmillerB, CarlsonJ, SvirskasR, et al. (2002) The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol 3: RESEARCH0084.
100. RiddleNC, MinodaA, KharchenkoPV, AlekseyenkoAA, SchwartzYB, et al. (2011) Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res 21 : 147–163.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle
- Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion
- Genetic Dissection of Photoreceptor Subtype Specification by the Zinc Finger Proteins Elbow and No ocelli
- GC-Rich DNA Elements Enable Replication Origin Activity in the Methylotrophic Yeast
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy