
WNK1/HSN2 Mutation in Human Peripheral Neuropathy Deregulates Expression and Posterior Lateral Line Development in Zebrafish ()
Hereditary sensory and autonomic neuropathy type 2 (HSNAII) is a rare pathology characterized by an early onset of severe sensory loss (all modalities) in the distal limbs. It is due to autosomal recessive mutations confined to exon “HSN2” of the WNK1 (with-no-lysine protein kinase 1) serine-threonine kinase. While this kinase is well studied in the kidneys, little is known about its role in the nervous system. We hypothesized that the truncating mutations present in the neural-specific HSN2 exon lead to a loss-of-function of the WNK1 kinase, impairing development of the peripheral sensory system. To investigate the mechanisms by which the loss of WNK1/HSN2 isoform function causes HSANII, we used the embryonic zebrafish model and observed strong expression of WNK1/HSN2 in neuromasts of the peripheral lateral line (PLL) system by immunohistochemistry. Knocking down wnk1/hsn2 in embryos using antisense morpholino oligonucleotides led to improper PLL development. We then investigated the reported interaction between the WNK1 kinase and neuronal potassium chloride cotransporter KCC2, as this transporter is a target of WNK1 phosphorylation. In situ hybridization revealed kcc2 expression in mature neuromasts of the PLL and semi-quantitative RT–PCR of wnk1/hsn2 knockdown embryos showed an increased expression of kcc2 mRNA. Furthermore, overexpression of human KCC2 mRNA in embryos replicated the wnk1/hsn2 knockdown phenotype. We validated these results by obtaining double knockdown embryos, both for wnk1/hsn2 and kcc2, which alleviated the PLL defects. Interestingly, overexpression of inactive mutant KCC2-C568A, which does not extrude ions, allowed a phenocopy of the PLL defects. These results suggest a pathway in which WNK1/HSN2 interacts with KCC2, producing a novel regulation of its transcription independent of KCC2's activation, where a loss-of-function mutation in WNK1 induces an overexpression of KCC2 and hinders proper peripheral sensory nerve development, a hallmark of HSANII.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003124
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003124
Summary
Hereditary sensory and autonomic neuropathy type 2 (HSNAII) is a rare pathology characterized by an early onset of severe sensory loss (all modalities) in the distal limbs. It is due to autosomal recessive mutations confined to exon “HSN2” of the WNK1 (with-no-lysine protein kinase 1) serine-threonine kinase. While this kinase is well studied in the kidneys, little is known about its role in the nervous system. We hypothesized that the truncating mutations present in the neural-specific HSN2 exon lead to a loss-of-function of the WNK1 kinase, impairing development of the peripheral sensory system. To investigate the mechanisms by which the loss of WNK1/HSN2 isoform function causes HSANII, we used the embryonic zebrafish model and observed strong expression of WNK1/HSN2 in neuromasts of the peripheral lateral line (PLL) system by immunohistochemistry. Knocking down wnk1/hsn2 in embryos using antisense morpholino oligonucleotides led to improper PLL development. We then investigated the reported interaction between the WNK1 kinase and neuronal potassium chloride cotransporter KCC2, as this transporter is a target of WNK1 phosphorylation. In situ hybridization revealed kcc2 expression in mature neuromasts of the PLL and semi-quantitative RT–PCR of wnk1/hsn2 knockdown embryos showed an increased expression of kcc2 mRNA. Furthermore, overexpression of human KCC2 mRNA in embryos replicated the wnk1/hsn2 knockdown phenotype. We validated these results by obtaining double knockdown embryos, both for wnk1/hsn2 and kcc2, which alleviated the PLL defects. Interestingly, overexpression of inactive mutant KCC2-C568A, which does not extrude ions, allowed a phenocopy of the PLL defects. These results suggest a pathway in which WNK1/HSN2 interacts with KCC2, producing a novel regulation of its transcription independent of KCC2's activation, where a loss-of-function mutation in WNK1 induces an overexpression of KCC2 and hinders proper peripheral sensory nerve development, a hallmark of HSANII.
Introduction
Hereditary sensory and autonomic neuropathies (HSAN) are rare inherited neuropathies predominantly characterized by sensory dysfunction associated with variable degrees of autonomous and motor involvement. HSANs were first classified in five distinct types according to clinical presentation of symptoms as well as age of onset and mode of inheritance 1. These distinct categories were later confirmed by identification of causative mutations by genome linkage studies, revealing heterogeneity amongst HSAN types both clinically and genetically. HSAN type 2 (HSANII, OMIM#201300) is of autosomal recessive inheritance and is characterized by an early onset sensory neuropathy, causing patients to lack all sensory modalities in a strictly peripheral glove-and-stocking distribution leading to a diagnosis in the first two decades of life 2. Other characteristics include a loss of tendon reflex, skin ulceration, Charcot joint, and spontaneous amputations while excluding motor involvement 3,4,5. In addition, upon sural nerve biopsy in affected patients, a reduction in the number of myelinated fibers is observed as well as a slight decrease in the number of non-myelinated fibers 6,7. In the absence of evidence suggesting degenerative changes in the peripheral nerves, HSANII is believed to be non-progressive and has been argued as being due to improper development 5. Despite there being published cases of HSANII since the last century, the mechanism leading to this disorder is still not understood.
Mutations restricted to an intron within the WNK1 (lysine deficient protein kinase 1) gene were found to be responsible for HSANII (location 12p13.33, gene/locus OMIM #605232). This sequence was at first attributed to a new gene-within-a-gene and named ‘HSN2’ for hereditary sensory neuropathy type 2 8 but it was later revealed to be an alternatively spliced exon of the serine/threonine kinase WNK1, nestled between exon 8 and 9 of the 28 exon gene 9. WNK1 (NCBI Gene ID: 65125; HGCN:14540) is one of four members of the with-no-lysine (K) kinases, characteristic among other serine/threonine kinases by a uniquely placed lysine involved in ATP binding. Each WNK member contains a well-conserved kinase domain and multiple coiled-coil domains as well as a host of proline-rich regions and potential SH3 domain binding-sites, pointing to an involvement in protein complex formation and modulation of signaling 10. A large section of the WNK1 gene, including exon HSN2, has no reported motifs suggesting any particular function. However, the isoform including the HSN2 exon, termed WNK1/HSN2, has been found to be selectively expressed in the nervous system whereas other isoforms of the WNK1 kinase are quite ubiquitously expressed in the CNS and other tissues 9,10. Within the neuron, WNK1/HSN2 is also sublocalized differently being found in the axon and cell body while isoforms lacking the HSN2 exon are confined to the cell body 9.
All mutations in the HSN2 exon reported to date are loss-of-function mutations, producing an early terminated mRNA which leads to a truncated protein 7,11. While a WNK1 knockout has proved to be lethal in mouse embryos, suggesting an important role in early development, selective knockout of the HSN2 exon has not been attempted thus far 12. The functions of WNK1 in the nervous system are not well understood. This kinase has been reported to interact with KCC2, potassium-chloride cotransporter type 2 (SLC12A5 gene for ‘solute carrier family 12, (potassium-chloride transporter) member 5’, NCBI Gene ID: 57468, HGCN:13818; 13, which is selectively expressed in neurons and participates in the regulation of the chloride gradient. WNK1 phosphorylation of KCC2 occurs in immature neurons but is absent in adult neurons 13,14, emphasizing a developmental role. Interestingly, KCC2 has been shown to regulate neurogenesis in the zebrafish (Danio rerio) spinal cord 15,16, which suggests it may also play a role in peripheral neurogenesis. We therefore used this simple model to investigate whether WNK1 is implicated, perhaps via KCC2, in the development of the peripheral nervous system of zebrafish.
Results
Wnk1 And Wnk1/hsn2 Are Expressed Throughout Embryonic Development
To investigate whether loss-of-function mutations in WNK1/HSN2 led to improper development of the peripheral nervous system, we used the zebrafish as it is a well-established model that is ideal for developmental biology since the first day post-fertilization roughly corresponds to the first trimester of mammalian development 17. It is also a model that has proven efficient in the study of functional genomics and pathogenesis of neurodegenerative disorders, with a relatively simple nervous system eliciting stereotyped responses 17,18,19,20. We first identified the zebrafish orthologs of the WNK1 kinase. Two separate loci were identified, named wnk1a (NCBI Gene ID: 100318736, ZFIN ID: ZDB-GENE-080917-49, chromosome 25) and wnk1b (NCBI Gene ID: 561159, ZFIN ID: ZDB-GENE-030131-2656, chromosome 4). These two genes were confirmed via Ensembl (Ensembl : ENSDARG00000078992). Only wnk1b conserves the HSN2 target exon (Figure 1A) and wnk1a appears to also be missing exons 11, 20, 21 and 22. Both copies have a split exon 10, and exons 11 to 13 of wnk1b are fused, which also appears in the Xenopus laevis ortholog sequence.

We next examined the developmental expression pattern of wnk1b. As the expression of the WNK1/HSN2 isoform had previously been assessed by Western blot in adult mouse tissue only 9, and there was no data available for its expression during embryogenesis. We first obtained an mRNA expression profile for both wnk1a and wnk1b by RT-PCR for zebrafish from the 16 cell stage to 7 days post-fertilization (dpf) (Figure 1B). Both orthologs were expressed early on with wnk1b expression increasing during the first few days whereas wnk1a expression was high from the start and maintained. The presence of the wnk1 and its wnk1/hsn2 isoform at the 16 cell stage (1.5 hpf) likely corresponds to a maternal transcript which leads early development prior to transcription of the zygotic genome at 3.5 hpf 21,22.
To localize the specific wnk1/hsn2 isoform within the nervous system, we performed whole-mount immunohistochemistry on 4 dpf zebrafish embryos using the previously described anti-HSN2 antibody 9. This revealed localization of the wnk1/hsn2 isoform (transcribed from the wnk1b gene) at the level of the posterior lateral line (PLL) neuromasts (Figure 1C) and not in the spinal cord. The wnk1/hsn2 protein was found within the two major neuromast cell types: hair cells and the support cells (inset, Figure 1C). This localization is consistent with HSANII to the extent that the neuropathy affects the peripheral sensory system and that the PLL is a peripheral mechanosensory system, albeit specific to aquatic animals.
Knockdown Of Wnk1/hsn2 Perturbs Posterior Lateral Line Formation
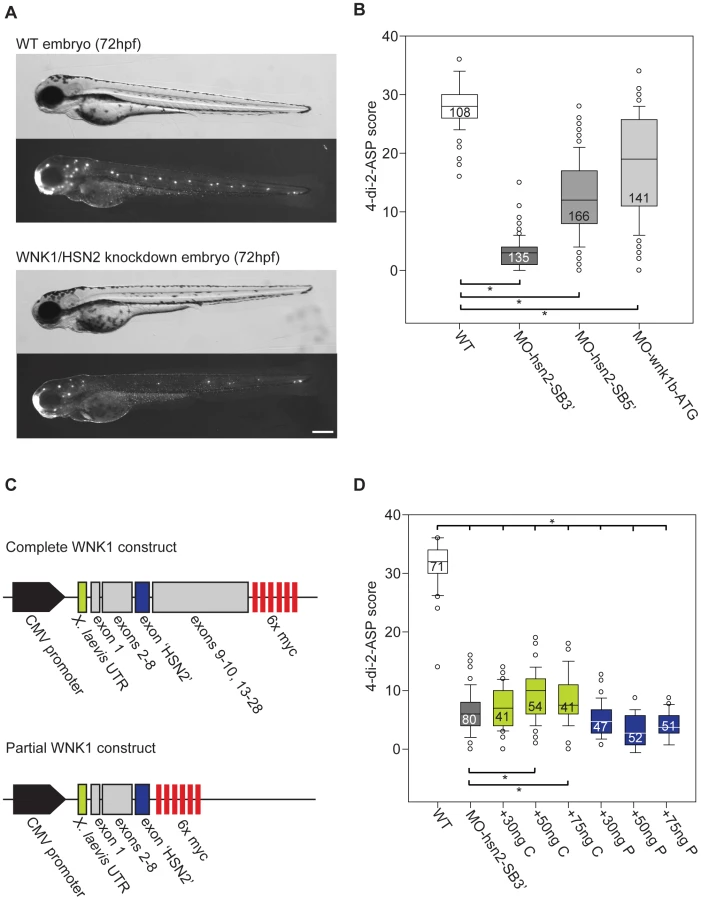
In order to replicate the pathogenic loss-of-function of the WNK1/HSN2 isoform linked with HSANII causative mutations, we designed antisense morpholino oligonucleotides (AMO) targeting the start codon of wnk1b (AMO targets, Figure 1A). We also designed AMOs targeting the splice junction sites of exon hsn2 of the wnk1b gene, MO-hsn2-SB5′ and MO-hsn2-SB3′, respectively targeting the splice donor and splice acceptor sites. As the wnk1/hsn2 protein was detected by immunohistochemistry at the level of the PLL, we started by observing this mechanosensory system upon knockdown. Knockdown embryos for all three conditions were morphologically indistinguishable from non-injected animals at 72 hpf but staining of the lateral line with the fluorescent vital dye 4-di-2-ASP revealed defects in the formation of the PLL (Figure 2A). In order to quantify this phenotype, we attributed a score to each PLL neuromast depending on fluorescence to account for both their presence and composition, as was done previously 23. The scores were attributed accordingly: Full, fluorescent neuromast = 2 points; smaller or dim neuromast = 1 point; absent neuromast = 0 point. As the data was non-parametric, medians values were used to compare groups. In the wild-type non-injected fish (PLL neuromasts mid-body to tail, n = 108 embryos) we obtained a median value of 28.0 for the 4-di-2-ASP score. All three knockdowns, although with varying efficiency, revealed a significantly lower score, with median values of 3.0, 12.0 and 19.0 for MO-hsn2-SB3′ (n = 135 embryos), MO-hsn2-SB5′ (n = 166 embryos) and MO-wnk1b-ATG (n = 141 embryos) respectively, when compared with wild-type embryos (one-way ANOVA with Dunn's multiple comparison, Figure 2B). We further confirmed the specificity of the knockdown phenotype by rescuing it with wild-type human WNK1 mRNA. Two constructs were assembled for the human sequence: a complete construct spanning exons 1 to 28 (but skipping small exons 11 and 12 which were unavailable) and a partial construct composed only of exons 1 to HSN2 (i.e. lacking exons 9 to 28, Figure 2C) tested over a range of concentrations (Figure S1). The complete construct was co-injected with the most efficient AMO, namely MO-hsn2-SB3′ and significantly alleviated the PLL defect phenotype, without however bringing it back to wild-type level, at concentrations of 50 and 75 ng/µl (Figure 2D green boxes). The partial construct proved unable to rescue the knockdown phenotype when co-injected at similar concentrations with the AMO and thus confirmed the predicted loss-of-function of WNK1 following HSANII truncating mutations in the HSN2 exon 7,11 (Figure 2D blue boxes).
To further characterize the defects in PLL formation, we examined the structure of individual neuromasts by knocking down wnk1/hsn2 in transgenic embryos expressing GFP under the Xenopus laevis neuron-specific beta-tubulin promoter Tg(NBT:MAPT-GFP) 24 which allowed us to visualize structural hair cells based on the presence of beta-tubulin as revealed by expression of GFP (Figure 3A). An effort was made to count all PLL neuromasts of observed embryos, disregarding terminal neuromasts found at the tip of the tail because they arise from fragmentation of the primordium at the end of migration, and not by deposition of pro-neuromasts along the dorsal midline 25. The number of structural hair cells per PLL neuromast for all three types of wnk1/hsn2 knockdown embryos was significantly lower than for non-injected transgenic embryos, with median values of 4.0, 6.0 and 5.0 hair cells per neuromast for MO-hsn2-SB3′ (n = 80 neuromasts, 38 embryos), MO-hsn2-SB5′ (n = 73 neuromasts, 19 embryos) and MO-wnk1b-ATG (n = 56 neuromasts, 16 embryos) respectively, when compared with wild-type embryos which had a median value of 10.0 hair cells per neuromast (n = 106 neuromasts, 20 embryos; non-parametric distributions, one-way ANOVA with Dunn's multiple comparison; Figure 3B). We then confirmed this decrease by looking at the number of functional hair cells revealed by the vital styryl dye FM-464FX 26. Knockdown for wnk1/hsn2 was obtained in transgenic embryos expressing GFP under the claudin-b promoter Tg(-8.0cldnb:lynEGFP), which allows membrane labeling of primordium cells as well as neuromast hair cells and support cells 27. The knockdown and non-injected transgenic embryos were then incubated in FM-464FX and observed under fluorescence, where whole neuromasts would be seen in green (GFP) and hair cells, in red fluorescence (FM-464FX) (Figure 3C). While the number of support cells (labeled only in green) did not seem to decrease, the number of functional hair cells per neuromast decreased in a similar fashion to what had been observed for structural hair cells, where knockdown embryos had median values of 0.0, 3.0 and 2.0 hair cells per neuromast for MO-hsn2-SB3′ (n = 21 neuromasts, 9 embryos), MO-hsn2-SB5′ (n = 69 neuromasts, 16 embryos) and MO-wnk1b-ATG (n = 62 neuromasts, 12 embryos) respectively, when compared with non-injected embryos which had an median value of 8.0 hair cells per neuromast (n = 85 neuromasts, 15 embryos; non-parametric distributions, one-way ANOVA with Dunn's multiple comparison; (Figure 3D). The PLL defect phenotype thus seemed to leave support cells unaffected, suggesting a problem in neural maturation with only the hair-cell-fated neuromast progenitors failing to become functional.

Wnk1/hsn2 Knockdown Embryos Overexpress Kcc2
The activity of the neuronal-specific KCC2 had recently been shown to be regulated by the WNK1 kinase, where phosphorylation decreased KCC2 activation 14. Because of this, we predicted the knockdown phenotype could increase the activity of the cotransporter, as it is usually downregulated by WNK1 kinases. In zebrafish, it has previously been shown that KCC2 (slc12a5 gene, Ensembl: ENSDARG00000078187, ZFIN ID: ZDB-GENE-080707-1) becomes expressed in parallel with neuronal maturation. Its delayed expression allows a timely reversal of the chloride gradient and is essential for appropriate neuronal differentiation 15. In the absence of an antibody detecting kcc2 specifically in zebrafish, we examined mRNA levels by RT - PCR. At 72 hpf, when the PLL defect phenotype is visible in wnk1/hsn2 knockdown embryos, we indeed found a higher expression of slc12a5 (Figure 4A). To determine if this overexpression was also a premature expression, which has been found to cause dendritic spine defects 28, we also looked at mRNA levels at 24 hpf and found early overexpression, shown for the most effective knockdown condition (Figure 4A).

Human Kcc2 Overexpression Replicates The Pll Phenotype
To confirm that KCC2 is implicated in the wnk1/hsn2 PLL phenotype, we overexpressed human KCC2 mRNA in WT embryos as was previously described 15. At 72 hpf, embryos showed a normal morphology, though some animals had a shorter tail (Figure 4B). Upon labeling of the PLL with 4-di-2-ASP, we observed defects similar to the ones of wnk1/hsn2 knockdown embryos and confirmed a decrease in hair cell number, both structurally (Figure 4C) and functionally (Figure 4D, black boxes) upon overexpression of KCC2.
If the increase in slc12a5 expression (coding for kcc2) following knockdown of wnk1b is indeed responsible for the loss of neuromasts, then we reasoned that knockdown of both slc12a5 and wnk1b should rescue the phenotype. First we examined the effect of knockdown of slc12a5 on its own using a previously described AMO (MO1-slc12a5, 29) for which knockdown is viable but leads to embryos with altered morphology, often exhibiting a shorter tail and curved spine (Figure 4B). Nonetheless, upon 4-di-2-ASP staining, these embryos had a structurally sound PLL, though with fewer neuromasts, which was probably due to their shorter length (score relative to WT, Figure 4D). Finally, we tested co-knockdown of wnk1b and slc12a5 and observed a partial rescue of the PLL defect phenotype, as visualized with 4-di-2-ASP (Figure 4D, green boxes), confirming that the KCC2 cotransporter is implicated in the establishment of the wnk1/hsn2 knockdown PLL phenotype.
Kcc2 In The Embryonic Posterior Lateral Line
As the presence of kcc2 has never been assessed in the zebrafish nervous system, we performed an in situ hybridization against slc12a5, revealing its expression in the hindbrain, in the rostral spinal cord and in neuromasts of 4dpf embryos (Figure 5A). Prior to kcc2 functional expression in the early zebrafish embryo, the chloride gradient is depolarizing due to the high chloride content 30. As a result, brief glycine application depolarizes the cells and evokes a Ca2+ transient 31. In contrast, we expected that if kcc2 is functionally expressed in neuromasts, the chloride content in its cells will be low and application of glycine will fail to evoke Ca2+ transients. We therefore loaded neuromasts with the Ca2+ indicator Rhod-2 AM and visualized their hair cells in 3–4 dpf transgenic Tg(tub:MAPT-GFP) embryos expressing GFP in axons. We observed that application of glycine onto these neuromasts (Figure 5C) failed to evoke Ca2+ transients (as shown in Figure 5B top image and traces; n = 4 neuromasts) whereas glutamate application did (Figure 5B middle image and traces), indicating that these neuromast cells were viable but unresponsive to glycine presumably due to the presence of kcc2 and a low intracellular chloride level. In contrast, application of glycine onto the primordium of 2dpf embryos visualized in Tg(-8.0cldnb:lynEGFP) 27 expressing GFP under the claudin-b promoter in cells composing the migrating primordium, progenitors of PLL neuromasts, and co-labeled with Rhod-2 AM evoked clear calcium transients (Figure 5B bottom image and traces; 6 cells in 2 primordia). This observation suggests a high chloride content in neuromast progenitor cells, the migrating primordium, much like the observations in spinal cord progenitors of equivalent stage zebrafish embryos 15,32. In summary, the lack of glycine evoked Ca2+ transients in neuromasts contrasted to their presence in the primordium and suggests that, as for spinal cord progenitors, kcc2 expression in neuromasts is functional and could be implicated in neural differentiation. This corroborates our previous results, showing an implication of KCC2 in WNK1/HSN2 knockdown phenotype, affecting the PLL.

Wnk1/hsn2 Knockdown Affects Pll Progenitors
Previous experiments in zebrafish reported that overexpression of KCC2 leads to impaired neurogenesis by perturbing neuronal maturation 15,16. KCC2 has also been reported to be involved in mammalian neural development as a premature overexpression disrupts development of the neural tube by diminishing neuronal differentiation, leading to mouse embryos with a thinner neural tube and abnormal body curvature 33. To characterize the effect of KCC2 overexpression on PLL progenitors, we injected Tg(-8.0cldnb:lynEGFP) embryos and observed the primordium in live embryos in order to assess its size after departure from the cephalic placode but before deposition of the first pro-neuromast. It was not possible to count each individual cell as the primordium is a highly motile structure that is constantly reorganizing its cells. In an effort to quantify the observed effect, we embedded live animals in agarose while positioning them on their side, allowing us an optimal view of the primordium. We then captured images and measured the surface area of the primordium as revealed by GFP expression. WNK1/HSN2 knockdown embryos, as well as embryos overexpressing human KCC2, showed a significantly smaller primordium (Figure 5D). As the size of primordium cells and organization seemed conserved for all conditions (close-up, Figure 5C), we conclude that an overexpression of KCC2, whether it be by injection or induced by wnk1/hsn2 knockdown, resulted in a lower number of PLL progenitors. While the size of the primordium was reduced both in wnk1/hsn2 knockdown and in KCC2 overexpressing embryos, the organization and size of primordium cells seemed to be conserved (Figure 5C).
Pll Defects Are Caused Independently Of Kcc2 Transporter Activity
KCC2 has been shown to have a role independent of its transporter activity, for example by influencing the development of dendritic spines through interaction with cytoskeleton protein 4.1 N 33,34, where loss 35 or premature expression 28 of KCC2 respectively induced abnormal morphology (lower number of functional spines) and an increase in dendritic spine density. Furthermore, phosphorylated KCC2 is found in neurons before the GABAergic response switch 36 and the implication of KCC2 in neuronal differentiation of the embryonic mouse neural tube was also shown to be independent of KCC2 activation and therefore independent of its chloride extruding function 33.
To determine whether the PLL defect phenotype resulting from KCC2 overexpression was due to its transporter function as a modulator of intracellular chloride concentration, we synthesized RNA for KCC2-C568A as this dominant-negative mutation has been shown to impair the chloride extruding function of the cotransporter and was used successfully as a control in the zebrafish spinal cord model 15. Here we show that overexpression of this inactive KCC2 mutant impairs proper PLL development in a similar manner to wild-type KCC2, as observed by 4-di-2-ASP staining (score, Figure 4E). We therefore suggest that loss of WNK1/HSN2 leads to an overexpression of KCC2 by a novel mechanism to regulate its transcription and that this overexpression impairs proper peripheral nervous system development in an activity-independent manner.
Discussion
In this study, we show that WNK1/HSN2 truncating mutations associated with HSANII lead to a loss-of-function of this kinase isoform which causes developmental defects in a relevant structure in zebrafish, by impairing PLL formation. By immunohistochemistry, we showed localization of this isoform in the neuromasts composing the PLL and presented evidence of early mRNA expression for both zebrafish WNK1 orthologs, suggesting a role in early development. We showed that knockdown of wnk1b resulted in a defect of the peripheral nervous system manifested by fewer PLL neuromasts each containing fewer hair cells, measured both by their structural presence and by functional assessment, and that this specific phenotype could be partially rescued upon co-injection of wild-type human WNK1. The low efficiency of the rescue for the wnk1/hsn2 knockdown phenotype by human WNK1 RNA injection could be due to the fact that the human and zebrafish sequences are only 47% identical. Additionally, the constructs were made with no regard to endogenous patterns of alternative splicing as the HSN2 exon is poorly characterized and splicing data is sparse. It is possible that it is inadequately processed in zebrafish and can therefore only be of limited use in rescuing the knockdown phenotype. By injection of a partial construct mimicking truncating mutations in exon HSN2 we also confirmed the loss-of-function of this isoform in HSANII.
We found an overexpression of neuronal cotransporter kcc2 (slc12a5 gene) in wnk1/hsn2 knockdown embryos, both at 72 hp and at 24 hpf. We replicated the PLL defect phenotype obtained in wnk1/hsn2 knockdown by overexpressing human KCC2 RNA in embryos, confirming a pathological link. To verify this link, we knocked down both wnk1/hsn2 and slc12a5 by co-injecting AMOs, which partially rescued the PLL defect phenotype, thereby validating that this overexpression led to improper PLL development.
The slc12a5 knockdown yielded morphologically abnormal embryos which still had a nicely developed PLL, though comprised of fewer neuromasts due to their shorter length. This observation is expected as KCC2 is known to be essential for proper neural development 15,36,37,38,39. Indeed, Kcc2 knockout in mice is embryonic lethal, causing defects in the development of the motor system leading to asphyxiation, as a reversal in GABAergic response necessary for proper neuronal maturation is never achieved 40. Knockdown of Kcc2 in neurons was also proven to compromise survival, by loss of its chloride extruding function 41. Both of these studies divulge a crucial for KCC2 in development and it is therefore not surprising to find abnormal morphology in embryos lacking a large proportion of their Kcc2. In contrast, overexpression of KCC2 leads to a neurogenic defect in the spinal cord of the early zebrafish embryo 15. Together, these observations support an important role of KCC2 regulation of the chloride gradient during development of the central nervous system.
We also demonstrated that kcc2 was localized to the peripheral nervous system (by in situ hybridization) at the level of the mature neuromast. Since KCC2 is known to be involved in neuronal maturation and proliferation, we assessed its effect on PLL progenitors. We found that embryos knocked down for wnk1/hsn2 (which overexpress kcc2) and embryos overexpressing human KCC2 had a smaller primordium, while maintaining normal cell size and organization, which led us to the conclusion that they contain fewer progenitor cells. However, the impact of this result on PLL formation is not clear. Indeed, the effect of primordium size on pro-neuromast deposition is a debated subject. It was previously reported 42 that ablation of up to two-thirds of the primordium leads to a defective PLL. This observed pattern is similar to the one observed in embryos lacking lef1, a major effector of Wnt signaling involved in the control of cxcr4b and cxcr7b, two chemokine receptors involved in PLL migration. The ablated and lef1-defficient primordium size is reduced after each deposition, and eventually disappears, having presumable run out of cells to deposit 42. Another study 43 has however discovered that a reduction of Notch activity gives rise to a smaller primordium, but the neuromasts deposited are of smaller size, rather than of regular size but fewer in number. This suggests a mechanism controlling the number of deposited pro-neuromast rather than one maintaining the size of deposits 43. The data was only acquired for the L1 pro-neuromast deposit and therefore it is possible that proliferative mechanisms taking place at the head of the migrating primordium 44, compensating for the deposits during migration, would be affected, leading to more severe defects further along in the PLL. While we did observe smaller primordia in wnk1/hsn2 knockdown and KCC2 overexpressing embryos, we can only suggest a possible role in progenitor proliferation, where as previously proposed, the deposition is triggered when the primordium reaches a threshold size 42. In this instance, progenitor proliferation would be affected at the placode and at the level of the mitotic head of the migrating primordium either by WNK1 as previously suggested (effect on proliferation, 45), or through interference of Notch signaling 43.
We therefore propose that loss of WNK1/HSN2 deregulates the levels of KCC2. Mechanisms controlling KCC2 expression are only beginning to be uncovered and due to a strikingly rapid turnover of the cotransporter at the cell membrane studies mostly looked at how activation influences transcription. For instance, it was previously shown that BDNF induces Egr4 expression, which rapidly activates the KCC2b promoter in immature neurons, increasing the expression of the KCC2 protein. In mature neurons, the BDNF/TrkB signaling pathway involving a downstream cascade implicating Shc/PRS-2 and PLC-gamma 46 was also found to reduce KCC2 expression, in an activity-dependent manner 47. As for the downregulation of KCC2, it has been observed upon functional loss, where various stresses induced tyrosine dephosphorylation, resulting in decreased levels of KCC2 protein and mRNAs 48. These results also suggest KCC2 transcription could be controlled by its levels of activation, where rapid inactivation leads to a decreased production of mRNAs.
We were also able to mimic the PLL defect phenotype upon injection of an inactive KCC2 mutant (C568A) although it was achieved with statistical difference from both the wild-type embryos and the ones overexpressing hKCC2. These results suggest that a novel regulation of transcription, independent of KCC2 activation, may contribute to the phenotype. It will be therefore important to consider other roles of KCC2 with regards to its implication in neuronal development. For instance, this cotransporter has been reported to play a morphogenic role in dendritic spine formation 28 and is known to interact with cytoskeleton-associated protein 4.1 N 35). This interaction has also been shown to be diminished for the KCC2-C568A mutant where an overexpression could not replicate aberrant actin and 4.1 N patterns observed upon overexpression of WT KCC2 33. This could explain why we could not obtain a PLL phenotype as severe following injection of KCC2-C568A in zebrafish as what is observed when embryos overexpress KCC2 (Figure 4E). Since proteins like 4.1 N anchor the cytoskeleton to the plasma membrane 49, interaction with KCC2, possibly regulated by WNK1 phosphorylation, could prove crucial at this level. HSANII mutations found in the KIF1a kinesin 50 could also affect transport of cargo along the microtubules or unloading at axonal tips.
Previous studies localizing KCC2 mRNAs in rat have been unable to find staining in the primary sensory neurons of the dorsal root ganglia (DRG) and of the trigeminal nucleus presumably because these neurons have depolarizing responses to GABA, where the high intracellular chloride concentration is maintained by expression of NKCC1 38. However, another KCC family member KCC3 is also expressed in neurons, some interneurons, as well as in the spinal cord and in radial glia-like cells 51. This cotransporter has been studied in the context of hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC), where causative mutations have been identified in SC12A6 (coding for KCC3) 52. Truncating as well as loss-of-function mutations have been reported to cause mis-trafficking of proteins, decreasing their plasma membrane expression 53. This neuropathy is characterized by progressive sensory-motor deficits, where axonal swelling can be observed in patients. Since it is also found in radial glia-like cells, a role for KCC3 in migration and proliferation has been proposed 51. Additionally, KCC3 has homologous regulatory sites to the ones found on KCC2, phosphorylated by WNK1 (T991 and T1048 in KCC3) 13, but it has been reported to be unable to interact with cytoskeleton-associated protein 4.1 N 35. Both KCC2 and KCC3 deregulation could therefore be involved in improper development of the peripheral sensory nervous, with KCC2 leading to HSANII pathogenesis.
This article presents the first findings of the molecular basis for HSANII. The zebrafish model we have developed by use of AMO technology exhibited defects in a peripheral sensory system (the PLL) which were apparent during embryonic development, similar to the clinical description of HSANII. Motor defects were also absent upon observation of motor neurons in our wnk1/hsn2 knockdown embryos, which is also a characteristic of HSANII (Figure S2). Likewise, overexpression of KCC2 was shown previously to affect spinal cord interneuron populations but not motoneurons or intrinsic (Rohon-Beard) sensory neurons 15,16 and wnk1/hsn2 was not detected in the spinal cord, consistent with a selective role in sensory lateral line development. We hypothesized that the mutations identified in the HSN2 exon of HSANII patients, producing a truncated protein, would lead to a loss-of-function of this WNK1 and have validated this by using an AMO targeting the start codon of the wnk1 gene, blocking translation of all isoforms in the zebrafish embryos. However, by modifying the splicing patterns by use of the splice blocking AMOs, we confirmed that loss of the hsn2 exon was enough to induce the pathogenic phenotype. It is important to point out that this model is a transient one, due to the use of AMO technology, and that it does not provide full knockdown efficiency. It is therefore possible that the phenotype is not as severe as it would be in knockout animals and in future studies a zebrafish knockout could be obtained by genome editing. We have also identified a pathway involving the KCC2 cotransporter as a downstream target of the WNK1/HSN2 isoform. This cotransporter has been linked to neural differentiation and its regulation by WNK1 has previously been reported, but the interaction between them has not been investigated. Our results suggest the HSN2 exon is critical for normal development to take place and it would be very interesting to understand how its loss influences KCC2 expression or affects WNK1 binding to the cotransporter.
Our results in the zebrafish indicate that KCC2 regulation by WNK1 is an important factor in promoting peripheral nerve development, which may be compromised in HSANII. Whether this is due to regulation of the chloride gradient and peripheral neurogenesis or in addition to a transport-independent KCC2 action in concert with related transporters remains to be determined.
Materials And Methods
Ethics Statement And Transgenic Animals
A colony of wild-type zebrafish (Danio rerio) was bred and maintained according to standard procedures 54. All experiments were performed in compliance with the guidelines of the Canadian Council for Animal Care and the Comité de déontologie de l'expérimentation sur les animaux (CDEA) of the University of Montreal. Embryos were anesthetized in 0.02% tricaïne (MS-222, Sigma) in Embryo medium prior to all experiments.
We used embryos from transgenic lines expressing green fluorescent protein (GFP) under various promoters as neuronal population markers.
Tg(-8.0cldnb:lynEGFP) : membrane-tethered EGFP(enhanced GFP) is under the claudinB promoter labeling the migrating lateral line primordial, the neuromast organs as well as the chain of interneuromast cells deposited during migration 27.
Tg(NBT:MAPT-GFP) : GFP is expressed under the Xenopus laevis neuron-specific beta-tubulin promoter 24.
Genomic Structure Of Wnk1 Orthologs In Zebrafish
The sequence of human WNK1 was used to find homolog sequences in GenBank, leading to the identification of the Xenopus laevis ortholog of WNK1. We then used this ortholog sequence to search the zebrafish assembly using the BLAT genome browser from UCSC (http://genome.ucsc.edu/). The identified genomic sequence from zebrafish was then analyzed, and exons were identified through EST alignments or comparative genomic techniques. cDNA sequences were reconstituted based on the predicted exons and ORFs from the predicted cDNAs were used to derived the predicted peptide sequences. Exons were numbered from 1–28 and the HSN2 name was conserved for the target exon present only in the zebrafish wnk1b (chromosome 4). Orthologous protein sequences were aligned using CLUSTALW and amino acid identity/similarity was calculated using MatGAT program v2.01.
Exons 11 to 13 are fused, as is the case with Fugu and Tetradon, and exon 10 has been split in two smaller exons. The human WNK1 and zebrafish wnk1b orthologs are 47.4% identical and 56.8% similar along the full length of the proteins. We also compared the HSN2 exon sequence, but since the human putative exon 8b sequence 9 contains several frameshifting mutations, the chimp sequence was used instead for comparison purposes. Chimp and zebrafish wnk1b HSN2-like peptide sequences are well conserved, being 54.7% similar and 38.2% identical.
Antisense Morpholino Oligonucleotides And Rna Injections
In order to obtain a knockdown of the wnk1/hsn2 isoform, we designed splice-block (SB) AMOs specific to the donor and acceptor splice sites of the HSN2 exon to interfere with pre-mRNA splicing (MO-hsn2-SB3′: 5′ - CGAGAACGAGTATTTCTAGGTACCA - 3′ and MO-hsn2-SB5′: 5′ - TGCAGTGACAATAACATACAGCATC - 3′). We also designed an AMO targeting the initiation codon of wnk1b, inhibiting protein translation from the only copy of the gene containing the HSN2 exon (MO-wnk1-ATG: 5′ - TTGGGATTTCCGATGACATCTTC - 3′) (Gene Tools, Philomath, OR).
To knockdown zebrafish kcc2 we used two AMOs targeting the initiation codon, the first of which had been previously used in another study (MO1-slc12a5 : 5′ - TGGATGTTGCATCTCCTGTGAACAT - 3′ from 29) and the second was designed according to the latest zebrafish genome assembly as a different target, confirming specificity of the resulting phenotype (MO2 - slc12a5 : 5′ – CTCCTTTGATCTCCAGTGTCTGCAT - 3′).
Human KCC2 mRNAs (hKCC2) were transcribed from the Nhe1-linearized pGEMHE-KCC2 and pGEMHE-KCC2-C568A constructs using the T7 polymerase with the mMESSAGE Machine T7 Kit (Ambion, Austin, TX) as described previously 15. Both constructs were injected at the same concentration known to cause an overexpression phenotype 15,16.
Human WNK1 constructs were assembled in the pCS2 vector with a Cytomegalovirus promoter and a Xenopus laevis beta-glotine UTR region. A partial construct (containing exon 1 to HSN2) and a complete construct (containing exon 1 to 28, but missing exons 11 and 12) were both flanked with 6 myc tags. Exon 1 was amplified from human genomic, as well as exon HSN2, while sequences from exons 2–9 and 10–28 were obtained from clones (CF142377 and BC141881 respectively). mRNAs were transcribed from the KpnI-linearized plasmids using the mMESSAGE Machine SP6 Kit (Ambion, Austin, TX).
All AMOs and mRNAs were diluted in nuclease-free water (Ambion) with 0.2% FastGreen vital dye (Sigma) to judge of injection volume. Injections were performed in 1–4 cell stage zebrafish eggs using a Picospritzer III (Parker Hannifin, Cleveland, OH, USA) pressure ejector.
Whole-Mount Immunohistochemistry
Immunohistochemistry was performed as previously described 15 against the HSN2 exon with the anti-HSN2 antibody previously used 9. The secondary antibody was a goat anti-rabbit Alexa Fluor 488 (Invitrogen). Imaging was performed using a compound fluorescence microscope (Nikon).
Reverse Transcription–Pcr
All RT–PCR were performed using the Expand Long Template enzyme kit (Roche) against control housekeeping gene GAPDH performed with a 1∶2 cDNA dilution to avoid saturation. All samples were run on a 1% agarose gel containing ethidium bromide.
Total RNA from embryos of different developmental stages was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA) and cDNA was synthesized using the RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas). Expression pattern of wnk1a and wnk1b was assessed using primer pairs amplifying the sequence between exon 1–8 and within exon HSN2 respectively.
wnk1a_exon1_f: CTACAAGGGACTGGATACGGAAACTAC
wnk1a_exon8_r: GAGCCTCGAGGATGGTCACTG
wnk1b_hsn2_f: GGGATGCCGGCTCAAAGATT
wnk1b_hsn2_r: TGATGGGACAAGGCAGGCTCGTG
In order to permit a comparison of levels of endogenous kcc2 in our various wnk1/hsn2 knockdown embryos, several precautions were taken in the RT-PCR protocol. Batches of injected embryos from each different group were obtained for the same clutch and staged. The same number of embryo was taken from each condition to perform total RNA extraction using the TRIzol reagent. RNA extraction as well as cDNA synthesis for each experiment was done in parallel, using master mixes whenever possible. Prior to this, we tested PCR parameters for kcc2 primers using only wild-type cDNA in order to make sure the amplification would stay within the exponential amplification segment of the reaction in conditions where an overexpression occurs. The RT-PCR reaction was first tested using wild-type cDNA to establish the optimal condition for annealing temperature, elongation time as well as number of amplification cycles for comparison between conditions using the specific primer pair targeting endogenous kcc2 (slc12a5). Forward primer (slc12a5_f): TTTCACCGAGGGCCACATTGACG. Reverse primer (slc12a5_r): TCCACCTCCACGCACAAGAAGGAC. All samples from each experiment, as well as the GAPDH controls were run on the same 1% agarose gel.
In Situ Hybridization
Hybridization was performed using sense and antisense probes designed against the zebrafish ortholog of KCC2 to view endogenous localization of slc12a5 mRNA. Embryos of 4dpf and 7dpf were processed for in situ hybridization using fluorescent FastRed as previously described 55 with minor modifications, allowing for conservation of the superficial lateral line structure.
Lateral Line Staining
The lateral line was labeled using the vital dye 4-(4-diethylaminostyryl)-N-methylpyridinium (4-di-2-ASP, Invitrogen) diluted to 0.5 mM in embryo medium. Embryos were dechorionated and staged at 72 hpf then incubated in the solution for 30 minutes at 28.5°C. They were then washed 3 times 10 minutes in fresh embryo medium and anesthetized before imaging on an epifluorescence dissection microscope (Olympus) equipped with a Flea2 CCD Camera (IEEE 1394, Point Grey Research Inc. Richmond, BC, Canada). This protocol, adapted from 56,57,58 allows for visualization of full neuromasts, as the dye gets incorporated into hair cells as well as support cells during a longer incubation period.
FM-464FX (Invitrogen), a styryl dye fixable analog, was also used as a vital dye for labeling functional hair cells. Embryos were dechorionated and staged at 72 hpf, then incubated for 1 minute in a 5 µM solution made from a diluted stock in DMSO. Embryos were then washed in embryo medium and anesthetized before being imaged by confocal microscopy.
Calcium Imaging
Calcium imaging experiments were done using live transgenic embryos. To visualize the lateral line primordium and the neuromasts we used the Tg(-8.0cldnb:lynEGFP) embryos and to visualize the innervations of hair cells, we used the Tg(NBT:MAPT-GFP) expressing GFP under the neuron-specific promoter. 2–4 dpf embryos were anaesthetized in 0.02% tricaine (MS-222, Sigma) diluted in Evans solution (134 mM NaCl, 2.9 mM KCl, 2.1 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.8, 290 mOsm). The embryos were then embedded in 2% low-melting-point agarose (Invitrogen) and placed on their sides in the recording chamber. Membrane-permeable Ca2+ indicator dye Rhod-2 AM (Invitrogen/Molecular Probes) was dissolved in DMSO with 20% Pluronic (Invitrogen/Molecular Probes) to yield a 10 mM stock solution and further diluted in Evans solution to a final concentration of 1 mM, as described previously 31. A small volume of the Ca2+ indicator was then pressure injected (Picospritzer III, General Valve Fairfield, N.J., USA) into the primordium or to the neuromast hair cell. Recordings were performed at room temperature in the presence of tricaine to block movement related Ca2+ transients 59 (Ashworth and Bolsover, 2002) and started recordings 60 min after the dye injection. Evoked Ca2+ transients were acquired for 20 minutes at 0.5 Hz (Volocity software, PerkinElmer) by confocal microscopy. To evoke calcium transients, glycine (1 M) or glutamate (100 mM, Na-glutamate, Sigma) were ionophoresed (MVCS-02, npi, Tamm, Germany) from a fine glass pipette (20–30 MΩ) 31. Background-corrected images were analyzed off-line with Volocity software and average changes in Ca2+ levels within regions of interest were calculated as ΔF/F, which is the ratio between the fluorescence change (ΔF) and the baseline fluorescence before stimulation (F). The images of the primordium presented in Figure 5 C were created by overlaying images taken from the green (GFP) and red (Rhod-2 AM) channels, while a bright field image of the neuromasts was added to better illustrate the location of the structure.
Confocal Microscopy And Analysis
Embryos were anesthetized in 0.02% tricaïne (MS-222) in embryo medium and embedded in 1% low melting point agarose. Imaging was performed on a Quorum Technologies spinning-disk confocal microscope (Quorum WaveX Technology Inc Guelph, On, Canada) mounted on an upright Olympus BX61W1 fluorescence microscope with water-immersion lenses. The setup was fitted with a Hamamatsu ORCA-ER camera and image acquisition was done with the Volocity software (Perkin-Elmer) and analyzed with the ImageJ software (NIH). Stacks were acquired at 1 µm thickness and assembled in ImageJ before analysis. Merged images were obtained in Volocity and exported as TIFF files to be used in figures. Images were resized, cropped and brightness was adjusted using Photoshop CS2 (Adobe), the figures were assembled in Illustrator CS2 (Adobe).
Statistical And Data Analysis
Data was plotted and analyzed using the Sigma Plot 11 software (Systat Software Inc., San Jose, CA, USA) and statistical significance was determined using one-way ANOVA combined with the Holm-Sidak method of comparison (normal distribution) or using Kruskall-Wallis one way ANOVA on ranks combined with Dunn's method of comparison (non-parametric distribution). Non-parametric data are presented using medians, data ranges in a box plot diagram. Each box features a central line representing the median value, where the box itself delineates 25–75% of the data range and error bars represent 10–90% of the data range. Outlying data points are represented as circles outside the box. Significance was established at p<0.05.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. DyckPJ (1993) Peripheral Neuropathy; Neuronal atrophy and degeneration predominantly affecting peripheral sensory neuropathy and autonomic neurons.
2. Auer-GrumbachM, MaukoB, Auer-GrumbachP, PieberTR (2006) Molecular genetics of hereditary sensory neuropathies. Neuromolecular Med 8 : 147–158.
3. OgryzloMA (1946) A familial peripheral neuropathy of unknown etiology resembling Morvan's disease. Can Med Assoc J 54 : 547–553.
4. JohnsonRH, SpaldingJM (1964) Progressive Sensory Neuropathy in Children. J Neurol Neurosurg Psychiatry 27 : 125–130.
5. MurrayTJ (1973) Congenital sensory neuropathy. Brain 96 : 387–394.
6. AxelrodFB, Gold-von SimsonG (2007) Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis 2 : 39.
7. KurthI Hereditary Sensory and Autonomic Neuropathy Type II.
8. LafreniereRG, MacDonaldML, DubeMP, MacFarlaneJ, O'DriscollM, et al. (2004) Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am J Hum Genet 74 : 1064–1073.
9. ShekarabiM, GirardN, RiviereJB, DionP, HouleM, et al. (2008) Mutations in the nervous system–specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest 118 : 2496–2505.
10. XuB, EnglishJM, WilsbacherJL, StippecS, GoldsmithEJ, et al. (2000) WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem 275 : 16795–16801.
11. RotthierA, BaetsJ, De VriendtE, JacobsA, Auer-GrumbachM, et al. (2009) Genes for hereditary sensory and autonomic neuropathies: a genotype-phenotype correlation. Brain 132 : 2699–2711.
12. ZambrowiczBP, AbuinA, Ramirez-SolisR, RichterLJ, PiggottJ, et al. (2003) Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci U S A 100 : 14109–14114.
13. RinehartJ, MaksimovaYD, TanisJE, StoneKL, HodsonCA, et al. (2009) Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138 : 525–536.
14. KahleKT, RinehartJ, de Los HerosP, LouviA, MeadeP, et al. (2005) WNK3 modulates transport of Cl - in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci U S A 102 : 16783–16788.
15. ReynoldsA, BrusteinE, LiaoM, MercadoA, BabiloniaE, et al. (2008) Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci 28 : 1588–1597.
16. CoteS, DrapeauP (2012) Regulation of spinal interneuron differentiation by the paracrine action of glycine. Dev Neurobiol 72 : 208–214.
17. KabashiE, ChampagneN, BrusteinE, DrapeauP (2010) In the swim of things: recent insights to neurogenetic disorders from zebrafish. Trends Genet 26 : 373–381.
18. BandmannO, BurtonEA (2010) Genetic zebrafish models of neurodegenerative diseases. Neurobiol Dis 40 : 58–65.
19. MathurP, GuoS (2010) Use of zebrafish as a model to understand mechanisms of addiction and complex neurobehavioral phenotypes. Neurobiol Dis 40 : 66–72.
20. KabashiE, BrusteinE, ChampagneN, DrapeauP (2011) Zebrafish models for the functional genomics of neurogenetic disorders. Biochim Biophys Acta 3 : 335–345.
21. AanesH, WinataCL, LinCH, ChenJP, SrinivasanKG, et al. (2011) Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res 21 : 1328–1338.
22. KimmelCB, BallardWW, KimmelSR, UllmannB, SchillingTF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203 : 253–310.
23. HarrisJA, ChengAG, CunninghamLL, MacDonaldG, RaibleDW, et al. (2003) Neomycin-induced hair cell death and rapid regeneration in the lateral line of zebrafish (Danio rerio). J Assoc Res Otolaryngol 4 : 219–234.
24. GoldmanD, HankinM, LiZ, DaiX, DingJ (2001) Transgenic zebrafish for studying nervous system development and regeneration. Transgenic Res 10 : 21–33.
25. GhysenA, Dambly-ChaudiereC (2007) The lateral line microcosmos. Genes Dev 21 : 2118–2130.
26. OuHC, RaibleDW, RubelEW (2007) Cisplatin-induced hair cell loss in zebrafish (Danio rerio) lateral line. Hear Res 233 : 46–53.
27. HaasP, GilmourD (2006) Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell 10 : 673–680.
28. FiumelliH, BrinerA, PuskarjovM, BlaesseP, BelemBJ, et al. (2012) An Ion Transport-Independent Role for the Cation-Chloride Cotransporter KCC2 in Dendritic Spinogenesis In Vivo. Cereb Cortex 17 : 17.
29. ZhangRW, WeiHP, XiaYM, DuJL (2010) Development of light response and GABAergic excitation-to-inhibition switch in zebrafish retinal ganglion cells. J Physiol 588 : 2557–2569.
30. BrusteinE, DrapeauP (2005) Serotoninergic Modulation of Chloride Homeostasis during Maturation of the Locomotor Network in Zebrafish. The Journal of Neuroscience 25 : 10607–10616.
31. BrusteinE, Saint-AmantL, BussRR, ChongM, McDearmidJR, et al. (2003) Steps during the development of the zebrafish locomotor network. J Physiol Paris 97 : 77–86.
32. BrusteinE, DrapeauP (2005) Serotoninergic modulation of chloride homeostasis during maturation of the locomotor network in zebrafish. J Neurosci 25 : 10607–10616.
33. HornZ, RingstedtT, BlaesseP, KailaK, HerleniusE (2010) Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci 31 : 2142–2155.
34. GauvainG, ChammaI, ChevyQ, CabezasC, IrinopoulouT, et al. (2011) The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci U S A 108 : 15474–15479.
35. LiH, KhirugS, CaiC, LudwigA, BlaesseP, et al. (2007) KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56 : 1019–1033.
36. SteinV, Hermans-BorgmeyerI, JentschTJ, HubnerCA (2004) Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 468 : 57–64.
37. DelpireE (2000) Cation-Chloride Cotransporters in Neuronal Communication. News Physiol Sci 15 : 309–312.
38. KanakaC, OhnoK, OkabeA, KuriyamaK, ItohT, et al. (2001) The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104 : 933–946.
39. PayneJA, RiveraC, VoipioJ, KailaK (2003) Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26 : 199–206.
40. HubnerCA, SteinV, Hermans-BorgmeyerI, MeyerT, BallanyiK, et al. (2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30 : 515–524.
41. PellegrinoC, GubkinaO, SchaeferM, BecqH, LudwigA, et al. (2011) Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol 589 : 2475–2496.
42. GambaL, CubedoN, LutfallaG, GhysenA, Dambly-ChaudiereC (2010) Lef1 controls patterning and proliferation in the posterior lateral line system of zebrafish. Dev Dyn 239 : 3163–3171.
43. MizoguchiT, TogawaS, KawakamiK, ItohM (2011) Neuron and sensory epithelial cell fate is sequentially determined by Notch signaling in zebrafish lateral line development. J Neurosci 31 : 15522–15530.
44. LaguerreL, GhysenA, Dambly-ChaudiereC (2009) Mitotic patterns in the migrating lateral line cells of zebrafish embryos. Dev Dyn 238 : 1042–1051.
45. SunX, GaoL, YuRK, ZengG (2006) Down-regulation of WNK1 protein kinase in neural progenitor cells suppresses cell proliferation and migration. J Neurochem 99 : 1114–1121.
46. RiveraC, VoipioJ, Thomas-CrusellsJ, LiH, EmriZ, et al. (2004) Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci 24 : 4683–4691.
47. LudwigA, UvarovP, SoniS, Thomas-CrusellsJ, AiraksinenMS, et al. (2011) Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J Neurosci 31 : 644–649.
48. WakeH, WatanabeM, MoorhouseAJ, KanematsuT, HoribeS, et al. (2007) Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci 27 : 1642–1650.
49. DenkerSP, BarberDL (2002) Ion transport proteins anchor and regulate the cytoskeleton. Curr Opin Cell Biol 14 : 214–220.
50. RiviereJB, RamalingamS, LavastreV, ShekarabiM, HolbertS, et al. (2011) KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 89 : 219–230.
51. ShekarabiM, Salin-CantegrelA, LaganiereJ, GaudetR, DionP, et al. (2011) Cellular expression of the K+-Cl − cotransporter KCC3 in the central nervous system of mouse. Brain Res 16 : 15–26.
52. HowardHC, MountDB, RochefortD, ByunN, DupreN, et al. (2002) The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32 : 384–392.
53. Salin-CantegrelA, RiviereJB, ShekarabiM, RasheedS, DacalS, et al. (2011) Transit defect of potassium-chloride Co-transporter 3 is a major pathogenic mechanism in hereditary motor and sensory neuropathy with agenesis of the corpus callosum. J Biol Chem 286 : 28456–28465.
54. WesterfieldM (1995) The Zebrafish Book : A Guide for the Laboratory Use of Zebrafish (Danio rerio).
55. JowettT (2001) Double in situ hybridization techniques in zebrafish. Methods 23 : 345–358.
56. CollazoA, FraserSE, MabeePM (1994) A dual embryonic origin for vertebrate mechanoreceptors. Science 264 : 426–430.
57. LedentV (2002) Postembryonic development of the posterior lateral line in zebrafish. Development 129 : 597–604.
58. HernandezPP, MorenoV, OlivariFA, AllendeML (2006) Sub-lethal concentrations of waterborne copper are toxic to lateral line neuromasts in zebrafish (Danio rerio). Hear Res 213 : 1–10.
59. AshworthR, BolsoverSR (2002) Spontaneous activity-independent intracellular calcium signals in the developing spinal cord of the zebrafish embryo. Brain Res Dev Brain Res 139 : 131–137.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy