
Mendelian and Non-Mendelian Regulation of Gene Expression in Maize
Transcriptome variation plays an important role in affecting the phenotype of an organism. However, an understanding of the underlying mechanisms regulating transcriptome variation in segregating populations is still largely unknown. We sought to assess and map variation in transcript abundance in maize shoot apices in the intermated B73×Mo17 recombinant inbred line population. RNA–based sequencing (RNA–seq) allowed for the detection and quantification of the transcript abundance derived from 28,603 genes. For a majority of these genes, the population mean, coefficient of variation, and segregation patterns could be predicted by the parental expression levels. Expression quantitative trait loci (eQTL) mapping identified 30,774 eQTL including 96 trans-eQTL “hotspots,” each of which regulates the expression of a large number of genes. Interestingly, genes regulated by a trans-eQTL hotspot tend to be enriched for a specific function or act in the same genetic pathway. Also, genomic structural variation appeared to contribute to cis-regulation of gene expression. Besides genes showing Mendelian inheritance in the RIL population, we also found genes whose expression level and variation in the progeny could not be predicted based on parental difference, indicating that non-Mendelian factors also contribute to expression variation. Specifically, we found 145 genes that show patterns of expression reminiscent of paramutation such that all the progeny had expression levels similar to one of the two parents. Furthermore, we identified another 210 genes that exhibited unexpected patterns of transcript presence/absence. Many of these genes are likely to be gene fragments resulting from transposition, and the presence/absence of their transcripts could influence expression levels of their ancestral syntenic genes. Overall, our results contribute to the identification of novel expression patterns and broaden the understanding of transcriptional variation in plants.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003202
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003202
Summary
Transcriptome variation plays an important role in affecting the phenotype of an organism. However, an understanding of the underlying mechanisms regulating transcriptome variation in segregating populations is still largely unknown. We sought to assess and map variation in transcript abundance in maize shoot apices in the intermated B73×Mo17 recombinant inbred line population. RNA–based sequencing (RNA–seq) allowed for the detection and quantification of the transcript abundance derived from 28,603 genes. For a majority of these genes, the population mean, coefficient of variation, and segregation patterns could be predicted by the parental expression levels. Expression quantitative trait loci (eQTL) mapping identified 30,774 eQTL including 96 trans-eQTL “hotspots,” each of which regulates the expression of a large number of genes. Interestingly, genes regulated by a trans-eQTL hotspot tend to be enriched for a specific function or act in the same genetic pathway. Also, genomic structural variation appeared to contribute to cis-regulation of gene expression. Besides genes showing Mendelian inheritance in the RIL population, we also found genes whose expression level and variation in the progeny could not be predicted based on parental difference, indicating that non-Mendelian factors also contribute to expression variation. Specifically, we found 145 genes that show patterns of expression reminiscent of paramutation such that all the progeny had expression levels similar to one of the two parents. Furthermore, we identified another 210 genes that exhibited unexpected patterns of transcript presence/absence. Many of these genes are likely to be gene fragments resulting from transposition, and the presence/absence of their transcripts could influence expression levels of their ancestral syntenic genes. Overall, our results contribute to the identification of novel expression patterns and broaden the understanding of transcriptional variation in plants.
Introduction
The maize species exhibits high levels of phenotypic variation, which is likely the result of both genetic and epigenetic variation [1]. Dissection of genetic and epigenetic variation may shed light on the understanding of phenotypic variation and provide tools to accelerate maize breeding. The maize genome has a complex organization with interspersed repetitive elements and genes [2]. The genomes of different maize inbreds can vary substantially due to single nucleotide polymorphisms [3], small insertions/deletions [4]–[5], gene copy number variation (CNV) and genomic presence-absence variation (PAV) [2], [6]–[7]. Transposable elements, discovered in maize by Barbara McClintock [8]–[9], comprise a significant portion of the maize genome [2], [10]–[13] and can contribute substantially to genomic variation among lines [14]–[17]. There are many examples illustrating the potential for transposons to capture and mobilize genes or gene fragments [14], [18]–[24]. In addition to genetic changes, there is also evidence for epigenetic variation among maize inbred lines. The epigenetic differences vary within maize populations and show relatively stable trans-generational inheritance [25]. These diverse forms of genetic and epigenetic variation likely interact to affect relative transcript abundance, which contributes to phenotypic variation among maize individuals.
Exploring transcriptome variation and elucidating the underlying mechanisms of transcriptional regulation may further our understanding of the molecular bases of phenotypic variation [26]–[27]. Several groups have used microarray profiling to compare the transcriptomes of maize inbreds [28]–[33]. A comparison of the F1 hybrids and the parents revealed that much of the parental variation resulted in additive expression with some rare examples of unexpected expression in the F1 [28], [33]. A recent RNA-seq based analysis of transcriptomic variation in 21 maize elite inbred lines found that a substantial number of genes showed presence/absence expression patterns [34].
Genetical genomics or expression quantitative trait loci (eQTL) mapping is an efficient method for understanding the genetic basis of transcriptome variation [26]–[27], [35]–[36]. eQTL mapping uses transcript abundance as a phenotypic trait and maps the genomic loci controlling the transcript abundance [35]. eQTL are generally classified as cis- or trans- depending on whether they are physically linked to the gene that is regulated or unlinked, respectively. Both cis- and trans-eQTLs have been identified in plants and while trans-eQTLs are more abundant, they generally explain less expression variation than cis-eQTLs [37]–[42]. Several eQTL mapping experiments have utilized microarrays to reveal the complexity of transcriptome variation and their underlying genetic regulators such as trans-eQTL hotspots in human, animals and plants [37], [39], [42]–[44]. eQTL mapping of transcriptome variation has also been employed directly to help dissect phenotypic variation [42], [45]–[46]. The analyses of transcriptome variation in segregating populations have generally focused on exploring how a single locus contributes variation to transcript abundance in a Mendelian fashion. However, there is also the potential for non-Mendelian segregation of gene expression levels [47].
RNA-based sequencing (RNA-Seq) provides several key advantages for transcriptome research including robust expression detection especially for lowly expressed genes, unprecedented access to the fine structure of the transcriptome, and powerful detection of all the transcripts not depending on the reference genome annotation [48]–[49]. Here, we employed RNA-Seq on shoot apices of a well-studied maize intermated RIL population derived from B73 and Mo17 (IBM) [50]. We characterized the relationship of transcriptional variation between the progeny population and the parents in detail to understand how the parental variation combines to affect transcript abundance. This analysis identified a number of genes that exhibit unexpected patterns of expression variation including paramutation-like segregation patterns and presence/absence expression patterns between progeny and parents. Meanwhile, global eQTL mapping, a pair-wise epistasis scan and co-expression analysis were conducted to dissect the possible factors underlying this variation.
Results
Global expression variation in maize was assessed using the intermated B73×Mo17 recombinant inbred line (IBM RIL) population [50]. The IBM RIL population provides higher genetic resolution than conventional RIL populations due to four generations of intermating before self-pollination (Figure 1A) [50]–[51]. RNA-seq was conducted on the shoot apices (4 mm cubic dissected tissue that includes the shoot apical meristem and several leaf primordia) of two-week old seedlings from the inbred lines B73 and Mo17, and 105 recombinant inbred lines (RILs) from the IBM population. In total, 3.47 billion reads (NCBI sequence read archive accession number-SRA054779) were obtained, trimmed for sequence quality and aligned to the B73 genome sequence v2 (AGPv2) [2]. For each genotype, an average of 23.5 million single end reads (93∼102 bp) were uniquely mapped to the annotated genes (Table S1). Based on the uniquely mapped reads, 28,603 genes were expressed in at least 10% of the RILs or at least one of the two parents at a false discovery rate of 0.05. A subset of 22,242 of these genes was expressed in both parents and in at least 90% of the RILs. Prior to further analysis, quantitative Real Time-PCR (qRT-PCR) was employed to assay the accuracy of the RNA-seq results by randomly selecting ten genes that exhibit a range of mean expression-levels. The qRT-PCR results largely confirmed the RNA-seq results, showing the accuracy of RNA-seq for RNA profiling () as in previous studies [48]–[49].

Variation in gene expression levels in RILs
A population of RILs is expected to segregate 1∶1 for the parental alleles and provides an opportunity to examine variation in transcript abundance within the RILs and the relationship between the population and the parents. We first focused on the expression levels of 22,242 genes that were detected in both parents and at least 90% of the IBM RILs. The mean expression levels in the RILs were similar to the mid-parent values for most genes (Figure 1B). Transgressive segregation, defined here as at least 10% of RILs exhibiting expression levels outside the parental range, was observed for 598 genes (2.6%). The other 21,644 (97.4%) genes have expression levels in the RILs that are within the parental range. The level of variation for gene expression levels in the RILs was significantly correlated with the difference between the two parents (Pearson's product-moment correlation: r = 0.728, P<2.2E-16; Figure 1C). The type of distribution for expression levels within the RIL population relative to the parents was assessed using a τ score [52]. We found that 4,822 (22%) genes fit bimodal distributions, 14,564 exhibited normality (65%) and the remaining 2,856 (13%) showed other unclassified distributions. Genes with little or no expression difference among the parents typically exhibited a normal distribution in the RILs (Figure 1D). However, many genes with large expression differences among the parents exhibited a bimodal distribution among the RILs (Figure 1D). These trends indicated that much of the variation in gene expression levels in the RILs is reflective of differences present between the parents.
Paramutation-like expression pattern in RILs
While the majority of genes exhibit expression patterns in the RILs that are quite predictable from the parental levels, there were a subset of genes (0.7%) that have average expression levels in the RILs that are greater than 2-fold different than the mid-parent, indicative of other potential patterns of expression variation. It is possible that some of these genes may have expression patterns similar to those observed for genes that are subject to paramutation such that the expression levels in all RILs would be similar to the expression level of one of the parents [53]. The distribution of expression patterns in the RILs was compared to the parental expression patterns for 8,269 out of 28,603 detected genes that have at least two-fold expression level difference between B73 and Mo17. There were 145 genes (86 of these genes are from the 22,242 genes expressed in both parents and 90% of the RILs) with paramutation-like expression patterns for the RILs in which one parent was within the expression distribution (two standard deviations from the population mean) of the RILs but the other parent had an expression level at least three standard deviations from the population mean (Figure 2A; Figure S2; Table S2). It is important to note that, while these genes exhibit patterns that are similar to those expected due to paramutation these genes may either be directly regulated by paramutation or be secondary targets that are influenced by another factor that is subject to paramutation. For many (80/145) of these genes one of the two parents had an expression level that was outside the range of all RIL genotypes. The expression levels of B73 and Mo17 relative to the population mean and standard deviation helps illustrate several trends observed for these genes (Figure 2A). The majority of these genes (124/145) had patterns in which the RILs were all expressed at levels similar to the lower parent as might be expected given that most examples of paramutation involve a paramutagenic allele that is expressed at lower levels than the paramutable allele (Figure 2B). The expression level for these genes was assessed in the F1 hybrid relative to the two parents (Figure S3). Well characterized examples of paramutation in maize include some examples of dominant expression in the F1 as well as other examples that do not exert effects until the F2 generation [54]–[57]. The genes that had high levels of expression in all RILs were expressed at additive levels in the F1. The genes that had expression levels similar to the lower parent included many examples of additive expression but also had a number of cases with partial to complete dominance in expression such that the F1 had levels more similar to the lower parent (Figure S3). Ten of the genes with paramutation-like patterns were selected for analysis in F2 individuals (Figure S4). Seven of the ten genes exhibited paramutation-like patterns in the F2 individuals and these include examples of both high and low expression.

Mapping the basis of expression level variation
The basis for the regulatory variation in transcript levels was examined using a high-resolution SNP genetic map of the IBM population based upon 7,856 high quality SNP markers derived from the RNA-seq data to perform eQTL analysis for the 22,242 genes that are expressed in both parents and at least 90% of the RILs. This approach is likely to capture much of the variation for gene expression that segregates in a Mendelian fashion but is less likely to capture the basis of variation for examples of gene expression such as those described above. A total of 30,774 eQTLs (α = 0.05) with a threshold logarithm of odds (LOD)> = 4.17 were identified for 19,304 genes, of which 5,303 (27.5%) were controlled only by a single cis-eQTL, 6,201 (32.1%) controlled by both cis- and trans-eQTLs and 7,800 (40.4%) only by trans-eQTLs. The 30,774 eQTLs include 11,504 (∼37%) cis-eQTLs and 19,270 (∼63%) trans-eQTLs (Figure 3A and Table S3). The number of eQTLs affecting the expression level of each gene ranged from zero to six. In general, cis-eQTLs tend to have larger effects than trans-eQTLs (Figure S5A). For example, 83.7% of cis-eQTLs account for at least 20% of the expression variation in contrast to only 12.7% of the trans-eQTL meeting this criterion. However, there are examples of trans-eQTLs that contribute substantially to expression variation. There were 133 trans-eQTLs that contribute at least 60% of the variation for expression of a target gene. The overall contribution of cis- and trans-eQTLs was heavily influenced by the level of expression variation in the parents (Figure S5B). The contribution of cis-eQTLs increased as the parental expression level became increasingly different. In addition, the amount of variation explained by the cis-eQTL also increased as the parental expression levels become more different (Figure S5C) while the amounts of variation explained by trans-eQTL decreased as the parental differences increased (Figure S5D). The proportion of cis- and trans-eQTL for the 598 genes exhibiting transgressive segregation was similar to the proportion of cis- and trans-eQTL for the global eQTL analysis, however, the genes with transgressive segregation were more often (37%) controlled by multiple eQTLs with opposite effects than all genes (27%).
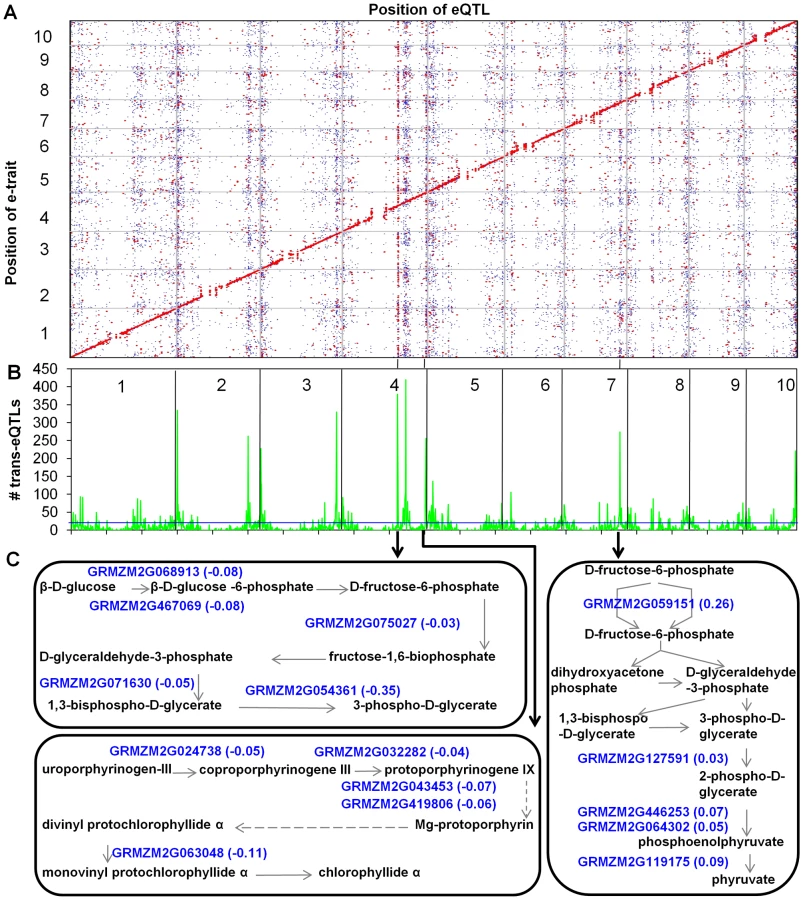
The genomic distribution of trans-eQTL was assessed in an attempt to identify potential trans-eQTL hotspots that might reflect substantial regulatory differences between B73 and Mo17. The analysis of trans-eQTL density in a 1 Mb (which is slightly larger than the average physical distance between adjacent markers with a recombination event) sliding window revealed 96 significant (P<0.01) trans-eQTL hotspots (Figure 3B and Table S4), including 10 major hotspots that contain at least 200 trans-eQTLs (Table 1). These hotspots have many more trans-eQTL than other genomic regions and in the majority (78%) of examples the target genes regulated at the trans-eQTL hotspots show a consistent pattern with significantly more target genes altered in expression in the same direction by the haplotype at the trans-eQTL hotspot (haplotype bias). More examples in which the B73 allele (49) at the trans-eQTL hotspot promoted higher expression of the target loci than the Mo17 allele (26) were identified. The lists of target genes regulated by each of the trans-eQTL hotspots were used to search for GO enrichments; 43% of the trans-eQTL hotspots target lists exhibited enrichments for at least one GO term (Table S5). We performed further analyses for the ten trans-eQTL hotspots that had at least 200 targets (Table 1). Nine of these ten trans-eQTL hotspots showed consistent haplotype bias (six for B73 and three for Mo17) and the targets for each of these hotspots had GO enrichments for at least one term. Multiple genes in the same MaizeCyc pathway [58] are observed to be co-regulated by the same trans-eQTL hotspot (Figure 3C, Table S6). These trans-eQTL hotspots may be due to functional differences in transcriptional regulators. At least in some cases it might be expected that differential expression of a regulator present at the trans-eQTL hotspot is the cause of the differences in trans-regulation.

Structural variants associated with the regulatory variation
To examine the influence of structural rearrangements-gene copy number variation (CNV) and genomic presence/absence variation (PAV) on gene expression, we compared our transcriptomic data for the 28,603 expressed genes with previous Comparative Genomic Hybridization (CGH) data [59]. We focused on the full set of 28,603 genes as the more limited set of 22,242 genes assessed for eQTL analysis required expression to be present in both parents while some of the PAV are expected to abolish expression in Mo17. There are 1,212 expressed genes with CNV/PAVs that affect the gene or flanking regions (Table S7). The structural rearrangements include copy number gains in B73 or Mo17 as well as PAV that are present in B73 but absent in the Mo17 genome. We might expect that copy number gains would lead to increased expression in the genotype with more copies while PAV would only be expressed in one genotype. There was evidence that this was true in many cases (Table S8 and Figure 4). eQTL mapping was conducted on these CNV/PAV-related genes and a total of 1,466 were identified for 1,009 genes, of which 704 (69.8%) were controlled by cis-eQTLs. The cis-eQTLs proportion of genes with CNV/PAVs nearby is significantly higher (P = 0.00) than those of all detected genes (Figure 4A). Noteworthy was the observation that 89.2% of these genes entirely within the PAV were controlled by cis-eQTLs, while ∼10% of these genes have trans-eQTLs, indicating that other regulators underlie the expression variation in addition to PAVs. There was also evidence for an enrichment of cis-acting variation when the CNV/PAV occurred in regions surrounding the gene. Nearly half (120/242) of the genes entirely within structural variants exhibit differential expression in B73 and Mo17. There were many examples in which the RIL genotype at the gene of interest was highly correlated with the expression difference (Figure 4B, 4C). Typically, the copy number of genes entirely within CNV/PAV regions positively correlated with the genes' expression (99 out of the 120 differentially expressed genes between the two parents) (Figure 4B). We also noted examples (21/120) in which a copy number gain was associated with lower expression in the parents (Figure 4C).

Unexpected patterns for presence/absence expression in the RILs
We were struck that a large proportion of genes were only detected in a subset of the RILs or parents. While there were 22,242 genes expressed in both parents and the RILs, there were an additional 6,361 genes that had detectable (False Discovery Rate-FDR>0.05) levels in at least 10% of the RILs or at least one of their parents. These 6,361 genes may include (a) some genes with very low expression levels that manage to cross the threshold of detectability in some samples but not others, (b) genes that are only expressed in one parent and that based on Mendelian segregation would therefore be expected to be expressed in only 50% of the RILs, and (c) genes with unusual regulatory mechanisms. We elected to impose a more restrictive set of filtering criteria for expression to limit the number of low-expressed genes near the detection threshold. Based on the alignment of RNA-seq reads to non-genic genomic regions, an RPKM of 1.03 corresponds to a FDR of 0.01 and 499 of the 6,361 genes have a RPKM value of ≧1.03 in at least 10% of the RILs or at least one of their parents. A substantial proportion of these genes (289/499) were expressed in only one of the parents and were observed in approximately 50% of the RILs (with the Chi-square test at the P value<0.01). The lack of expression in one parent and half of the RILs may reflect differences in genome content or regulatory variation. eQTL analysis of these genes revealed that 186 (64%) of these genes had cis-eQTL that explained >20% of the expression variation and 54 of these genes intersect with CNV/PAVs. However, there were also 92 (32%) of these genes that had evidence for at least one strong trans-eQTL with R2>20%. In total, eQTLs could explain more than 20% of the expression variation of 273 of these 289 genes (96.1%).
The other 210 of these genes exhibited unexpected patterns of expression that could be classified into four groups (Table S9). The type I pattern included 40 genes that were expressed in both parents but were not detected (RPKM = 0) in over 10% of the RILs. The type II pattern included 19 genes that were not detected (RPKM = 0) in the parents but were detected in at least 10% of the RILs. The type III patterns include genes that were expressed in one parent but not the other and had expression in very few RILs (type IIIA – 66 genes) or the majority of the RILs (type IIIB – 85 genes) (Figure 5A). A subset of genes (2 type I genes and 19 type III genes) with unexpected expression patterns also exhibited paramutation-like expression patterns. These unexpected patterns of expression detected by RNA-seq were validated for the majority of genes tested (43/55) using RT-PCR on a subset of the RIL genotypes (Figure S6). In addition, the same type of expression patterns could be observed in an independent set of B73×Mo17 F2 individuals for all the six tested genes (Figure 5B). These RT-PCR assays confirmed that the unexpected segregation patterns for presence or absence of gene expression observed in the RILs are reproducible. Further, genomic PCR was employed to assess if the expression presence/absence transcript variation might be attributable to differences in genome content. We found that genes exhibiting presence/absence transcript variation could be amplified from genomic DNA of each of the IBM RILs that were tested (Figure S7), indicating that the difference in expression was not due to segregation for genomic presence of the sequence. For each of the four patterns, the proportion of RILs expressing a gene was compared to the mean expression level in genotypes that express the gene (Figure 5C). Some of these genes are quite highly expressed and there is a substantial range in the number of genotypes with expression. To further distinguish the genes with unexpected expression patterns from the genes with very low expression levels that manage to cross the threshold of detectability, we examined the maximum expression levels, population mean RPKM and the standard deviations in the population of the genes with unexpected expression patterns in comparison to all detected genes expressed in more than 90% of the RILs. Although the maximum expression levels, population mean RPKMs of genes with unexpected expression patterns are slightly lower than those of all expressed genes, the differences are not significant (Figure S8). Importantly, the standard deviation of expression levels of genes with unexpected expression patterns is similar to that of all other genes (Figure S8).

The observation that there were many examples in which the proportions of RILs with detected expression was close to 25% or 75% (Figure 5C) may suggest that multiple genetic factors play interaction roles underlying the unexpected expression patterns for some of these genes. To test this hypothesis, a genome-wide epistasis scan with all possible pair-wise marker interactions was employed to search for evidence of two-locus interactions that control expression for genes that were detected in approximately one-quarter or three-quarters of the RILs. If two different loci are both required to achieve expression of a gene, these loci could both be present in one parent (type III) or could have one functional locus in either parent (type II). In these examples we would expect 25% of the RILs to exhibit expression of the gene. There are 28 type IIIA and 10 type II genes with expression in only ∼25% of the RILs using Chi-square test with the p-value<0.01 as the cut-off. A genome-wide scan for two-locus interactions that control the variation of expression for these 38 genes found that 92% of these could be explained by a two locus interaction (Figure S9A, S9B). In half of the cases in which a two-locus interaction explained a significant proportion of the expression variation we found that one of the two loci mapped in cis to the gene itself. We could also envision a scenario in which two different loci are required for loss of expression of a gene and this would be expected to result in expression in 75% of the RILs. There are 71 type IIIB and 28 type I genes that are expressed in ∼75% of the RILs and for 91% of these genes the pattern of presence/absence can be explained by a two-locus interaction, including 12 examples in which one of the two loci maps in cis to the gene itself (Figure S9C, S9D). This suggests that a significant subset of the genes with unexpected patterns of presence-absence for expression can be explained by two-locus interactions.
Non-syntenic genes enriched in the genes with unexpected expression patterns
The genes that exhibit presence/absence expression patterns in progeny relative to their parents were further characterized. As a group, these genes with unexpected expression patterns were enriched for single copy genes, and for low copy number gene families relative to all maize genes (Table 2). The FGS (Filtered Gene Set) genes of maize represent an attempt to identify higher confidence gene models and remove gene fragments and transposon-derived sequences [2]. However, there are likely a number of gene fragments and transposon-derived sequences still present within the FGS. Comparative genomic localization can provide more confidence in syntenic genes as “real” genes [60]. Only 36/210 genes with presence/absence expression patterns are in syntenic locations relative to other grass species (Figure 5D). This is a smaller proportion than expected based on the finding that 67.5% of all FGS genes are located in syntenic positions. It is worth noting that while the genes with unexpected patterns are enriched for non-synteny there is a subset of these genes that do have synteny and likely represent functional genes (Table S9). Annotation of the syntenic genes with unexpected presence/absence expression patterns reveal a variety of putative functions such as serine threonine protein kinase, electron transport sco1 family protein and basic leucine-zipper 44 protein, but there is no evidence for GO enrichments within this set of genes.

Genes with unexpected expression patterns are likely to be transposon-related genes
The 174 genes with unexpected segregation patterns that are non-syntenic with other grass species may represent insertions of these genes or gene fragments in the maize genome. To test the hypothesis, the genomic regions surrounding these genes were examined for enrichment of specific classes of repetitive sequences (Figure S10). Over one-third (65) of the 174 genes had a CACTA-like element within 20 kb and these include examples of all types of unexpected expression patterns. This is significant (P = 0.00) enrichment of CACTA-like transposable elements surrounding these genes relative to the expected genomic frequency (Figure 6A). The 65 genes with CACTA-like sequences nearby (3.20 exons) and the other 145 genes with unexpected segregation patterns (3.10 exons) tended to have fewer exons (P = 0.00) than the average exon number (4.88 exons) of all maize genes (Figure 6B). These features, less exons, non-syntenic genomic localization and CACTA-like element enrichment, suggest that many of these genes may be gene fragments that were captured and transposed by CACTA-like transposons.

Transposon-related genes with unexpected expression patterns could regulate the expression of their ancestral syntenic genes
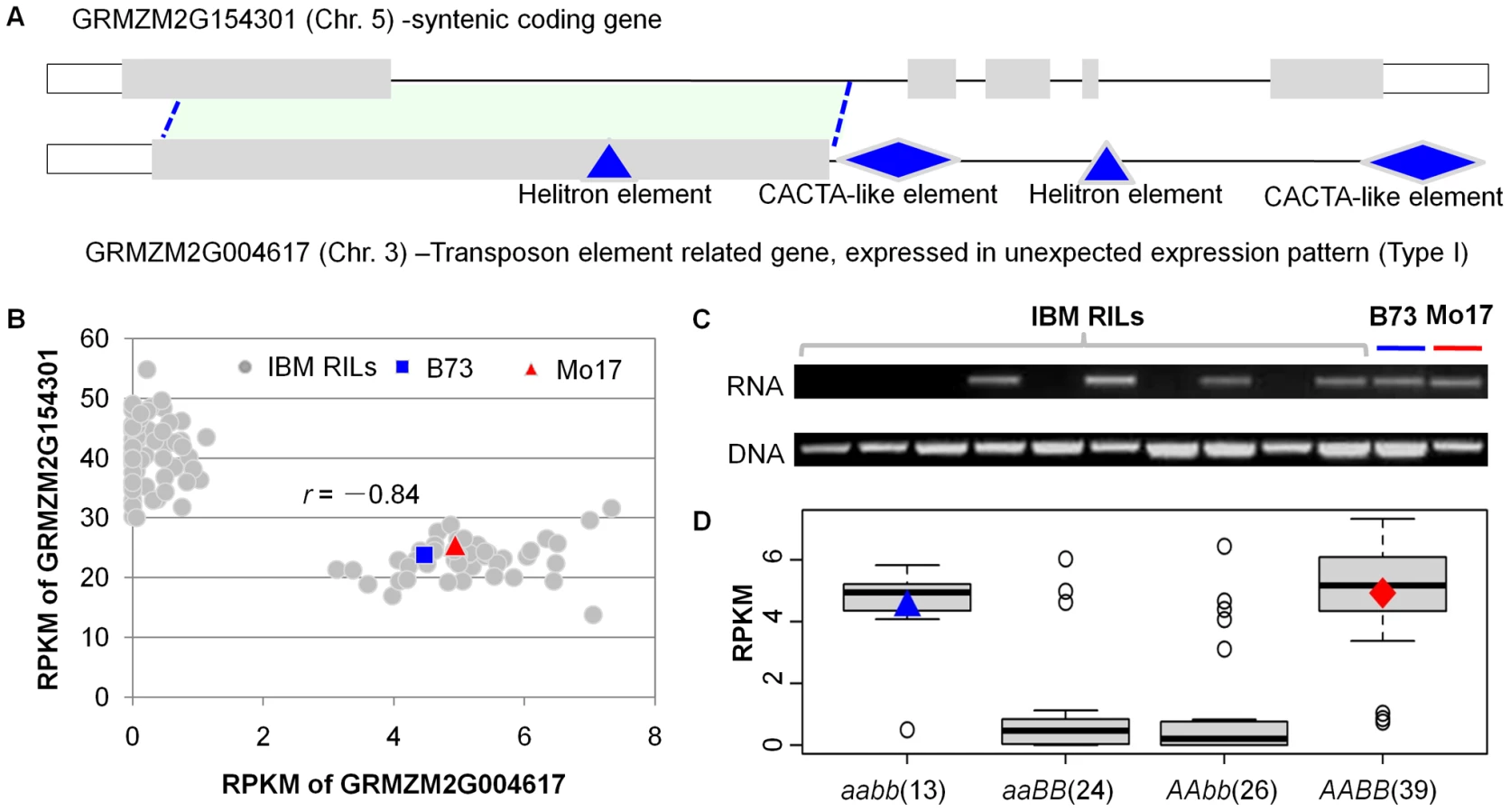
We proceeded to assess whether the non-syntenic gene fragments with presence/absence expression might affect the regulation of homologous full-length syntenic genes (ancestral syntenic genes) elsewhere in the maize genome. All 174 of the non-syntenic genes were homologous to at least a portion of another maize gene (E value<1.0E-10). The correlation between the expression level of each of these genes and the other homologous full-length sequences (possible ancestral syntenic genes) was assessed in the RIL population. There were 25 examples in which the presence/absence expression patterns of the non-syntenic genes were correlated with transcript abundance for ancestral syntenic genes (Table S10). For example, the presence/absence expression of a gene fragment located on chromosome 3 was highly correlated with the abundance of a transcript from its ancestral syntenic gene annotated as an Erwinia Induced Protein 1 located on chromosome 5 (Figure 7A). A comparison of the expression levels for the two sequences revealed an inverse correlation such that the presence/absence of transcripts from the gene fragment correlated with low or high expression of the ancestral syntenic gene (Figure 7B). However, the presence/absence of transcripts from the transposed fragment does not result from genomic differences among RILs because according to the genomic PCR amplifications this gene fragment exists in all tested RILs (Figure 7C). The expression pattern of gene GRMZM2G004617 was also identified to be controlled by two-locus interaction (Figure 7D). Many (20) of the other 25 examples involve similar negative correlations between presence/absence of a gene fragment and abundance of a full-length transcript (Table S10). These examples provide evidence for the ability of transposed gene fragments to influence transcript abundance of their ancestral syntenic genes.
Discussion
We used RNA-Seq to profile the shoot apex transcriptome variation within the maize IBM RIL population and to compare this variation to the parental B73 and Mo17 transcriptomes. In our study, we revealed that: (i) Much of the variation (the population mean, the coefficient of variation) in gene expression levels in progeny is reflective of differences present among the parents; (ii) A genome-wide search for paramutation-like expression identified 145 genes with paramutation-like patterns in the progeny; (iii) Multiple genes in a pathway are regulated in the same direction by a trans-eQTL hotspot, indicative of transcriptional regulators; (iv) CNV/PAVs could be either positively or negatively correlated with expression level variation; (v) A set of 210 genes were identified that exhibited unexpected presence/absence expression patterns within the RILs relative to the parents; and (vi) These genes with unexpected presence/absence expression patterns in the RILs likely include functional genes as well as transposed gene fragments that may contribute to regulatory variation of their ancestral syntenic genes. These findings provide an insightful understanding of the mechanisms that contribute to transcriptome variation in maize populations. We will discuss the identification of trans-eQTL hotspots and the implications for the unexpected segregation patterns of gene expression.
Trans-eQTL hotspots
The analysis of eQTLs allows for the dissection of the genomic regions that affect transcript abundance. Cis-regulatory eQTL reflect regulatory variation that is tightly linked to a gene and affects the allelic expression levels. In contrast, trans-eQTL reflects regulatory variation at unlinked genomic positions. The analysis of all trans-eQTL can reveal trans-eQTL hotspots, also known as trans-eQTL clusters, which are genomic regions that affect the expression of many unlinked loci [39], [61]. These trans-eQTLs are thought to reflect differences in gene regulation that may be important for phenotypic variation [39], [41]–[42], [44].
Due to the limitations of mapping resolution, the identified trans-eQTL hotspots could result from the presence of a single causal regulatory factor (pleiotropic effects) or several tightly linked loci that affect transcript levels of different genes (genetic coupling) [62]. In addition, each trans-eQTL hotspot is relatively large (∼1 Mb) and will likely include the targets of the hotspot itself as well as several other trans-eQTLs that only regulate a small number of genes. Most of the trans-eQTL hotspots identified in our study showed significant haplotype effect bias, which means the haplotype of one parent could increase expression levels of significantly more target genes than expected. The hotspots with haplotype effect bias are more likely to reflect “master regulators”, while some of the others may be a result of genetic linkage, even though we had already taken gene density into account. It might be expected that variation in an important regulatory locus may result in variation for transcript levels for a number of genes that share related GO annotation or are present in the same biochemical pathway. Here, the expression level of genes involved in these pathways were found to be consistently altered in the same direction by trans-eQTL hostpots, which implies that pathway variation may exhibit genetic variation underlying the phenotypic variation among different elite inbred lines.
The regulatory variation provided by the trans-eQTL could be the result of differences in the expression level for a regulator located within the trans-eQTL (a cis-eQTL) or it could be the result of a qualitative variant for a gene located within the genomic region. If the cause of the trans-eQTL hotspot is a cis-regulatory variant then we would expect to find a cis-eQTL located within the trans-eQTL that is highly correlated with the expression level of the target genes. The analysis of these cis-eQTLs located within the trans-eQTL hotspot did not find enrichment for transcription factors. However, we did identify transcription factors or other putative regulatory genes. These candidate genes provide a potential avenue for future research to understand the basis of regulatory variation in maize (Table S11).
Transgressive segregation for eQTL
The majority of genes behave in a manner that is predictable based on the expression levels of the parents. In general, genes with relatively little expression variation in the parental genotypes exhibit a normal distribution of expression levels centered on the parental levels in the offspring and the genes with variation between the parents exhibit a bimodal distribution in the offspring. Our results showed that for the majority of genes expression trait variation is mainly caused by additive effects, which differs from the results observed in Arabidopsis, and rice where non-additive gene action was the more common form of regulating transcript accumulation [39]–[42].
However, a portion of genes exhibit transgressive segregation in the RILs such that at least 10% of the RILs exhibit expression levels outside the parental range. The proportion of transgressive segregation for expression traits was small (2%) compared with the levels reported in other species [39]–[42]. The measurement of eQTL for many genes at once provides an opportunity to assess the potential causes of transgressive segregation. One likely cause of transgressive segregation would be the presence of multiple trans-eQTL including examples in which both parental haplotypes promote expression. For example, if a single gene has two trans-eQTLs for which the B73 allele promotes higher expression and two other trans-eQTLs in which the Mo17 allele promotes higher expression then one might expect to observe a number of RILs with expression levels that are higher or lower than the parental values due to segregation of these trans-eQTLs. Indeed, we found that the 598 genes with transgressive segregation tended to have higher numbers of trans-eQTL than the other genes and that these frequently included a mixture of B73/Mo17 favorable alleles for the underlying gene expression trait.
Unexpected patterns of gene expression in off-spring relative to parents
While the majority of genes behaved in predictable fashions in the RILs relative to parents and had variation that could be attributed to eQTL there were some genes with unexpected expression patterns. We focused our analysis on a couple of subsets of these genes including genes with paramutation-like pattern of expression and genes with unexpected patterns of presence/absence of the transcripts.
When two parents exhibit variation in a trait it would be expected that off-spring would exhibit a similar range of variation. However, we found a number of genes for which none of the recombinant off-spring had expression levels similar to one of the parents. This is an apparent violation of Mendel's principle of segregation and might be reminiscent of paramutation. Paramutation describes instances in which there is communication between two alleles that are present in a heterozygote [53], [63]–[65]. The paramutable allele can be altered to behave more like a paramutagenic allele. Most of the examples of paramutation have been described in maize [64]. These examples include a variety of stabilities and behaviors [64] but are often sensitive to mutations in the same genes [65]–[67]. It has been hypothesized that paramutation will affect numerous other genes but that these other examples may not have been noted due to the lack of observable phenotypes. A recent study in tomato identified several transcripts that had expression patterns in RIL genotypes that were not indicative of the parental levels and could indicate paramutation [47]. We searched for examples of genes that had expression patterns that might be expected to result from paramutation. There were 145 examples of genes for which all of the RILs had expression similar to one of the parents while the other parent had a unique expression pattern. The majority (55%) of these genes represent examples in which the RILs all had expression levels similar to the lower expressing parent. The fact that these patterns were observed in RILs that have been subjected to >6 generations of inbreeding would suggest that these patterns of expression are relatively stable. While we do not have evidence to show direct interaction of the alleles in the heterozygote, we propose that the expression patterns observed for many of these 145 genes are the result of paramutation-like phenomena. Our analysis of expression in a RIL population relative to the parents suggests that paramutation-like mechanisms may contribute to regulatory variation for a number of maize loci. The analysis of F2 individuals provided further evidence for paramutation-like patterns for seven of the ten genes tested. It is possible that some of these examples may reflect spontaneous mutation or epimutation in the specific B73 and Mo17 individuals that were used for this study and these may account for the lack of validation for some examples. We also examined our dataset for genes whose expression was only detectable in a subset of the RIL population or at least one of the parents. Nearly 500 genes with various patterns of segregation for the presence/absence of transcripts were identified using a relatively stringent (FDR = 0.01) expression threshold. If the threshold for detection was relaxed (FDR = 0.05), the number of genes with segregation for presence/absence of transcripts increased to 4,689. These results suggest the presence of substantial qualitative as well as quantitative variation for the maize transcriptome following segregation. We further evaluated these genes to begin to understand the causes and consequences for this variation.
The most likely cause for variation in presence/absence of a transcript would be examples in which one parent expresses a gene and the other parent does not. In these instances we would expect approximately 50% of the RIL progeny to exhibit expression of the gene. Over half (289/489) of the genes with segregation for the presence of transcripts exhibit this type of pattern. This pattern could be caused by a strong cis-regulatory variant or actual difference in genome content such as PAV [6], [59]. The mapping of regulatory variation for these 289 genes revealed that many of them can be attributed to variation mapping to the location of the gene itself and likely reflect sequence differences in regulatory regions or content variation. Alternatively, the presence/absence of a transcript could reflect a strong trans-regulatory variant and a subset of the genes do exhibit trans-eQTL. This set of genes with expression in one parent and roughly 50% of RILs are expected based on previous studies of maize genome content variation and regulatory variation [68].
Many of the genes with segregation for the presence of transcripts exhibit other, unexpected, patterns of expression. These include genes that are expressed in both parents but a few RILs, genes expressed in neither parent but many of the RILs and other patterns. These segregation patterns are not expected to result from traditional single, gene segregation. We did not find evidence that there was segregation for the presence/absence of these genes within the genomic DNA of progeny. It is quite possible that many of these unexpected patterns of segregation for transcript presence reflect epigenetic or small-RNA based regulatory mechanisms. For instance, an example from tomato illustrates that a miRNA present in one of the parents can become detectably expressed in all the hybrids and their progeny [47]. In addition, there are examples of molecular dominance in siRNA levels and DNA methylation in Arabidopsis F1 plants [69]–[70]. It will be important to further understand the mechanisms that generate these unexpected patterns of segregation to understand the inheritance of traits in RIL populations.
There is a growing appreciation for the qualitative variation among the genomes and transcriptomes of maize inbreds. Inbreds of maize can have substantial variation for gene content [6]–[7], [59], [71]. These inbreds can also have substantial variation for the presence of transcripts [29], [34]. The F1 genotypes will contain the full set of genes found in both parents and generally tend to express this full set leading to a potential contribution to heterosis [72]. In this study, we showed that the RILs can also vary in transcriptome content relative to the parental genotypes. This leads to questions about the functional consequences of variation in transcriptome content. Many of the studies on genome content and variation in transcriptome content have found that the variable genes are under-represented for syntenic genes with functional annotations. Consistently, we found that only 36 of the 210 genes with unexpected patterns of segregation for expression were located in syntenic chromosomal positions. The variation for the presence of expression for these genes may directly impact phenotypes. The other 174 genes include a number of inserted sequences relative to gene order in other grass species. The maize genome is known to be littered with gene fragments that have been captured and mobilized by transposons [14], [22]–[23], [73]. In many cases, the presence of these gene fragments is variable among maize genotypes [14]–[15], [74] and can contribute to novel transcripts [24]. Here we provide evidence that the presence/absence of transcripts from these gene fragments can act to modulate the expression level of the full-length parent gene. This suggests that some of the qualitative variation for gene fragment transcripts acts to provide a trans-acting regulator for the full-length gene and suggests a mechanism for the origin of selectable variation in expression level for single genes.
Materials and Methods
Plant materials
A maize IBM (Intermated B73×Mo17) RIL population derived from the cross of the inbred lines B73 and Mo17 [50] was used to assess segregation of gene expression. At least 10 seedlings per genotype of 105 IBM RILs and their parents were planted in a single growth chamber. A randomized block design was employed with three replicates. The order of the flats within each block was rotated daily to minimize the effects of local environmental variation. Fourteen days after planting, at least 6 healthy seedlings were harvested and a 4 mm cubic tissue including the shoot apex were dissected and pooled for each genotype-replication combination. After separately grinding tissue from each genotype-replication pool in liquid nitrogen, RNA was extracted using the TRIzol and Qiagen RNeasy mini kit following the manufacturer's instructions.
RNA–seq and bioinformatic analysis
The three replicate RNA samples of each genotype were pooled with barcoding. RNA sequencing libraries were prepared and sequenced using the Illumina Hi-Seq2000 with 103–110 cycles. The resulting sequencing data were trimmed and aligned to the B73 reference genome v2 (AGPv2) [2] by Data2Bio (http://www.data2bio.com/). The majority (69–80%) of the trimmed reads were uniquely mapped and 94% of mapped reads were located in annotated gene regions. The uniquely-mapped reads were further analyzed for SNPs and read counts per genes in the RILs and their parents. RPKM values were determined using Cufflinks v0.9.3 (http://cufflinks.cbcb.umd.edu/) based on the uniquely mapped reads of each genotype. The AGP v2 5b maize genome annotation was used as a reference, while maximum intron length = 60,000 bp and the quartile normalization option were employed. To establish a threshold for detectable expression, we conducted global permutation tests with 10,000 randomly selected non-genic fragments from B73 RNA-seq data [75]. We found the RPKMs were 0.055, 1.03, 2.02 and 5.41 as cutoffs for gene expression at different significant levels of FDR = 0.05, 0.01, 0.005 and 0.001, respectively. For the initial analyses, a transcript presence/absence was assessed using a threshold of 0.055 RPKM. For the more stringent analysis of unexpected segregation patterns a threshold of 1.03 RPKM was employed and gene presence required values >1.03 and absence required a value of 0.0. Intermediate values were not assigned presence or absence calls.
Global expression analysis
The 22,242 genes expressed in more than 90% of the RILs and the parents were used to interrogate the global expression variation. The population mean and coefficient of variation of gene expression levels were summarized for the attributes of the RIL population, whereas the absolute value of log2 of the expression-level in B73 divided by the level in Mo17 was used for the expression fold change between B73 and Mo17. The Kolmogorov-Smirnov test was applied to judge whether the expression levels of genes fit a normal distribution in the RIL population. The τ statistic, introduced by Bessarabova et al. [52], was employed to distinguish between one-modal (normal) and bimodal distributions. We simulated 10,000 normal distribution data (μ = 0, σ = 1), each containing 105 numbers, to obtain the global threshold of τ = 3.24 (P = 0.01). We treated the expression levels, which did not fit either normal or bimodal distribution, as unclassified distribution. The relationship between coefficient of variation and abs(log2(B73/Mo17)), and the relationship between τ value and abs(log2(B73/Mo17)) of the variation of global gene expression were assessed by Pearson's product-moment correlation analysis in R (http://www.R-project.org).
Ten randomly selected genes with expression-level (RPKM) ranging from 0.05 to 2552.91 were selected to validate the expression profiling accuracy of RNA-seq by quantitative RT-PCR (qRT-PCR) using the same RNA samples as the ones used for RNA-seq. For qRT-PCR, cDNA samples ware amplified using the iQ SYBR Green Supermix on the CFX96 Real-Time PCR detection system (Bio-Rad, Hercules, CA). Each PCR reaction contained 25 µl of reagent, consisting of 5 µl cDNA; 12.5 µl of the iQ SYBR Green Supermix; 2.5 µl of nuclease-free water; and 5 µl of the forward and reverse primers (1 µM stock). The qRT-PCR conditions included an initial incubation at 95°C for 3 min, followed by 40 cycles of 95°C for 10 sec, 58°C for 20 sec, and 72°C for 25 sec.
To test the expression pattern of the paramutation-like genes, we examined gene expression in the shoot apex from 18 individuals from an F2 population derived from a cross between B73 and Mo17. The F2 individuals, Mo17 and B73 were grown in a growth chamber using similar conditions as those used to obtain the RNA-seq data from the RIL population. RNA samples from the shoot apex were isolated from 2-week old seedlings and reverse-transcribed into the first strand cDNAs for the qRT-PCR quantification. Ten randomly selected paramutation-like genes were examined for the relative quantitation of expression level in the F2 individuals and their parents. qRT-PCR was performed with the SYBR Green master mix according to the manufacturer's instructions (Applied Biosystems, Carlsbad, California). Three replicates were conducted to calculate the average and standard deviation of expression levels. The 2−ΔΔCT method was employed to calculate the relative quantitation of expression levels with the housekeeping gene Actin as the endogenous control and B73 as the reference genotype.
To validate the unexpected expression patterns we conducted two experiments. In the first experiment, we replanted 10 IBM RIL genotypes, using the same growth conditions as used in the RNA-seq experiment, with 10 plants per genotype and sampled the shoot apices of the seedlings 14 days after planting. RNA was isolated from at least 6 healthy plants per genotype. In the second experiment, we tested the expression variation of genes with unexpected expression patterns in 18 individuals from an F2 population derived from a cross between B73 and Mo17. A total of 55 genes with unexpected expression patterns were randomly selected for validation. RT-PCR was conducted using a Touchdown PCR program [76]. Two cycling phrases were set for the Touchdown PCR program: the TM reduced from 72°C to 62°C by 1°C every successive cycle in the first phrase with 10 cycles, while 25 other cycles were used for the amplification in the second phrase with TM = 62°C. Thus, 35 cycles were conducted. We also conducted genomic DNA PCR amplifications on the same RILs with the Touchdown PCR program on 8 randomly selected genes with unexpected expression patterns to check whether the extraordinary expression occurred only at the transcript level. The concentration of the template cDNA and DNA was 10 ng/µl for all the validations of RT-PCR and genomic PCR. All primer information can be found in Table S12.
To examine the expression patterns in hybrids of B73 and Mo17 for the paramutation-like genes, we dissected shoot apices from 10 plants from B73, Mo17 and their reciprocal hybrids, isolated RNA and conducted RNA-seq. For this experiment, the plants were grown in the same growth chamber conditions used for the original RNA-seq experiment, using consistent protocols for sampling, library preparation, RNA-seq and analysis.
For the analyses of attributes of genes with unexpected presence/absence expression patterns, we downloaded the gene family information of the whole B73 gene set from EnsemblPlants (http://plants.ensembl.org/index.html). Gene family relationships were constructed through EnsemblCompara GeneTrees by using the phylogenetic approach [77]. The syntenic information of maize genes was obtained from the CoGe database (http://genomevolution.org/CoGe/).
Transposon enrichment effect analyses
We annotated 20 Kb of flanking sequence for the genes with unexpected expression patterns (Type I, Type II and Type IIIA and Type IIIB) in 5 Kb windows as a fragment Bin by RepeatMasker (http://repeatmasker.org). As controls, 210 genes were randomly selected and 10,000 permutations were conducted. Then, we annotated the adjacent fragments from 5 Kb upstream and downstream for all the FGS and summarized the number of all the different kinds of transposon-like sequences in the adjacent fragment of genes.
eQTL mapping
Data2Bio (http://www.data2bio.com/) identified 648,230 putative SNPs in 28,603 genes (72% of all maize genes) using RNA-Seq reads from the RILs and their parents. High quality unique SNP markers with minimal missing data in the RILs were selected, grouped and integrated into chromosomes before constructing the genetic map. Maximum Likelihood Estimation with minimal threshold LOD score = 3.0 by JoinMap 4.0 [78] was employed to construct a high-resolution genetic map. The expression-levels of 22,242 genes were treated as expression traits (e-traits) for the global gene eQTL mapping. The genetic determinants controlling variation in e-traits were mapped via composite interval mapping [79]–[80] with a walking speed of 1 cM in the procedure of SRmapqtl and Zmapqtl of QTL cartographer [81]. A global permutation with 1000 randomly selected e-traits×1000 replicates were done as a representative null distribution of 1,000,000 maximum likelihood ratio test (LRT) statistics. A global permutation threshold as the significant cutoff of eQTL mapping was obtained at a significance level of 0.05, giving a likelihood ratio test value of 19.23, which corresponds to a Logarithm of Odds (LOD) score of 4.17. The range with a 1.0 LOD drop on each side from the LOD peak point was selected as the confidence interval. If two adjacent peaks overlap in less than 10 cM, we treated them as one eQTL. A global permutation of randomly distribution of trans-eQTLs along the whole maize genome was performed to find the threshold of trans-eQTLs hotspots. One thousand of the maximum number of trans-eQTL scattering in 1 Mb genomic region of each permutation were obtained to compute the cutoff of hotspots. Further, we took gene density into account to rule out the gene number factor for the identification of trans-eQTL hotspots. For global trans-eQTLs hotspots, the cutoff (#_trans-eQTLs/(Mb×#_genes)) was 1.25. The GO enrichments and the pathway enrichments of the regulated genes by hotspots were conducted using BiNGO plugin [82] in Cytoscape [83] based on the annotation information from AgriGO [84] and MaizeCyc database [58], respectively.
Epistasis scan of the transcriptomic variations of genes with unexpected expression patterns
The epistasis scan with all possible pairwise marker interactions for the genes with unexpected expression patterns was conducted with a generalized linear model. We employed an α-level of 0.05 (P<2.1E-06), which was adjusted by following the suggestion of dividing the α-level by the number of possible independent pairwise interactions among recombinant blocks [85].
Relating CNVs to transcriptome variation
We obtained genomic variation information between B73 and Mo17 from Springer et al. 2009. The formula of CGH signal abundance of B73 and Mo17 of log2(Mo17/B73) were used to classify different CGH types [59]. The segments with a peak at log2(Mo17/B73) = 0 were simply classified as B = M, while the segments with a peak at log2(Mo17/B73) = 20.43 were classified as B73<Mo17_SNP. B = M_int represents segments with an intermediate value between 0 and 20.43. Mo17>B73_CNV shows segments that are predicted to occur in more copies in Mo17 than in B73. B>M_CNV indicates segments containing significantly more copies in B73 than in Mo17, while B>M_int represents segments having intermediate more copies in B73 than in Mo17. B>M_PAV shows segments present in B73 but absent in Mo17. Of these genomic variants we mainly focused on CGH segments B>M_int, B>M_CNV, B>M_PAV and M>B_CNV for the relationship analyses between genomic variation and transcriptome variation in the maize IBM RIL population. First, we coordinated genes with CGH segments by coding scripts to compare the coordinates of genes (according to the annotation of the maize reference genome AGPv2) with the CGH segments. Four main relationships could be obtained as genes entirely within CGH segments, genes intersecting CGH segments, genes in regions having multiple CGH segments, and other. Second, we filtered expressed genes and CGH segments. We limited the analysis to the expressed genes, which we defined as those displaying a normalized expression value (RPKM) of at least 1.03 (corresponding to 21 reads per gene, FDR = 0.01) in more than 40% of the samples. Further, we considered the pair-wise datasets between genes and CNVs only if genes were expressed in at least 40 samples for each inferred genotype (B73 and Mo17) in the RIL population. Finally, we conducted eQTL mapping of genes with CNVs nearby, for the inference of associations between structural variation and expression levels.
RNA–seq data availability
The raw RNA-seq data on shoot apices of the IBM RIL population used in this study were submitted to NCBI's Sequence Read Archive (SRA) with accession number SRA055066 and will be released to public after approval of publication. The transcriptome profiling data were also deposited in MaizeGDB (http://www.maizegdb.org/).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ChenZJ (2007) Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol 58 : 377–406.
2. SchnablePS, WareD, FultonRS, SteinJC, WeiF, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326 : 1112–1115.
3. GoreMA, ChiaJM, ElshireRJ, SunQ, ErsozES, et al. (2009) A first-generation haplotype map of maize. Science 326 : 1115–1117.
4. BhattramakkiD, DolanM, HanafeyM, WinelandR, VaskeD, et al. (2002) Insertion-deletion polymorphisms in 3′ regions of maize genes occur frequently and can be used as highly informative genetic markers. Plant Mol Biol 48 : 539–547.
5. LaiJ, LiR, XuX, JinW, XuM, et al. (2010) Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42 : 1027–1030.
6. BelóA, BeattyMK, HondredD, FenglerKA, LiB, et al. (2010) Allelic genome structural variations in maize detected by array comparative genome hybridization. Theor Appl Genet 120 : 355–367.
7. Swanson-WagnerRA, EichtenSR, KumariS, TiffinP, SteinJC, et al. (2010) Pervasive gene content variation and copy number variation in both maize and its undomesticated progenitor. Genome Res 20 : 1689–1699.
8. McClintockB, NeurosporaI (1945) Preliminary observations of the chromosomes of Neurospora crassa. Am J Bot 1945 : 671–678.
9. McClintockB (1953) Induction of instability at selected loci in maize. Genetics 38 : 579–599.
10. SanMiguelP, TikhonovA, JinYK, MotchoulskaiaN, ZakharovD, et al. (1996) Nested retrotransposons in the intergenic regions of the maize genome. Science 274 : 765–768.
11. SanMiguelP, BennetzenJL (1998) Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Annals Bot 82 : 37–44.
12. MeyersBC, TingeySV, MorganteM (2001) Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res 11 : 1660–1676.
13. MessingJ, BhartiAK, KarlowskiWM, GundlachH, KimHR, et al. (2004) Sequence composition and genome organization of maize. Proc Natl Acad Sci USA 101 : 14349–14354.
14. LaiJ, LiY, MessingJ, DoonerHK (2005) Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc Natl Acad Sci USA 102 : 9068–9073.
15. MorganteM, BrunnerS, PeaG, FenglerK, ZuccoloA, et al. (2005) Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet 37 : 997–1002.
16. DoonerHK, HeL (2008) Maize genome structure variation: interplay between retrotransposon polymorphisms and genic recombination. Plant Cell 20 : 249–258.
17. TenaillonMI, HuffordMB, GautBS, Ross-IbarraJ (2011) Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol Evol 3 : 219–229.
18. JinYK, BennetzenJL (1994) Integration and nonrandom mutation of a plasma membrane proton ATPase gene fragment within the Bs1 retroelement of maize. Plant Cell 6 : 1177–1186.
19. KapitonovVV, JurkaJ (2001) Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA 98 : 8714–8719.
20. JiangN, BaoZ, ZhangX, EddySR, WesslerSR (2004) Pack-MULE transposable elements mediate gene evolution in plants. Nature 431 : 569–573.
21. GuptaS, GallavottiA, StrykerGA, SchmidtRJ, LalSK (2005) A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudogenes. Plant Mol Biol 57 : 115–127.
22. BennetzenJL (2005) Transposable elements, gene creation and genome rearrangement in flowering plants. Curr Opin Genet Dev 15 : 621–627.
23. LiQ, LiL, DaiJR, LiJS, YanJB (2009) Identification and characterization of CACTA transposable elements capturing gene fragments in maize. Chin Sci Bull 54 : 642–651.
24. BarbagliaAM, KlusmanKM, HigginsJ, ShawJR, HannahLC, et al. (2012) Gene capture by helitron transposons reshuffles the transcriptome of maize. Genetics 190 : 965–975.
25. EichtenSR, Swanson-WagnerRA, SchnableJC, WatersAJ, HermansonPJ, et al. (2011) Heritable epigenetic variation among maize inbreds. PLoS Genet 7: e1002372 doi:10.1371/journal.pgen.1002372.
26. HollandJB (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10 : 156–161.
27. KliebensteinD (2009) Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu Rev Plant Biol 60 : 93–114.
28. Swanson-WagnerRA, JiaY, DeCookR, BorsukLA, NettletonD, et al. (2006) All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci USA 103 : 6805–6810.
29. StuparRM, SpringerNM (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 173 : 2199–2210.
30. GuoM, RupeMA, YangX, CrastaO, ZinselmeierC, et al. (2006) Genome-wide transcript analysis of maize hybrids: allelic additive gene expression and yield heterosis. Theor Appl Genet 113 : 831–845.
31. MeyerS, PospisilH, ScholtenS (2007) Heterosis associated gene expression in maize embryos 6 days after fertilization exhibits additive, dominant and overdominant pattern. Plant Mol Biol 63 : 381–391.
32. UzarowskaA, KellerB, PiephoHP, SchwarzG, IngvardsenC, et al. (2007) Comparative expression profiling in meristems of inbred-hybrid triplets of maize based on morphological investigations of heterosis for plant height. Plant Mol Biol 63 : 21–34.
33. StuparRM, GardinerJM, OldreAG, HaunWJ, ChandlerVL, et al. (2008) Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol 8 : 33.
34. HanseyCN, VaillancourtB, SekhonRS, de LeonN, KaepplerSM, et al. (2012) Maize (Zea mays L.) genome diversity as revealed by RNA-sequencing. PLoS ONE 7: e33071 doi:10.1371/journal.pone.0033071.
35. DamervalC, MauriceA, JosseJM, de VienneD (1994) Quantitative trait loci underlying gene product variation: a novel perspective for analyzing regulation of genome expression. Genetics 137 : 289–301.
36. JoosenRV, LigterinkW, HilhorstHW, KeurentjesJJ (2009) Advances in genetical genomics of plants. Curr Genomics 10 : 540–549.
37. SchadtEE, MonksSA, DrakeTA, LusisAJ, CheN, et al. (2003) Genetics of gene expression surveyed in maize, mouse and man. Nature 422 : 297–302.
38. DeCookR, LallS, NettletonD, HowellSH (2006) Genetic regulation of gene expression during shoot development in Arabidopsis. Genetics 172 : 1155–1164.
39. WestMA, KimK, KliebensteinDJ, van LeeuwenH, MichelmoreRW, et al. (2007) Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175 : 1441–1450.
40. JordanMC, SomersDJ, BanksTW (2007) Identifying regions of the wheat genome controlling seed development by mapping expression quantitative trait loci. Plant Biotechnol J 5 : 442–453.
41. PotokinaE, DrukaA, LuoZ, WiseR, WaughR, et al. (2008) Gene expression quantitative trait locus analysis of 16 000 barley genes reveals a complex pattern of genome-wide transcriptional regulation. Plant J 53 : 90–101.
42. WangJ, YuH, XieW, XingY, YuS, et al. (2010) A global analysis of QTLs for expression variations in rice shoots at the early seedling stage. Plant J 63 : 1063–1074.
43. Swanson-WagnerRA, DeCookR, JiaY, BancroftT, JiT, et al. (2009) Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 326 : 1118–1120.
44. HollowayB, LuckS, BeattyM, RafalskiJA, LiB (2011) Genome-wide expression quantitative trait loci (eQTL) analysis in maize. BMC Genomics 12 : 336.
45. WentzellAM, RoweHC, HansenBG, TicconiC, HalkierBA, et al. (2007) Linking metabolic QTLs with network and cis-eQTLs controlling biosynthetic pathways. PLoS Genet 3: e162 doi:10.1371/journal.pgen.0030162.
46. MoscouMJ, LauterN, SteffensonB, WiseRP (2011) Quantitative and qualitative stem rust resistance factors in barley are associated with transcriptional suppression of defense regulons. PLoS Genet 7: e1002208 doi:10.1371/journal.pgen.1002208.
47. ShivaprasadPV, DunnRM, SantosBA, BassettA, BaulcombeDC (2011) Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J 31 : 257–266.
48. WangZ, GersteinM, SnyderM (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 : 57–63.
49. MontgomerySB, SammethM, Gutierrez-ArcelusM, LachRP, IngleC, et al. (2010) Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464 : 773–777.
50. LeeM, SharopovaN, BeavisWD, GrantD, KattM, et al. (2002) Expanding the genetic map of maize with the intermated B73×Mo17 (IBM) population. Plant Mol Biol 48 : 453–461.
51. CandelaH, HakeS (2008) The art and design of genetic screens: maize. Nat Rev Genet 9 : 192–203.
52. BessarabovaM, KirillovE, ShiW, BugrimA, NikolskyY, et al. (2010) Bimodal gene expression patterns in breast cancer. BMC Genomics Suppl 1: S8.
53. ChandlerVL (2007) Paramutation: from maize to mice. Cell 126 : 641–645.
54. BrinkRA (1956) A genetic change associated with the R locus in maize which is directed and potentially reversible. Genetics 41 : 872–889.
55. BrinkRA (1973) Paramutation. Annu Rev Genet 7 : 129–152.
56. Mittelsten ScheidO, AfsarK, PaszkowskiJ (2003) Formation of stable epialleles and their paramutation-like interaction in tetraploid Arabidopsis thaliana. Nature Genet 34 : 450–454.
57. StokesTL, RichardsEJ (2002) Induced instability of two Arabidopsis constitutive pathogen-response alleles. Proc Natl Acad Sci USA 99 : 7792–7796.
58. MonacoMK, SenTZ, RenL, SchaefferM, AmarasingheV, et al. (2012) MaizeCyc: Metabolic Networks in Maize. Plant & Animal Genome XX: 858–858.
59. SpringerNM, YingK, FuY, JiT, YehCT, et al. (2009) Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet 5: e1000734 doi:10.1371/journal.pgen.1000734.
60. SchnableJC, FreelingM (2011) Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6: e17855 doi:10.1371/journal.pone.0017855.
61. GiladY, RifkinSA, PritchardJK (2008) Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet 24 : 408–415.
62. SinghND, ShawKL (2012) On the scent of pleiotropy. Proc Natl Acad Sci USA 109 : 5–6.
63. BrinkRA, MikulaB (1958) Plant color effects of certain anomalous forms of the Rr allele in maize. Z Ind Abst Vererb 89 : 94–102.
64. ChandlerVL, StamM (2004) Chromatin conversations: mechanisms and implications of paramutation. Nat Rev Genet 5 : 532–544.
65. ErhardKFJr, HollickJB (2011) Paramutation: a process for acquiring trans-generational regulatory states. Curr Opin Plant Biol 14 : 210–216.
66. DorweilerJE, CareyCC, KuboKM, HollickJB, KermicleJL, et al. (2000) Mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell 12 : 2101–2118.
67. Arteaga-VazquezMA, ChandlerVL (2010) Paramutation in maize: RNA mediated trans-generational gene silencing. Curr Opin Genet Dev 20 : 156–163.
68. SchlattlA, AndersS, WaszakSM, HuberW, KorbelJO (2011) Relating CNVs to transcriptome data at fine resolution: assessment of the effect of variant size, type, and overlap with functional regions. Genome Res 21 : 2004–2013.
69. GroszmannM, GreavesIK, AlbertynZI, ScofieldGN, PeacockWJ, et al. (2011) Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci USA 108 : 2617–2622.
70. GreavesIK, GroszmannM, YingH, TaylorJM, PeacockWJ, et al. (2012) Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci USA 109 : 3570–3575.
71. FuH, DoonerHK (2002) Intraspecific violation of genetic colinearity and its implications in maize. Proc Natl Acad Sci U S A 99 : 9573–9578.
72. SpringerNM, StuparRM (2007) Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 19 : 2391–2402.
73. BrunnerS, FenglerK, MorganteM, TingeyS, RafalskiA (2005) Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17 : 343–360.
74. DuC, FefelovaN, CaronnaJ, HeL, DoonerHK (2009) The polychromatic Helitron landscape of the maize genome. Proc Natl Acad Sci USA 106 : 19916–19921.
75. LiP, PonnalaL, GandotraN, WangL, SiY, et al. (2009) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42 : 1060–1067.
76. KorbieDJ, MattickJS (2008) Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc 3 : 1452–1456.
77. VilellaAJ, SeverinJ, Ureta-VidalA, HengL, DurbinR, et al. (2009) EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19 : 327–335.
78. Van Ooijen JW (2006) JoinMap 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands.
79. ZengZB (1993) Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc Natl Acad Sci USA 90 : 10972–10976.
80. ZengZB (1994) Precision mapping of quantitative trait loci. Genetics 136 : 1457–1468.
81. Basten CJ, Weir BS, Zeng ZB. 2004. QTL Cartographer Version 1.17. Department of Statistics, North Carolina State University, Raleigh NC.
82. MaereS, HeymansK, KuiperM (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21 : 3448–3449.
83. ShannonP, MarkielA, OzierO, BaligaNS, WangJT, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13 : 2498–2504.
84. DuZ, ZhouX, LingY, ZhangZ, SuZ (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38: W64–70.
85. HollandJB, PortyankoVA, HoffmannDL, LeeM (2002) Genomic regions controlling vernalization and photoperiod responses in oat. Theor Appl Genet 105 : 113–126.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy