
Mutational Spectrum Drives the Rise of Mutator Bacteria
Understanding how mutator strains emerge in bacterial populations is relevant both to evolutionary theory and to reduce the threat they pose in clinical settings. The rise of mutator alleles is understood as a result of their hitchhiking with linked beneficial mutations, although the factors that govern this process remain unclear. A prominent but underappreciated fact is that each mutator allele increases only a specific spectrum of mutational changes. This spectrum has been speculated to alter the distribution of fitness effects of beneficial mutations, potentially affecting hitchhiking. To study this possibility, we analyzed the fitness distribution of beneficial mutations generated from different mutator and wild-type Escherichia coli strains. Using antibiotic resistance as a model system, we show that mutational spectra can alter these distributions substantially, ultimately determining the competitive ability of each strain across environments. Computer simulation showed that the effect of mutational spectrum on hitchhiking dynamics follows a non-linear function, implying that even slight spectrum-dependent fitness differences are sufficient to alter mutator success frequency by several orders of magnitude. These results indicate an unanticipated central role for the mutational spectrum in the evolution of bacterial mutation rates. At a practical level, this study indicates that knowledge of the molecular details of resistance determinants is crucial for minimizing mutator evolution during antibiotic therapy.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003167
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003167
Summary
Understanding how mutator strains emerge in bacterial populations is relevant both to evolutionary theory and to reduce the threat they pose in clinical settings. The rise of mutator alleles is understood as a result of their hitchhiking with linked beneficial mutations, although the factors that govern this process remain unclear. A prominent but underappreciated fact is that each mutator allele increases only a specific spectrum of mutational changes. This spectrum has been speculated to alter the distribution of fitness effects of beneficial mutations, potentially affecting hitchhiking. To study this possibility, we analyzed the fitness distribution of beneficial mutations generated from different mutator and wild-type Escherichia coli strains. Using antibiotic resistance as a model system, we show that mutational spectra can alter these distributions substantially, ultimately determining the competitive ability of each strain across environments. Computer simulation showed that the effect of mutational spectrum on hitchhiking dynamics follows a non-linear function, implying that even slight spectrum-dependent fitness differences are sufficient to alter mutator success frequency by several orders of magnitude. These results indicate an unanticipated central role for the mutational spectrum in the evolution of bacterial mutation rates. At a practical level, this study indicates that knowledge of the molecular details of resistance determinants is crucial for minimizing mutator evolution during antibiotic therapy.
Introduction
Despite their increased load of deleterious mutations [1], [2], mutator strains of bacteria are isolated routinely in laboratory and clinical settings [3]–[8]. Theory [9]–[11] and experiments [5], [12], [13] explain these observations as a consequence of genetic hitchhiking, whereby mutator alleles reach high frequency by being co-selected with linked beneficial mutations. Mutator evolution is therefore dependent on the absence of horizontal gene transfer [14] and the availability of adaptive mutations with substantial fitness effects [11] – conditions frequently met during adaptation to a host or during antibiotic therapy, which have been invoked to explain the prevalence of mutators among pathogenic bacteria [15]–[17].
Whereas genetic hitchhiking provides a satisfactory mechanism to explain how mutator bacteria can be selected, the precise mechanistic details of this process are still a matter of research. Some authors emphasize that mutator fixation involves many consecutive hitchhiking events, prompted by frequent environmental shifts [9], [18], [19] or by the concurrence of multiple beneficial mutations [10], [11]. Other authors, in contrast, consider mutator success as primarily the result of a single step in which one hitchhiking event takes mutator frequency from rareness to fixation [20]. Despite this theoretical effort, two major observations remain to be accounted for. First, although hitchhiking probability is predicted to increase with the extent of adaptive opportunity offered by the environment (i.e., the number and effects of available beneficial mutations) [14], [20], , selection experiments with comparable adaptive opportunity report contrasting frequencies for mutator emergence [4], [12], [22], [23]. Second, mutators of different strength are predicted to emerge according to the degree of adaptive opportunity [14], [21]; nonetheless, both clinical and laboratory observations show a marked bias towards strong mutators, particularly those caused by defects in the mismatch repair system (MMR) [17], [24]. To explain these deviations, it is argued that mutator alleles of other mutator strengths might be under-represented due to putative fitness disadvantages, and that MMR mutator mutants might be over-represented because they can also be selected for their high recombination rates [24].
An additional, previously unrecognized factor is that each mutator exhibits its own mutational spectrum, that is, mutation rate increase is limited to characteristic types of mutations [25]. This bias stems from the type of mutation avoidance mechanism that is altered in each mutator genotype; depending on which mechanism is affected, its failure will lead to a preferential increase in specific types of transitions, transversions or frameshifts [25]. In line with previous suggestions [12], [26], we hypothesized that if mutators and wild-type strains have differential access to specific beneficial mutations, they might produce mutants with different fitness levels, which could influence their evolutionary dynamics. Mutator alleles that on average generate stronger beneficial mutations will have a better chance to achieve fixation; conversely, those that more often produce weaker adaptive mutations will have limited spread. The fixation probability of a mutator allele would thus depend not only on its mutation rate, but also on its mutational spectrum.
Here we test these predictions by characterizing beneficial mutations from wild-type and knockout strains of the mutT (ΔmutT) and mutY (ΔmutY) antimutator genes of Escherichia coli. mutT-defective strains show a strong mutator phenotype, leading specifically to A·T→C·G transversions [25], whereas mutY defects lead to a moderate mutator phenotype that increases G·C→T·A transversions [25]. Both mutators were reported to arise spontaneously in evolution experiments with E. coli [4], [12]. Since beneficial mutations are exceedingly rare and difficult to detect, our experimental design focused on mutations that confer antibiotic resistance, which are particularly suitable for this kind of study. We show that according to their mutational spectra, wild-type and mutator strains can generate distinct fitness distributions for antibiotic-resistant mutants. Notably, these dissimilarities can significantly alter the competitive abilities of mutators against the wild-type strain, as measured in direct competition experiments. Furthermore, using computer simulation of a simple population genetics model, we show that the hitchhiking dynamics is highly sensitive to average fitness deviations of the beneficial mutations with which mutator alleles hitchhike.
Results/Discussion
Colony size polymorphism of antibiotic-resistant mutants
To test whether mutational spectrum differences translate into significant fitness differences, beneficial mutations were selected by plating cultures of wild-type, ΔmutT and ΔmutY strains in three antibiotics: rifampicin, streptomycin and tetracycline. Rifampicin - and streptomycin-resistant mutants showed marked colony size polymorphism (Figure 1A). Resistance to these antibiotics arises readily through alterations in their targets, the RNA polymerase [27] and the 30S ribosomal subunit [28], respectively. Since many alterations can affect the function of these essential machineries to different degrees, it is unsurprising that resistance mutants show growth differences. Consistent with the predictions of our hypothesis, wild-type and mutator strains displayed distinct colony size polymorphism in each antibiotic (Figure 1A). To test whether access to beneficial mutations with varying selection coefficients was the only factor responsible for differences among strains, we used strains with an insertion bearing a tetracycline-resistance gene and its constitutive repressor [29]. As the probability of achieving resistance to high tetracycline concentrations through point mutation in E. coli is negligible (mutant frequency <10−9), resistance arises here only through loss-of-function mutations, which relieve the repression. All of these loss-of-function mutations can be considered equivalents, and no fitness differences are thus anticipated among tetracycline-resistant mutants. Indeed, these mutants showed little growth polymorphism (Figure 1A). Small differences, apparently independent of the mutator background, probably reflect phenotypic lag or other sources of phenotypic variability.

Distribution of fitness effects on plate of antibiotic-resistant mutants
To quantify the fitness distribution of the mutants generated by each mutator, we randomly selected 42 independent resistant colonies from each combination of genotype and antibiotic, and measured growth rate as a proxy for Darwinian fitness. It is important to remark that we are dealing with the fitness distribution after selection, not the intrinsic fitness distribution; and so it should be understood in what follows. In rifampicin, the distribution of mutants generated by the ΔmutY strain is shifted toward higher growth rates compared with both those of wild-type and ΔmutT strains (Figure 1B, centre), whereas in streptomycin the situation is the reverse (Figure 1B, right) (n = 42, P<0.0001, Kolmogorov-Smirnov's one-sided two-sample test, in all cases). These results suggested that G·C→T·A substitutions in the rpoB gene (the characteristic transversion increased in this mutator) produced alleles encoding a high-fitness rifampicin-resistant RNA polymerase; coincidentally, the same transversion generated rpsL alleles encoding a low-fitness streptomycin-resistant ribosomal protein S12. Similar reasoning could be applied to the ΔmutT strain results, which preferentially raises the A·T→C·G transversion. In the case of tetracycline, no significant differences were found between wild-type and any mutator strains (Figure 1B, left) (n = 42, P>0.18, Kolmogorov-Smirnov's two-sided two-sample test).
Characterization of the genetic basis of antibiotic resistance
To confirm our interpretation, we randomly picked 10 colonies from each combination of antibiotic and strain, sequenced their rpoB and rpsL genes, and measured growth rates (Figure 2). Several G·C→T·A substitutions found in rpoB can explain the higher fitness of rifampicin-resistant ΔmutY mutants, supporting our hypothesis. The highest-fitness class (Figure 1B, centre) is likely to be composed of V146F mutants, the fastest-growing mutant detected (Figure 2A); other high-fitness mutations that resulted from this transversion were H526N and S531Y. The fitness distribution in rifampicin-resistant ΔmutT mutants can be explained, at least in part, by an A·T→C·G substitution that produces the low-fitness mutation Q513P. Among streptomycin-resistant mutants, idiosyncratic transversions in rpsL similarly help to explain the fitness differences. The low fitness of streptomycin-resistant ΔmutY mutants probably reflects predominance of the P90Q mutation (8/10 mutants tested), whereas the high fitness of ΔmutT mutants might be due to prevalence of the K42T mutation (9/10). As a brief remark, 3/10 of the streptomycin-resistant mutants in the wild-type background carry the substitution P90L. This mutation is known to prevent growth in the absence of the antibiotic [28], and offers a suggestive example of how spectra can determine the access to mutations with differences not only in fitness, but also in other related properties.

The biochemical bases of the fitness cost of both rifampicin and streptomycin resistance have been discussed elsewhere. The costs of rpoB mutations are explained by the impairment of the transcription activity of the RNA polymerase [30], [31]. Similarly, it has been long established that rpsL streptomycin resistance mutants exhibit hyperaccurate translation, resulting in a slower rate of protein synthesis and consequently, in a slower growth rate [32].
Average fitness of antibiotic-resistant mutator genotypes
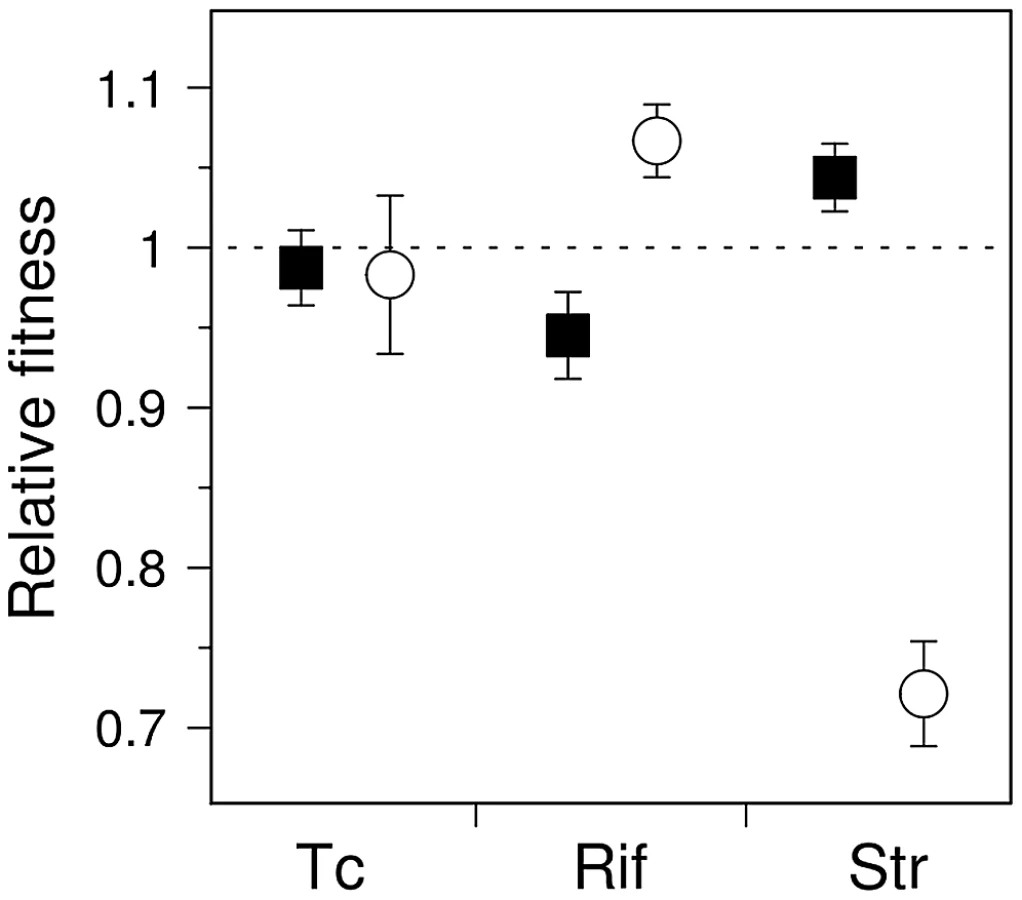
It is tempting to assume that if the fitness distribution of a given mutator is altered compared to that of wild-type, the average fitness of that mutator will change accordingly. These distributions nonetheless have distinct shapes and degrees of overlap (Figure 1B); it is thus of interest to determine the extent to which these differences translate into an overall fitness difference between each mutator and the wild-type genotypes. Direct competition experiments between mutators and wild-type strains in rifampicin and streptomycin showed significant differences in mean fitness (Figure 3) (n = 4, P<0.012, Mann-Whitney U-test, one-sided, all cases), confirming the predictions made by visual examination of Figure 1A.
Mutational spectrum effect on hitchhiking dynamics
Our data provide evidence that in a specific environment, distinct mutators generate their own fitness distribution among newly-arising mutants, which can influence their competitive ability. Theory predicts the fixation probability of a mutator allele to be dependent on the selection coefficient of the driver allele with which the mutator hitchhikes [11], [20]. According to our results, this coefficient can vary substantially depending on the mutational spectrum; the mutational spectrum should thus have some effect on the hitchhiking dynamics. To estimate how large this effect could be, we used simple computer simulations based on previous studies [10], [11], [20]. Briefly, we simulated the basic scenario of a non-mutator bacteria population growing in batch culture, where only one beneficial mutation is needed to achieve full adaptation. Mutators are generated at a constant rate, and produce the adaptive mutation with a 100-fold higher probability than the wild-type strain. Once the mutation is fixed in either background, the simulation ends. The mutational spectrum effect (σ) was introduced as a multiplicative factor to modify the selection coefficient (s) of the driver allele only on the mutator background (see Material and Methods).
The simulations showed that σ exerts a modest influence on the establishment of mutator genotypes bearing the adaptive mutation (i.e., on the probability that they escape random drift) (Figure 4). This effect is not surprising, as the probability of a beneficial mutation surviving drift is approximately 2s [33]. In contrast, σ had a notable effect on the fate of established genotypes en route to fixation. In the absence of mutational spectrum effects (σ = 1), a mutator genotype bearing the adaptive mutation can only succeed if it reaches fixation before any adapted wild-type bacteria escapes drift; it will otherwise always be outcompeted due to its increased deleterious mutation load (Figure 5, upper row). When the average s of the driver allele is lower on the mutator background (σ<1), there are no qualitative changes. Success is further hindered because drift is more intense and time to fixation is longer, extending the period available for the establishment and subsequent selective sweep of an adapted wild-type strain. In contrast, when σ>1 a threshold appears above which the population dynamics switches. This threshold is determined by the value of σ that offsets the increased deleterious load of mutator genotypes. Above this value, the adapted mutator is fitter than its wild-type counterpart, and therefore needs only to escape drift to reach fixation (Figure 5, lower row); as a consequence, fixation probability rises sharply (Figure 4). It is worth noting that, since the deleterious load is as small as the order of magnitude of the mutation rate [33], only a slight spectrum-dependent fitness advantage is needed to substantially increase mutator success.


Remarkably, the non-linear response of fixation probability to σ implies that previous models could have been underestimating the likelihood of mutator success by several orders of magnitude. This is clearly illustrated in Figure 4 where a change from σ = 0.56 to σ = 1.33, which is equivalent to a change in relative fitness [37] from w = 0.96 to w = 1.03, represents a ∼196-fold increase in fixation probability.
Conclusions
Selection of mutators has been understood exclusively in terms of the increased number of beneficial mutations they generate [5], [10]. Here we show that not only the number but also the type of these linked mutations are relevant. Our results indicate that mutator alleles can bias the average selection coefficient of the beneficial alleles with which they hitchhike. Besides, they suggest that the magnitude of this effect can easily be sufficient to drastically modify their probabilities to reach fixation. The only requirement for this bias is that the locus or loci under selection produce mutants with some variability for fitness. This is a fairly permissive condition, likely to be satisfied in several adaptive scenarios. Examples in which resistant mutants with varying degrees of fitness are commonly found include bacteriophage [34] and antibiotic resistance [35], considered major drivers of mutator evolution [8], [15]. It will be of interest to determine whether the observed relative abundance of each mutator is explained, at least in part, by the effect of its mutational spectrum on successive mutations undergone during adaptation. In the context of clinical infections, future work should address the extent to which antibiotic therapies show different propensities to select for mutator bacteria. Since mutators are recognized as a risk factor for treatment failure [15]–[17], this knowledge could help to improve the design of safer therapeutic strategies.
The fact that even slight mutational spectrum effects markedly alter hitchhiking, together with the apparent commonness of circumstances that potentially allow this to happen, lead us to conclude that the mutational spectrum is a major factor in the evolution of mutators in laboratory and clinical populations of bacteria [3]–[8], as well as in certain cancers [36].
Materials and Methods
Strains and culture conditions
Strains are E. coli MG1655 derivatives, obtained from Dr. I. Matic [29]. Bacteria were grown on Luria broth (LB) or LB agar plates (37°C). Antibiotics used were tetracycline (15 mg/L), rifampicin (100 mg/L) and streptomycin (100 mg/L). Incubation time was 24 h, except for streptomycin plates (42 h).
Growth rate assays
Overnight cultures of each strain were plated on each antibiotic at appropriate dilutions to ensure low colony density (∼50/plate). After incubation, independent colonies were picked at random (by proximity to an arbitrary point) and resuspended in saline solution. Population size (N) was estimated from viable counts by subsequent dilution and plating. Assuming that each colony originated from a single cell, generation number was calculated as log2N and the growth rate expressed as number of generations per hour. Note that this measurement integrates growth rate over all growth phases.
Competition experiments
Overnight cultures of each mutator and wild-type strain were mixed at a ratio based on their mutation rates, plated on antibiotic, and allowed to compete for 24 or 42 h. Initial competitor frequency was calculated by estimating the resistant-mutant frequency of each overnight culture. To distinguish them from the wild-type bacteria, mutators carried antibiotic resistance markers [29], which entailed no significant fitness cost in these conditions (n = 4, P>0.28, Mann-Whitney U-test, two-sided, both cases). After incubation, agar plates were washed in saline solution and the final mutator∶wild-type ratio obtained by plating on LB agar and selective media. Relative fitness was estimated using a standard formula [37].
Computer simulation
(i) General description of the algorithm
We simulated the serial passage of an asexual population, originated from a single wild-type cell, in a laboratory environment in which only one beneficial mutation is available. The overall scheme is the same as in Tenaillon et al. [11], and can be summarized in two phases: (1) population growth, which includes differential reproduction and mutation during a number of generations, and (2) a random sampling process. Programming was carried out using the R programming language [38] (source code available from www.free-bit.org/public/acouce/).
Genotypes are defined by three loci: B, D and M. The first locus codes for the absence (b = 0) or presence (b = 1) of the beneficial mutation, the second locus controls the number of accumulated deleterious mutations (d), and the last encodes the absence (m = 0) or presence (m = 1) of a mutator allele with a 100-fold increase in mutation rate. In this model, the wild-type genotype, for example, will be unequivocally denoted by 000. In each generation, non-mutator individuals reproduce deterministically in accordance with the absolute fitness (rbd0) defined by their genotypes (bd0) as:

(ii) Modeling the mutational spectrum effect
As we showed experimentally, the mutational spectrum can affect the average selection coefficient of the beneficial mutation with which a mutator allele ‘hitches a ride’. To implement this effect, we defined the the absolute fitness of mutators (bd1) as:

Care has been taken to ensure that equation (2) is only applied to adapted mutator genotypes (1d1) generated from a mutator background (0d1), but not to those generated in the rare but non-negligible case of an already adapted wild-type background (1d0) mutating the M locus.
(iii) Modeling transitions among genotypes
In each generation, after reproduction, all types of mutations are implemented by generating Poisson-distributed pseudorandom numbers [40], according to the population size of each genotype and a defined set of mutation rates (which act as the parameter of the Poisson distribution). Wild-type mutation rates were chosen to be in the range of estimated values for E. coli, 10−7 for beneficial mutations [41], 10−4 for deleterious mutations [42] and 5×10−6 for acquiring a mutator genotype [1]. Lethal mutation was introduced at a rate of 10−5, based on previous reports [10], [11]. Back-mutation was denied. One order of magnitude variation on these parameters showed no qualitative changes relative to the results shown here.
Zdroje
1. BoeL, DanielsenM, KnudsenS, PetersenJB, MaymannJ, et al. (2000) The frequency of mutators in populations of Escherichia coli. Mutat Res 448 : 47–55.
2. FunchainP, YeungA, StewartJL, LinR, SlupskaMM, et al. (2000) The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154 : 959–970.
3. LeClercJE, LiB, PayneWL, CebulaTA (1996) High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274 : 1208–1211.
4. SniegowskiPD, GerrishPJ, LenskiRE (1997) Evolution of high mutation rates in experimental populations of E. coli. Nature 387 : 703–705.
5. MaoEF, LaneL, LeeJ, MillerJH (1997) Proliferation of mutators in a cell population. J Bacteriol 179 : 417–422.
6. OliverA, CantónR, CampoP, BaqueroF, BlázquezJ (2000) High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 : 1251–1253.
7. RichardsonAR, YuZ, PopovicT, StojiljkovicI (2002) Mutator clones of Neisseria meningitidis in epidemic serogroup A disease. Proc Natl Acad Sci USA 99 : 6103–6107.
8. PalC, MaciaMD, OliverA, SchacharI, BucklingA (2007) Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 450 : 1079–1081.
9. LeighEG (1970) Natural selection and mutability. Am Nat 104 : 301–305.
10. TaddeiF, RadmanM, Maynard-SmithJ, ToupanceB, GouyonPH, et al. (1997) Role of mutator alleles in adaptive evolution. Nature 387 : 700–702.
11. TenaillonO, ToupanceB, Le NagardH, TaddeiF, GodelleB (1999) Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152 : 485–493.
12. Notley-McRobbL, SeetoS, FerenciT (2002) Enrichment and elimination of mutY mutators in Escherichia coli populations. Genetics 162 : 1055–1062.
13. ShaverAC, DombrowskiPG, SweeneyJY, TreisT, ZappalaRM, et al. (2002) Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics 162 : 557–566.
14. TenaillonO, Le NagardH, GodelleB, TaddeiF (2000) Mutators and sex in bacteria: Conflict between adaptive strategies. Proc Natl Acad Sci USA 97 : 10465–10470.
15. BlázquezJ (2003) Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin Infect Dis 37 : 1201–1209.
16. LabatF, PradillonO, GarryL, PeuchmaurM, FantinB, et al. (2005) Mutator phenotype confers advantage in Escherichia coli chronic urinary tract infection pathogenesis. FEMS Immunol. Med Microbiol 44 : 317–321.
17. OliverA, MenaA (2010) Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin Microbiol Infect 16 : 798–808.
18. TravisJMJ, TravisER (2002) Mutator dynamics in fluctuating environments. Proc Biol Sci 269 : 591–597.
19. TanakaMM, BergstromCT, LevinBR (2003) The evolution of mutator genes in bacterial populations: the roles of environmental change and timing. Genetics 164 : 843–854.
20. WylieCS, GhimCM, KesslerD, LevineH (2009) The fixation probability of rare mutators in finite asexual populations. Genetics 181 : 1595–1612.
21. AndreJ, GodelleB (2006) The evolution of mutation rate in finite asexual populations. Genetics 172 : 611–626.
22. MaharjanR, SeetoS, Notley-McRobbL, FerenciT (2006) Clonal adaptive radiation in a constant environment. Science 313 : 514–517.
23. TenaillonO, Rodríguez-VerdugoA, GautRL, McDonaldP, BennettAF, et al. (2012) The molecular diversity of adaptive convergence. Science 335 : 457–461.
24. DenamurE, MaticI (2006) Evolution of mutation rates in bacteria. Mol Microbiol 60 : 820–827.
25. MillerJH (1996) Spontaneous mutators in bacteria: insights into pathways of mutagenesis and repair. Annu Rev Microbiol 50 : 625–643.
26. MacLeanRC, HallAR, PerronGG, BucklingA (2010) The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nat Rev Genet 11 : 405–414.
27. GaribyanL, HuangT, KimM, WolffE, NguyenA, et al. (2003) Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair 2 : 593–608.
28. TimmsAR, SteingrimsdottirH, LehmannAR, BridgesBA (1992) Mutant sequences in the rpsL gene of Escherichia coli B/r: mechanistic implications for spontaneous and ultraviolet light mutagenesis. Mol Gen Genet 232 : 89–96.
29. BjedovI, DasguptaCN, SladeD, Le BlastierS, SelvaM, et al. (2007) Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 176 : 1431–1440.
30. HallAR, IlesJC, MacLeanRC (2002) The fitness cost of rifampicin resistance in Pseudomonas aeruginosa depends on demand for RNA polymerase. Genetics 187 : 817–822.
31. ReynoldsMG (2000) Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156 : 1471–1481.
32. Kurland C, Hughes D, Ehrenberg M (1996) Limitations of translational accuracy. In: Neidhardt F.C III, et al., editors. Escherichia coli and Salmonella: Cellular and molecular biology. ASM Press; Washington, DC: 1996. pp. 979–1004.
33. Kimura M (1994) Population genetics, molecular evolution, and the neutral theory: selected papers (University of Chicago Press)
34. BohannanBJ, LenskiRE (2000) Linking genetic change to community evolution: insights from studies of bacteria and bacteriophage. Ecol Lett 3 : 362–377.
35. AnderssonDI, HughesD (2010) Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Micro 8 : 260–271.
36. PrindleMJ, FoxEJ, LoebLA (2010) The mutator phenotype in cancer: molecular mechanisms and targeting strategies. Curr. Drug Targets 11 : 1296–1303.
37. LenskiRE, RoseMR, SimpsonSC, TadlerSC (1991) Long-term experimental evolution in Escherichia coli I. Adaptation and divergence during 2,000 generations. Am Nat 138 : 1315–1341.
38. R Development Core Team (2011) R: A language and environment for statistical computing, reference index version 2.13.0. (R Foundation for Statistical Computing, Vienna).
39. de VisserJA, ZeylCW, GerrishPJ, BlanchardJL, LenskiRE (1999) Diminishing returns from mutation supply rate in asexual populations. Science 283 : 404–406.
40. AhrensJH, DieterU (1982) Computer generation of Poisson deviates from modified normal distributions. ACM Trans Math Softw 8 : 163–179.
41. RozenDE, de VisserJAGM, GerrishPJ (2002) Fitness effects of fixed beneficial mutations in microbial populations. Curr Biol 12 : 1040–1045.
42. TrindadeS, PerfeitoL, GordoI (2010) Rate and effects of spontaneous mutations that affect fitness in mutator Escherichia coli. Philos Trans R Soc Lond B Biol Sci 365 : 1177–1186.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy