
Genetic Determinants Influencing Human Serum Metabolome among African Americans
Most contemporary GWAS studies have achieved increased power by increasing the size of the discovery sample to tens of thousands of individuals. An alternative approach for detecting the effects of novel loci is to measure phenotypes that more immediately reflect the effects of gene function. The metabolome consists of a collection of small molecules resulting from a variety of cellular and biologic processes, which can be considered intermediate phenotypes proximal to gene function. Here, we report a genome-wide association study identifying nineteen genetic loci influencing untargeted metabolomes traits among African Americans in the Atherosclerosis Risk in Communities (ARIC) Study. Fourteen genes mapped within nineteen loci, including twelve enzyme-encoding genes (KLKB1, SIAE, CPS1, NAT8, ACE, GATM, ACY3, ACSM2B, THEM4, ADH4, UGT1A and TREH), a transporter gene (SLC6A13) and a polycystin protein gene (PKD2L1). In addition, four potential disease-associated paths were identified, including two direct longitudinal predictive relationships: NAT8 with N-acetylornithine, N-acetyl-1-methylhistidine and incident chronic kidney disease, and TREH with trehalose and incident diabetes. These results highlight the value of using phenotypes proximal to gene function to promote novel gene discovery.
Published in the journal:
. PLoS Genet 10(3): e32767. doi:10.1371/journal.pgen.1004212
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004212
Summary
Most contemporary GWAS studies have achieved increased power by increasing the size of the discovery sample to tens of thousands of individuals. An alternative approach for detecting the effects of novel loci is to measure phenotypes that more immediately reflect the effects of gene function. The metabolome consists of a collection of small molecules resulting from a variety of cellular and biologic processes, which can be considered intermediate phenotypes proximal to gene function. Here, we report a genome-wide association study identifying nineteen genetic loci influencing untargeted metabolomes traits among African Americans in the Atherosclerosis Risk in Communities (ARIC) Study. Fourteen genes mapped within nineteen loci, including twelve enzyme-encoding genes (KLKB1, SIAE, CPS1, NAT8, ACE, GATM, ACY3, ACSM2B, THEM4, ADH4, UGT1A and TREH), a transporter gene (SLC6A13) and a polycystin protein gene (PKD2L1). In addition, four potential disease-associated paths were identified, including two direct longitudinal predictive relationships: NAT8 with N-acetylornithine, N-acetyl-1-methylhistidine and incident chronic kidney disease, and TREH with trehalose and incident diabetes. These results highlight the value of using phenotypes proximal to gene function to promote novel gene discovery.
Introduction
The power to detect genetic effects for complex traits is influenced by, among other things, the study sample size and the effect size of a particular locus. Most contemporary genome-wide association studies (GWAS) have achieved increased power by increasing the size of the discovery sample to tens of thousands of individuals [1]. Besides expanding the sample size, focusing on variants with large effects is an alternative strategy for novel gene discovery. The human metabolome consists of a collection of small molecules resulting from a variety of cellular and biologic processes, the activity of which is regulated by coordinated enzyme action [2]. In addition, as metabolites reflect multiple metabolic and physiological activities in the body, they hold promise to discover intermediate traits between gene action and disease processes [3].
GWASs of known risk factor phenotypes of clinical disease, such as cholesterol or urate levels, have shown that genetic association with functional intermediate traits, as opposed to the clinical endpoint itself, are often more highly powered and may provide information into the biological mechanism of disease [4]–[7]. Untargeted metabolomic approaches simultaneously measure numerous known and unknown metabolites present in a study sample. Recent studies combining genetics and metabolomics have identified multiple common variant-metabolite associations with large effect sizes in populations of European ancestry, and provided new functional insights into common complex disease. [8]–[11]. African ancestry-derived populations have higher levels of genetic variation and population substructure, and lower levels of linkage disequilibrium (LD) compared to European ancestry-derived populations, so studies in African-Americans may lead to identification of new genes or variants and fine map of existing loci [12]–[14]. To date, no such study has been conducted in African Americans, a population that bears a disproportionate burden of disease, such as cardiovascular disease, diabetes and chronic kidney disease [15]–[17]. Our goal here is to identify common genetic variations influencing the human metabolome in African Americans among the Atherosclerosis Risk in Communities (ARIC) Study in order to reveal novel pathways underlying disease etiology and possible avenues of disease prevention and treatment.
Results
A total of 308 known serum metabolites including 83 amino acids, 16 carbohydrates, 9 cofactors and vitamins, 7 energies, 136 lipids, 12 nucleotides, 25 peptides and 20 xenobiotics (Table S1) were included and a set of 2,341,704 common autosomal SNPs were tested in 1,260 African Americans (demographics in Table S2) for each metabolite levels. Nineteen significant (p-value<1.6×10−10 after correction for multiple testing) common variant-metabolite associations were identified (locus association summaries are presented in Table 1, regional association plots and quantile-quantile plots are presented in Figures S1 and S2, respectively), including 13 novel loci which have not been reported in previous metabolomics studies. Depending on the particular metabolite, these loci were associated with 7–50% of the difference in metabolite levels per allele (average at 25%), and the variance explained ranged from 4% to 20%.
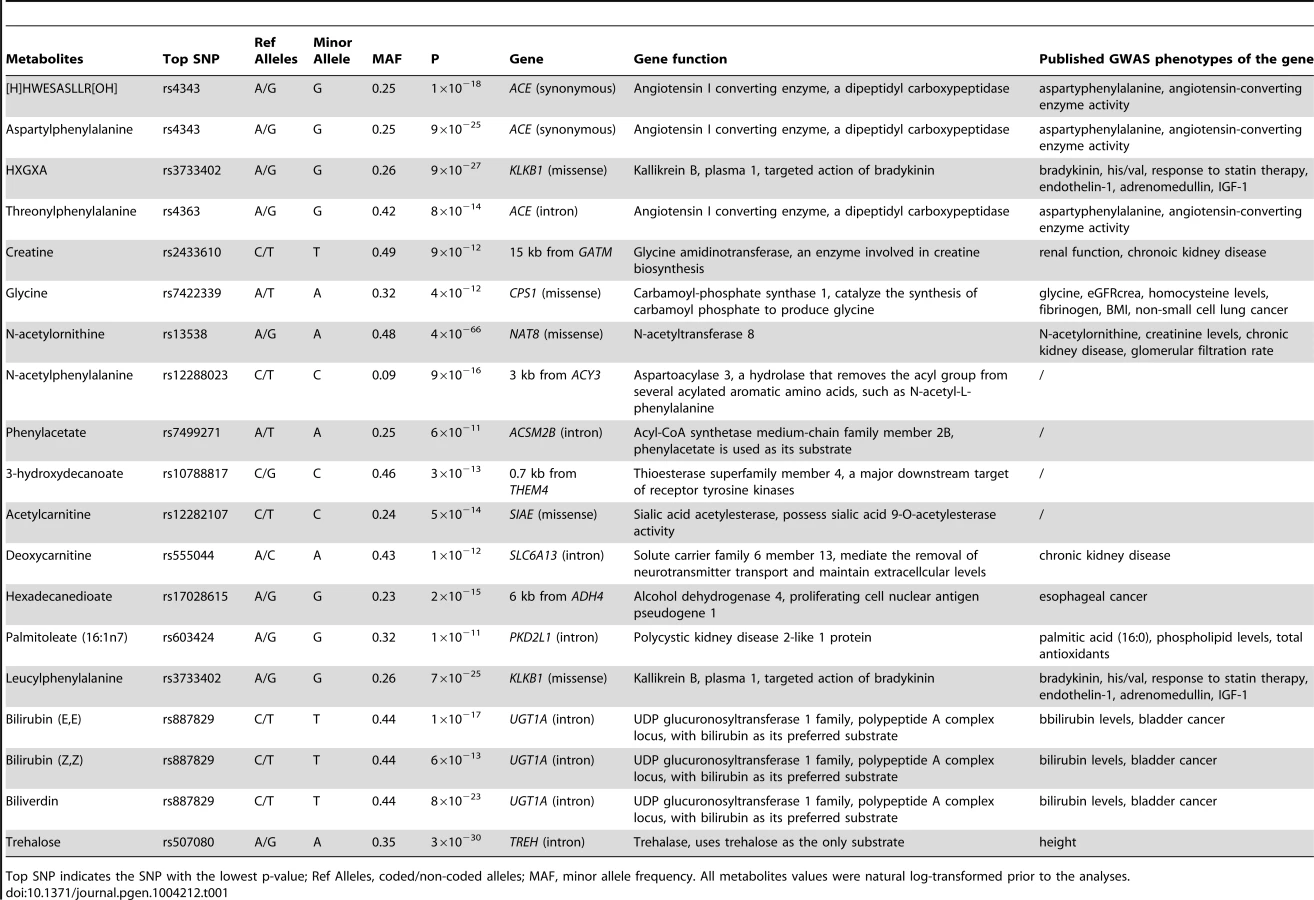
Fourteen genes were mapped within the nineteen significant genetic loci; eight of them encode enzymes that catalyze the reaction of the corresponding metabolite as a substrate or product (gene names shown in red in Figure 1). Four of the associated loci contained non-synonymous substitutions in four enzyme-encoding genes (KLKB1, SIAE, CPS1, and NAT8). The other significant loci consist of eight other enzyme-encoding genes (ACE, GATM, ACY3, ACSM2B, THEM4, ADH4, UGT1A, and TREH), a transporter gene (SLC6A13) and a polycystin protein gene (PKD2L1). Two protease-encoding genes, ACE and KLKB1, showed pleiotropic effects on multiple oligopeptide metabolites, and the UDP-glucuronosyltransferases gene, UGT1A, contributed to the levels of several bile pigments (Figure 1).

Nineteen significant common variant-metabolite associations were compared with previously published SNP-metabolite associations in Caucasians [10]. Eleven out of nineteen metabolites were shared between the published study and the data presented here, and six of them showed the same significant SNP-metabolite associations in both ethnicities (Table 2). A CPS1-glycine association was reported in the Caucasion metabolomic GWAS, but the sentinel SNP was different (r2<0.5) from that reported here (Table 2). A CPS1-glycine association was also reported in a recent genetic study for glycine metabolism among Caucasians [18]. The other four shared metabolites had different signals in African-Americans when compared to Caucasians (Table S3).

We identified a missense mutation in NAT8 (rs13538) that was significantly associated with N-acetylornithine levels (p = 4.0×10−66). A recent biochemical study has shown that NAT8 catalyzed the N-acetylation of cysteine conjugates [19]. We next asked whether the presumed specificity of NAT8's function could be used to identify the identity of any unknown metabolites by analyzing its effect on 294 unknown metabolites. Two metabolites, X-11333 and X-11787 reached our a priori defined level of significance (p = 1.0×10−61 and p = 2.5×10−25, respectively). By targeted mass spectroscopy, X-11333 was determined to be N-acetyl-1-methylhistidine (Figure S3), a type of N-acetyl amino acid; and X-11787 was an isoform of either hydroxy leucine or isoleucine, as reported previously [20].
Among nineteen metabolites that reached genome-wide significance, we identified four potential disease-associated paths among African Americans for cardiovascular disease, chronic kidney disease (CKD) and diabetes, including two direct longitudinal associations (Figure 2, detailed estimates in Table S4). As described above, a missense mutation in NAT8 (rs13538), a known susceptibility locus for chronic kidney disease [21], was significantly associated with N-acetylornithine and N-acetyl-1-methylhistidine levels. We identified a pronounced relationship of both N-acetylornithine and N-acetyl-1-methylhistidine levels with kidney function, whereby higher levels of of N-acetylornithine and N-acetyl-1-methylhistidine were related to lower eGFR (p = 9.0×10−13 and 1.6×10−21; respectively) and higher risk of incident CKD after 19 average years of follow-up among 1,921 African Americans (demographics in Table S5, HR = 1.64, p = 0.003 and HR = 1.34, p = 0.03, respectively). However, the longitudinal associations with the metabolites were attenuated and no longer significant after further adjusting for eGFR (data not shown). Finally, trehalose levels were significantly associated with TREH gene variation. Trehalose can be cleaved to two molecules of glucose. In this study, trehalose levels were significantly associated with glucose levels (p = 2.9×10−17), and showed a 1.34 fold increased risk of incident diabetes after an average 7 years of follow-up (p = 2.0×10−5) in a sample of 1,430 ARIC African Americans (demographics in Table S5). With further adjustment of glucose levels, trehalose levels persisted to show an apparent association with incident diabetes, although the effect size was lessened (HR = 1.16, p = 0.02).

Discussion
By combining high-throughput metabolomic and genomic technologies, we identified nineteen common variant-metabolite associations among African Americans with p-values ranging from 6.0×10−11 to 4.0×10−66. We inferred the structure of an unknown metabolite to be N-acetyl-1-methylhistidine using knowledge of the associated gene's function and targeted mass spectroscopy. We further established potential novel disease-associated pathways for cardiovascular disease risk factors, CKD and diabetes. The results offer new evidence about the genetic impact on metabolites and disease among African Americans, which advance our understanding of disease causation and progression.
Most loci identified by GWA studies of complex disease traits contribute relatively small effects and the variance explained remains modest [14], [22], [23]. Thus, contemporary GWAS are shifting focus to phenotypes that more immediately reflect the effects of gene action. For example, although the effect sizes of genetic loci related to coronary heart disease (CHD) are relatively small (OR from 1.08 to 1.47) [24]–[26], loci related to plasma triglyceride and cholesterol levels explained a meaningful proportion of the variance (9–13%) [4]. The human metabolome, the ultimate downstream product of gene and environment interaction, holds the promise to identify genes that directly reflect gene action with large effects sizes [8], [10], [27]. Our results show relatively large effect sizes of nineteen identified genetic loci related to human metabolome among African Americans (average at 25% shift per allele copy). In addition, the majority of identified loci (15/19) are located in or near genes, and these loci explained up to 20% of the variance of each trait.
Twelve out of fourteen genes that were significantly associated with metabolite levels were enzyme-encoding genes, including four genes involved in disease-associated processes. These data underscore the important role of enzyme activity and regulation in controlling metabolite levels. As metabolite levels are closely related to disease process, to understand whether the underlying mutations detected here lead to gain-of-function or loss-of-function for these enzyme-encoding genes offers new opportunities for disease treatment and prevention (e.g. design an antagonist/agonist of the gene as a drug candidate). The majority of the gene-metabolite associations are consistent with the gene's known function, but the direction of effect of the coded allele does not provide direct evidence as to whether or not the variant represents gain of function or loss of function. Future investigation of the functional impact of the underlying causal variants is critical and is an area of intense research.
NAT8 is expressed mainly in the kidney and liver [28], but its function is not fully understood. Several previous, seemingly unrelated, observations have found that mutations in N-acetyltransferase 8 (NAT8), are contributed to N-acetylornithine levels, creatinine levels, kidney function and CKD [10], [21], [29], [30]. Our results show that an amino acid substitution in NAT8 is related to N-acetylornithine, N-acetyl-1-methylhistidine and eGFR, which in-turn influence risk to incident CKD. These findings provide evidences that N-acetylation plays a role in the development of CKD [10].
Trehalose is a food ingredient with the ability to prevent protein denaturation [31]. Because of its ability to inhibit lipid and protein misfolding, trehalose has become a potential therapeutic in neurodegenerative studies [32], [33]. Animal safety studies concluded that trehalose is safe for use as an ingredient in consumer products [34], and it is now widely used in food and cosmetics. Here, we report that trehalose levels are regulated by TREH, which encodes the trehalase enzyme which hydrolyzes trehalose to two glucose molecules. In addition, we show that trehalose is associated with glucose levels and the onset of incident diabetes.
Environment factors, in addition to and interacting with genetic factors, (e.g. dietary intake) explain part of the variability of human metabolome. Follow-up investigations of the interactions between the genes identified here and possible environment factors are likely to provide new insight into the understanding of disease etiology and its metabolism. For example, alcohol dehydrogenase 4 (ADH4) contributes to esophageal squamous-cell carcinoma (ESCC) through an interaction with alcohol consumption [35]. Here, we reported that ADH4 is associated with hexadecanedioate levels, a metabolite with an antitumor activity [36]. Moreover, studies have shown that coffee consumption is associated with lower bilirubin levels [37] and UGT1A is contributed to bilirubin levels as well [10]. Our data show that mutations in UGT1A are associated with the levels of several bile pigments. Thus, future investigations of genes related to metabolite levels with environment interaction are of interest.
Untargeted metabolomics approaches measure numerous known and unknown metabolites presented in a sample simultaneously. Since the chemical identities for unknown metabolites have not been elucidated, previous GWAS on metabolomic traits largely ignored unknown metabolites for the analysis. In our study, we show an example of unknown metabolite identification (i.e. X-11333) by combining GWAS results (i.e. NAT8) with existing knowledge about the function of the gene product (i.e. N - acetylation). A recent study has used GWAS results and Gaussian graphical modeling to predict unknown metabolite identities [38]. These two examples demonstrate the feasibility for unknown compounds structure identification by combing genetic and metabolomics information.
Limitation of this study warrants consideration. To our knowledge, the ARIC study is the only cohort with serum metabolome measurements in African-Americans, so it is unlikely to find an independent sample for replication. In our study, the SNP-metabolite associations identified were compared with the results from a published study in Caucasians [10] as a surrogate replication. Six distinct SNP-metabolite associations were replicated out of eleven shared metabolites, indicating homogeneous genetic effects on several metabolites regardless of ethnicities. Differences in the site frequency spectrum between African-Americans and Caucasians and lower LD in African-Americans may explain the lack of significant association at the other loci. As a consequence of lack of replication, the proportion of variance explained by the SNPs was reported from the discovery sample, which may be an over-estimate. Future studies are needed to replicate our findings in independent samples of African-Americans. Despite limitations, the data presented here have important strength. Previously published GWAS on human metabolites estimate only cross-sectional relationships between metabolites and clinical endpoints. In contrast, the data presented here originate from a large, well-defined, longitudinal cohort study, allowing establishments of longitudinal predictive relationships.
In summary, we report here the first genome-wide association study of untargeted metabolome in African-Americans. The genetic variant-metabolite associations along with the disease path reported here will continue to be improved with further use of contemporary omics technologies. Our study highlights the value of utilizing omics studies in deeply phenotyped individuals to provide new insights into gene function, disease etiology and epidemiology.
Methods
Study Population
The Atherosclerosis Risk in Communities (ARIC) study is a longitudinal cohort study designed to ascertain the etiology and predictors of cardiovascular disease (CVD). The ARIC study enrolled 15,792 middle-aged adults from four U.S. communities (Forsyth County, NC; Jackson, MS; suburbs of Minneapolis, MN; and Washington County, MD) between 1987–89 and followed by four completed visits with each approximately three years apart, in 1987–89, 1990–92, 1993–95, and 1996–98. In general, each visit included interviews and a physical examination. A detailed description of the ARIC study design and methods was published elsewhere [39]. Metabolomic profiles were measured in baseline serum from 1,977 African-Americans selected from the Jackson, MS field center. Participants were excluded if they did not give consent for use of DNA information.
Assessment of Metabolomic Profiles
Metabolite profiling was completed in June 2010 using fasting serum samples which had been stored at −80° since collection at the baseline examination in 1987–1989. In total, detection and quantification of 602 metabolites was completed by Metabolon Inc. (Durham, USA) using an untargeted, gas chromatography-mass spectrometry and liquid chromatography-mass spectrometry (GC-MS and LC-MS)-based metabolomic quantification protocol [40], [41]. Prior to the analyses presented here, a rigorous assessment of the metabolomic data was done. Metabolites were excluded if: 1) more than 50% of the samples had values below the detection limit; or 2) they had unknown chemical structures, except for X-11333 and X-11787 which were followed-up as part of more detailed NAT8 investigations. After this assessment, a total of 308 named metabolites were included in the present study. Structural identifications for X-11333 and X-11787 were proposed using a mass spec-based structural approach, including targeted accurate mass and MSn fragmentation with accurate mass [41].
Genotyping and Imputation
In the present study, common (minor allele frequency, MAF≥5%) autosomal single-nucleotide polymorphisms (SNPs) were genotyped on the Affymetrix 6.0 chip and were imputed to 2,341,704 SNPs based on a panel of cosmopolitan reference haplotypes from HapMap CEU and YRI. MACH v1.0 was used to do imputation and allele dosage information was summarized in the imputation results. SNPs were excluded before imputation if they had no chromosomal location, were monomorphic, had a call rate <95%, or had a Hardy-Weinberg equilibrium p-value<10−5. For each SNP, the ratio of the observed versus expected variance of the dosage served as a measure of imputation quality.
Genome-Wide Association Analyses
A total of 308 metabolites were included in this analysis. Metabolite levels below the detectable limit of the assay were imputed with the lowest detected value for that metabolite in all samples, and all metabolites values were natural log-transformed prior to the analyses. Linear regressions and an additive genetic model were applied to each metabolite, adjusting for age, sex and the first 10 principal components. The significant threshold was defined as a p-value<1.6×10−10 (5.0×10−8/308) based on Bonferroni correction. SNPs with MAF<5% were excluded. Quantile-quantile (QQ) plots were generated for each analysis to illustrate the distribution of the observed and expected p-values for all eligible SNPs. Regional plots showing LD and the location of nearby genes (if any) were generated for the top ranking SNPs for each metabolite. If more than one significant SNP clustered at a locus, the SNP with the smallest p-value was reported as the sentinel marker. All analyses were performed using ProbABEL and R (www.r-project.org). The identified sentinel SNPs were further compared with the metabolite-SNP association from the KORA and TwinsUK studies [10] using their public GWAS server (http://metabolomics.helmholtz-muenchen.de/gwa/index.html) and other published GWA studies through NHGRI GWAS Catalog (http://www.genome.gov/gwastudies/).
Disease Association Analyses
Analyses included all African-American samples with metabolomic data were conducted to estimate the association between genome-wide significant metabolite levels and relevant clinical risk factors and endpoints, including incident chronic kidney disease and incident type 2 diabetes. Nine associations, including six cross-sectional associations with clinical risk factors and three longitudinal associations with clinical endpoints, were tested. In each analysis, metabolite levels were natural log-transformed. The cross-sectional associations were assessed using linear regression with adjustment for age and gender. Longitudinal associations with disease endpoints were estimated using Cox proportional hazards models adjusting for age, gender, systolic blood pressure (SBP), antihypertensive medication use, diabetes, high-density lipoprotein, low-density lipoprotein, current smoking and prevalent CHD for incident the CKD analysis; and age, gender, SBP, antihypertensive medication use, body mass index, total cholesterol for the incident type 2 diabetes analysis. The proportional hazards assumption was examined and not rejected using the methods developed by Grambsch and Therneau [42]. Covariates were measured at baseline (1987–1989) and The Chronic Kidney Disease Epidemiology Collaboration equation was applied to estimate glomerular filtration rate (eGFRCKD-EPI) [43]. For the disease association analyses, the significant threshold was defined as p<0.005 using Bonferroni correction (0.05/9) and the analyses were performed using R (www.r-project.org).
Supporting Information
Zdroje
1. PsatyBM, O'DonnellCJ, GudnasonV, LunettaKL, FolsomAR, et al. (2009) Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet 2 : 73–80.
2. GermanJB, HammockBD, WatkinsSM (2005) Metabolomics: building on a century of biochemistry to guide human health. Metabolomics 1 : 3–9.
3. SuhreK, GiegerC (2012) Genetic variation in metabolic phenotypes: study designs and applications. Nat Rev Genet 13 : 759–769.
4. TeslovichTM, MusunuruK, SmithAV, EdmondsonAC, StylianouIM, et al. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466 : 707–713.
5. SurakkaI, WhitfieldJB, PerolaM, VisscherPM, MontgomeryGW, et al. (2012) A genome-wide association study of monozygotic twin-pairs suggests a locus related to variability of serum high-density lipoprotein cholesterol. Twin Res Hum Genet 15 : 691–699.
6. DupuisJ, LangenbergC, ProkopenkoI, SaxenaR, SoranzoN, et al. (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42 : 105–116.
7. KottgenA, AlbrechtE, TeumerA, VitartV, KrumsiekJ, et al. (2013) Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet 45 : 145–154.
8. GiegerC, GeistlingerL, AltmaierE, Hrabe de AngelisM, KronenbergF, et al. (2008) Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 4: e1000282.
9. SuhreK, WallaschofskiH, RafflerJ, FriedrichN, HaringR, et al. (2011) A genome-wide association study of metabolic traits in human urine. Nat Genet 43 : 565–569.
10. SuhreK, ShinSY, PetersenAK, MohneyRP, MeredithD, et al. (2011) Human metabolic individuality in biomedical and pharmaceutical research. Nature 477 : 54–60.
11. KettunenJ, TukiainenT, SarinAP, Ortega-AlonsoA, TikkanenE, et al. (2012) Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet 44 : 269–276.
12. FrazerKA, BallingerDG, CoxDR, HindsDA, StuveLL, et al. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449 : 851–861.
13. CampbellMC, TishkoffSA (2008) African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu Rev Genomics Hum Genet 9 : 403–433.
14. ManolioTA, CollinsFS, CoxNJ, GoldsteinDB, HindorffLA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461 : 747–753.
15. GoAS, MozaffarianD, RogerVL, BenjaminEJ, BerryJD, et al. (2013) Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127: e6–e245.
16. Tarver-CarrME, PoweNR, EberhardtMS, LaVeistTA, KingtonRS, et al. (2002) Excess risk of chronic kidney disease among African-American versus white subjects in the United States: a population-based study of potential explanatory factors. J Am Soc Nephrol 13 : 2363–2370.
17. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention.
18. XieW, WoodAR, LyssenkoV, WeedonMN, KnowlesJW, et al. (2013) Genetic variants associated with glycine metabolism and their role in insulin sensitivity and type 2 diabetes. Diabetes 62 : 2141–2150.
19. Veiga-da-CunhaM, TytecaD, StroobantV, CourtoyPJ, OpperdoesFR, et al. (2010) Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J Biol Chem 285 : 18888–18898.
20. ZhengY, YuB, AlexanderD, ManolioTA, AguilarD, et al. (2013) Associations between metabolomic compounds and incident heart failure among African Americans: the ARIC Study. Am J Epidemiol 178 : 534–542.
21. KottgenA, PattaroC, BogerCA, FuchsbergerC, OldenM, et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat Genet 42 : 376–384.
22. HindorffLA, SethupathyP, JunkinsHA, RamosEM, MehtaJP, et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106 : 9362–9367.
23. ParkJH, GailMH, WeinbergCR, CarrollRJ, ChungCC, et al. (2011) Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc Natl Acad Sci U S A 108 : 18026–18031.
24. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 : 661–678.
25. SamaniNJ, ErdmannJ, HallAS, HengstenbergC, ManginoM, et al. (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357 : 443–453.
26. ErdmannJ, GrosshennigA, BraundPS, KonigIR, HengstenbergC, et al. (2009) New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 41 : 280–282.
27. IlligT, GiegerC, ZhaiG, Romisch-MarglW, Wang-SattlerR, et al. (2010) A genome-wide perspective of genetic variation in human metabolism. Nat Genet 42 : 137–141.
28. OzakiK, FujiwaraT, NakamuraY, TakahashiE (1998) Isolation and mapping of a novel human kidney - and liver-specific gene homologous to the bacterial acetyltransferases. J Hum Genet 43 : 255–258.
29. ChambersJC, ZhangW, LordGM, van der HarstP, LawlorDA, et al. (2010) Genetic loci influencing kidney function and chronic kidney disease. Nat Genet 42 : 373–375.
30. TinA, ColantuoniE, BoerwinkleE, KottgenA, FranceschiniN, et al. (2013) Using multiple measures for quantitative trait association analyses: application to estimated glomerular filtration rate. J Hum Genet 58 : 461–6.
31. JainNK, RoyI (2009) Effect of trehalose on protein structure. Protein Sci 18 : 24–36.
32. TanakaM, MachidaY, NiuS, IkedaT, JanaNR, et al. (2004) Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10 : 148–154.
33. DaviesJE, SarkarS, RubinszteinDC (2006) Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet 15 : 23–31.
34. RichardsAB, KrakowkaS, DexterLB, SchmidH, WolterbeekAP, et al. (2002) Trehalose: a review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem Toxicol 40 : 871–898.
35. WuC, KraftP, ZhaiK, ChangJ, WangZ, et al. (2012) Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat Genet 44 : 1090–1097.
36. YouYJ, KimY, NamNH, BangSC, AhnBZ (2004) Alkyl and carboxylalkyl esters of 4′-demethyl-4-deoxypodophyllotoxin: synthesis, cytotoxic, and antitumor activity. Eur J Med Chem 39 : 189–193.
37. CasigliaE, SpolaoreP, GinocchioG, AmbrosioGB (1993) Unexpected effects of coffee consumption on liver enzymes. Eur J Epidemiol 9 : 293–297.
38. KrumsiekJ, SuhreK, EvansAM, MitchellMW, MohneyRP, et al. (2012) Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet 8: e1003005.
39. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol 129 : 687–702.
40. OhtaT, MasutomiN, TsutsuiN, SakairiT, MitchellM, et al. (2009) Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol Pathol 37 : 521–535.
41. EvansAM, DeHavenCD, BarrettT, MitchellM, MilgramE (2009) Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem 81 : 6656–6667.
42. GrambschPM, TherneauTM (1994) Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 81 : 515–526.
43. LeveyAS, StevensLA, SchmidCH, ZhangYL, CastroAF3rd, et al. (2009) A new equation to estimate glomerular filtration rate. Ann Intern Med 150 : 604–612.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Worldwide Patterns of Ancestry, Divergence, and Admixture in Domesticated Cattle
- Genome-Wide DNA Methylation Analysis of Human Pancreatic Islets from Type 2 Diabetic and Non-Diabetic Donors Identifies Candidate Genes That Influence Insulin Secretion
- Genetic Dissection of Photoreceptor Subtype Specification by the Zinc Finger Proteins Elbow and No ocelli
- GC-Rich DNA Elements Enable Replication Origin Activity in the Methylotrophic Yeast
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy