
Tbx2 Controls Lung Growth by Direct Repression of the Cell Cycle Inhibitor Genes and
Vertebrate organ development relies on the precise spatiotemporal orchestration of proliferation rates and differentiation patterns in adjacent tissue compartments. The underlying integration of patterning and cell cycle control during organogenesis is insufficiently understood. Here, we have investigated the function of the patterning T-box transcription factor gene Tbx2 in lung development. We show that lungs of Tbx2-deficient mice are markedly hypoplastic and exhibit reduced branching morphogenesis. Mesenchymal proliferation was severely decreased, while mesenchymal differentiation into fibrocytes was prematurely induced. In the epithelial compartment, proliferation was reduced and differentiation of alveolar epithelial cells type 1 was compromised. Prior to the observed cellular changes, canonical Wnt signaling was downregulated, and Cdkn1a (p21) and Cdkn1b (p27) (two members of the Cip/Kip family of cell cycle inhibitors) were strongly induced in the Tbx2-deficient lung mesenchyme. Deletion of both Cdkn1a and Cdkn1b rescued, to a large degree, the growth deficits of Tbx2-deficient lungs. Prolongation of Tbx2 expression into adulthood led to hyperproliferation and maintenance of mesenchymal progenitor cells, with branching morphogenesis remaining unaffected. Expression of Cdkn1a and Cdkn1b was ablated from the lung mesenchyme in this gain-of-function setting. We further show by ChIP experiments that Tbx2 directly binds to Cdkn1a and Cdkn1b loci in vivo, defining these two genes as direct targets of Tbx2 repressive activity in the lung mesenchyme. We conclude that Tbx2-mediated regulation of Cdkn1a and Cdkn1b represents a crucial node in the network integrating patterning information and cell cycle regulation that underlies growth, differentiation, and branching morphogenesis of this organ.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003189
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003189
Summary
Vertebrate organ development relies on the precise spatiotemporal orchestration of proliferation rates and differentiation patterns in adjacent tissue compartments. The underlying integration of patterning and cell cycle control during organogenesis is insufficiently understood. Here, we have investigated the function of the patterning T-box transcription factor gene Tbx2 in lung development. We show that lungs of Tbx2-deficient mice are markedly hypoplastic and exhibit reduced branching morphogenesis. Mesenchymal proliferation was severely decreased, while mesenchymal differentiation into fibrocytes was prematurely induced. In the epithelial compartment, proliferation was reduced and differentiation of alveolar epithelial cells type 1 was compromised. Prior to the observed cellular changes, canonical Wnt signaling was downregulated, and Cdkn1a (p21) and Cdkn1b (p27) (two members of the Cip/Kip family of cell cycle inhibitors) were strongly induced in the Tbx2-deficient lung mesenchyme. Deletion of both Cdkn1a and Cdkn1b rescued, to a large degree, the growth deficits of Tbx2-deficient lungs. Prolongation of Tbx2 expression into adulthood led to hyperproliferation and maintenance of mesenchymal progenitor cells, with branching morphogenesis remaining unaffected. Expression of Cdkn1a and Cdkn1b was ablated from the lung mesenchyme in this gain-of-function setting. We further show by ChIP experiments that Tbx2 directly binds to Cdkn1a and Cdkn1b loci in vivo, defining these two genes as direct targets of Tbx2 repressive activity in the lung mesenchyme. We conclude that Tbx2-mediated regulation of Cdkn1a and Cdkn1b represents a crucial node in the network integrating patterning information and cell cycle regulation that underlies growth, differentiation, and branching morphogenesis of this organ.
Introduction
The development of organs and organisms depends on the precise control of the progression through and the exit from the cell cycle to achieve appropriate patterns of proliferation and differentiation in time and space. Progression through the cell cycle is regulated predominantly by a series of serine/threonine kinases, the cyclin-dependent kinases (CDKs) that link proliferative signals with mechanical aspects of cell duplication. CDK function is controlled by a variety of mechanisms, including a group of molecules that inhibits CDK activity by complex formation. These CDK inhibitors (CKIs) have been categorized into two families, the Cip/Kip (Cdkn1) family with three members in mammals (Cdkn1a, Cdkn1b, Cdkn1c (also known as p21, p27 and p57)), that inhibit all kinases involved in G1/S transition, and the Ink4 (Cdkn2) family with four mammalian members (Cdkn2a, Cdkn2b, Cdkn2c, Cdkn2d (also known as p16/p19ARF, p15, p18, p19)) that specifically inhibit Cdk4 and Cdk6. Biochemical and cell culture experiments have identified CKIs as primary effectors of signaling pathways that control cell cycle exit, an event critical for differentiation. Expression or stability of CKIs is reduced in tumors, and deletion of six of the seven family members leads to organ hyperplasia and increased tumor susceptibility. In contrast to the obvious relevance of CKIs in tissue homeostasis, their role in development of tissues and organs, and the transcriptional mechanisms that mediate their precise temporal and spatial expression in the embryo have been much less well defined. This may relate to functional redundancy between family members as well as to the complexity of their regulatory modules (for reviews on CKIs see [1]–[3]).
T-box (Tbx) genes encode an evolutionary conserved family of transcription factors that regulate patterning and differentiation processes during vertebrate development [4]. Tbx2 and Tbx3 are two closely related members of the Tbx2-subfamily that are required in the development of numerous organs during mammalian embryogenesis including the heart, the palate, the limbs, and the liver [5]–[10]. In these contexts, these two transcriptional repressors mainly seem to regulate cell fate decisions and differentiation. However, in vitro studies indicated a role for Tbx2 and Tbx3 in the progression of the cell cycle [11]–[13]. Expression of Tbx2 and Tbx3 is upregulated in a number of tumors including those of the breast, pancreas, liver and bladder, and in melanomas, and both genes can function as immortalizing agents to bypass senescence, i.e. escape irreversible growth arrest (for reviews see [14], [15]). In cell culture assays, this phenomenon is mediated by transcriptional repression of Cdkn1a and Cdkn2a [12], [13], [16], [17]. Although often speculated (e.g. [6]), the relevance for this molecular function in a developmental context has remained unclear. Intriguingly, mice analyzed in our lab that were mutant for Tbx2, showed severely hypoplastic lungs, pointing to a possible role of this T-box factor in the regulation of proliferation and/or differentiation during development of this organ.
The architecture of the mammalian lung arises from a complex developmental program in which the tight orchestration of proliferation and differentiation processes assures the formation of an appropriately sized organ with a correct distribution of differentiated cell types for air-conduction and gas-exchange. In the mouse, the conducting airways develop from two primary buds that emerge from the ventral wall of the foregut endoderm at embryonic day (E) 9.5 by iterative rounds of stereotyped outgrowth and branching until E16.5. The gas-exchange units, the alveoli, only arise subsequently to this pseudoglandular stage from the terminal buds until late in postnatal life. Normal morphogenesis and patterning of the bronchial tree critically depends on the underlying mesenchyme that is derived from the splanchnic mesoderm. This mesenchyme is a source of signals that mediate proliferation of epithelial precursors, and direct their correct spatial differentiation. It also gives rise to a number of different cell types, including parabronchial and vascular smooth muscle cells, lipocytes, fibrocytes and endothelial cells. In turn, epithelial signals from the endoderm but also from the mesothelium maintain proliferation of mesenchymal precursors, closing a reciprocal signaling loop that directs outgrowth of the distal epithelial buds (for a recent review see [18]).
Previous work reported the expression of Tbx2 and Tbx3 in the mesenchymal compartment of the developing lung, but a functional significance has not been assigned to this expression [19], [20]. Here, we show by loss - and gain-of-function experiments in the mouse that Tbx2 is required and sufficient to maintain proliferation and inhibit differentiation in the mesenchymal compartment of the developing lung. Expression, organ culture and biochemical assays identify the cell cycle inhibitors encoded by the Cdkn1a and Cdkn1b genes as direct targets of Tbx2 repressive activity in this developmental program in vivo. Lung growth was substantially rescued by genetically limiting Cdkn1a and Cdkn1b expression in Tbx2-deficient mice, indicating that suppression of these genes is a critical function of Tbx2 in the control of organ growth during development.
Results
Tbx2-deficient mice exhibit hypoplastic lungs
Mice homozygous for a null allele of Tbx2 (Tbx2cre) that is maintained on an NMRI outbred background survive embryogenesis but die shortly after birth due to a cleft palate [9], [21]. Morphological and histological examination of mutant embryos at E18.5 revealed hypoplastic lungs that frequently manifested with alveolar haemorrhages. Air was present in the bronchial network but the lung was poorly inflated. Lobulation was normal but all four right lung lobes and the left lung lobe were reduced in size; the tissue appeared thickened. The weight of the mutant lung was reduced to approx. 50% of that of the littermate control whereas the liver and the spleen were unaffected excluding a general growth retardation problem (Figure 1A–1C). At E16.5, the mutant lung was visibly smaller and haemorrhagic. Its weight was reduced to 33% of the wildtype level (Figure 1D–1F). No obvious difference in morphology, histology and weight of the lung between wildtype and Tbx2-deficient embryos was observed at E14.5 (Figure 1G–1I).

To evaluate whether the decreased size of Tbx2-deficient lungs after E14.5 relates to a reduction in branching morphogenesis, we explanted E11.5 lung rudiments and analyzed their (2-dimensional) outgrowth after 6 days of culture. Whole-mount in situ hybridization analysis for expression of the epithelial tip marker gene Id2 showed an almost 3-fold reduction of branching endpoints in the Tbx2-mutant lung explants suggesting that epithelial branching morphogenesis is indeed severely hampered by loss of Tbx2 (Figure 1J–1L). However, reduction of branching morphogenesis was restricted to the late phase of lung outgrowth as revealed by non-significant changes of the number of branching endpoints in Tbx2-deficient cultures at 2 and 4 days (Figure S1). We conclude that Tbx2 is required to maintain normal branching morphogenesis and growth of the developing lung after E14.5.
Decrease in proliferation of mesenchymal progenitor cells
Lung growth during the pseudoglandular stage is driven by branching morphogenesis of the distal lung buds. This, in turn, relies on rapid proliferation of the precursor cells in the bud epithelium and its underlying mesenchyme. Reduced size of Tbx2-deficient lungs could therefore relate to increased apoptosis and/or to decreased proliferation of distal epithelial and mesenchymal tissue compartments as shown for other models of lung hypoplasia [22]. Terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) staining revealed that apoptosis was absent both in wildtype and mutant lungs at E14.5 and E16.5 but was increased in Tbx2-deficient lungs at E18.5 indicating a late contribution to the hypoplasia of this organ (Figure 2A and 2A′, 2D and 2D′, 2G and 2G′).

Analysis of 5-bromo-2′-deoxyuridine (BrdU) incorporation showed that the epithelial and mesenchymal tissue compartments of the lung were highly proliferative irrespective of the genotype at E14.5 (Figure 2B–2C). However, at E16.5 the BrdU labeling index showed a highly significant reduction from 29.6+/−1.5% in the wildtype to 16.0+/−3.4% in the mutant mesenchyme, and a significant reduction from 19.7+/−1.6% in the wildtype to 14.2+/−4.7% in the mutant distal lung epithelium (marked by expression of SRY-box containing protein (Sox)9 [23], [24]) while the proximal lung epithelium (marked by expression of Sox2 [25], [26]) or a control tissue (the diaphragm) were unaffected (Figure 2E–2F, Figure S2). At E18.5, proliferation as indicated by the BrdU labeling index was dramatically decreased in the whole lung to levels similar in wildtype and mutant embryos (Figure 2H–2I). Thus, Tbx2 is required to maintain normal proliferation of the mesenchyme and distal epithelium of the lung in a narrow temporal window.
Premature differentiation of the Tbx2-deficient lung mesenchyme
As proliferation and differentiation are often inversely correlated, we next investigated the occurrence of changes in the differentiation patterns of both mesenchyme and epithelium in Tbx2-deficient lungs at E14.5, E16.5 and E18.5 to cover the period before, around and after the histological and cellular defects were apparent in the mutant (Figure 3). At all analyzed stages a normal distribution of networks of endomucin (Emcn)-positive endothelial cells [27] was present throughout the mutant lung. In the mutant mesenchyme, transgelin (Tagln)-positive smooth muscle cells were restricted to the proximal airways as in the wildtype. It has recently been shown that expression of S100 calcium binding protein A4 (S100a4) marks fibroblasts that are highly proliferative and express low levels of extracellular matrix proteins indicating the precursor character of these cells [28]–[30]. We found that in E14.5 wildtype lungs all mesenchymal cells that were positive for S100a4 also incorporated BrdU confirming the proliferative character of this cell type (Figure S3). In the Tbx2-deficient lung, expression of S100a4 was completely abolished. Fibronectin (Fn) and periostin (Postn), extracellular matrix proteins that are secreted by mature fibrocytes at low levels in proximal airways in the wildtype [30]–[32], were expressed throughout the mesenchyme starting from E14.5 (Fn) and E16.5 (Postn) in the mutant lung (Figure 3). This suggests that Tbx2 is required to maintain the precursor state of a subpopulation of future fibrocytes in the lung mesenchyme.

We next investigated whether these mesenchymal changes are accompanied by alterations in proximal-distal patterning of the respiratory tree and cell differentiation in the epithelium during development in Tbx2-deficient lungs (Figure S4). In the wildtype lung, Sox2 was expressed in the trachea and proximal airways and was excluded from distal endoderm at all analyzed stages. Sox9 was expressed in the distal tip endoderm and excluded proximally at E14.5 and E16.5. At E18.5, Sox9 was downregulated distally and reactivated in the mesenchyme of the proximal airways possibly indicating onset of cartilage formation in this region. Expression of keratin 14 (Krt14, also known as cytokeratin 14) in basal cells in the trachea, of tubulin, beta 4A class IVA (Tubb4a) in ciliated cells, and of secretoglobin, family 1A, member 1 (Scgb1a1, also known as CC10 and uteroglobin) in secretory or Clara cells was activated at E16.5 and maintained at E18.5. Krt14 was also found in myofibroblasts surrounding the proximal airways at E16.5. Surfactant associated protein C (Sftpc1, also known as SP-C) was expressed in alveolar epithelial cells type II (AEC2) from E16.5. Expression of podoplanin (Pdpn) and aquaporin 5 (Aqp5) was activated in AEC1 at E18.5. All of these markers (described in [18]) were appropriately activated and maintained in Tbx2-deficient lungs with the exception of Pdpn and Aqp5 that showed reduced expression levels at E18.5. We conclude that mesenchymal loss of Tbx2 does not affect proximal-distal patterning of the lung epithelium. Reduced or delayed differentiation of AEC1 from AEC2 may relate to the loss of appropriate signaling from the prematurely differentiated mesenchyme.
Coexpression of T-box genes in the developing lung mesenchyme
Phenotypic changes of Tbx2-deficient lungs were confined to the late phase of branching morphogenesis suggesting a narrow temporal window of expression and/or activity of this gene. Alternatively, Tbx2 may act redundantly with other T-box genes during early lung development. In fact, previous work reported expression of Tbx2 as well as of Tbx3, Tbx4 and Tbx5 in the pulmonary mesenchyme [20], [33]. To assess the comparative temporal expression patterns of these genes during lung development, we performed in situ hybridization analysis on sagittal sections of the lung (Figure 4). We observed coexpression of Tbx2 and Tbx3 in the mesenchymal compartment from E10.5 to E14.5. Expression of Tbx3 declined sharply after this stage, whereas Tbx2 was maintained at high levels at subsequent embryonic stages. Coexpression of Tbx4 and Tbx5 was found between E10.5 to E16.5 in the lung mesenchyme. Hence, late onset of phenotypic changes in Tbx2-deficient lungs may relate to functional redundancy with the closely related Tbx3 gene during the initial phase of branching morphogenesis. This notion is supported by the finding that mice homozygous for a null allele of Tbx3 exhibit lungs morphologically and histologically indistinguishable from the wildtype at E14.5, shortly before these mice die ([8] and data not shown). Since mice with more than two mutant alleles of Tbx2 and Tbx3 die around E9.5 due to cardiac defects [10], analysis of the functional redundancy of the two genes in early lung development was not possible with the mouse lines available to us.

De-repression of cell cycle inhibitors in Tbx2-deficient lung mesenchyme
An antisense oligonucleotide approach with cultured lung rudiments and more recently conditional gene targeting demonstrated a requirement for mesenchymal Tbx4 and Tbx5 in the regulation of pulmonary branching morphogenesis [19], [33]. Tbx4 and Tbx5 genetically interact with Fgf10 during lung growth and branching, and may direct transcriptional activation of Fgf10 that encodes a potent growth factor in the lung but also in other developmental contexts [33], [34]. Given the molecular nature of Tbx2 and Tbx3 as transcriptional repressors, Tbx2 and Tbx3 may compete with Tbx4 and Tbx5 for binding to conserved DNA-binding sites in the promoter of Fgf10, similar to the antagonistic control of Nppa expression in the heart by Tbx5 and Tbx2/Tbx3 [35].
To test this hypothesis and determine the molecular changes underlying the lung phenotype, we analyzed components as well as targets of bone morphogenetic protein (Bmp)-, fibroblast growth factor (Fgf), sonic hedgehog (Shh) and wingless-related MMTV integration site (Wnt) pathways that collectively confer outgrowth and branching morphogenesis of the respiratory tree [18]. To accurately identify expression changes we used quantitative RT-PCR of whole lung extracts at different developmental stages. We started our analysis with lungs at E16.5, when morphological, histological and proliferation defects were fully apparent (Figure 5A, grey bars). At this stage, we observed a significant downregulation of components of the Bmp pathway such as Bmp4 and Bmp receptor (Bmpr)2 as well as the Bmp target gene homeobox, msh-like (Msx)1 [36]. Bmp2 and Bmpr1a expression, however, was not significantly altered. Expression of Shh was markedly reduced but not accompanied by decreased intracellular signaling as revealed by almost normal expression of the target gene patched (Ptch)1 [37]. Wnt ligands Wnt2 and Wnt5a were strongly reduced in their expression as was the target of the canonical (Ctnnb1-dependent) sub-branch of Wnt signaling, Axin2 [38]. Unexpectedly, no changes in Fgf pathway components were found. Fgf10 expression was at wildtype level as was the receptor Fgfr2 and the known Fgf target ets variant gene 4 (Etv4, also known as Pea3) [39]. At E14.5, i.e. prior to the observed phenotypic changes, components and targets of Shh-, Fgf - and Bmp-activity were unchanged in their expression. The canonical Wnt target gene Axin2 and the non-canonical ligand Wnt5a were strongly and Wnt2 expression was slightly reduced in mutant lungs (Figure 5A, black bars). In situ hybridization analysis showed that downregulation of Wnt2, Wnt5a and Axin2 was confined to the mesenchymal compartment of E14.5 Tbx2−/− lungs (Figure 5B). These data suggest, that Tbx2 does not counteract the transcriptional activation of Fgf10 transcription and Fgf signaling by Tbx4/Tbx5 but targets canonical Wnt signaling in the lung mesenchyme, what, in turn, may secondarily affect Bmp signaling.

Next, we analyzed expression of cell cycle regulators potentially involved in proliferation control of lung mesenchyme (Figure 5C, grey bars). Among the tested cell cycle activators cyclin-dependent kinase (Cdk)1 and cyclin D (Ccnd)1 showed significant reduction whereas Ccnd2 and Ccnd3 expression was unchanged at E16.5. As Ccnd1 has been described as target of canonical Wnt signaling [40], its reduced expression may relate to the observed downregulation of this pathway. The cell cycle inhibitors Cdkn1a, Cdkn1c, Cdkn2a and Cdkn2d were unchanged whereas Cdkn1b was upregulated more than 7 times in the mutant at this stage. At E14.5, all cell cycle regulators were unaffected except Cdkn1b and Cdkn1a that were upregulated 4 and 3.5 times, respectively, in the Tbx2-deficient lung (Figure 5C, black bars). Expression of Tbx3 was unaltered at both analyzed stages excluding a compensatory upregulation of this gene in the Tbx2-mutant background (Figure 5C). In situ hybridization and immunofluorescence analyses confirmed strong upregulation of Cdkn1a/Cdkn1b mRNA and Cdkn1a/Cdkn1b protein both in the mesenchymal and in the distal epithelial compartment of E14.5 Tbx2−/− lungs (Figure 5D). These results argue that reduced proliferation in the mesenchyme and distal epithelium (that are probably secondary to altered mesenchymal signals) of E16.5 Tbx2-deficient lungs may be caused by de-repression of cell cycle inhibitors Cdkn1a and Cdkn1b. Decreased (canonical) Wnt signaling in Tbx2-deficient lungs may reflect an independent branch of Tbx2 activity, or may merely present a secondary consequence of de-repression of cell cycle inhibitors.
Repression of Cdkn1a and Cdkn1b by Tbx2 is direct and contributes to lung growth
To unravel the contribution of increased expression of Cdkn1a and Cdkn1b to the growth deficit of Tbx2-deficient lungs, we ablated the two genes in the mutant background. Compound Tbx2;Cdkn1a and Tbx2;Cdkn1b mutants, respectively, exhibited lungs that were morphologically indistinguishable from the Tbx2-single mutant organ. In contrast, triple Tbx2;Cdkn1a;Cdkn1b mutants exhibited visibly larger lungs at E18.5 (Figure 6A). To quantify the observed changes, we determined the relative lung weight (lung weight to body weight ratios, normalized to that of Tbx2+/− control embryos) of the different compound mutants. Statistical analysis did not detect significant weight changes between Tbx2−/−;Cdkn1a−/− and Tbx2−/−;Cdkn1b−/− lungs, whereas the increase in weight in Tbx2−/−;Cdkn1a−/−;Cdkn1b−/− lungs was highly significant (Figure 6B). Although Tbx2−/−;Cdkn1a−/−;Cdkn1b−/− lungs reached 80% of the control weight, the difference remained significant indicating an incomplete rescue. This suggests that the combined de-repression of Cdkn1a and Cdkn1b accounts predominantly but not completely for hypoplasia of Tbx2-deficient lungs.

Since the individual deletion of Cdkn1a and Cdkn1b in the Tbx2-mutant background did not lead to even a partial rescue of growth, we tested for the presence of a compensatory mechanism by analyzing expression of Cdkn1a and Cdkn1b, respectively, by quantitative RT-PCR analysis on mRNA of E16.5 (compound) mutant lungs (Figure 6C). Cdkn1a expression was increased 2-fold in Cdkn1b−/− lungs and 9-fold in Tbx2−/;−Cdkn1b−/− lungs whereas Cdkn1b was increased 2-fold in Cdkn1a−/− lungs and 3-fold in Tbx2−/−;Cdkn1a−/− lungs at this stage. Thus, either gene shows a compensatory upregulation upon loss of the other gene.
Binding sites for TBX2 within the Cdkn1a promoter have recently been described in cell culture experiments [12] whereas Cdkn1b has not been recognized as a direct target of Tbx2 repressive activity before. In silico analysis of the mouse Cdkn1a and Cdkn1b genes identified a consensus DNA-binding site for T-box proteins (T-box binding element (TBE): AGGTGTGA) [41] in the Cdkn1a promoter and two putative TBEs in the Cdkn1b locus. The first element (AGGTGTGTG) was detected 3 kbp upstream of the start codon, the second element with the reverse complementary sequence CACACCT was localized within an intron of that gene (Figure S5). ChIP experiments with E15.5 lung tissue revealed in vivo binding of Tbx2 to the known TBE in the Cdkn1a locus and to the 5′ located but not the intronic TBE in the Cdkn1b gene (Figure 6D) compatible with the notion that Cdkn1a and Cdkn1b represent direct targets of Tbx2 repressive activity in the lung mesenchyme.
It has previously been shown that Cdkn1a expression is elevated on inactivation of endogenous Tbx2 in the murine B16 melanoma and the human MCF-7 breast cancer cell line [12]. Using the previously published conditions [12], we downregulated Tbx2 in both cell lines using a Tbx2-specific siRNA approach. Immunofluorescence analysis showed that in the non-silencing control nuclear Tbx2 protein was present in all cells whereas Cdkn1b was not detected. In contrast, in cells treated with the Tbx2-specific siRNA Tbx2 expression was extinguished in almost all cells examined whereas Cdkn1b expression was strongly upregulated in the cytoplasm (in MCF-7 cells) and in the nucleus (in B16 melanoma cells) (Figure S6). This further supports that Cdkn1b similar to Cdkn1a is a true target of Tbx2.
Maintenance of Tbx2 expression prevents terminal differentiation of lung fibrocytes
To further evaluate the mechanistic role of Tbx2 in the lung mesenchyme, we additionally employed an in vivo gain-of-function approach. For this, we crossed the Tbx2cre line and an HprtTBX2-allele, that was generated by integration of a bicistronic transgene-cassette containing the human TBX2 ORF followed by IRES-GFP in the ubiquitously expressed X-chromosomal Hypoxanthine guanine phosphoribosyl transferase (Hprt) locus [10], [42] to maintain TBX2 expression in its endogenous domains including the lung mesenchyme. Male (Tbx2cre/+;HprtTBX2/y) embryos were not recovered after E12.5 most likely due to cardiac defects. In contrast, female (Tbx2cre/+;HprtTBX2/+) embryos, which exhibit a mosaic expression due to random X-chromosome inactivation, survived embryogenesis and puberty. Lungs of E18.5 Tbx2cre/+;HprtTBX2/+ embryos were slightly bigger than those of control littermates and showed a looser tissue organization (Figure 7A). Apoptosis was not detected in either genotype, but Tbx2cre/+;HprtTBX2/+ lungs exhibited a strong increase of proliferation in the mesenchyme as shown by the BrdU assay (Figure 7B). Notably, Western blot analysis of lungs of E18.5 Tbx2cre/+;HprtTBX2/+ embryos showed that transgenic TBX2 expression did not reach unphysiologically high levels (Figure S7).

Branching morphogenesis is downregulated after E16.5 concomitant with the shut-down of signaling pathways involved in epithelial-mesenchymal tissue interactions at the distal lung buds. Therefore, increased proliferation in Tbx2cre/+;HprtTBX2/+ lungs may relate to continued branching by maintained activity of these signaling pathways. Morphological inspection did not detect changes of branching between E12.0 wildtype and Tbx2cre/+;HprtTBX2/+ lung rudiments cultured for 6 days (Figure S8). Furthermore, RT-PCR analysis found unchanged expression of targets of Shh (Ptch1), Fgf (Etv4), Bmp (Msx1) and canonical Wnt (Axin2) pathways in Tbx2cre/+;HprtTBX2/+ lungs at E18.5 showing that Tbx2 is not sufficient to induce these pathways, thus, branching morphogenesis (Figure 7C). However, when testing cell cycle regulators in this assay, we detected a selective downregulation of Cdkn1a and Cdkn1b in Tbx2cre/+;HprtTBX2/+ lungs showing that Tbx2 is not only required but also sufficient to repress expression of Cdkn1a and Cdkn1b (Figure 7D).
To evaluate long-term consequences of prolonged TBX2 expression in the lung mesenchyme, we analyzed Tbx2cre/+;HprtTBX2/+ mice at postnatal day (P) 40, a stage when they were present in the expected numbers. Although Tbx2cre/+;HprtTBX2/+ mice were visibly smaller than their littermate controls at this stage, the relative lung mass was increased by a factor of 1.27 (Figure 8A and 8B). Immunofluorescence for GFP and TBX2 expression on lung sections confirmed the widespread expression of the transgene in the mesenchymal compartment of P40 Tbx2cre/+;HprtTBX2/+ mice. Histological analysis by haematoxylin and eosin staining uncovered clusters of tissue thickenings, and alveolar air spaces were surrounded by multiple cell layers in these transgenic lungs. Histological staining for keratin and collagen (Masson's trichrome) did not detect changes in the transgenic lung, excluding the possibility that tissue thickening is caused by excessive deposition of extracellular matrix (Figure 8C). Analysis of BrdU incorporation showed that the lung tissue was highly proliferative in the transgenic animals at P40 (Tbx2cre/+;HprtTBX2/+: 31.0%±5.2, control: 2.1%±0.8) (Figure 8D). Apoptosis as detected by TUNEL staining was similarly absent from control and transgenic lungs (Figure 8E).
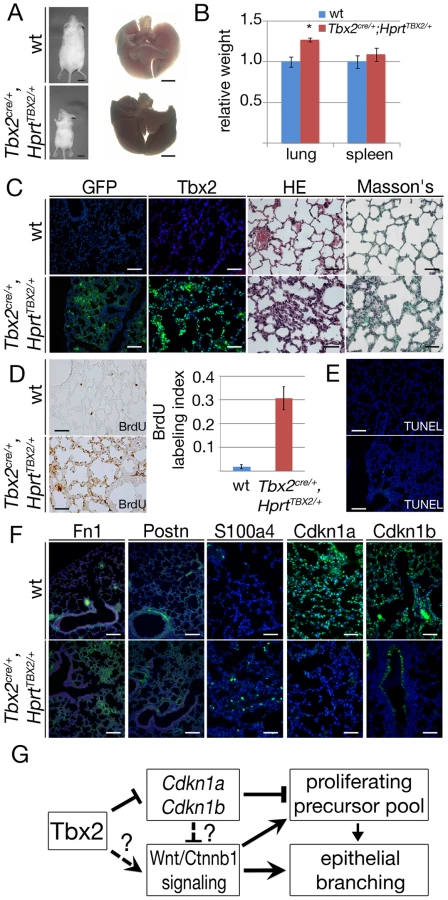
Analysis of cell differentiation by immunofluorescence of marker proteins showed normal presence of lung epithelial cell types, of endothelial cells, and of mesenchymal smooth muscle cells around the proximal airways of Tbx2cre/+;HprtTBX2/+ lungs (Figure S9). Fn1 and Postn deposition in the extracellular matrix was augmented, and S100a4-positive cells were increased in number. Expression of the cell cycle inhibitors Cdkn1a and Cdkn1b was dramatically downregulated in the mesenchymal compartment (Figure 8F). Together these findings indicate that prolonged expression of TBX2 maintains mesenchymal proliferation at a high level. While a part of these mesenchymal cells differentiate into ECM-producing cell-types, a substantial fraction retains a S100a4-positive precursor character.
To determine the contribution of reduced expression of Cdkn1a and Cdkn1b to the observed histological, immunohistochemical and molecular changes in Tbx2cre/+;HprtTBX2/+ mice, we additionally analyzed Cdkn1a−/−;Cdkn1b−/− mice at P40 using a similar panel of assays. Double mutant lungs, normalized against the increased body weight, were significantly larger than lungs of their littermates (Figure S10A, S10B). Histological analysis did not find changes in the tissue organization (Figure S10C) but Cdkn1a−/−;Cdkn1b−/− lungs exhibited a 5-fold increase of proliferation as shown by the BrdU assay compared to the wildtype. Apoptosis was unaffected (Figure S10D, S10E). Fn1 and Postn deposition in the extracellular matrix was normal and immature fibroblasts (S100a4) were absent as in the wildtype. Immunofluorescence analysis of Cdkn1a and Cdkn1b confirmed that both proteins were completely absent in the Cdkn1a−/−;Cdkn1b−/− lung (Figure S10F). Hence, Cdkn1a−/−;Cdkn1b−/− mice do not feature the histological and cellular changes seen in Tbx2cre/+;HprtTBX2/+ mice, but exhibit increased lung mass due to increased proliferation. We conclude that downregulation of Cdkn1a and Cdkn1b mediates the pro-proliferative effects of Tbx2 overexpression to a large degree but may not account for changes in tissue architecture and cell differentiation.
Discussion
Branching morphogenesis and growth of the lung requires the coordination of cellular behaviors of its epithelial and mesenchymal tissue compartments. Here, we have identified Tbx2 as a crucial mesenchymal factor that maintains the mesenchymal signaling center for epithelial branching morphogenesis. We suggest that Tbx2 promotes mesenchymal proliferation and inhibits terminal differentiation partly via direct transcriptional repression of cell cycle inhibitor genes. Irrespective of its precise mode, Tbx2 additionally maintains canonical Wnt signaling in the mesenchyme, which, in turn, may account for maintenance of epithelial growth and branching at the distal tips of the lung buds (Figure 8G).
Tbx2 directly represses cell cycle regulators in the lung mesenchyme
Cdkn1a, Cdkn1b together with Cdkn1c constitute the Cip/Kip family of CKIs that inhibit cell cycle progression by binding to and inhibition of a broad range of cyclin-CDK complexes via a shared N-terminal cyclin-CDK binding domain. Cdkn1 activity correlates with cell cycle exit and differentiation, and is, thus, under tight control of anti-mitogenic signals. In tissue homeostasis, expression and activity of CKIs is regulated by a large number of molecular mechanisms including protein binding and posttranslational modification that affect cyclin/CDK binding as well as stability and degradation of CKIs (for a review see [43]). Gene targeting experiments have unambiguously shown that all three members are important players in tissue homeostasis and cancer (for a review see [2]) whereas Cdkn1c is the only CKI to be uniquely required for embryonic development [44], [45]. However, additional congenital defects have been described in mice lacking more than one member of this gene family pointing to redundant functions in some but not all developmental processes (see e.g. [46], [47].
To exert a precise timing of cell cycle exit and differentiation in development, expression of Cdkn1 genes must be tightly controlled on the transcriptional level. In fact, Cdkn1a and Cdkn1c have specific patterns of expression in development that correlate with terminal differentiation of multiple cell lineages including skeletal muscle, cartilage, skin, and nasal epithelium. In contrast, Cdkn1b expression appears more widespread (for a review see [2]. Cell culture experiments identified Cdkn1a as a transcriptional target of p53 [48], [49] whereas the transcriptional regulation of Cdkn1c is mediated by factors that play critical roles during embryogenesis such as Notch/Hes1, MyoD and p73 [50]–[52]. To our knowledge, the in vivo relevance of these regulatory modules has remained unclear. Interestingly, previous efforts were largely directed towards the identification of transcriptional activators of Cdkn1 genes, and the possibility that these genes are subject to negative regulation in vivo, i.e. that activation of expression in a certain cell type results from attenuation or abolition of a prior transcriptional repression, was neglected.
Here, we have shown that Cdkn1a and Cdkn1b are derepressed in the pulmonary mesenchyme in Tbx2-deficient mice prior to other molecular changes, that Cdkn1a and Cdkn1b are repressed upon ectopic expression of TBX2 in mature lung mesenchyme, and that deletion of Cdkn1a and Cdkn1b largely rescued the growth defects of Tbx2-deficient lungs. Furthermore, we identified by ChIP analysis Tbx2 binding to Cdkn1a and Cdkn1b loci in the developing lung. Together, our genetic and biochemical analyses provide evidence that Cdkn1a and Cdkn1b are subject to direct repression by Tbx2 and are crucial downstream mediators of this gene in the mesenchymal compartment of the developing lung. In turn, it is the first clear evidence, that Tbx2 directly regulates cell cycle control genes in a developmental context in vivo. Intriguingly, ChIP-seq analysis of genomic binding of Tbx3 in cardiomyocytes in vivo, identified a large number of loci with binding peaks containing a variant TBE [53]. Tbx3 and Tbx2 are closely related family members that recognize the same DNA binding site. Re-inspection of this data set identified binding peaks of Tbx3 in both the Cdkn1a and Cdkn1b loci. In fact, the DNA-element used in our ChIP analysis precisely mapped to a major Tbx3 peak in the promoter of the Cdkn1a locus which contained additional less conserved TBEs, whereas the DNA element used for our Cdkn1b ChIP located closely to a minor peak (Figure S11). This together with enhanced expression of Cdkn1a and Cdkn1b in melanoma and breast cancer cell lines depleted of endogenous Tbx2 [12], [13], [17] (and this study), indicates that Tbx3 and the closely related Tbx2 protein occupy DNA sites in the Cdkn1a and Cdkn1b loci in other cell types and may regulate these genes in other developmental contexts as well.
It should be noted that changes of Tbx2 did not only (inversely) affect proliferation in the lung mesenchyme but directly correlated with the precursor state of at least one mesenchymal sub-population, S100a4-positive fibroblasts. Although Cdkn1-mediated cell cycle arrest has been associated with cellular differentiation in different developmental contexts [44], [54], [55], we did not observe differentiation defects in lungs double mutant for Cdkn1a and Cdkn1b. This may indicate that in this developmental context negative control of cell differentiation by Tbx2 is not mediated by repression of Cdkn1 and Cdkn1b. Changes of Tbx2 expression did not affect differentiation of other mesenchyme-derived cell types including smooth muscle cells. This may indicate that Tbx2 does not control differentiation of these cell types, or it may simply reflect the fact that these cell types differentiate prior to E14.5 when Tbx3 expression is downregulated and Tbx2 is uniquely required. In the future, it will be interesting to study the relation between mesenchymal proliferation and differentiation in mice deficient for both Tbx2 and Tbx3, which are likely to act redundantly throughout the pseudoglandular stage until E14.5.
Tbx2 maintains canonical Wnt signaling in the lung mesenchyme
Combined deletion of Cdkn1a and Cdkn1b function in Tbx2-deficient embryos restored lung growth largely but not completely suggesting that additional factors or pathways may act downstream of Tbx2 to mediate mesenchymal proliferation. Our RT-PCR analysis of signaling pathways relevant for branching morphogenesis did not detect changes of Fgf and Shh signaling but uncovered reduced activity of canonical Wnt and Bmp signaling. Notably, we detected decreased expression of Wnt components and signaling as early as E14.5 in the pulmonary mesenchyme, whereas Bmp4 expression and Bmp signaling was unchanged at that stage suggesting a secondary mode of change of the latter. As Bmp4 was shown to act as an autocrine signal for distal endoderm proliferation [56], reduced expression may contribute to the reduced proliferation in the distal endoderm at E16.5 in Tbx2-deficient lungs.
A number of studies have implicated different Wnt genes in lung development. Mice deficient for the non-canonical Wnt ligand gene Wnt5a, which is expressed in the distal lung mesenchyme, exhibit increased cell proliferation in both epithelium and mesenchyme with a resulting expansion of the distal lung and increased lung size [57]. Wnt2 is a canonical Wnt ligand robustly expressed in the mesenchyme of the developing lung. Wnt2, in cooperation with Wnt2b, is essential for specification of the respiratory lineage in the anterior foregut endoderm [58]. Later, Wnt2 acts upstream of Fgf10 and the critical transcription factor myocardin to regulate early airway smooth muscle cell differentiation in the multipotent lung mesenchyme [59]. Finally, Wnt7b, a canonical ligand expressed in the pulmonary epithelium stimulates embryonic lung growth by increasing proliferation in both tissue compartments of the developing lung without affecting the differentiation patterns [22]. Furthermore, tissue-specific deletion of the unique signaling mediator of the canonical pathway, Ctnnb1, in the epithelium led to defects in proximal-distal differentiation of airway epithelium [60] whereas mesenchymal deletion of Ctnnb1 resulted in hypoplasia due to reduced epithelial and mesenchymal proliferation [61].
Maintained differentiation of airway smooth muscle cells but decreased proliferation in the epithelial and mesenchymal compartments during the late phase of branching morphogenesis is compatible with the idea that loss of Tbx2 affects the canonical Wnt pathway in the mesenchyme triggered by the epithelial Wnt7b signal. The growth-promoting effect of this pathway may at least partly be mediated by activation of the pro-proliferative gene Ccnd1 that was previously recognized as a target of Wnt signaling [62]. This is compatible with the finding that proliferation defects observed in Tbx2-mutant lungs at E16.5 coincide with a strong decline of expression of this gene at this stage. However, the significance of downregulation of Wnt2 and Wnt5a in the Tbx2-deficient lung remains unclear. We assume that it provides only a minor contribution to the observed changes.
At present, we cannot distinguish whether changes of Wnt signaling activity are secondary to cell cycle exit and/or upregulation of Cdkn1a and Cdkn1b or represent an independent branch of Tbx2 transcriptional activity in the lung mesenchyme. Unfortunately, the recovery of mice triple mutant for Tbx2, Cdkn1a and Cdkn1b for analysis of signaling pathways at E14.5 is extremely inefficient. The finding that constitutive expression of Tbx2 in the lung mesenchyme of adult mice did not increase canonical Wnt signaling, suggests that Tbx2 is not sufficient to activate this pathway. However, Tbx2 may be required for repression of an inhibitor of Wnt signaling to maintain this pathway during branching morphogenesis. The relevance of the control of canonical Wnt signaling by Tbx2 in the lung mesenchyme will be addressed in future experiments.
Materials and Methods
Ethics statement
All animal work conducted for this study was approved by H. Hedrich, state head of the animal facility at Medizinische Hochschule Hannover and performed according to German legislation.
Mice and genonotyping
Mice carrying a null allele of Cdkn1a (Cdkn1atm1Tyj, synonym Cdkn1a−) [63], a null allele of Cdkn1b (Cdkn1btm1Mlf, synonym: Cdkn1b−) [64] or a null allele of Tbx2 (Tbx2tm1.1(cre)Vmc, synonyms: Tbx2−, Tbx2cre) [21], and mice with integration of the human TBX2 gene in the Hprt locus (Hprttm2(CAG-TBX2,-EGFP)Akis, synonym: HprtTBX2) [10] were maintained on an outbred (NMRI) background. For timed pregnancies, vaginal plugs were checked in the morning after mating; noon was taken as embryonic day (E) 0.5. Pregnant females were sacrificed by cervical dislocation; embryos were harvested in phosphate-buffered saline, decapitated, fixed in 4% paraformaldehyde overnight, and stored in 100% methanol at −20°C before further use. Genomic DNA prepared from yolk sacs or tail biopsies was used for genotyping by polymerase chain reaction (PCR). For primers and conditions see Table S2.
Histological analysis and immunofluorescence
Embryos were embedded in paraffin and sectioned to 5 µm. For histological analyses, sections were stained with haematoxylin and eosin (HE), Masson's trichrome (Masson's) and picrosirius red (Sirius red) following standard protocols. For the detection of antigens, antigen retrieval was performed using citrate-based antigen unmasking solution (H-3300, Vector Laboratories Inc). Sections were pressure-cooked for 5 min and signal amplification was performed with the Tyramide Signal Amplification (TSA) system (NEL702001KT, Perkin Elmer LAS) or the DAB substrate kit (SK-4100, Vector Laboratories Inc). The following primary antibodies were used: rabbit anti-mouse E-cadherin (gift from Rolf Kemler, MPI for Immunobiology and Epigenetics, Freiburg/Germany) [65], rabbit polyclonal antibody against GFP (1∶200, sc-8334, Santa Cruz), mouse monoclonal antibody against GFP (1∶200, 11814460001, Roche), monoclonal antibody against alpha smooth muscle actin, Cy3-conjugate (1∶200, C 6198, Sigma), monoclonal antibody against alpha smooth muscle actin, FITC-conjugate (1∶200, F3777, Sigma), rabbit polyclonal against SM22a (transgelin, 1∶200, ab14106, Abcam), rat monoclonal antibody against endomucin (1∶2, gift from Dietmar Vestweber, MPI for Molecular Medicine, Münster/Germany) [66], rabbit polyclonal antibodies against Tbx2 (1∶100, ab33298, Abcam), Cdkn1a (1∶200, sc-397, SantaCruz), Cdkn1b (1∶200, 554069, BD Biosciences), uteroglobin (1∶200, ab40873, Abcam), cytokeratin14 (1∶200, ab7800, Abcam), Tubb4a (1∶100, ab11315, Abcam), prosurfactant protein C (1∶200, ab40879, Abcam), Sox2 (1∶100, ab97959, Abcam), Sox9 (1∶200, ab5535, Millipore), aquaporin5 (1∶100, ab92320, Abcam), hamster monoclonal against podoplanin (1∶50, ab11936, Abcam) and mouse monoclonal against BrdU (1∶100, 1170376, Roche). For immunofluorescent stainings on adult sections or double immunofluorescent stainings with two primary mouse antibodies the Biotinylated Mouse on Mouse (M.O.M.) Anti-Mouse Ig Reagent (Vector laboratories) was used.
Organ culture
For analysis of branching morphogenesis E11.5 or E12.0 lung rudiments were dissected and kept on Transwell permeable 0.4-µm pore size, PET 6-well plates (Corning) supplied with DMEM supplemented with 10% fetal calf serum (Biowest), 2 mM Glutamax, 100 units/ml Penicillin, 100 µg/ml Streptomycin (Gibco). Lungs were cultivated at 37°C and 5% CO2 for 2 to 6 days and the number of branching endpoints was counted.
Cell culture and siRNA
Human MCF-7 breast adenocarcinoma cell line was cultured in RPMI 1640 with Glutamax (Gibco) supplemented with 10% FBS, MEM non-essential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), 10 µg/ml human insulin (Roche) and 100 units/ml Penicillin, 100 µg/ml Streptomycin (Gibco). Mouse B16 melanoma cells were cultured in RPMI 1640 with glutamax, supplemented with 10% FBS and 100 units/ml Penicillin, 100 µg/ml Streptomycin.
Downregulation of TBX2 or Tbx2 was achieved by siRNA exactly as recently described [12].
In situ hybridization analysis
Whole-mount in situ hybridization was performed following a standard procedure with digoxigenin-labeled antisense riboprobes [67]. Stained specimens were transferred in 80% glycerol prior to documentation. In situ hybridization on 10 µm paraffin sections was done essentially as described [68]. For each marker at least three independent specimens were analyzed.
Proliferation and apoptosis assays
Cell proliferation in embryonic and adult lungs was investigated by detection of incorporated 5-bromo-2′-deoxyuridine (BrdU) similar to published protocols [69]. At least nine sections from three individual embryos per genotype and stage were used for quantification. Statistical analysis was performed using the two-tailed Student's t-test. Data were expressed as mean ± standard deviation. Differences were considered significant when the P-value was below 0.05.
For detection of apoptotic cells in 5 µm paraffin sections of embryos, the terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) assay was performed as recommended by the manufacturer (Serologicals Corp.) of the ApopTag kit used.
Semi-quantitative reverse transcription PCR
Total RNA was extracted from dissected lungs with RNAPure reagent (Peqlab). RNA (500 ng) was reverse transcribed with RevertAid H Minus reverse transcriptase (Fermentas). For semiquantitative PCR, the number of cycles was adjusted to the mid-logarithmic phase. Quantification was performed with Quantity One software (Bio-Rad). Assays were performed at least twice in duplicate, and statistical analysis was done as previously described [9]. For primers and PCR conditions see Table S3.
Chromatin immunoprecipitation (ChIP) assays
2ChIP was performed essentially as previously described [70]. Dissected E15.5 lung tissue was treated with 4% paraformaldehyde overnight. The DNA-containing supernatants were incubated overnight with anti-Tbx2 antibodies and collected on protein G beads. Cross-linked products were reversed by cooking for 15 min, treated with Proteinase K and RNAse H at 56°C for 30 min and the immunoprecipitated DNA was purified. Primers for PCR amplification were 5′-CCGAGAGGTGTGAGCCGC-3′ (Cdkn1a-f1) and 5′ - GTCATCCACCTGCCGCGG-3′ (Cdkn1a-r1); 5′-GGCTTAGATTCCCAGAGGG-3′ (Cdkn1af2) and 5′-TTCTGGGGACACCCACTGG-3′ (Cdkn1a-r2) for the Cdkn1a promoter and 5′ - CAAGTTCAGTAAACTAAGTAGG-3′ (Cdkn1b-f1) and 5′ - GCACATATGTGGACAAACTCG-3′ (Cdkn1b-r1) for the 5′-T-site in the Cdkn1b promoter. For the intron located T-site 5′-ATATACCTTCTACAGACATAGC-3′ (Cdkn1b-f2) and 5′ - GCTTTTGACTAGAGTCTTATGG-3′ (Cdkn1b-r2) primers were used. Primers for the negative control region were 5′-CTCTGAAACTCGAACAGGCC-3′ (ncr-f1) and 5′ - ACTCTGAATTGGATTCCTAGC-3′ (ncr-r1).
Image analysis
Sections were photographed using a Leica DM5000 microscope with a Leica DFC300FX digital camera. Whole mount specimens were photographed on a Leica M420 microscope with a Fujix digital camera HC-300Z. Images were processed in Adobe Photoshop CS3.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SherrCJ, RobertsJM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13 : 1501–1512.
2. VidalA, KoffA (2000) Cell-cycle inhibitors: three families united by a common cause. Gene 247 : 1–15.
3. BessonA, DowdySF, RobertsJM (2008) CDK inhibitors: cell cycle regulators and beyond. Dev Cell 14 : 159–169.
4. NaicheLA, HarrelsonZ, KellyRG, PapaioannouVE (2005) T-box genes in vertebrate development. Annu Rev Genet 39 : 219–239.
5. DavenportTG, Jerome-MajewskaLA, PapaioannouVE (2003) Mammary gland, limb and yolk sac defects in mice lacking Tbx3, the gene mutated in human ulnar mammary syndrome. Development 130 : 2263–2273.
6. HarrelsonZ, KellyRG, GoldinSN, Gibson-BrownJJ, BollagRJ, et al. (2004) Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development 131 : 5041–5052.
7. SuzukiA, SekiyaS, BuscherD, Izpisua BelmonteJC, TaniguchiH (2008) Tbx3 controls the fate of hepatic progenitor cells in liver development by suppressing p19ARF expression. Development 135 : 1589–1595.
8. LudtkeTH, ChristoffelsVM, PetryM, KispertA (2009) Tbx3 promotes liver bud expansion during mouse development by suppression of cholangiocyte differentiation. Hepatology 49 : 969–978.
9. ZirzowS, LudtkeTH, BronsJF, PetryM, ChristoffelsVM, et al. (2009) Expression and requirement of T-box transcription factors Tbx2 and Tbx3 during secondary palate development in the mouse. Dev Biol 336 : 145–155.
10. SinghR, HoogaarsWM, BarnettP, GrieskampT, RanaMS, et al. (2012) Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell Mol Life Sci 69 : 1377–1389.
11. JacobsJJ, KeblusekP, Robanus-MaandagE, KristelP, LingbeekM, et al. (2000) Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet 26 : 291–299.
12. PrinceS, CarreiraS, VanceKW, AbrahamsA, GodingCR (2004) Tbx2 directly represses the expression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cancer Res 64 : 1669–1674.
13. VanceKW, CarreiraS, BroschG, GodingCR (2005) Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res 65 : 2260–2268.
14. LuJ, LiXP, DongQ, KungHF, HeML (2010) TBX2 and TBX3: the special value for anticancer drug targets. Biochim Biophys Acta 1806 : 268–274.
15. AbrahamsA, ParkerMI, PrinceS (2009) The T-box transcription factor Tbx2: its role in development and possible implication in cancer. IUBMB Life 62 : 92–102.
16. LingbeekME, JacobsJJ, van LohuizenM (2002) The T-box repressors TBX2 and TBX3 specifically regulate the tumor suppressor gene p14ARF via a variant T-site in the initiator. J Biol Chem 277 : 26120–26127.
17. HoogaarsWM, BarnettP, RodriguezM, CloutDE, MoormanAF, et al. (2008) TBX3 and its splice variant TBX3 + exon 2a are functionally similar. Pigment Cell Melanoma Res 21 : 379–387.
18. MorriseyEE, HoganBL (2010) Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev Cell 18 : 8–23.
19. Cebra-ThomasJA, BromerJ, GardnerR, LamGK, SheipeH, et al. (2003) T-box gene products are required for mesenchymal induction of epithelial branching in the embryonic mouse lung. Dev Dyn 226 : 82–90.
20. ChapmanDL, GarveyN, HancockS, AlexiouM, AgulnikSI, et al. (1996) Expression of the T-box family genes, Tbx1–Tbx5, during early mouse development. Dev Dyn 206 : 379–390.
21. AanhaanenWT, BronsJF, DominguezJN, RanaMS, NordenJ, et al. (2009) The Tbx2+ primary myocardium of the atrioventricular canal forms the atrioventricular node and the base of the left ventricle. Circ Res 104 : 1267–1274.
22. RajagopalJ, CarrollTJ, GusehJS, BoresSA, BlankLJ, et al. (2008) Wnt7b stimulates embryonic lung growth by coordinately increasing the replication of epithelium and mesenchyme. Development 135 : 1625–1634.
23. LiuY, HoganBL (2002) Differential gene expression in the distal tip endoderm of the embryonic mouse lung. Gene Expr Patterns 2 : 229–233.
24. OkuboT, KnoepflerPS, EisenmanRN, HoganBL (2005) Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development 132 : 1363–1374.
25. GontanC, de MunckA, VermeijM, GrosveldF, TibboelD, et al. (2008) Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev Biol 317 : 296–309.
26. IshiiY, RexM, ScottingPJ, YasugiS (1998) Region-specific expression of chicken Sox2 in the developing gut and lung epithelium: regulation by epithelial-mesenchymal interactions. Dev Dyn 213 : 464–475.
27. MorganSM, SamulowitzU, DarleyL, SimmonsDL, VestweberD (1999) Biochemical characterization and molecular cloning of a novel endothelial-specific sialomucin. Blood 93 : 165–175.
28. LawsonWE, PolosukhinVV, ZoiaO, StathopoulosGT, HanW, et al. (2005) Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med 171 : 899–907.
29. PaxsonJA, ParkinCD, IyerLK, MazanMR, IngenitoEP, et al. (2009) Global gene expression patterns in the post-pneumonectomy lung of adult mice. Respir Res 10 : 92.
30. Kaarteenaho-WiikR, PaakkoP, SormunenR (2009) Ultrastructural features of lung fibroblast differentiation into myofibroblasts. Ultrastruct Pathol 33 : 6–15.
31. GompertsBN, StrieterRM (2007) Fibrocytes in lung disease. J Leukoc Biol 82 : 449–456.
32. ShimazakiM, NakamuraK, KiiI, KashimaT, AmizukaN, et al. (2008) Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 205 : 295–303.
33. AroraR, MetzgerRJ, PapaioannouVE (2012) Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet 8: e1002866 doi:10.1371/journal.pgen.1002866.
34. AgarwalP, WylieJN, GalceranJ, ArkhitkoO, LiC, et al. (2003) Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 130 : 623–633.
35. HabetsPE, MoormanAF, CloutDE, van RoonMA, LingbeekM, et al. (2002) Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev 16 : 1234–1246.
36. MaedaR, KobayashiA, SekineR, LinJJ, KungH, et al. (1997) Xmsx-1 modifies mesodermal tissue pattern along dorsoventral axis in Xenopus laevis embryo. Development 124 : 2553–2560.
37. GoodrichLV, JohnsonRL, MilenkovicL, McMahonJA, ScottMP (1996) Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 10 : 301–312.
38. JhoEH, ZhangT, DomonC, JooCK, FreundJN, et al. (2002) Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22 : 1172–1183.
39. MunchbergSR, SteinbeisserH (1999) The Xenopus Ets transcription factor XER81 is a target of the FGF signaling pathway. Mech Dev 80 : 53–65.
40. TetsuO, McCormickF (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398 : 422–426.
41. KispertA, HerrmannBG (1993) The Brachyury gene encodes a novel DNA binding protein. EMBO J 12 : 3211–3220.
42. LucheH, WeberO, Nageswara RaoT, BlumC, FehlingHJ (2007) Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol 37 : 43–53.
43. StarostinaNG, KipreosET (2012) Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol 22 : 33–41.
44. ZhangP, LiegeoisNJ, WongC, FinegoldM, HouH, et al. (1997) Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387 : 151–158.
45. YanY, FrisenJ, LeeMH, MassagueJ, BarbacidM (1997) Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 11 : 973–983.
46. HolsbergerDR, BucholdGM, LealMC, KiesewetterSE, O'BrienDA, et al. (2005) Cell-cycle inhibitors p27Kip1 and p21Cip1 regulate murine Sertoli cell proliferation. Biol Reprod 72 : 1429–1436.
47. OkahashiN, MuraseY, KosekiT, SatoT, YamatoK, et al. (2001) Osteoclast differentiation is associated with transient upregulation of cyclin-dependent kinase inhibitors p21(WAF1/CIP1) and p27(KIP1). J Cell Biochem 80 : 339–345.
48. el-DeiryWS, TokinoT, VelculescuVE, LevyDB, ParsonsR, et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75 : 817–825.
49. GartelAL, TynerAL (1999) Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res 246 : 280–289.
50. BlintE, PhillipsAC, KozlovS, StewartCL, VousdenKH (2002) Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci U S A 99 : 3529–3534.
51. GeorgiaS, SolizR, LiM, ZhangP, BhushanA (2006) p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol 298 : 22–31.
52. VaccarelloG, FigliolaR, CramerottiS, NovelliF, MaioneR (2006) p57Kip2 is induced by MyoD through a p73-dependent pathway. J Mol Biol 356 : 578–588.
53. van den BoogaardM, WongLY, TessadoriF, BakkerML, DreizehnterLK, et al. (2012) Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest 122 : 2519–2530.
54. TanakaH, YamashitaT, AsadaM, MizutaniS, YoshikawaH, et al. (2002) Cytoplasmic p21(Cip1/WAF1) regulates neurite remodeling by inhibiting Rho-kinase activity. J Cell Biol 158 : 321–329.
55. ZhangP, WongC, DePinhoRA, HarperJW, ElledgeSJ (1998) Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev 12 : 3162–3167.
56. EblaghieMC, ReedyM, OliverT, MishinaY, HoganBL (2006) Evidence that autocrine signaling through Bmpr1a regulates the proliferation, survival and morphogenetic behavior of distal lung epithelial cells. Dev Biol 291 : 67–82.
57. LiC, XiaoJ, HormiK, BorokZ, MinooP (2002) Wnt5a participates in distal lung morphogenesis. Dev Biol 248 : 68–81.
58. GossAM, TianY, TsukiyamaT, CohenED, ZhouD, et al. (2009) Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev Cell 17 : 290–298.
59. GossAM, TianY, ChengL, YangJ, ZhouD, et al. (2011) Wnt2 signaling is necessary and sufficient to activate the airway smooth muscle program in the lung by regulating myocardin/Mrtf-B and Fgf10 expression. Dev Biol 356 : 541–552.
60. ShuW, GuttentagS, WangZ, AndlT, BallardP, et al. (2005) Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Dev Biol 283 : 226–239.
61. YinY, WhiteAC, HuhSH, HiltonMJ, KanazawaH, et al. (2008) An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev Biol 319 : 426–436.
62. ShtutmanM, ZhurinskyJ, SimchaI, AlbaneseC, D'AmicoM, et al. (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 96 : 5522–5527.
63. BrugarolasJ, ChandrasekaranC, GordonJI, BeachD, JacksT, et al. (1995) Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 377 : 552–557.
64. FeroML, RivkinM, TaschM, PorterP, CarowCE, et al. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 85 : 733–744.
65. YoungP, BoussadiaO, HalfterH, GroseR, BergerP, et al. (2003) E-cadherin controls adherens junctions in the epidermis and the renewal of hair follicles. EMBO J 22 : 5723–5733.
66. BrachtendorfG, KuhnA, SamulowitzU, KnorrR, GustafssonE, et al. (2001) Early expression of endomucin on endothelium of the mouse embryo and on putative hematopoietic clusters in the dorsal aorta. Dev Dyn 222 : 410–419.
67. WilkinsonDG, NietoMA (1993) Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol 225 : 361–373.
68. MoormanAF, HouwelingAC, de BoerPA, ChristoffelsVM (2001) Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem 49 : 1–8.
69. BussenM, PetryM, Schuster-GosslerK, LeitgesM, GosslerA, et al. (2004) The T-box transcription factor Tbx18 maintains the separation of anterior and posterior somite compartments. Genes Dev 18 : 1209–1221.
70. BraunsteinM, RoseAB, HolmesSG, AllisCD, BroachJR (1993) Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev 7 : 592–604.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy