
Dot1-Dependent Histone H3K79 Methylation Promotes Activation of the Mek1 Meiotic Checkpoint Effector Kinase by Regulating the Hop1 Adaptor
During meiosis, accurate chromosome segregation relies on the proper interaction between homologous chromosomes, including synapsis and recombination. The meiotic recombination checkpoint is a quality control mechanism that monitors those crucial events. In response to defects in synapsis and/or recombination, this checkpoint blocks or delays progression of meiosis, preventing the formation of aberrant gametes. Meiotic recombination occurs in the context of chromatin and histone modifications, which play crucial roles in the maintenance of genomic integrity. Here, we unveil the role of Dot1-dependent histone H3 methylation at lysine 79 (H3K79me) in this meiotic surveillance mechanism. We demonstrate that the meiotic checkpoint function of Dot1 relies on H3K79me because, like the dot1 deletion, H3-K79A or H3-K79R mutations suppress the checkpoint-imposed meiotic delay of a synapsis-defective zip1 mutant. Moreover, by genetically manipulating Dot1 catalytic activity, we find that the status of H3K79me modulates the meiotic checkpoint response. We also define the phosphorylation events involving activation of the meiotic checkpoint effector Mek1 kinase. Dot1 is required for Mek1 autophosphorylation, but not for its Mec1/Tel1-dependent phosphorylation. Dot1-dependent H3K79me also promotes Hop1 activation and its proper distribution along zip1 meiotic chromosomes, at least in part, by regulating Pch2 localization. Furthermore, HOP1 overexpression bypasses the Dot1 requirement for checkpoint activation. We propose that chromatin remodeling resulting from unrepaired meiotic DSBs and/or faulty interhomolog interactions allows Dot1-mediated H3K79-me to exclude Pch2 from the chromosomes, thus driving localization of Hop1 along chromosome axes and enabling Mek1 full activation to trigger downstream responses, such as meiotic arrest.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003262
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003262
Summary
During meiosis, accurate chromosome segregation relies on the proper interaction between homologous chromosomes, including synapsis and recombination. The meiotic recombination checkpoint is a quality control mechanism that monitors those crucial events. In response to defects in synapsis and/or recombination, this checkpoint blocks or delays progression of meiosis, preventing the formation of aberrant gametes. Meiotic recombination occurs in the context of chromatin and histone modifications, which play crucial roles in the maintenance of genomic integrity. Here, we unveil the role of Dot1-dependent histone H3 methylation at lysine 79 (H3K79me) in this meiotic surveillance mechanism. We demonstrate that the meiotic checkpoint function of Dot1 relies on H3K79me because, like the dot1 deletion, H3-K79A or H3-K79R mutations suppress the checkpoint-imposed meiotic delay of a synapsis-defective zip1 mutant. Moreover, by genetically manipulating Dot1 catalytic activity, we find that the status of H3K79me modulates the meiotic checkpoint response. We also define the phosphorylation events involving activation of the meiotic checkpoint effector Mek1 kinase. Dot1 is required for Mek1 autophosphorylation, but not for its Mec1/Tel1-dependent phosphorylation. Dot1-dependent H3K79me also promotes Hop1 activation and its proper distribution along zip1 meiotic chromosomes, at least in part, by regulating Pch2 localization. Furthermore, HOP1 overexpression bypasses the Dot1 requirement for checkpoint activation. We propose that chromatin remodeling resulting from unrepaired meiotic DSBs and/or faulty interhomolog interactions allows Dot1-mediated H3K79-me to exclude Pch2 from the chromosomes, thus driving localization of Hop1 along chromosome axes and enabling Mek1 full activation to trigger downstream responses, such as meiotic arrest.
Introduction
During the specialized meiotic cell cycle, two rounds of chromosome segregation follow a single phase of DNA replication dividing the number of chromosomes by half to generate haploid gametes. One of the hallmarks of meiosis is the complex interaction between homologous chromosomes (homologs) involving synapsis and recombination. During meiotic prophase I, homologs find each other, get aligned and finally closely associate along their entire length (synapsis) in the context of the synaptonemal complex (SC). The SC is a tripartite structure composed of two lateral elements (LEs), connected by transverse filaments, which constitute the central region. The chromatin of both sister chromatids of each homolog is organized in loops attached at their base to each of the LEs [1], [2]. In budding yeast, the Red1 and Hop1 proteins localize to the LEs [3], whereas the Zip1 protein is a major component of the SC central region [4], [5]. Concomitant with SC development, meiotic recombination takes place. Meiotic recombination initiates with programmed double-strand breaks (DSBs) introduced by Spo11 and accessory proteins [6]. Meiotic DSBs are preferentially repaired using an intact non-sister chromatid resulting in physical connections between homologs (chiasmata), which promote proper chromosome segregation.
Accurate distribution of chromosomes to the progeny is essential for generation of functional gametes; thus, meiotic cells are endowed with a meiosis-specific surveillance mechanism, the so-called pachytene checkpoint or meiotic recombination checkpoint, which contributes to faithful chromosome segregation. In response to defects in meiotic recombination and/or chromosome synapsis, the pachytene checkpoint is triggered and blocks or delays exit from prophase of meiosis I to prevent aberrant chromosome segregation and the formation of aneuploid meiotic products [7], [8].
This evolutionary-conserved quality-control mechanism operates from yeast to mammals. In S. cerevisiae, the meiotic recombination checkpoint responding to unrepaired resected DSBs shares the same sensors with the DNA damage checkpoint operating in vegetative cells, including the Mec1/Ddc2 kinase, Rad24 and the 9-1-1 complex [9]–[11]. However, the Rad9 adaptor and the Rad53 checkpoint kinase are dispensable for this meiotic checkpoint. On the contrary, given the special chromosomal context where meiotic recombination takes place, the meiosis-specific axial chromosomal components Red1 and Hop1 act as adaptors between the upstream sensors and the downstream Mek1 meiotic effector kinase, which, like Rad53, is hyperphosphorylated upon checkpoint activation [12], [13]. In turn, the meiotic cell cycle delay is imposed by inhibition of crucial regulators of meiosis I progression, including the cyclin-dependent kinase Cdc28, the transcription factor Ndt80 and the polo-like kinase Cdc5 [14]–[19]. Budding yeast meiotic mutants, such as zip1 (defective in SC and crossover formation) or dmc1 (defective in the strand invasion step of interhomolog recombination), are invaluable genetic tools to activate the meiotic recombination checkpoint.
Meiotic recombination as well as detection and signaling of recombination intermediates by the checkpoint machinery occur in the context of chromatin; therefore, histone posttranslational modifications are expected to play important roles on these processes [20]. For example, Set1-dependent H3K4 methylation is linked to meiotic DSB formation [21], [22]. On the other hand, the Dot1 histone methyltranferase, which targets H3K79 [23]–[25], is largely dispensable for unperturbed meiosis, but is essential for meiotic checkpoint function. Mutation of DOT1 relieves the meiotic prophase arrest of zip1 and dmc1 mutants resulting in defective meiotic products [26]. Dot1 is also involved in several aspects of the DNA damage response in vegetative yeast cells and the DOT1L mammalian homolog also plays crucial cellular functions [27]. However, the molecular mechanisms underlying the meiotic checkpoint function of Dot1 are unknown.
Here, we investigated the role of Dot1-dependendent H3K79 methylation in zip1-induced checkpoint activation. By manipulation of Dot1 catalytic activity and levels, we found that the extent of H3K79 trimethylation correlates with the strength of checkpoint-imposed meiotic delay. We demonstrate that while the meiotic defects of a synapsis and recombination-deficient zip1 mutant are correctly sensed by Mec1-Ddc2 in the absence of H3K79me, activation of the downstream effector kinase Mek1 is impaired. We dissected the Mek1 phosphorylation events and found that Dot1 promotes its Hop1-dependent dimerization and auto-phosphorylation. Finally, we show that the effect of Dot1-dependent H3K79me on Hop1 localization is exerted, at least in part, by excluding Pch2 from the chromosomes. Our results indicate that constitutive methylation of H3K79 by Dot1 is required for proper chromosomal recruitment of Hop1 to relay the checkpoint signal to Mek1 in response to meiotic defects.
Results
Histone H3K79 methylation regulates the meiotic recombination checkpoint
Dot1 catalyzes the mono-, di - and tri-methylation of histone H3K79 (Figure 1A) by a non-processive mechanism [27], [28] and plays a crucial role in the meiotic recombination checkpoint [26]. Notably, we found that overall levels of H3K79me do not significantly change upon meiosis induction (Figure 1A, compare vegetative and meiotic cells; Figure S1A) or upon meiotic checkpoint activation (Figure 1A, compare wild type and zip1 meiotic cells; Figure S1A). Moreover, H3K79me meiotic levels were not significantly altered in the spo11 mutant, lacking meiotic recombination [6], or in other mutants defective in the meiotic recombination checkpoint, such as rad24, pch2 and ddc2 [10], [11], [29] (Figure S1B). Therefore, to determine whether regulation of the meiotic recombination checkpoint by Dot1 relies on H3K79me, we generated and analyzed H3-K79R and H3-K79A mutants, in which the lysine 79 targeted by Dot1 cannot be methylated (Figure 1A). Importantly, like dot1, both methylation-site mutants suppressed the pronounced checkpoint-imposed meiotic delay of the zip1 mutant (Figure 1B). In an otherwise wild-type background, DOT1 deletion has no or little meiotic effects and spore viability is high [26], [30]; likewise, the H3-K79R and H3-K79A single mutants showed wild-type levels of spore viability (Figure 1C), suggesting that H3K79me is dispensable in unperturbed meiosis. However, similar to zip1 dot1, spore viability was strongly reduced in zip1 H3-K79R and zip1 H3-K79A (Figure 1C), indicating that the defects conferred by zip1 persist in the double mutants despite their wild-type kinetics of meiotic progression. Thus, Dot1-dependent H3K79me is essential for meiotic recombination checkpoint function.
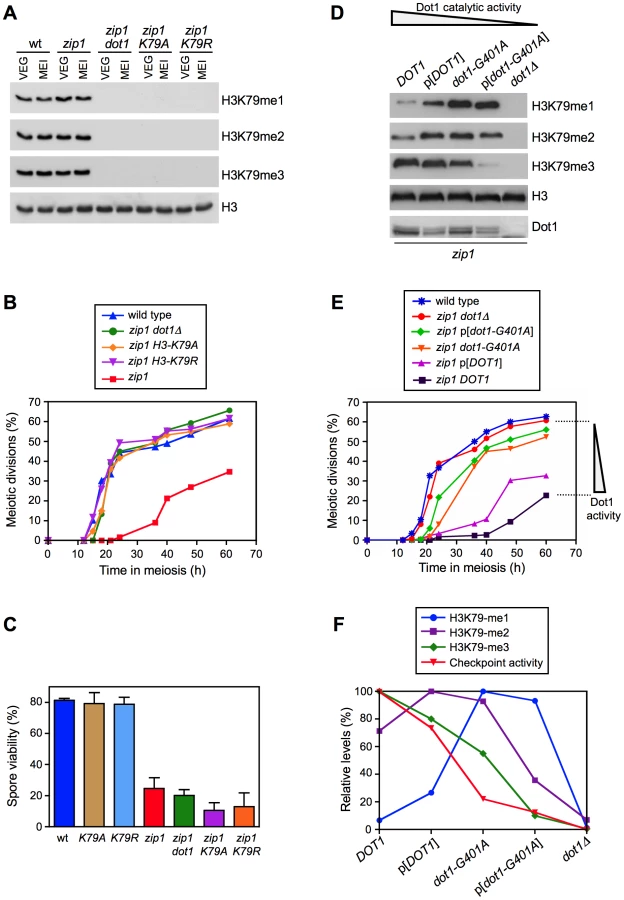
To further investigate the regulation of the meiotic checkpoint by H3K79me, we monitored checkpoint function in zip1 diploid strains exhibiting gradually decreased Dot1 activity. In order to generate this set of strains, we used the combination of the dot1-G401A allele, which confers partial catalytic activity [28], with the expression of DOT1 (or dot1-G401A) from a plasmid, which results in lower protein levels (Figure 1D; [31]). Analysis of H3K79-me1, -me2 and -me3 levels in meiotic cells confirmed a gradually reduced Dot1 activity following this order: DOT1>p[DOT1]>dot1-G401A>p[dot1-G401A]>dot1Δ, as manifested by progressively reduced H3K79-me3 and, conversely, progressively increased H3K79-me1 (Figure 1D). Interestingly, meiotic checkpoint activity, monitored as the ability to impose the zip1 meiotic delay, also showed a gradual decrease mirroring the drop in Dot1 catalytic function (Figure 1E). Quantification of the relative levels of each H3K79 methylation state revealed a marked correlation between H3K79-me3 and checkpoint function (Figure 1F). Thus, the status of H3K79 methylation modulates the meiotic recombination checkpoint, with the H3K79-me3 form being the most relevant to sustain the checkpoint response.
Dot1 is required for activation of the Mek1 effector kinase
Next, we sought to determine where in the meiotic recombination checkpoint pathway Dot1-dependent H3K79me is acting. We first analyzed checkpoint sensor function by monitoring the formation of zip1-induced Ddc2-GFP foci [11]. Formation of Ddc2 foci was not disrupted in the absence of Dot1 (Figure 2A), suggesting that H3K79me is not required for the ability of Mec1-Ddc2 to detect meiotic recombination intermediates. Upon checkpoint activation, the Mek1 effector kinase forms nuclear foci that can be detected both on chromosome spreads ([9]; see below) and in live meiotic cells (Figure 2A). Strikingly, we found that the zip1 mutant accumulated multiple discrete Mek1-GFP foci during meiotic prophase, whereas most zip1 dot1 cells displayed a diffuse Mek1 nuclear signal and only occasional foci were observed (Figure 2A) indicating that Dot1 promotes checkpoint-induced association of Mek1 to meiotic chromosomes (see below).

Mek1 is activated by phosphorylation in mutants that trigger the meiotic recombination checkpoint, including zip1 [12], [14], [32], [33]; therefore, we followed Mek1 phosphorylation throughout meiosis in wild-type, zip1 and zip1 dot1 cells using Phos-tag gels (Figure 2B). In the wild type, Mek1 was weakly and transiently activated during the peak of meiotic prophase in this strain background (around 12–15 h). In contrast, Mek1 was hyperactivated in zip1 cells as evidenced by the presence of additional, more persistent, and stronger phosphorylated forms. However, Mek1 hyperactivation was not observed in the zip1 dot1 double mutant; like in wild type, only a weak and transient phosphorylated form was detected. To rule out the possibility that the difference between zip1 and zip1 dot1 were due to their different kinetics of meiotic progression (zip1 exhibits a marked delay that is bypassed in zip1 dot1; Figure 1B), we monitored Mek1 phosphorylation in ndt80 pachytene-arrested cells. As presented in Figure 2C, zip1-induced hyperphosphorylation of Mek1 was severely impaired in the absence of Dot1.
In summary, these results place Dot1 function upstream of Mek1 in the meiotic recombination checkpoint pathway and indicate that, whereas Mec1/Ddc2 act independently of H3K79 methylation to sense meiotic defects, Dot1 is required for checkpoint-induced activation of Mek1.
Autophosphorylation of Mek1 depends on Dot1
In ndt80-arrested cells, using high-resolution Phos-tag gels, we were able to resolve several zip1-induced shifted forms of Mek1 above the basal band (Figure 3A–3D). Phosphatase treatment eliminated all band shifts indicating that they represent distinct phosphorylated forms (Figure 3A). We used different mek1 versions carrying specific mutations, as well as mutants in upstream components of the checkpoint pathway, in order to determine the contribution of different phosphorylation events to the observed checkpoint-induced Mek1 forms in zip1 ndt80 cells (Figure 3E). Mek1 phosphorylation was completely abolished in the hop1 mutant, lacking a LE-component meiotic checkpoint adaptor [3], [34], [35] (Figure 3B) and in the spo11 mutant, which does not initiate recombination [36] (Figure 3C). However, in the absence of Dot1, only the upper phosphorylated bands were eliminated (Figure 3B–3D, white arrowheads), but the form immediately above the basal Mek1 band remained intact (Figure 3B–3D, black arrowhead). Interestingly, this moderately-shifted form was reduced in mec1 cells and virtually disappeared in mec1 tel1 and rad24 tel1 mutants (Figure 3C and 3D, black arrowhead), suggesting that it arises from Mec1/Tel1-dependent phosphorylation. On the other hand, the kinase-dead mek1-K199R allele, as well as the autophosphorylation-defective mek1-T327A and mek1-T331A mutants [33], specifically lacked the upper bands displaying the stronger mobility shift, suggesting that they result from Mek1 autophosphorylation (Figure 3D, white arrowheads). In contrast, the Mek1 form immediately above the basal band (i.e., resulting from Mec1/Tel1 action) remained invariable in those mek1 mutants (Figure 3D, black arrowhead). Thus, interestingly, the zip1 dot1 mutant showed a similar pattern to that of zip1 mek1-K199R, zip1 mek1-T327A or zip1 mek1-T331A (Figure 3D), strongly suggesting that Dot1 is mainly required for Mek1 autophosphorylation, but not for its Mec1/Tel1-dependent phosphorylation (Figure 3E).

It has been proposed that dimerization of Mek1 promotes its function, likely by facilitating in trans autophosphorylation [33], [37]. Thus, we hypothesized that Dot1 could be required for Mek1 dimerization. Importantly, we found that GST-driven forced dimerization of Mek1 restored its full phosphorylation even in the absence of Dot1, although Mek1 activation was not maintained at late time points (Figure 2B). Consistently, expression of GST-MEK1 in zip1 dot1 strains conferred a brief, but significant, meiotic delay (Figure 2D). As previously reported, the zip1 GST-MEK1 mutant was completely halted (Figure 2D) [37], and we found that this block was accompanied by the persistent hyperphosphorylation of GST-Mek1 (Figure 2B). The permanent or transient arrest conferred by GST-Mek1 in zip1 or zip1 dot1, respectively, was completely relieved when inactive kinase (GST-mek1-K199R) or autophosphorylation-defective (GST-mek1-T327A) versions were introduced (Figure 2D), confirming that in GST-MEK1 strains, meiotic progression was slowed down by forced Mek1 activation and not by another unrelated cause. To further support this conclusion, we monitored another downstream molecular marker of pachytene checkpoint activation, such as the inhibition of the production of the Cdc5 polo-like kinase [14], [18]. As expected, whereas induction of Cdc5 was delayed in zip1 cells, the zip1 dot1 double mutant displayed wild-type kinetics of Cdc5 production (Figure 2B). Strikingly, consistent with the kinetics of meiotic progression (Figure 2D), expression of GST-MEK1 in zip1 dot1 cells restored a significant delay in Cdc5 induction. Furthermore, Cdc5 production was severely impaired in the arrested zip1 GST-MEK1 strain (Figure 2B). In summary, these observations indicate that artificial dimerization of Mek1 partially overcomes Dot1 requirement for Mek1 activation and further supports the conclusion that Dot1 function promotes Mek1 autophosphorylation.
Dot1 is required for localization and activation of the Hop1 meiotic checkpoint adaptor
It has been reported that activated Hop1 promotes Mek1 dimerization via a C-terminal domain [33], [38]; therefore, we investigated whether the effect of Dot1 on Mek1 phosphorylation was mediated by Hop1. First, we studied Hop1 localization on chromosome spreads of ndt80-arrested zip1 and zip1 dot1 strains. As previously described [3], Hop1 displayed a predominantly linear staining along the lateral elements of zip1 chromosomes. In contrast, only short stretches of Hop1 could be detected in the zip1 dot1 mutant, which showed a predominating Hop1 punctate pattern (Figure 4A and 4B, left panel). Consistent with our observations in live cells (Figure 2A), we also detected a marked reduction of Mek1 chromosomal foci in zip1 dot1, compared to the zip1 single mutant (Figure 4A and 4B, right panel). In addition, we also analyzed Hop1 localization in zip1 and zip1 dot1 live meiotic cells expressing HOP1-GFP. In line with the aberrant distribution on spreads, we observed that Hop1-GFP signal was weaker and less continuous in zip1 dot1 cells. (Figure 4C and 4D; Video S1). This discontinuous localization of Hop1 does not result from a pronounced alteration of overall chromosome structure, because the SC lateral component Red1 [3] displayed a linear distribution in both zip1 and zip1 dot1 strains (Figure S2). On the other hand, the dot1 single mutant only showed a modest decrease of Hop1-GFP signal compared with the wild type (see Figure 7C and 7D below). Thus, upon zip1-induced checkpoint activation, Dot1 enables proper loading or maintenance of Hop1 onto chromosomes.


Since Mec1/Tel1-dependent phosphorylation of Hop1 at defined S/T-Q motifs is required for Mek1 activation and localization [34], we examined zip1-induced Hop1 phosphorylation in the absence of Dot1, by monitoring its gel mobility shift. As shown in Figure 4E, the zip1 dot1 mutant displayed a severe defect in Hop1 phosphorylation, similar to the zip1 mec1 and zip1 spo11 mutants also analyzed as controls (Figure 4E and Figure S3). Even after long overexposure of the gels, only a barely visible phosphorylated form of Hop1 could be detected in the absence of Dot1 (Figure 4E).
These observations suggest that the defect in Mek1 autophosphorylation observed in the absence of Dot1 stems from impaired Hop1 function. To confirm this notion, we overexpressed HOP1 from a high-copy plasmid in zip1 dot1 cells. As shown in Figure 5, whereas the zip1 dot1 mutant transformed with empty vector showed defective Mek1 localization and activation, HOP1 overexpression in zip1 dot1 restored Mek1 chromosomal foci (Figure 5A), Mek1 phosphorylation (Figure 5B), and reestablished a substantial meiotic delay (Figure 5C). We found that Hop1 overproduction also conferred a slight reduction in the efficiency of meiotic progression in the wild type (Figure 5C) and further enhanced the zip1 meiotic delay, as expected from the strong hyperphosphorylation of Mek1 (Figure 5B and 5C). Notably, in all cases (wild type, zip1 or zip1 dot1), the further delay in meiotic progression imposed by high levels of Hop1 was suppressed by the absence of Mek1 (Figure 5C), proving that it was caused from amplified pachytene checkpoint signaling and not from an unrelated cause.

H3K79me is required for Mek1 and Hop1 phosphorylation and localization
We have shown that, like dot1, mutation of H3K79 to non-methylatable residues completely bypasses the checkpoint-induced meiotic delay of zip1 (Figure 1B). On the other hand, we have revealed that, in zip1 cells, Dot1 orchestrates Hop1 and Mek1 activation and chromosomal distribution (Figure 2, Figure 3, Figure 4, and Figure 5). To confirm that Hop1 and Mek1 checkpoint functions are also directly regulated by H3K79me, and not by another possible methyltransferase-independent function of Dot1, we examined their phosphorylation and localization in the zip1 H3-K79R and zip1 H3-K79A mutants. We found that, indeed, these histone point mutants phenocopy the dot1 defects in Mek1 foci formation (Figure 6A and 6B) and Mek1 autophosphorylation (Figure 6D; Figure S4). Likewise, the zip1 H3-K79R and zip1 H3-K79A mutants resemble dot1 in the impaired Hop1 chromosomal distribution (Figure 6A and 6C; Figure S5) and checkpoint-induced phosphorylation (Figure 6E; Figure S4).

Thus, taken together, our results indicate that, upon meiotic recombination checkpoint triggering, Dot1-dependent H3K79 methylation promotes proper chromosomal localization and activation of Hop1, which in turn, is required to sustain Mek1 autophosphorylation and the ensuing checkpoint response.
H3K79me partially controls Hop1 chromosomal localization via Pch2
Previous studies have shown that whereas in the zip1 mutant the Pch2 meiotic checkpoint protein is detected only in the nucleolar (rDNA) region, in the zip1 dot1 double mutant Pch2 is distributed throughout all chromatin [26]. To confirm that the regulation of Pch2 localization by Dot1 depends on the histone H3 methyltransferase activity, we analyzed Pch2 distribution on spread meiotic chromosomes of the zip1 H3-K79R and zip1 H3-K79A mutants. Although global Pch2 protein levels remained fairly invariable in the different mutants (Figure S4), we found that, like in zip1 dot1, Pch2 mislocalized to chromatin outside the rDNA in zip1 H3-K79R and zip1 H3-K79A strains (Figure 7A), suggesting that H3K79me excludes Pch2 from chromosomes.
Several lines of evidence support a role for Pch2 in promoting the turnover of Hop1 from meiotic chromosomes, at least in unperturbed meiosis [29], [39], [40]; therefore, it was possible that the reduced localization of Hop1 in the absence of Dot1 could stem from the action of the Pch2 protein aberrantly present at chromosomal locations removing Hop1 from zip1 chromosomes. To investigate this possibility, we monitored Hop1 localization in zip1 dot1 pch2 strains. Interestingly, we found that deletion of PCH2 alleviated to some extent the defective Hop1 localization pattern of zip1 dot1, although it did not fully restore the high and continuous Hop1 levels present in zip1 (Figure 7B–7D). To determine whether the increased abundance of Hop1 along chromosomes in zip1 dot1 pch2 restores the checkpoint-induced delay we analyzed meiotic divisions and Mek1 phosphorylation (Figure 7E–7F). We found that the checkpoint was still impaired in the zip1 dot1 pch2 triple mutant because, like the zip1 dot1 and the zip1 pch2 double mutants, it displayed wild-type kinetics of meiotic progression (Figure 7E) and defective Mek1 activation (Figure 7F), implying a more complex contribution of Pch2's function to the pachytene checkpoint response (see Discussion).
In summary, these observations indicate that in the zip1 mutant, methylation of H3K79 by Dot1 controls proper chromosomal distribution of Hop1 by maintaining Pch2 confined in the nucleolar region. The fact that Hop1 localization is still partially impaired in the zip1 dot1 pch2 triple mutant suggests that Dot1 may also regulate Hop1 chromosomal recruitment by a Pch2-independent mechanism (Figure 8).

Discussion
Previous studies have shown that Dot1 is important for the pachytene checkpoint, but the molecular mechanism underlying such function remained unclear. Here, we provide evidence that methylation of H3K79 by Dot1 contributes to the meiotic recombination checkpoint response by enabling proper Hop1 chromosomal recruitment, which, in turn is a requisite for Mek1 activation by autophosphorylation.
We demonstrate that the function of Dot1 in the meiotic recombination checkpoint specifically relies on the methylation of H3K79, since the non-methylatable H3-K79A and H3-K79R mutations confer essentially the same meiotic phenotypes as the lack of Dot1. Moreover, by modulating Dot1 catalytic activity, we found that high levels of the H3K79-me3 are required for full checkpoint activation raising the possibility that this methylation state is particularly critical for promoting the proper localization of the Hop1 meiotic checkpoint adaptor (see below).
In mitotic cells, methylated histones are well-known chromatin marks for recognition of DSBs by checkpoint adaptors. In S. cerevisiae, the Rad9 adaptor is recruited to DSB sites by H3K79me [41], [42], whereas in S. pombe, which lacks H3K79me, the recruitment of the Crb2 adaptor relies on H4K20me [43]. In mammalian cells, the Rad9 and Crb2 homolog 53BP1 appears to recognize both H3K79me and H4K20me [44]–[46]. All these DNA damage checkpoint adaptors (Rad9, Crb2 and 53BP1) contain tandem tudor domains that mediate the interaction with the methylated histones. Rad9, Crb2 and 53BP1 also possess BRCT motifs; in fact, the recognition of DSBs by Rad9 and Crb2 in S. cerevisiae and S. pombe, respectively, is also mediated by their binding to phosphorylated histone H2A (hereafter γH2AX) via the BRCT domains [47], [48]. However, the Hop1 meiotic checkpoint adaptor lacks either tudor or BRCT motifs and contains a HORMA domain likely involved in protein-protein interactions [49], raising the possibility that its chromosomal recruitment can be mediated by different mechanisms.
As mentioned before, in DNA damaged vegetative cells, Rad9 function depends both on H3K79me and γH2AX [47], [50]–[52]; however, the relevance of both histone modifications appears to be different in meiotic cells. Dot1-dependent H3K79me is crucial for checkpoint function, at least in Zip1-deficient cells, because deletion of DOT1 (or mutation of H3K79) results in complete bypass of the zip1 meiotic block. In contrast, an H2A-S129* mutant, lacking the four C-terminal amino acids of histone H2A including the SQ phosphorylation site [53], has no defect in the zip1-induced checkpoint (Figure S6A). Moreover, like in both single mutants, meiotic progression and spore viability are essentially normal in the dot1 H2A-S129* double mutant (Figure S6A, S6B).
We show here that Dot1 is required for Mek1 and Hop1 activation in meiotically-challenged cells, but in addition to the checkpoint function, Mek1 and Hop1 promote the repair of meiotic DSBs by Dmc1-dependent interhomolog recombination [34], [37], [38], [54]. Consistent with this function, in the absence of Dmc1, Dot1 prevents the repair of DSBs by Rad54-dependent sister-chromatid recombination, which is controlled, at least in part, by inhibitory phosphorylation of Rad54 by Mek1 [26], [54]. In principle, it could be possible that impaired Hop1/Mek1 function in the absence of Dot1 could induce an alternative intersister recombination pathway resulting in meiotic progression because of the disappearance of the meiotic defects initially triggering the checkpoint. However, deletion of DOT1 alleviates the meiotic arrest of zip1 rad54 and dmc1 rad54 mutants, where intersister repair is impaired, strongly suggesting that Dot1 performs a bona-fide meiotic checkpoint function [26]. The fact that, unlike Mek1 and Hop1, the Dot1 protein is dispensable in otherwise unperturbed meiosis implies the H3K79me is mostly relevant to signal defects when meiotic chromosome metabolism is disturbed (i.e., zip1 or dmc1 mutants). Consistent with this notion, Hop1 localization on zip1 chromosomes is dramatically altered in the absence of Dot1, but it is only slightly reduced in the dot1 single mutant as compared with the wild type (Figure 7C and 7D).
In other studies, activation of the Mek1 effector meiotic kinase has been monitored either by a slight electrophoretic mobility shift [32], [34] or by using an anti-phospho-Ser/Thr Akt substrate antibody, which specifically recognizes phosphorylation of Mek1 at T327 [33], [37], [55]. However, those assays do not permit one to delineate the different events contributing to Mek1 activation. Here, by using high-resolution Phos-tag gels, we identify several phosphorylated Mek1 forms and dissect the genetic requirements for sequential Mek1 activation. Our findings support a model (Figure 3E; Figure 8) in which the presence of unrepaired DSBs and/or unsynapsed chromosomes results in the initial phosphorylation of Mek1 by the redundant action of Mec1/Tel1. This priming phosphorylation is followed by autophosphorylation of Mek1 at T327 and T331 leading to full Mek1 activation supporting the checkpoint response. We found that Dot1 is chiefly required for this last step, which is mediated by Mek1 dimerization promoted by the Hop1 C-terminal domain [38]. Thus, the altered localization of Hop1 on zip1 dot1 chromosomes likely explains the defect in Mek1 autophosphorylation. Interestingly, GST-mediated forced dimerization of Mek1 bypasses Dot1 requirement for its activation; however, this activation is only transient in the absence of Dot1, suggesting that proper chromosome axis architecture is required for maintenance of Mek1 activity.
We found that global levels of H3K79me do not significantly change in response to the meiotic defects of the zip1 mutant, but this methylation is critical for the checkpoint response. The nature of the signal that triggers the meiotic checkpoint in zip1 is still unclear. Like in mammals [56], the existence of a synapsis checkpoint in yeast has also been proposed [7], [55], [58]. Nevertheless, Dot1 is also required for the meiotic cell cycle arrest of the dmc1 mutant that accumulates unrepaired DSBs [26], indicating that H3K79me is also involved in the response to meiotic DSBs. It has been reported that, under certain conditions, DSBs are efficiently repaired in zip1 mutants [37] implying that the signal triggering the checkpoint could be different. However, Ddc2 foci marking the presence of recombination intermediates are detected in zip1 [11] (Figure 2A), consistent with at least some DSBs remaining unrepaired in zip1 mutants [57], [59], [60] sufficient to induce the checkpoint. Alternatively, or in addition, Mec1-Ddc2 may also sense defects in structural aspects of interhomolog interactions resulting from the lack of the central region of the SC [39]. In any case, independently of the nature of the signal triggering the meiotic checkpoint response(s), the question of how a constitutive histone mark, such as H3K79me, contributes to Hop1-mediated Mek1 activation specifically in challenged meiosis remains to be elucidated. In the DNA damage response in vegetative yeast cells or somatic mammalian cells it has been proposed, though never proven, that chromatin remodeling in the vicinity of DNA lesions may locally expose constitutive marks (i.e., H3K79me, H4K20me) supporting the recruitment of DNA damage checkpoint adaptors to activate the checkpoint [44], [45]. In meiotic cells, the DSB metabolism is linked to the special architecture of the chromosome axis [61]. Therefore, we envision that unrepaired DSBs and/or defects in interhomolog connections may provoke chromatin conformational changes unmasking H3K79me capable to drive proper Hop1 distribution along the axes, enabling its activation by Mec1 to elicit the downstream checkpoint events including Mek1 full activation by autophosphorylation (Figure 8).
Although it is formally possible that H3K79me may directly facilitate Hop1 recruitment to some extent, we provide evidence indicating that the control of Hop1 chromosomal distribution by H3K79me is substantially driven by regulation of the Pch2 protein. Pch2 was initially discovered as a meiotic checkpoint protein required for the zip1-induced meiotic arrest [29], but more recent studies have shown that Pch2 impacts multiple aspects of meiotic chromosome dynamics [55], [62]–[64]. In particular, Pch2 acts as a negative regulator of Hop1 chromosomal abundance [39], [40]. In wild-type pachytene chromosomes, Pch2 localizes to the unsynapsed rDNA region (nucleolus) and also along synapsed chromosomes [29], [40]. In contrast, Pch2 is solely detectable at the nucleolar region in the zip1 mutant [29]; remarkably, in the absence of H3K79me, Pch2 is redistributed throughout all chromatin of zip1 nuclei (Figure 7A). We hypothesize that, as a consequence of the synapsis defects of zip1, the H3K79me mark becomes exposed functioning as an anti-binding signal for Pch2, thus permitting the extensive Hop1 distribution found on zip1 chromosomes (Figure 8). In the absence of Dot1 (or H3K79me), the presence of chromosomal Pch2 triggers the removal of Hop1 and the consequent defect in Mek1 activation. The reduced global levels of Hop1 detected in zip1 dot1 (Figure S3 and Figure 6E) are also consistent with a higher protein turnover.
Interestingly, like in zip1 dot1, the synapsis checkpoint is still completely defective in the zip1 dot1 pch2 triple mutant, despite the partial restoration of Hop1 localization. Since the excess of Hop1 induced by other means, such as HOP1 overexpression, but in the presence of Pch2, does confer a meiotic delay in zip1 dot1 and restores Mek1 phosphorylation (Figure 5), it is conceivable that nucleolar Pch2 performs an additional downstream function in Mek1 activation (Figure 8) and/or that the excess of Hop1 in the absence of Pch2 is not correctly assembled on chromosome axes to support checkpoint activation. In fact, the zip1 pch2 mutant itself is also checkpoint deficient. Future studies will address these intriguing possibilities.
Dot1/DOT1L is structurally conserved throughout evolution from budding yeast to worms, flies, mice and humans; therefore, it is possible that members of the Dot1 family play similar roles in Metazoa. DOT1L is essential in mammals [65] functioning in embryogenesis, hematopoiesis and cardiac development [27]; however, much less is known about the impact of mammalian DOT1L in the DNA damage response. It would be interesting to determine whether, like the yeast counterpart, Dot1 orthologs are involved in meiotic checkpoint control in higher eukaryotes.
Materials and Methods
Yeast strains and plasmids
Yeast strains genotypes are listed in Table 1. All the strains are in the BR1919 background [66]. Gene deletions were made using a PCR-based approach [67], [68] except for dot1::URA3, zip1::LYS2 and ndt80::LEU2, which were previously described [19], [26], [29]. MEK1-13myc, MEK1-GFP and HOP1-GFP were made by a PCR approach [68]. The C-terminally tagged Mek1-13myc and Mek1-GFP proteins are functional because spore viability of homozygous tagged wild-type diploids was similar to that of untagged strains and, in addition, they supported the checkpoint-induced delay of a hop2 mutant. In zip1 HOP1-GFP strains the meiotic block was less tight, but Hop1-GFP displayed a localization pattern indistinguishable from that of the untagged protein (Figure S5); therefore, we used the native GFP fluorescence for quantitation of Hop1 localization. N-terminal tagging of Pch2 with three copies of the HA epitope has been previously described [29]. Strains carrying DOT1 or dot1-G401A at its genomic locus or in the pRS315 vector (plasmids pRS315-DOT1 and pFvL54, respectively) were described [28], [31]. The H3-K79A and H3-K79R strains are deleted for all genomic copies of the histone H3-H4 encoding genes (HHT1-HHF1 and HHT2-HHF2) and express different versions of H3 from centromeric plasmids carrying either the hht2(K79A)-HHF2 or hht2(K79R)-HHF2 mutant genes (pFvL87 and pFvL88, respectively). The mek1-T327A, mek1-T331A, mek1-K199R mutations, as well as the GST-MEK1 construct were introduced as described [33], using plasmids kindly provided by N. Hollingsworth (Stony Brook University, NY). The high-copy HOP1 plasmid was also described [69]. Strains harboring the hta1-S129* and hta2-S129* mutations lacking the last four amino acids of the C-terminal tail of histone H2A including the serine 129 phosphorylated by Mec1/Tel1 [53] were made using plasmids pJHA16 and pJHA17 (provided by J. Downs, University of Sussex) following a pop-in/pop-out strategy. For meiotic time courses, strains were grown in 2×SC (3.5 ml) for 20–24 hours, then transferred to YPDA (2.5 ml) and incubated to saturation for additional 8 hours. Cells were harvested, washed with 2% potassium acetate (KAc), resuspended into 2% KAc (10 ml) and incubated at 30°C with vigorous shaking to induce meiosis and sporulation. Both YPDA and 2% KAc were supplemented with 20 mM adenine and 10 mM uracil. The culture volumes were scaled-up when needed.

Western blotting and analysis of Mek1 phosphorylation
TCA cell extracts from 5–10 ml of sporulating cultures were processed as described [14]. To resolve the phosphorylated forms of Mek1 or Mek1-GFP, 10% or 7% gels (acrylamide∶bisacrylamide 29∶1), respectively, containing 37.5 µM Phos-tag (Wako Chemicals) and 75 µM MnCl2 were used. Gels were run on ice at 100 volts in a MiniProtean3 (Bio-Rad) for 3 h. After running, gels were washed with 1 mM EDTA before transfer to PVDF membranes.
For dephosphorylation assays, total TCA cell extracts solubilized in Laemmli buffer were diluted 10 times with phosphatase buffer supplemented with 1 mM MnCl2. Diluted extracts were treated with 2000 units of lambda phosphatase (New England Biolabs) for 30 min at 30°C. As control, a similar aliquot of the diluted extract was incubated under the same conditions but without adding phosphatase. Samples were re-precipitated with 20% TCA, washed with acetone, boiled in Laemmli buffer and loaded in Phostag gels.
The following antibodies were used: rabbit polyclonal anti-Mek1 (1∶1000 dilution) [11] and anti-Dot1 (1∶2000 dilution) [31]. Rabbit polyclonal anti-H3K79-me1 (ab2886; 1∶1000 dilution), anti-H3K79-me2 (ab3594; 1∶2000 dilution), anti-H3K79-me3 (ab2621; 1∶2000 dilution), and anti-histone H3 (ab1791 : 1∶5000) were from Abcam. Rabbit polyclonal anti-Hop1 (1∶2000 dilution) [3], was from S. Roeder (Yale University). Anti-Cdc5 (sc-6733; 1∶1000 dilution) was from Santa Cruz Biotechnology. Mouse monoclonal anti-HA (12CA5; 1∶2000 dilution) was from Roche. Anti-phosphoglycerate kinase (PGK) (A-6457, 1∶10000 dilution) was from Molecular Probes. The ECL or ECL+ reagents were used for detection. The signal was captured on film and/or with a ChemiDoc XRS (Bio-Rad) system and quantified with the Quantity One software (Bio-Rad).
Cytology
Immunofluorescence of chromosome spreads was performed essentially as described [29]. To detect Mek1-myc and Mek1-GFP, mouse monoclonal anti-myc (clone 4A6, Millipore) and mouse monoclonal anti-GFP (JL-8, Clontech) antibodies, respectively, were used at 1∶200 dilution. Rabbit polyclonal anti-Red1 and anti-Hop1 antibodies (gifts from S. Roeder) have been previously described [3], [5]. Anti-mouse and/or anti-rabbit AF-488 and AF-594 conjugated secondary antibodies (Molecular Probes) were used at 1∶200 dilution. Images were captured with a Nikon Eclipse 90i fluorescence microscope controlled with the MetaMorph software and equipped with an Orca-AG (Hammamatsu) CCD camera and a PlanApo VC 100×1.4 NA objective.
Whole cell images were captured with an Olympus IX71 fluorescence microscope equipped with a personal DeltaVision system (Applied Precision), a CoolSnap HQ2 (Photometrics) camera and a 100× UPLSAPO 1.4 NA objective. Exposure times were 800 ms, 400 ms and 300 ms for Ddc2-GFP, Mek1-GFP, and Hop1-GFP, respectively. Stacks of 20 planes at 0.2 µm intervals were captured. Maximum intensity projections of deconvolved images were generated with the SoftWoRx 5.0 software (Applied Precision). Quantification of GFP signals in the projections of individual nuclei was performed with the Image J software (http://rsb.info.nih.gov/ij/). Background signal was subtracted using the Otsu's or the Renyi's entropy threshold methods in Image J. To outline the contour of the cells in the representative whole-cell images presented, an overlay of the DIC image with 15–20% transparency over the GFP signal is shown.
Other techniques
To analyze meiotic nuclear divisions, cells were fixed in 70% ethanol, washed in PBS and stained with 1 µg/µl DAPI for 15 minutes at room temperature. At least 300 cells of every strain were scored at each time point. Analyses of meiotic kinetics were repeated several times; representative time courses are shown. Spore viability was determined by tetrad dissection. To calculate the statistical significance of differences a two-tailed Student t-test was used. P-values were calculated using the GraphPad Prism 4.0 software. P<0.01 was considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RoederGS (1997) Meiotic chromosomes: it takes two to tango. Genes Dev 11 : 2600–2621.
2. ZicklerD, KlecknerN (1999) Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33 : 603–754.
3. SmithAV, RoederGS (1997) The yeast Red1 protein localizes to the cores of meiotic chromosomes. J Cell Biol 136 : 957–967.
4. DongH, RoederGS (2000) Organization of the yeast Zip1 protein within the central region of the synaptonemal complex. J Cell Biol 148 : 417–426.
5. SymM, EngebrechtJA, RoederGS (1993) ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72 : 365–378.
6. KeeneyS (2001) Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol 52 : 1–53.
7. MacQueenAJ, HochwagenA (2011) Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell Biol 21 : 393–400.
8. RoederGS, BailisJM (2000) The pachytene checkpoint. Trends Genet 16 : 395–403.
9. HongEJ, RoederGS (2002) A role for Ddc1 in signaling meiotic double-strand breaks at the pachytene checkpoint. Genes Dev 16 : 363–376.
10. LydallD, NikolskyY, BishopDK, WeinertT (1996) A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383 : 840–843.
11. RefolioE, CaveroS, MarconE, FreireR, San-SegundoPA (2011) The Ddc2/ATRIP checkpoint protein monitors meiotic recombination intermediates. J Cell Sci 124 : 2488–2500.
12. BailisJM, RoederGS (2000) Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell 101 : 211–221.
13. WanL, de los SantosT, ZhangC, ShokatK, HollingsworthNM (2004) Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell 15 : 11–23.
14. AcostaI, OntosoD, San-SegundoPA (2011) The budding yeast polo-like kinase Cdc5 regulates the Ndt80 branch of the meiotic recombination checkpoint pathway. Mol Biol Cell 22 : 3478–3490.
15. ChuS, HerskowitzI (1998) Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell 1 : 685–696.
16. LeuJY, RoederGS (1999) The pachytene checkpoint in S. cerevisiae depends on Swe1-mediated phosphorylation of the cyclin-dependent kinase Cdc28. Mol Cell 4 : 805–814.
17. PakJ, SegallJ (2002) Role of Ndt80, Sum1, and Swe1 as targets of the meiotic recombination checkpoint that control exit from pachytene and spore formation in Saccharomyces cerevisiae. Mol Cell Biol 22 : 6430–6440.
18. SourirajanA, LichtenM (2008) Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev 22 : 2627–2632.
19. TungKS, HongEJ, RoederGS (2000) The pachytene checkpoint prevents accumulation and phosphorylation of the meiosis-specific transcription factor Ndt80. Proc Natl Acad Sci U S A 97 : 12187–12192.
20. BrachetE, SommermeyerV, BordeV (2011) Interplay between modifications of chromatin and meiotic recombination hotspots. Biol Cell 104 : 51–69.
21. BordeV, RobineN, LinW, BonfilsS, GeliV, et al. (2009) Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J 28 : 99–111.
22. SollierJ, LinW, SoustelleC, SuhreK, NicolasA, et al. (2004) Set1 is required for meiotic S-phase onset, double-strand break formation and middle gene expression. EMBO J 23 : 1957–1967.
23. FengQ, WangH, NgHH, Erdjument-BromageH, TempstP, et al. (2002) Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol 12 : 1052–1058.
24. NgHH, FengQ, WangH, Erdjument-BromageH, TempstP, et al. (2002) Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev 16 : 1518–1527.
25. van LeeuwenF, GafkenPR, GottschlingDE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109 : 745–756.
26. San-SegundoPA, RoederGS (2000) Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell 11 : 3601–3615.
27. NguyenAT, ZhangY (2011) The diverse functions of Dot1 and H3K79 methylation. Genes Dev 25 : 1345–1358.
28. FrederiksF, TzourosM, OudgenoegG, van WelsemT, FornerodM, et al. (2008) Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol 15 : 550–557.
29. San-SegundoPA, RoederGS (1999) Pch2 links chromatin silencing to meiotic checkpoint control. Cell 97 : 313–324.
30. LuiDY, Peoples-HolstTL, MellJC, WuHY, DeanEW, et al. (2006) Analysis of close stable homolog juxtaposition during meiosis in mutants of Saccharomyces cerevisiae. Genetics 173 : 1207–1222.
31. CondeF, OntosoD, AcostaI, Gallego-SanchezA, BuenoA, et al. (2010) Regulation of tolerance to DNA alkylating damage by Dot1 and Rad53 in Saccharomyces cerevisiae. DNA Repair 9 : 1038–1049.
32. Cartagena-LirolaH, GueriniI, ViscardiV, LucchiniG, LongheseMP (2006) Budding Yeast Sae2 is an In Vivo Target of the Mec1 and Tel1 Checkpoint Kinases During Meiosis. Cell Cycle 5 : 1549–1559.
33. NiuH, LiX, JobE, ParkC, MoazedD, et al. (2007) Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol 27 : 5456–5467.
34. CarballoJA, JohnsonAL, SedgwickSG, ChaRS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132 : 758–770.
35. WolteringD, BaumgartnerB, BagchiS, LarkinB, LoidlJ, et al. (2000) Meiotic segregation, synapsis, and recombination checkpoint functions require physical interaction between the chromosomal proteins Red1p and Hop1p. Mol Cell Biol 20 : 6646–6658.
36. KeeneyS, GirouxCN, KlecknerN (1997) Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88 : 375–384.
37. WuHY, HoHC, BurgessSM (2010) Mek1 kinase governs outcomes of meiotic recombination and the checkpoint response. Curr Biol 20 : 1707–1716.
38. NiuH, WanL, BaumgartnerB, SchaeferD, LoidlJ, et al. (2005) Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol Biol Cell 16 : 5804–5818.
39. BornerGV, BarotA, KlecknerN (2008) Yeast Pch2 promotes domainal axis organization, timely recombination progression, and arrest of defective recombinosomes during meiosis. Proc Natl Acad Sci U S A 105 : 3327–3332.
40. JoshiN, BarotA, JamisonC, BornerGV (2009) Pch2 links chromosome axis remodeling at future crossover sites and crossover distribution during yeast meiosis. PLoS Genet 5: e1000557 doi:10.1371/journal.pgen.1000557..
41. GrenonM, CostelloeT, JimenoS, O'ShaughnessyA, FitzgeraldJ, et al. (2007) Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast 24 : 105–119.
42. WysockiR, JavaheriA, AllardS, ShaF, CoteJ, et al. (2005) Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol 25 : 8430–8443.
43. SandersSL, PortosoM, MataJ, BahlerJ, AllshireRC, et al. (2004) Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119 : 603–614.
44. BotuyanMV, LeeJ, WardIM, KimJE, ThompsonJR, et al. (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127 : 1361–1373.
45. HuyenY, ZgheibO, DitullioRAJr, GorgoulisVG, ZacharatosP, et al. (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432 : 406–411.
46. WakemanTP, WangQ, FengJ, WangXF (2012) Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J 31 : 2169–2181.
47. HammetA, MagillC, HeierhorstJ, JacksonSP (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 8 : 851–857.
48. SandersSL, AridaAR, PhanFP (2010) Requirement for the phospho-H2AX binding module of Crb2 in double-strand break targeting and checkpoint activation. Mol Cell Biol 30 : 4722–4731.
49. HunterN (2008) Hop1 and the meiotic DNA-damage response. Cell 132 : 731–732.
50. CondeF, RefolioE, Cordon-PreciadoV, Cortes-LedesmaF, AragonL, et al. (2009) The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 182 : 437–446.
51. JavaheriA, WysockiR, Jobin-RobitailleO, AltafM, CoteJ, et al. (2006) Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc Natl Acad Sci U S A 103 : 13771–13776.
52. TohGW, O'ShaughnessyAM, JimenoS, DobbieIM, GrenonM, et al. (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5 : 693–703.
53. DownsJA, LowndesNF, JacksonSP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408 : 1001–1004.
54. NiuH, WanL, BusyginaV, KwonY, AllenJA, et al. (2009) Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol Cell 36 : 393–404.
55. HoHC, BurgessSM (2011) Pch2 acts through Xrs2 and Tel1/ATM to modulate interhomolog bias and checkpoint function during meiosis. PLoS Genet 7: e1002351 doi:10.1371/journal.pgen.1002351..
56. WojtaszL, CloutierJM, BaumannM, DanielK, VargaJ, et al. (2012) Meiotic DNA double-strand breaks and chromosome asynapsis in mice are monitored by distinct HORMAD2-independent and -dependent mechanisms. Genes Dev 26 : 958–973.
57. BishopDK (1994) RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell 79 : 1081–1092.
58. HochwagenA, AmonA (2006) Checking your breaks: surveillance mechanisms of meiotic recombination. Curr Biol 16: R217–228.
59. BornerGV, KlecknerN, HunterN (2004) Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117 : 29–45.
60. StorlazziA, XuL, SchwachaA, KlecknerN (1996) Synaptonemal complex (SC) component Zip1 plays a role in meiotic recombination independent of SC polymerization along the chromosomes. Proc Natl Acad Sci U S A 93 : 9043–9048.
61. PanizzaS, MendozaMA, BerlingerM, HuangL, NicolasA, et al. (2011) Spo11-accessory proteins link double-strand break sites to the chromosome axis in early meiotic recombination. Cell 146 : 372–383.
62. FarmerS, HongEJ, LeungWK, ArgunhanB, TerentyevY, et al. (2012) Budding yeast Pch2, a widely conserved meiotic protein, is involved in the initiation of meiotic recombination. PLoS ONE 7: e39724 doi:10.1371/journal.pone.0039724..
63. VaderG, BlitzblauHG, TameMA, FalkJE, CurtinL, et al. (2011) Protection of repetitive DNA borders from self-induced meiotic instability. Nature 477 : 115–119.
64. ZandersS, AlaniE (2009) The pch2Δ mutation in baker's yeast alters meiotic crossover levels and confers a defect in crossover interference. PLoS Genet 5: e1000571 doi:10.1371/journal.pgen.1000571..
65. JonesB, SuH, BhatA, LeiH, BajkoJ, et al. (2008) The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4: e1000190 doi:10.1371/journal.pgen.1000190..
66. RockmillB, RoederGS (1990) Meiosis in asynaptic yeast. Genetics 126 : 563–574.
67. GoldsteinAL, McCuskerJH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15 : 1541–1553.
68. LongtineMS, McKenzieA3rd, DemariniDJ, ShahNG, WachA, et al. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 : 953–961.
69. HollingsworthNM, PonteL (1997) Genetic interactions between HOP1, RED1 and MEK1 suggest that MEK1 regulates assembly of axial element components during meiosis in the yeast Saccharomyces cerevisiae. Genetics 147 : 33–42.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy