
Delineating a Conserved Genetic Cassette Promoting Outgrowth of Body Appendages
The acquisition of the external genitalia allowed mammals to cope with terrestrial-specific reproductive needs for internal fertilization, and thus it represents one of the most fundamental steps in evolution towards a life on land. How genitalia evolved remains obscure, and the key to understanding this process may lie in the developmental genetics that underpins the early establishment of the genital primordium, the genital tubercle (GT). Development of the GT is similar to that of the limb, which requires precise regulation from a distal signaling epithelium. However, whether outgrowth of the GT and limbs is mediated by common instructive signals remains unknown. In this study, we used comprehensive genetic approaches to interrogate the signaling cascade involved in GT formation in comparison with limb formation. We demonstrate that the FGF ligand responsible for GT development is FGF8 expressed in the cloacal endoderm. We further showed that forced Fgf8 expression can rescue limb and GT reduction in embryos deficient in WNT signaling activity. Our studies show that the regulation of Fgf8 by the canonical WNT signaling pathway is mediated in part by the transcription factor SP8. Sp8 mutants elicit appendage defects mirroring WNT and FGF mutants, and abolishing Sp8 attenuates ectopic appendage development caused by a gain-of-function β-catenin mutation. These observations indicate that a conserved WNT-SP8-FGF8 genetic cassette is employed by both appendages for promoting outgrowth, and suggest a deep homology shared by the limb and external genitalia.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003231
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003231
Summary
The acquisition of the external genitalia allowed mammals to cope with terrestrial-specific reproductive needs for internal fertilization, and thus it represents one of the most fundamental steps in evolution towards a life on land. How genitalia evolved remains obscure, and the key to understanding this process may lie in the developmental genetics that underpins the early establishment of the genital primordium, the genital tubercle (GT). Development of the GT is similar to that of the limb, which requires precise regulation from a distal signaling epithelium. However, whether outgrowth of the GT and limbs is mediated by common instructive signals remains unknown. In this study, we used comprehensive genetic approaches to interrogate the signaling cascade involved in GT formation in comparison with limb formation. We demonstrate that the FGF ligand responsible for GT development is FGF8 expressed in the cloacal endoderm. We further showed that forced Fgf8 expression can rescue limb and GT reduction in embryos deficient in WNT signaling activity. Our studies show that the regulation of Fgf8 by the canonical WNT signaling pathway is mediated in part by the transcription factor SP8. Sp8 mutants elicit appendage defects mirroring WNT and FGF mutants, and abolishing Sp8 attenuates ectopic appendage development caused by a gain-of-function β-catenin mutation. These observations indicate that a conserved WNT-SP8-FGF8 genetic cassette is employed by both appendages for promoting outgrowth, and suggest a deep homology shared by the limb and external genitalia.
Introduction
Development of the external genitalia is a crucial aspect of mammalian evolution that enables internal fertilization, a pivotal step towards land invasion. All therian mammals including metatherians develop external genitalia around their urogenital outlets. In mice, development of the embryonic anlage of external genitalia, the genital tubercle (GT), is identical in both sexes before androgen-mediated penile masculinization which occurs around embryonic day 16. The early androgen-independent phase of GT development is achieved through coordinated growth and patterning of cloacal endoderm-derived urethral epithelium (UE), mesoderm-derived para-cloacal mesenchyme (PCM) and the ventral ectoderm, which results in a cone-like structure with a ventral-medial positioned urethra surrounded by GT mesenchyme within an ectodermal epithelial capsule.
The development of the GT as an unpaired body appendage, is often compared to that of the paired-type appendages, the limbs [1], [2]. Despite their anatomical and functional differences, the morphogenesis of both structures appears to involve similar genetic controls. Regulatory genes/pathways including canonical WNT signaling [3], [4], HH signaling [5]–[7], BMP signaling [8], [9] and Hox genes [10]–[14] are essential for the development of both appendages. Some have suggested that the regulatory mechanisms common to both might be evolutionarily linked [11], [15], [16].
The first step in appendage outgrowth is the establishment of an independent proximodistal developmental axis apart from the main body axis. This process requires precise regulation from instructive signals, which often come from a distal signaling center. In addition to promoting and maintaining outgrowth, these signals also provide directional information that determines the orientation and shape of future structures. Moreover, genes required for subsequent patterning and differentiation are often regulated by the distal signaling center. For example, during limb development, the initiation and continuous outgrowth of the limb bud rely on growth factors secreted from a strip of ectodermal epithelium, termed the apical ectodermal ridge (AER), positioned at the distal edge of limb bud (Figure 1A and 1E). Fibroblast Growth Factors (FGFs) are crucial signals provided by the AER, as FGF-soaked beads can replace the AER to induce limb outgrowth [17]–[20]. Furthermore, the AER FGF signals are obligatory to maintain a positive feedback loop involving SHH and Gremlin that coordinates patterning and growth of the limb [21]–[23].

The early GT is built by two mesenchymal swellings at either side of the cloacal membrane, which is later joined by a third outgrowth anterior to the cloacal membrane (Figure 1B–1D). Unlike limb, subsequent outgrowth of the GT has to accommodate a continuous extension of the cloacal endoderm, which forms the epithelial lining of the future urethral tube. This unique pattern of GT development suggests that the centrally located distal cloacal endoderm (later the distal urethral epithelium or dUE, marked in red in Figure 1B–1D, also shown by Fgf8 in situ in Figure 1F–1H) is a strategic place for GT outgrowth. Consistently, an earlier study demonstrated that Fgf8 is expressed in a strip of cells located at the distal-most part of the cloacal endoderm right below the ventral ectoderm (Figure 1B inset). Subsequent functional analyses revealed that physically removing Fgf8-expressing cells or application of neutralizing FGF8 antibody could abolish GT growth in organ culture, and this growth inhibition could be reversed by adding FGF8-soaked beads [24].
Along with these studies, we uncovered that activity of the canonical WNT-β-catenin pathway is restricted to the Fgf8-expressing distal cloacal endoderm and later the dUE [4]. We found that abolishing β-catenin (β-Cat) in the cloacal endoderm caused GT agenesis/reduction, whereas ectopic activation of the same pathway resulted in GT over-development. Interestingly, the site and level of WNT signaling activity positively correlated with Fgf8 expression and the extent of outgrowth in both limbs and GT. These findings illustrated a parallel between dUE and AER signaling during appendage outgrowth. However, the exact mechanisms and functional relevance for this WNT-Fgf8 regulation remain to be elucidated.
Recently, two independent studies reported a surprising observation that abolishing Fgf8 [25], [26], or simultaneously abolishing Fgf4 and Fgf8 [26], in the cloacal endoderm does not affect GT outgrowth or GT-specific gene expression. These results questioned the relevance of FGF signals in external genitalia development and challenged the view that GT formation is organized and maintained by the dUE, which further suggested that the mechanisms underpinning limb and GT outgrowth are indeed divergent [2].
Herein, we adopted comprehensive genetic approaches to address the inductive signals in GT development, in comparison with that of the limb. We sought to define the role for FGF signaling and dUE-expressed Fgf8 in the GT, and explore mechanisms upstream of FGF activation in the dUE of the GT and AER of the limb. Results from our analyses revealed a remarkably conserved Wnt-Sp8-Fgf8 genetic circuitry that is crucial for proximodistal outgrowth of both paired - and unpaired-type appendages in mice.
Results
FGF Responsiveness Is Indispensable for GT Development
Previous studies demonstrated that inactivating one or two FGF ligands did not affect genital development [25], [26]. We reasoned that the extensive genetic redundancy among FGF ligands might have undermined the power of these experiments. Therefore, we sought to re-evaluate the function of FGF signaling in GT development by conditionally abolishing FGF receptors. Fgfr1 and Fgfr2 are the only FGF receptors expressed in the developing GT [27]. Both Fgfr1 and Fgfr2IIIc are expressed in the para-cloacal mesenchyme (PCM) during GT outgrowth [27], and Fgfr2IIIb is expressed in both the PCM and the urethral epithelium (UE) [27], [28]. To abolish all FGF responsiveness in the developing GT, we employed an Msx2rtTA;tetO-Cre system which enables doxycycline (Dox)-inducible gene ablation in both the PCM and the UE [29], [30]. Dox was given to pregnant females on embryonic day (E) 9.5 and E10.5 to induce Cre-mediated recombination of the Fgfr1f/f [31] and Fgfr2f/f [32] alleles. The phenotypes of Msx2rtTA;tetO-Cre;Fgfr1f/f;Fgfr2f/f mutant embryos [hereafter referred to as GT-Fgfr1/2-double conditional knockouts (cKO), or dcKO] were analyzed by scanning electron microscopy (SEM). GT-Fgfr1;r2-dcKO genital tubercles were underdeveloped compared to their littermate controls at all stages examined starting from E11.5, when a clear tubercle structure could first be detected (Figure 2B). At E12.5, the dcKO GTs showed a clear deficiency in proximodistal outgrowth, as they were much shorter than the controls (data not shown). At E15.5, the mutant GTs were smaller in size, deformed (Figure 2D) and lacked the characteristic mesenchymal patterning present in controls (Figure S1A–S1B). To further explore the cellular basis for the reduction in GT size, we analyzed proliferation and cell-death in the GT of these mutants. We performed phospho-histone H3 (PHH3) staining on E11.0 coronal GT sections and counted the number of PHH3+ cells in a fixed area. A 28% reduction in PHH3-positive cell number in the genital mesenchyme of the dcKOs was observed (Figure S1C-S1E, n≥10, p = 0.0017). We also performed TUNEL analyses but did not observe any differences in the number of apoptotic cells between control and dcKO mutant GTs (data not shown).

An examination of genes known to mediate genital tubercle initiation revealed alterations in normal gene expression patterns as early as E11.5. Bmp4, Wnt5a and Msx1 were expressed in the PCM in control GTs (Figure 2E and 2G, and Figure S1F), and their expression was barely detectable in dcKO GTs (Figure 2F and 2H, and Figure S1G). Msx2 was expressed in both the PCM and UE in controls (Figure S1H). In the GT-Fgfr1;r2-dcKOs, Msx2 expression was absent in the PCM and downregulated in the UE (Figure S1I). Moreover, UE expression of Shh was also downregulated in the dcKOs (Figure 2I–2H). In contrast, dUE-Fgf8 expression remained unchanged in the GT-Fgfr1;r2-dcKOs (Figure 2K–2L), suggesting that maintenance of Fgf8 expression is independent of FGFR1 and FGFR2 during GT development. Collectively, these data demonstrate that FGF signaling is obligatory for promoting proximodistal GT outgrowth and for maintaining genital-specific gene expression.
FGF Targets Genital Mesenchyme during GT Outgrowth
The Msx2rtTA;tetO-Cre system mediates gene deletion in both the cloacal endoderm as well as the PCM. Therefore, it is not clear whether changes observed in the aforementioned Fgfr1;r2-dcKOs were direct results of compromised FGF responsiveness in the PCM, or secondary to loss of FGF receptors in the cloacal endoderm. Thus, we tested the requirement for FGF responsiveness in these two compartments by using a previously characterized endoderm-specific ShhEGFPCre allele [4], [33], and a PCM-specific Dermo1Cre allele [4], [32], respectively. ShhEGFPCre/+;Fgfr1f/f;Fgfr2f/f (UE-Fgfr1;r2-dcKO) and Dermo1Cre/+;Fgfr1f/f;Fgfr2f/f (PCM-Fgfr1;r2-dcKO) mutants were generated and their GTs analyzed. Intriguingly, GTs from UE-Fgfr1;r2-dcKOs did not exhibit any morphological abnormalities in the early outgrowth phase, and their size and shape were comparable to stage-matched controls (Figure S2A–S2D). Histological analysis revealed normal patterning of the genital mesenchyme in UE-Fgfr1;r2-dcKOs embryos (Figure S2E and S2F). The only phenotype observed in these mutants was the abnormal maturation of urethral epithelium similar to what was observed in Fgfr2IIIb mutants [28], which will be discussed in a separate manuscript. Furthermore, regulatory genes including Msx2, Bmp4, Wnt5a and Fgf8, were also properly expressed in these UE-Fgfr1;r2-dcKO embryos (Figure S2G–S2J).
In contrast, the GTs of PCM-Fgfr1;r2-dcKO were clearly smaller than their littermate controls (Figure S3A–S3B). Further analyses revealed that the distal GT mesenchymal expression of P-ERK1/2, a previously established FGF target gene [26], was also downregulated in these mutants (Figure S3C–S3D). In addition, PHH3 staining on E11.5 embryos revealed a 20% reduction in mitotic figure number in the PCM-Fgfr1;r2-dcKOs (Figure S3E–S3G). Collectively, these data indicate that the main target for FGF signaling during GT outgrowth is the PCM. A similar requirement for FGF signaling in the limb mesenchyme has been described previously [34].
FGF8 Is Sufficient to Regulate GT Development
Fgf8 is normally expressed in the distal-posterior cloacal endoderm at E10.5, and then in the dUE through E11.5–E14.5. We sought to determine whether the PCM could respond to dUE-expressed Fgf8 in vivo. We generated a conditional Fgf8 overexpressor mouse line by knocking the Fgf8 full-length cDNA (Accession: BC048734) into the ubiquitously expressed Rosa26 locus, preceded by a floxed transcriptional stop cassette (R26Fgf8). This design allows ectopic Fgf8 expression upon Cre-mediated recombination (Figure 3A). We used a tamoxifen (Tm)-inducible ShhCreERT2 allele [4], [33] and generated ShhCreERT2/+;R26Fgf8 gain of function (GOF) mutants (UE-R26Fgf8-GOF), to achieve UE-specific Fgf8 overexpression upon Tm treatment at E10.5. The Cre expression domain of this ShhCreERT2 allele recapitulates endogenous Shh expression, which includes all cloacal endodermal cells. Eight hours after Tm administration, we noted a clear upregulation (arrowhead in Figure 3C) and an anterior expansion (arrow in Figure 3C) of Fgf8 expression in the cloacal endoderm evidenced by whole-mount in situ hybridization. The ectopic expression in the anterior cloacal endoderm (arrow in Figure 3C and 3E) was the result of ectopic Fgf8 expression from the R26Fgf8 allele. Notably, a concurrently augmented Bmp4 expression was evident in the PCM (insets in Figure 3B and 3C). Sixteen hours after the initial tamoxifen injection, we observed a 19% increase in mitotic index in the PCM (Figure S4A–S4C, n = 10, p = 0.009). Consistently, the mutant GTs were larger than controls at E12.5 (Figure S4D–S4E) and E14.5 (Figure 3H–3I), respectively. These findings clearly demonstrate that endodermally expressed FGF8 can mediate gene expression and promote cell proliferation in the neighboring PCM.

Interestingly, we noted that endogenous Fgf8 expression was differentially regulated in the UE-R26Fgf8-GOF mutants. Sixteen hours after Tm treatment, we observed a distinct downregulation of Fgf8 expression in the GOF mutant dUE (arrowhead in Figure 3E) that persisted 24 (Figure 3G) and 48 hours after treatment (data not shown). It is also noteworthy that transcription from the R26 locus was much weaker than that from the endogenous Fgf8 locus, evidenced by the faint signal in the anterior cloacal endoderm (indicated by arrows in Figure 3C and 3E). Collectively, these findings demonstrate that the developing GT is sensitive to changes in FGF dosage, and a feedback loop is deployed to ensure proper signaling activity when misregulation occurs.
To compare the function of FGF8 in the limb and GT, we mated the R26Fgf8 allele with a transgenic Msx2-Cre line [35], which confers Cre expression in the forming and mature AER and the ventral limb ectoderm. As expected, the AER-R26Fgf8-GOF mutants exhibited excessive limb growth and developed extra digits (asterisk in Figure 3J), an enlarged calcaneus bone (arrowhead in Figure 3K), and ectopic skeletal elements (arrows in Figure 3J–3K, and Figure S4G) in both forelimbs and hindlimbs. These overgrowth phenotypes are similar but more severe than what has been observed in the AER-Fgf4-GOF embryos [36], and further support the concept that FGF8 plays a pivotal role in promoting the outgrowth of both appendages.
FGF8 Is the Main Signal Output for the Canonical WNT Pathway in the Distal Signaling Epithelia of the Appendages
Our previous work has shown that the WNT-β-catenin signaling pathway and Fgf8 expression are tightly coupled in the distal signaling centers both in the limb and in the GT [4]. The above finding that Fgf8 was repressed by its own overexpression was in sharp contrast to our previous observation in the UE-β-Cat-GOF mutants where ectopic up-regulation Fgf8 was sustained in the UE [4]. This suggested that the canonical WNT pathway plays a key role in controlling the Fgf8 auto-regulatory feedback loop. Together, these observations suggest that the WNT-Fgf8 regulatory relationship is essential for appendage formation, and prompted us to test whether loss of Fgf8 was the critical event causing appendage reduction in embryos deficient in β-Cat in the AER and the UE. Therefore, we attempted to restore Fgf8 expression in β-Cat loss of function (LOF) embryos and analyze its effect on appendage formation. We generated ShhEGFPCre/+; β-Catf/f;R26Fgf8/+ (UE-β-Cat-LOF;R26Fgf8) mutants and analyzed their GT development. In contrast to absence of the GT in the UE-β-Cat-LOF embryos (Figure 4B), a cone-shaped tubercle structure was readily discernible in compound mutants carrying the R26Fgf8 allele (Figure 4A–4C). To determine whether this rescued structure bears GT characteristics, we performed both histological and gene expression analyses. Hematoxylin and Eosin (H&E) stained E12.0 transverse GT sections showed that the morphology of the rescued GT closely resembled that of the controls with the urethra properly positioned at the ventral side of the GT (Figure 4D). In addition, Hoxa13 and Hoxd13, both markers of the GT, were expressed in the rescued genital tubercles (Figure 4E and 4F). Altogether, these data indicate that restoring FGF8 rescued GT agenesis caused by β-catenin deficiency. It is noteworthy that this rescue of β-Cat-cKO by FGF8 is confined to the GT, as other caudal malformations observed in the β-Cat-cKO including failed cloaca septation and defective tail formation, were still present in the UE-β-Cat-LOF;R26Fgf8 embryos (data not shown).

To test whether forced Fgf8 expression can also rescue limb deficiency caused by β-Cat ablation, we generated Msx2-Cre; β-Catf/f (AER-β-Cat-LOF) and Msx2-Cre; β-Catf/f;R26Fgf8/+ (AER-β-Cat-LOF;R26Fgf8) mutants. Consistent with a previous investigation [3], all autopod elements, radius, and distal two-thirds of the ulna were missing from the forelimbs of AER-β-Cat-LOFs (Figure 4I), whereas the humerus was thinner and lacked the deltoid tuberosity (Figure 4I compared to G, arrow). In contrast, the LOF embryos with the R26Fgf8 allele developed normal humeri with the deltoid tuberosity (Figure 4K, arrow). The radius was evident and the ulna was longer and thicker (Figure 4K). Moreover, several small pieces of alcian blue-stained cartilage were observed distal to the ulna, indicating the presence of autopod rudiments (arrowhead and inset in Figure 4K). The hindlimbs of the AER-β-Cat-LOFs were largely absent except for a small remnant of the pelvic girdle (Figure 4J), whereas the R26Fgf8-expressing β-Cat-LOF embryos developed near normal pelvic girdles and femurs (Figure 4L) along with one or two ectopic cartilages (asterisk in Figure 4L). These phenotypes were consistently observed in all AER-β-Cat-LOF;R26Fgf8 embryos (n = 10), and indicated that exogenously supplying FGF8 can partially restore distal limb structures lost in the AER-β-Cat-LOF embryos. Collectively, these data suggest that FGF8 can promote outgrowth of both the GT and the limb in the absence of canonical epithelial WNT activity, suggesting that it functions as a downstream effector of WNT signaling during limb and GT outgrowth.
Growth-Promoting Role for SHH Is Independent of FGF during Appendage Outgrowth
The positive feedback loop involving FGFs and SHH plays a critical role in appendage outgrowth, as evidenced by the down-regulation in Fgf expression in both the AER [21], [22], [35], [37] and the dUE [5], [6] of Shh-KOs, which display reductions in both appendages. We next examined whether forced Fgf8 expression could also rescue the severe appendage deficiencies caused by the absence of SHH. We generated ShhCreERT2/CreERT2;R26Fgf8/+ (Shh-KO;UE-R26Fgf8) embryos, which allowed us to induce Fgf8 expression in the UE of Shh null mutants (ShhCreERT2 allele is also a null allele). However, we detected neither tubercle formation (Figure S5B), nor Hoxa13 or Hoxd13 expression (Figure S5C and S5D) in the cloacal region of these compound mutants at E12.5, 48 hours after Tm treatment. We also generated Msx2-Cre; R26Fgf8/+; ShhCreERT2/CreERT2 (Shh-KO;AER-R26Fgf8) mutants, to test whether sustaining Fgf8 expression in the AER of Shh mutants can restore limb development. We carefully analyzed four Shh-KO embryos carrying both Msx2-Cre and R26Fgf8 alleles, and found no evidence of more advanced limb development (Figure S5G and S5H), compared to thirteen Shh-KO mutants without R26Fgf8 alleles (Figure S5E and S5F).Together, these results indicated that although Shh and Fgf8 expression are interdependent during appendage outgrowth, their function in promoting appendage outgrowth is independent and non-redundant.
GT-Specific Gene Expression Depends on dUE-FGF Signals
We next analyzed GT gene expression in UE-β-Cat-LOF; R26Fgf8 mutants to further interrogate the genetic networks underlying GT development and identify downstream targets of dUE signaling. Fgf8 expression was detected in the distal cloacal endoderm in controls (Figure 5A), but was absent in UE-β-Cat-LOF mutant (Figure 5B) at E10.5. The R26Fgf8-expressing LOF embryos, on the other hand, showed very weak Fgf8 expression throughout the cloacal endoderm (arrow in Figure 5C). This low-expression was consistent with what we observed in the UE - R26Fgf8-GOF embryos, suggesting that these Fgf8 transcripts were transcribed from the R26 locus. Expression of Bmp4 and Wnt5a was normally detected in the PCM (Figure 5D and Figure S6A), absent in the UE-β-Cat-LOFs (Figure 5E and Figure S6B), and partially restored by ectopically supplying Fgf8 expression from the R26Fgf8 allele (Figure 5F and Figure S6C). The SHH pathway was also compromised in the β-Cat-LOF mutants. Shh (Figure 5H) and Ptch1 expressions (Figure S6E) were markedly decreased in the cloacal endoderm and the PCM, respectively. On the other hand, in the LOF embryos with the R26Fgf8 allele, the expression of Shh was partially restored in the UE (Figure 5I) and the PCM expressed Ptch1 restored to a level comparable to controls (Figure 5F).

Combined, these data indicate that most genes differentially regulated in the UE-β-Cat-LOF mutants were responsive to FGF8 induction, suggesting that their expression is controlled by the dUE-FGF signals. However, we found that Sp8, a transcription factor normally expressed in the cloacal endoderm (Figure 5J), was lost in the UE of UE-β-Cat-LOF mutant (Figure 5K), and did not respond to FGF8 supplementation (Figure 5L).
SP8 Is Required for Appendage Outgrowth
Sp8 is expressed throughout the cloacal endoderm and later in the UE (Figure 6A), overlapping with the Fgf8-expressing dUE during GT development. Previous studies have implicated SP8 in the transcriptional regulation of Fgf8 expression in the mouse commissural plate [38] and the chick limb ectoderm [39]. These findings prompted us to explore its role in mediating the WNT-Fgf8 pathway. We first examined Sp8 expression in the UE-β-Cat-LOF and GOF-mutants (for GOF analyses, we used a previously established β-CatΔex3 allele which produces stabilized β-catenin upon Cre-mediated recombination [40]). We found that Sp8 expression was reduced in the β-Cat-LOF UE (Figure 6B) and increased in the β-CatΔex3 GOF UE (Figure 6C). Similarly, robust Sp8 expression was also detected in the AER (Figure 6D), markedly reduced in the AER - β-Cat-LOF mutants (Figure 6E), and upregulated in the β-CatΔex3 GOF mutants where β-catenin activity was ectopically augmented in the limb ectoderm (Figure 6F, inset showing ventral view of a hindlimb). These data indicated that Sp8 is downstream of Wnt-β-catenin signaling in the UE and the limb ectoderm.
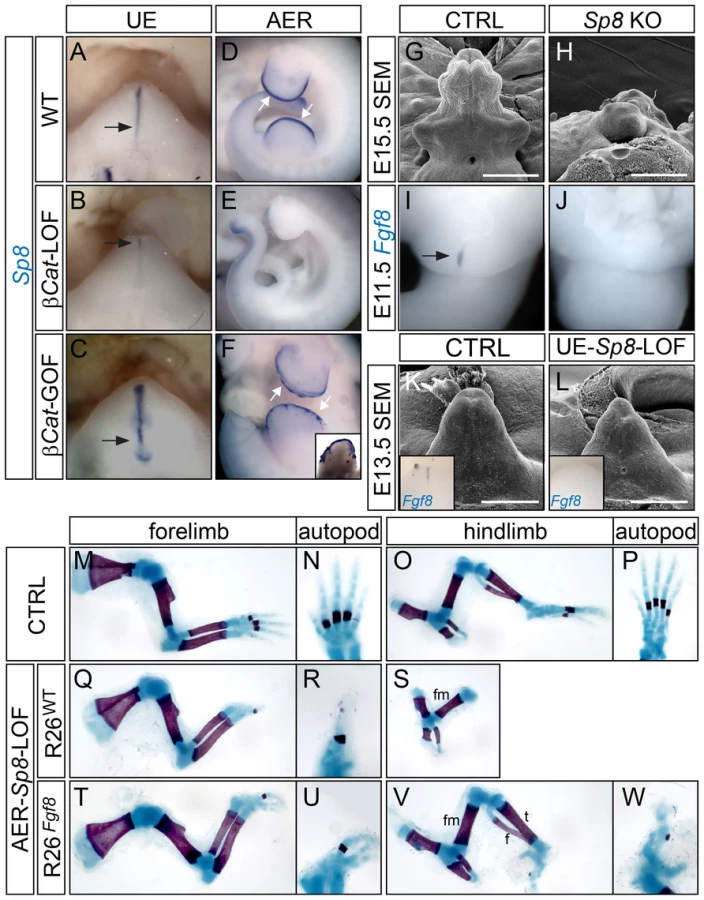
Next, we examined the GT phenotype of Sp8-null (KO) mutants. We found that 14/36 mutant embryos examined between E12.5–E15.5 exhibited GT agenesis (Figure 6H), while the rest demonstrated a range of GT defects including deformation, hypoplasia and proximal hypospadias (data not shown). Fgf8 expression was completely absent in the cloacal endoderm in all Sp8 KOs examined at E11.5 (Figure 6J, n = 9). Notably, these embryos also exhibited other caudal malformations such as deformed perineum, anal channel and tails, raising the concern whether the observed GT defects were secondary to earlier cloacal or neural tube malformations. Thus, we employed a conditional Sp8 null allele with the ShhCreERT2 line to generate UE-Sp8-LOF mutants, and induced Sp8 deletion by Tm treatment around the time of GT initiation (E10.5). GTs from the UE-Sp8-LOF mutants were smaller than their age-matched controls, especially at the distal tip (Figure 6L). We also found a clear reduction in Fgf8 expression in these mutants by in situ analyses (inset in Figure 6L). Consistently, the expression domains of Wnt5a (Figure S7B) and Msx2 (Figure S7D), both maintained by FGF8 from the neighboring dUE, were reduced.
Limb truncation, attributed to a failure to form the AER and consequently loss of Fgf8 expression, has been previously described in Sp8−/− embryos [41]. We generated AER-Sp8-LOFs using the Msx2-Cre line, and these mutants also showed a defect in limb outgrowth as evidenced by a loss of distal structures (Figure 6Q–6S). The stylopod and zeugopod developed normally in the forelimbs, while typically only one abnormal digit formed in these mutants (Figure 6Q–6R). The tibia and fibula were either missing or severely truncated, and no autopod was observed in the hindlimbs (Figure 6S). Notably, these limb defects can also be partially rescued by over expression of Fgf8. Skeleton preparation of E18.5 embryos showed that the AER-Sp8-LOF embryos carrying R26Fgf8 allele developed three digits in the forelimb (Figure 6U), and full-length tibia and fibula along with several irregular autopod elements in the hindlimbs (Figure 6V–6W). Altogether, these data indicated that Sp8 is required to maintain Fgf8 expression and appendage outgrowth in the distal signaling center of both the limb and GT.
Sp8 Mediates WNT-Induced Fgf8 Expression
To test whether Sp8 is in the same genetic pathway with β-catenin and Fgf8, we sought to determine whether SP8 mediates WNT-induced Fgf8 expression in the dUE and AER. We generated compound mutants using the β-CatΔex3 allele, which we have previously shown to activate Fgf8 expression in both the UE and the limb ectoderm, together with the floxed Sp8 allele and the corresponding Cre lines. We compared Fgf8 expression in embryos with one β-CatΔex3 allele and different numbers of functional Sp8 alleles by real-time RT-PCR and in situ hybridization. We found that the level of WNT-induced Fgf8 expression in the UE positively correlated with the number of functioning Sp8 alleles (Figure 7A–7E). Removing one wild type Sp8 allele caused a twofold reduction in Fgf8 expression, whereas ablating both wild type Sp8 alleles reduced Fgf8 expression by more than three-fold (Figure 7E). These results were further verified by Fgf8 in situ hybridization (Figure 7A–7D).

Similarly, deleting both Sp8 alleles abolished the ectopic Fgf8 expansion in the limb ectoderm of Msx2-Cre; β-CatΔex3/+ (AER-β-Cat-GOF) embryos (Figure 7F–7H), and consequently attenuated the polysyndactyly phenotype caused by constitutively active canonical WNT signaling (Figure 7I–7O). With two wild type Sp8 alleles, AER-β-Cat-GOF mutants developed an average of 6.8 digits in the forelimbs, and 6.1 digits in the hindlimbs (Figure 7J and 7M). In thirty percent of the embryos, ectopic limb in the flank ectoderm and ventral ectoderm were detected (Figure S8A and S8B). In comparison, AER-β-Cat-GOF;Sp8-null mutants only developed 4.9 digits in the forelimbs and 4.2 digits in the hindlimbs (Figure 7K and 7N, 28% and 31% reduction compared to AER-β-Cat GOFs, respectively). In addition, no extra limbs were observed at ectopic locations. Collectively, these results indicate that SP8 is responsible, at least in part, for the WNT-induced activation of Fgf8 expression during appendage outgrowth.
To test whether augmented Sp8 expression alone can induce Fgf8 overexpression, we generated a R26Sp8 allele to conditionally overexpress Sp8 using the same strategy as for the R26Fgf8 line (Figure 8A). Mice carrying ShhEGFPCre or Msx2-Cre alleles were used to generate corresponding UE-R26Sp8-GOF and AER-R26Sp8-GOF embryos. Overexpression of Sp8 in the UE and AER was confirmed by in situ hybridization (Figure 8C, and data not shown). However, unlike Fgf8 - or β-Cat-GOF embryos, Sp8-GOF embryos showed normal development in both appendages (Figure 8E and 8I). Fgf8 expression in the dUE and the AER of the corresponding Sp8-GOF mutant was also comparable to their wild type counterparts (Figure S9A–S9D).

We also crossed the R26Sp8 allele into the UE - or AER-β-Cat-LOF mutants to test whether forced Sp8 expression can bypass epithelial β-catenin to induce Fgf8 expression and initiate/maintain appendage outgrowth. We carefully analyzed six compound mutants and did not observe phenotypic rescue in either the GT (Figure 8F) or the limb (Figure 8J–8M). In addition, no Fgf8 induction was detected in the dUE of the corresponding β-Cat-LOF mutants carrying R26Sp8 allele (Figure 8G). All of these results indicate that SP8 by itself is insufficient to activate Fgf8 expression, and suggest that SP8 is a facilitator of WNT-mediated Fgf8 activation during appendage formation.
Discussion
In this study, we investigated the molecular cascade that regulates distal signaling centers in appendage development. Our study elicited a conservative genetic circuitry involving WNT-β-catenin signaling, the transcription factor Sp8, and the growth factor Fgf8, that underpins proximodistal outgrowth of limbs and external genitalia.
FGF8 Mediates FGF Signaling in GT Outgrowth
Our work provides in vivo evidence that FGF signaling is indispensible for early GT outgrowth. These findings are consistent with results from organ culture studies that inhibition of FGF signaling caused an arrest in GT development [24]. Our data demonstrates that the PCM's ability to respond to an FGF signal is essential for normal early genital tubercle outgrowth, as removing FGF receptors from the PCM caused impaired cell proliferation and perturbed normal gene expression patterns which led to severe GT reduction.
The obligatory role for FGF signaling during tubercle morphogenesis underscores the importance of identifying FGF ligands important for GT development. Our data are consistent with the views of Haraguchi et. al. [24], suggesting that dUE-expressed Fgf8 plays a key role in promoting GT outgrowth. Fgf8 is expressed at the correct time and place to signal to the PCM, which expresses both Fgfr1 and Fgfr2. This is in conflict with recent data by Seifert et. al. [25] in which they suggested that a normal GT could develop in the absence of Fgf8 expression. They did not detect FGF8 protein, which led to the conclusion that the Fgf8 mRNA may be present but not actively translated by the UE. They proposed that the ventral ectoderm may be an alternative source of other FGF ligands. In contrast, our results indicate that FGF8 protein can be made by the cloacal endoderm/UE as we demonstrated that a weakly-expressed R26Fgf8 allele can profoundly alter cell proliferation and gene expression in the neighboring PCM, and rescue GT agenesis in β-catenin mutants. In addition, our observation that the endogenous Fgf8 promoter is repressed upon forced Fgf8 overexpression revealed that the developing GT is equipped with mechanisms that can fine-tune the level of FGF8 signaling. All of these data indicate that the GT is not only responsive but also sensitive to FGF8 during normal development. The inability to detect FGF8 in the dUE by IHC is likely caused by the low expression level of Fgf8. We found that in E10.5 mouse embryos, robust AER Fgf8-expression was observed 2 hours after incubation with Alkaline Phosphatase substrates following standard in situ hybridization procedures, whereas dUE-expression was not observed until 12–18 hours later. In addition, the extensive genetic redundancy among FGFs has to be carefully considered. A recent study demonstrated ectopic Fgf4 expression in the dUE of Fgf8-cKO embryos, and ectopic Fgf3 expression in the dUE of Fgf4;Fgf8-dcKO embryos [26]. Both FGF4 and FGF3 can efficiently induce mitogenic activity when paired with FGFR1 or FGFR2 in vitro [42]. These observations indicate that not only FGFs endogenous to the dUE can compensate for the loss of Fgf8, other FGFs can also be ectopically activated to fulfill the requirement for FGF signaling. Activities from these ectopic FGFs could well explain the lack of GT phenotype in Fgf8-cKO mutants. Intriguingly, induction of Fgf3 and Fgf4 was not observed in either β-Cat - or Sp8-cKOs (Figure S10A–S10D), while the dUE expression of Fgf9 was downregulated in Sp8-cKOs (Figure S10E–S10F). These results suggest that the upregulation of these compensatory FGF factors also requires WNT and SP8. Alternatively, the hypothesis that FGFs can be produced by the ventral ectoderm is plausible [25]. However, one has to keep in mind that the GT is built and patterned around the UE. Most regulatory genes expressed distally including Msx1, Msx2, Wnt5a and Lef1, showed peri-dUE expression but not sub-ectodermal expression. Consistently, we have shown that the expression of these genes is orchestrated by UE-specific WNT and FGF signaling. Considering all the available evidence, we conclude that FGF8 produced by the dUE is most likely the endogenous ligand that mediates FGF responses during GT development.
Notably, the GT phenotype of the UE-Fgfr1;r2-dcKO embryos is less severe than what was observed previously in Fgfr2IIIb-KOs [28]. This difference is likely attributable to the method of gene ablation since in the Fgfr2IIIb-KO embryos, Fgfr2-IIIb is abolished from not only the UE, but also from the ventral ectoderm. The underdeveloped phenotype described in Fgfr2IIIb-KO embryos reflects a deficiency from ectodermal FGF responsiveness as it can be phenocopied by conditional ablation of both Fgfr1 and Fgfr2 using the ectodermally restricted Msx2-Cre allele (Lin et. al., unpublished data).
Implication of a Conserved WNT-SP8-FGF8 Pathway in Appendage Outgrowth
We have demonstrated a conserved WNT-SP8-FGF8 pathway in the distal signaling epithelia that functions to promote proximodistal outgrowth of both limbs and genitalia. We and others have shown that the canonical WNT pathway is a master molecular switch in the signaling epithelia during appendage formation, as epithelial WNT activation is not only necessary but also sufficient to induce Sp8 and Fgf8 expression and appendage outgrowth. FGF8 is the downstream signal output for the WNT pathway during this process as it acts directly on recipient mesenchymal cells to promote cell proliferation and establish patterns of gene expression. This genetic hierarchy has been supported by our observation that forced overexpression of Fgf8, even at a level much lower than endogenous expression, can bypass the requirement for epithelial WNT-β-catenin signaling to activate gene expression and initiate/maintain appendage outgrowth. Notably, in both appendages, the rescue of β-catenin deficiency by ectopic Fgf8 expression is only partial. This could be due to either weak Fgf8 expression from the R26 locus compared to the endogenous level of expression, or the possibility that in addition to regulating Fgf8 expression, canonical WNT signaling is also required for the formation of the AER structure, independent of FGF signaling [20]. The regulation of Fgf8 by the canonical WNT-β-catenin signaling pathway is in part mediated by SP8, as Sp8 expression is regulated by WNT signaling in both limb ectoderm and the UE, and is necessary for Fgf8 expression and subsequent appendage formation. However, unlike WNT activity and Fgf8 expression, the expression domain of Sp8 is not restricted to the AER and dUE but also includes the limb ectoderm [41] as well as proximal UE (Figure 6A), suggesting that SP8 is a permissive but not inductive factor for the establishment of Fgf8 expression. In support of this notion, overexpressing Sp8 in the limb ectoderm or UE did not cause any perturbation in Fgf8 expression or appendage formation. In addition, forced Sp8 expression failed to rescue Fgf8 expression and appendage outgrowth in the epithelial β-Cat-LOF mutants.
Notably, even with both Sp8 alleles mutated, the dUE and AER Fgf8 expression in the β-Cat-GOF mutants is still higher than in the controls. This finding apparently is counterintuitive to the obligatory role for SP8 in maintaining Fgf8 expression during normal outgrowth processes. One possible explanation is that the level of WNT signaling induced by the stabilized β-catenin protein from the β-CatΔex3 allele might be too high, and not subjected to endogenous regulatory mechanisms. This could potentially trigger ectopic events that lead to Fgf8 overexpression. It is also noteworthy that the other members of SP/KLF transcription factor family Sp6, was upregulated in the β-Cat-GOF mutants (Figure S11). Sp6 is expressed in the cloacal endoderm and the AER in early appendage development (Figure S11B and Figure S6A, respectively), and loss of Sp6 causes abnormal AER formation and Fgf8 expression [43]. Similar to Sp8, the expression of Sp6 is also downstream of the canonical WNT signaling pathway but independent of FGFs in the AER [43]. The exact function of SP6 in regulating GT development and dUE-Fgf8 expression remains to be determined. However, it is plausible that high levels of SP6 induced by ectopic WNT signaling might compensate for the absence of SP8 in the regulation of Fgf8 expression.
The exact molecular mechanisms through which Fgf8 expression is regulated by SP8 and β-catenin, and how Sp8 expression is regulated by WNT-β-catenin remain to be determined. Direct binding of the β-catenin/LEF1 complex to cis-regulatory elements within the Fgf8 promoter has been reported in dental epithelial cell lines [44] and nephron progenitors [45], suggesting that this WNT-Fgf8 pathway also functions in the development of other organs involving epithelial-mesenchymal interactions. In our preliminary studies, we identified several novel LEF binding sites around Fgf8 and Sp8 genes (data not shown). The functional relevance of these binding sites in GT and limb development will be the goal of future investigations.
Although the distal signaling cascades appear to be similar in both appendages, the upstream events leading to their initiation are likely different. In the limb, the ectoderm is the site for both Wnt3 expression [3], [46], and the induction of canonical Wnt downstream targets as evidenced by TOPGAL activity [47]. On the other hand, the establishment of the dUE signaling center in the cloacal endoderm appears to involve signal transduction between the cloacal endoderm and the ventral ectoderm. Both TOPGAL and Fgf8 expression are confined to the endodermal cells adjacent to the genital ectoderm [4], [24], and the contact between ectoderm and endoderm appears to be a prerequisite for Fgf8 induction and GT initiation [25]. In support of this notion, candidate WNT ligands responsible for activating canonical WNT signaling in the dUE, Wnt3 and Wnt7a, are both expressed in the genital ectoderm [4], [26].
Altogether, our results demonstrate extensive parallels between genetic networks regulating the outgrowth of both the limb and GT. These findings strongly support the notion initially brought up by two pivotal studies on the role of Hox genes in appendage development [10], [11], that mammalian GT development appears to be achieved through co-option of the limb outgrowth program, i.e. the mammalian GT and limb share deep homology [48], [49].
Materials and Methods
Construction of R26Fgf8 and R26Sp8 Alleles
Clones containing full length Fgf8 (BC048734) and Sp8 (BC082582) cDNAs were obtained from Invitrogen (Carlsbad, CA). The cDNAs were released and subcloned into the NotI site of the pBigT vector [50]. The insert containing either Fgf8 or Sp8 cDNA and a floxed transcription stop cassette was released by PacI/AscI double digestion and cloned into pRosa26-PA [51]. The targeting construct was linearized by SwaI and subjected to electroporation (performed by ES cell core at Washington University). The ES cells were screened for recombination by PCR and southern blotting. Five out of seventy-two clones were positive for recombination for R26Fgf8 allele, and six out of sixty clones were positive for R26Sp8 allele. Positive clones were expanded, karyotyped and used for blastocyst injection. For both lines, at least three chimeras were able to pass the knock-in alleles through germline transmission, and the phenotypes resulting from expression of the knock-in alleles were identical. For all experiments described in the manuscript, three embryos with the same genotype were examined if not otherwise specified.
Animal Maintenance and Treatments
All animals were maintained according to NIH guidelines and in compliance with animal protocol approved by Washington University. Msx2rtTA [29], Sp8f/f and Sp8−/− [41], tetO-Cre [52], β-Catf/f [53], β-Catex3/ex3 [40], ShhCreERT2 and ShhEGFPCre [33], Msx2-Cre [35], Fgfr1f/f [31], Dermo1Cre and Fgfr2f/f [32] alleles were previously described. Tamoxifen treatment and doxycycline treatment were performed as previously described [30].
Real-time RT–PCR
For all experiments, we used three independent biological samples from each genotype. Each sample contained a pool of RNA isolated from two E12.5 GTs with the corresponding genotype. The results were analyzed using the delta-CT method. Expression of the corresponding genes was normalized to that of housekeeping gene Rpl7.
Whole-Mount In Situ Hybridization
Whole mount in situ hybridization was performed using a standard protocol. The probes were previously described [4].
Anatomical and Histological Analyses
Paraformaldehyde (4%)-fixed embryos were dehydrated and embedded in paraffin. Five-micron transverse GT sections were generated using a standard microtome. Scanning electron microscopy analyses were performed as previously described [4].
Skeleton Preparation
Skeleton preparation was performed as previously described [54].
Accession Numbers
The Genbank accession numbers for genes included in the manuscript are as follows: Fgf8 (NM_010205.2), Sp8 (NM_177082.4), β-catenin (NM_007614.3), Fgfr1 (NM_010206.2), Fgfr2 (NM_010207.2), Shh (NM_009170.3).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. YamadaG, SuzukiK, HaraguchiR, MiyagawaS, SatohY, et al. (2006) Molecular genetic cascades for external genitalia formation: an emerging organogenesis program. Dev Dyn 235 : 1738–1752.
2. CohnMJ (2011) Development of the external genitalia: conserved and divergent mechanisms of appendage patterning. Dev Dyn 240 : 1108–1115.
3. BarrowJR, ThomasKR, Boussadia-ZahuiO, MooreR, KemlerR, et al. (2003) Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev 17 : 394–409.
4. LinC, YinY, LongF, MaL (2008) Tissue-specific requirements of beta-catenin in external genitalia development. Development 135 : 2815–2825.
5. HaraguchiR, MoR, HuiC, MotoyamaJ, MakinoS, et al. (2001) Unique functions of Sonic hedgehog signaling during external genitalia development. Development 128 : 4241–4250.
6. PerritonCL, PowlesN, ChiangC, MaconochieMK, CohnMJ (2002) Sonic hedgehog signaling from the urethral epithelium controls external genital development. Dev Biol 247 : 26–46.
7. ChiangC, LitingtungY, LeeE, YoungKE, CordenJL, et al. (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383 : 407–413.
8. SuzukiK, BachillerD, ChenYP, KamikawaM, OgiH, et al. (2003) Regulation of outgrowth and apoptosis for the terminal appendage: external genitalia development by concerted actions of BMP signaling [corrected]. Development 130 : 6209–6220.
9. DunnNR, WinnierGE, HargettLK, SchrickJJ, FogoAB, et al. (1997) Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev Biol 188 : 235–247.
10. DolleP, Izpisua-BelmonteJC, BrownJM, TickleC, DubouleD (1991) HOX-4 genes and the morphogenesis of mammalian genitalia. Genes Dev 5 : 1767–1767.
11. KondoT, ZakanyJ, InnisJW, DubouleD (1997) Of fingers, toes and penises. Nature 390 : 29.
12. ZakanyJ, Fromental-RamainC, WarotX, DubouleD (1997) Regulation of number and size of digits by posterior Hox genes: a dose-dependent mechanism with potential evolutionary implications. Proc Natl Acad Sci U S A 94 : 13695–13700.
13. MorganEA, NguyenSB, ScottV, StadlerHS (2003) Loss of Bmp7 and Fgf8 signaling in Hoxa13-mutant mice causes hypospadia. Development 130 : 3095–3109.
14. WarotX, Fromental-RamainC, FraulobV, ChambonP, DolleP (1997) Gene dosage-dependent effects of the Hoxa-13 and Hoxd-13 mutations on morphogenesis of the terminal parts of the digestive and urogenital tracts. Development 124 : 4781–4791.
15. CobbJ, DubouleD (2005) Comparative analysis of genes downstream of the Hoxd cluster in developing digits and external genitalia. Development 132 : 3055–3067.
16. SpitzF, GonzalezF, PeichelC, VogtTF, DubouleD, et al. (2001) Large scale transgenic and cluster deletion analysis of the HoxD complex separate an ancestral regulatory module from evolutionary innovations. Genes Dev 15 : 2209–2214.
17. NiswanderL, TickleC, VogelA, BoothI, MartinGR (1993) FGF-4 replaces the apical ectodermal ridge and directs outgrowth and patterning of the limb. Cell 75 : 579–587.
18. CohnMJ, Izpisua-BelmonteJC, AbudH, HeathJK, TickleC (1995) Fibroblast growth factors induce additional limb development from the flank of chick embryos. Cell 80 : 739–746.
19. FallonJF, LopezA, RosMA, SavageMP, OlwinBB, et al. (1994) FGF-2: apical ectodermal ridge growth signal for chick limb development. Science 264 : 104–107.
20. SunX, MarianiFV, MartinGR (2002) Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature 418 : 501–508.
21. NiswanderL, JeffreyS, MartinGR, TickleC (1994) A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature 371 : 609–612.
22. LauferE, NelsonCE, JohnsonRL, MorganBA, TabinC (1994) Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell 79 : 993–1003.
23. ScherzPJ, HarfeBD, McMahonAP, TabinCJ (2004) The limb bud Shh-Fgf feedback loop is terminated by expansion of former ZPA cells. Science 305 : 396–399.
24. HaraguchiR, SuzukiK, MurakamiR, SakaiM, KamikawaM, et al. (2000) Molecular analysis of external genitalia formation: the role of fibroblast growth factor (Fgf) genes during genital tubercle formation. Development 127 : 2471–2479.
25. SeifertAW, YamaguchiT, CohnMJ (2009) Functional and phylogenetic analysis shows that Fgf8 is a marker of genital induction in mammals but is not required for external genital development. Development 136 : 2643–2651.
26. MiyagawaS, MoonA, HaraguchiR, InoueC, HaradaM, et al. (2009) Dosage-dependent hedgehog signals integrated with Wnt/beta-catenin signaling regulate external genitalia formation as an appendicular program. Development 136 : 3969–3978.
27. SatohY, HaraguchiR, WrightTJ, MansourSL, PartanenJ, et al. (2004) Regulation of external genitalia development by concerted actions of FGF ligands and FGF receptors. Anat Embryol (Berl) 208 : 479–486.
28. PetiotA, PerritonCL, DicksonC, CohnMJ (2005) Development of the mammalian urethra is controlled by Fgfr2-IIIb. Development 132 : 2441–2450.
29. LinC, YinY, ChenH, FisherAV, ChenF, et al. (2009) Construction and characterization of a doxycycline-inducible transgenic system in Msx2 expressing cells. Genesis 47 : 352–359.
30. LinC, YinY, VeithGM, FisherAV, LongF, et al. (2009) Temporal and spatial dissection of Shh signaling in genital tubercle development. Development 136 : 3959–3967.
31. TrokovicR, TrokovicN, HernesniemiS, PirvolaU, Vogt WeisenhornDM, et al. (2003) FGFR1 is independently required in both developing mid - and hindbrain for sustained response to isthmic signals. EMBO J 22 : 1811–1823.
32. YuK, XuJ, LiuZ, SosicD, ShaoJ, et al. (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130 : 3063–3074.
33. HarfeBD, ScherzPJ, NissimS, TianH, McMahonAP, et al. (2004) Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118 : 517–528.
34. YuK, OrnitzDM (2008) FGF signaling regulates mesenchymal differentiation and skeletal patterning along the limb bud proximodistal axis. Development 135 : 483–491.
35. SunX, LewandoskiM, MeyersEN, LiuYH, MaxsonREJr, et al. (2000) Conditional inactivation of Fgf4 reveals complexity of signalling during limb bud development. Nat Genet 25 : 83–86.
36. LuP, MinowadaG, MartinGR (2006) Increasing Fgf4 expression in the mouse limb bud causes polysyndactyly and rescues the skeletal defects that result from loss of Fgf8 function. Development 133 : 33–42.
37. ChiangC, LitingtungY, HarrisMP, SimandlBK, LiY, et al. (2001) Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev Biol 236 : 421–435.
38. SaharaS, KawakamiY, Izpisua BelmonteJC, O'LearyDD (2007) Sp8 exhibits reciprocal induction with Fgf8 but has an opposing effect on anterior-posterior cortical area patterning. Neural Dev 2 : 10.
39. KawakamiY, EstebanCR, MatsuiT, Rodriguez-LeonJ, KatoS, et al. (2004) Sp8 and Sp9, two closely related buttonhead-like transcription factors, regulate Fgf8 expression and limb outgrowth in vertebrate embryos. Development 131 : 4763–4774.
40. HaradaN, TamaiY, IshikawaT, SauerB, TakakuK, et al. (1999) Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 18 : 5931–5942.
41. BellSM, SchreinerCM, WaclawRR, CampbellK, PotterSS, et al. (2003) Sp8 is crucial for limb outgrowth and neuropore closure. Proc Natl Acad Sci U S A 100 : 12195–12200.
42. OrnitzDM, XuJ, ColvinJS, McEwenDG, MacArthurCA, et al. (1996) Receptor specificity of the fibroblast growth factor family. J Biol Chem 271 : 15292–15297.
43. TalamilloA, DelgadoI, NakamuraT, de-VegaS, YoshitomiY, et al. (2010) Role of Epiprofin, a zinc-finger transcription factor, in limb development. Dev Biol 337 : 363–374.
44. WangXP, O'ConnellDJ, LundJJ, SaadiI, KuraguchiM, et al. (2009) Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development 136 : 1939–1949.
45. ParkJS, MaW, O'BrienLL, ChungE, GuoJJ, et al. (2012) Six2 and Wnt Regulate Self-Renewal and Commitment of Nephron Progenitors through Shared Gene Regulatory Networks. Dev Cell 23 : 637–651.
46. RoelinkH, NusseR (1991) Expression of two members of the Wnt family during mouse development–restricted temporal and spatial patterns in the developing neural tube. Genes Dev 5 : 381–388.
47. DasGuptaR, FuchsE (1999) Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 126 : 4557–4568.
48. ShubinN, TabinC, CarrollS (1997) Fossils, genes and the evolution of animal limbs. Nature 388 : 639–648.
49. ShubinN, TabinC, CarrollS (2009) Deep homology and the origins of evolutionary novelty. Nature 457 : 818–823.
50. SrinivasS, WatanabeT, LinCS, WilliamCM, TanabeY, et al. (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1 : 4.
51. SorianoP (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21 : 70–71.
52. PerlAK, WertSE, NagyA, LobeCG, WhitsettJA (2002) Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A 99 : 10482–10487.
53. BraultV, MooreR, KutschS, IshibashiM, RowitchDH, et al. (2001) Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128 : 1253–1264.
54. LinC, FisherAV, YinY, MaruyamaT, VeithGM, et al. (2011) The inductive role of Wnt-beta-Catenin signaling in the formation of oral apparatus. Dev Biol 356 : 40–50.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy