
Mutation of the Diamond-Blackfan Anemia Gene in Mouse Results in Morphological and Neuroanatomical Phenotypes
The ribosome is an evolutionarily conserved organelle essential for cellular function. Ribosome construction requires assembly of approximately 80 different ribosomal proteins (RPs) and four different species of rRNA. As RPs co-assemble into one multi-subunit complex, mutation of the genes that encode RPs might be expected to give rise to phenocopies, in which the same phenotype is associated with loss-of-function of each individual gene. However, a more complex picture is emerging in which, in addition to a group of shared phenotypes, diverse RP gene-specific phenotypes are observed. Here we report the first two mouse mutations (Rps7Mtu and Rps7Zma) of ribosomal protein S7 (Rps7), a gene that has been implicated in Diamond-Blackfan anemia. Rps7 disruption results in decreased body size, abnormal skeletal morphology, mid-ventral white spotting, and eye malformations. These phenotypes are reported in other murine RP mutants and, as demonstrated for some other RP mutations, are ameliorated by Trp53 deficiency. Interestingly, Rps7 mutants have additional overt malformations of the developing central nervous system and deficits in working memory, phenotypes that are not reported in murine or human RP gene mutants. Conversely, Rps7 mouse mutants show no anemia or hyperpigmentation, phenotypes associated with mutation of human RPS7 and other murine RPs, respectively. We provide two novel RP mouse models and expand the repertoire of potential phenotypes that should be examined in RP mutants to further explore the concept of RP gene-specific phenotypes.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003094
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003094
Summary
The ribosome is an evolutionarily conserved organelle essential for cellular function. Ribosome construction requires assembly of approximately 80 different ribosomal proteins (RPs) and four different species of rRNA. As RPs co-assemble into one multi-subunit complex, mutation of the genes that encode RPs might be expected to give rise to phenocopies, in which the same phenotype is associated with loss-of-function of each individual gene. However, a more complex picture is emerging in which, in addition to a group of shared phenotypes, diverse RP gene-specific phenotypes are observed. Here we report the first two mouse mutations (Rps7Mtu and Rps7Zma) of ribosomal protein S7 (Rps7), a gene that has been implicated in Diamond-Blackfan anemia. Rps7 disruption results in decreased body size, abnormal skeletal morphology, mid-ventral white spotting, and eye malformations. These phenotypes are reported in other murine RP mutants and, as demonstrated for some other RP mutations, are ameliorated by Trp53 deficiency. Interestingly, Rps7 mutants have additional overt malformations of the developing central nervous system and deficits in working memory, phenotypes that are not reported in murine or human RP gene mutants. Conversely, Rps7 mouse mutants show no anemia or hyperpigmentation, phenotypes associated with mutation of human RPS7 and other murine RPs, respectively. We provide two novel RP mouse models and expand the repertoire of potential phenotypes that should be examined in RP mutants to further explore the concept of RP gene-specific phenotypes.
Introduction
Ribosomes are responsible for constructing the myriad proteins required for the function of each individual cell. Ribosomes themselves, which consist of small (40S) and large (60S) subunits, are assembled from about 80 different ribosomal proteins (RPs) along with four species of rRNA synthesized in the nucleolus [1], [2]. RPs fall broadly into two categories; the RPL proteins that make up the large ribosomal subunit, and the RPS proteins that constitute the small subunit. Mutations in both RPL and RPS genes have been implicated in a set of shared phenotypic characteristics in invertebrates. This is best illustrated by the approximately 50 Drosophila melanogaster Minute mutants, a collection of ribosomal gene mutations that are characterized by developmental delay, short thin bristles, growth retardation, reduced fertility and recessive lethality [3].
In vertebrates, a phenotypic overlap among RP mutants occurs similar to that seen in Drosophila Minute mutants, however additional phenotypic complexity is emerging, in which mutation or suppression of RPs results in some phenotypes that depend upon which gene is mutated. This is highlighted by a recent study in which 20 different RP genes were targeted using morpholino antisense oligos in the zebrafish Danio rerio [4]. Whereas some phenotypes were shared among knockdowns, such as hypoplasia of the yolk sac, others were gene-specific. For instance, knockdown of rps15 resulted in an enlarged 4th ventricle of the brain, whereas knockdown of rpl35a caused a sharply bent tail, and targeting of rps29 produced an enlarged lens [4]. A similar picture of phenotypic overlap paired with gene-specific phenotypes is emerging in mice. For example, mutations in Rps19 and Rps20 result in increased epithelial pigmentation, a ventral belly spot, small body size, and a reduction in red blood cell count [5]. Mice with mutations in Rpl24 also have ventral belly spots and body size reduction, but they present with additional retinal abnormalities and skeletal defects [6]. Mice harboring the Rpl27a sooty foot allele share an epidermal hyperpigmentation phenotype with Rps19 and Rps20 mutants, but display the additional feature of cerebellar ataxia [7]. These distinct features of individual RP mutant phenotypes suggest that vertebrate RPs may have unique, tissue-specific functions and/or tissue-specific expression levels. Indeed, the broad spectrum of distinct phenotypes that have been characterized in a relatively small number of mammalian RP mutants also hints at possible extra-ribosomal functions of RPs.
In humans, mutations in RPs have been implicated in hematopoetic disorders, most notably Diamond-Blackfan anemia (DBA), which has been attributed to mutations in RPS19, RPS26, RPS24, RPS17, RPS10, RPS7, RPL35a, RPL26, RPL11, and RPL5 [8]–[14]. While anemia is a shared phenotype among all patients carrying these various gene mutations, some specific attributes are associated with individual genes. For example, mutations in RPL5 are associated with cleft palate and anomalies of the thumb and heart, whereas isolated thumb malformations are predominant in patients carrying mutations in RPL11 [11].
In this paper, we further examine the role of ribosomes in mammalian development by investigating Rps7. Through analysis of two new ENU-induced mouse mutants of Rps7, montu (Mtu) and zuma (Zma), we show that mutation of Rps7 impairs ribosomal biogenesis, resulting in variable lethality and reduced body size accompanied by abnormal skeletal, melanocyte, eye, and central nervous system (CNS) development. While many of these phenotypes have previously been associated with mutation of murine RP genes, the findings of overt brain malformations and behavioral abnormalities are novel. Similar to mutation of other RP genes in the mouse, the penetrance of the Rps7-associated phenotypes is affected by genetic background and the overt phenotypes are suppressed by TRP53 deficiency. These mutants provide the first mouse models of Rps7 disruption and increase our understanding of the phenotypic consequences of mammalian RP mutations.
Results
Identification and genetic mapping of montu and zuma
The montu (Mtu) mouse was identified in a large-scale ENU mutagenesis program [15], [16] exhibiting dominant inheritance of a ventral belly spot, kinked tail, and reduced body weight (Figure 1A, 1B). Linkage analysis was performed using a cross of the Mtu founder (BALB/c OlaHsd background) with C3H/HeH. Analysis of genotypes of 104 offspring demonstrated linkage to a 74 Mb region of proximal chromosome 12 that was further refined to a 3.43 Mb critical region (Figure S1A, S1C). DNA sequencing of exons and flanking regions of 17 candidate genes using genomic DNA from four affected animals and controls identified a single heterozygous sequence variant occurring only in affected mice within exon 6 of Rps7, which encodes a 194 amino acid ribosomal protein, RPS7 (or S7e, the eukaryotic specific homolog of the yeast S7A and S7B). The identified Rps7 variant, c.574T>G (NM_011300), encodes a Gly substitution of a highly conserved Val residue (p.V156G; NP_035430) (Figure 1C, Figure S2A). Subsequent sequence analysis of 91 animals from a C3H/HeH congenic Mtu colony showed 100% concordance between the c.574T>G mutation and the affected phenotypes. Analysis of Mtu animals demonstrated variable penetrance on a mixed background, however 100% penetrance of the tail kink, belly spot, and small body size phenotypes when assessed in the C3H/HeH congenic colony. We also observed a dominant lethality phenotype with incomplete penetrance, 72% viability in F1 BALB/c:C3H mice (N = 251) and 26% viability in the C3H/HeH congenic colony (N = 260).

The zuma (Zma) mouse was independently identified as part of a sensitized ENU screen designed to identify mutations that increased the severity of neural crest defects observed in Sox10 haploinsufficient mice (Sox10LacZ/+), a well characterized neural crest mutant which presents with a high frequency of small, white belly spots [17]. Affected backcross mice (BALB/cJ×C57BL/6J) from the Zma pedigree exhibited large white belly spots, tail kinks, and reduced body size. Linkage analysis of twelve affected N1 mice initially detected linkage of Zma to a region of chromosome 12 overlapping where the Mtu mutation was localized (Figure S1B, S1C). Subsequent sequencing of Rps7 in Zma mice revealed a heterozygous A to C point mutation in exon 7 of Rps7, predicted to cause substitution of a conserved amino acid (p.Y177S; c.637A>C) (Figure 1C, Figure S2A). Genotyping analysis of Zma mice outcrossed to C57BL/6J to establish a congenic colony showed that the c.637A>C mutation was observed in 100% of affected mice. We initially observed incompletely penetrant phenotypes in N2 heterozygous Zma mice on a mixed BALB/cJ; C57Bl/6J background, where Zma/+ mice showed 74% viability, 76% belly spots, and 90% tail kinks (N = 46). However, heterozygosity for the Zma allele rapidly changed to a completely penetrant, lethal phenotype during outcrossing onto C57BL/6J, and no Zma/+ mice were observed at N4. This contrasted with the phenotype we observed during establishment of a congenic C3H/HeJ Zma colony; on this genetic background, the N3 generation exhibited a low frequency of belly spotting and tail kinks (4% and 0%, respectively; N = 41) yet no lethality, as Zma heterozygotes were observed at the expected frequency through N6. The comparably mild phenotype we observed in the congenic C3H/HeJ Zma colony suggests that the Mtu allele may exert more severe phenotypic effects than the Zma allele. This hypothesis is supported by the observations that Mtu/+ mice presented with reduced viability, fully penetrant belly spots, tail kinks, and small body size on a predominantly C3H/He genetic background, while Zma/+ mice on a similar background were observed at the expected frequency and were generally indistinguishable in phenotype from their+/+littermates.
Sequencing of exons 6 and 7 of Rps7 in 9 inbred strains (A/J, AKR/J, BALB/c, C57BL6/J, C3H/HeJ, CBA, DBA, LP/J, and 101) confirmed that the point mutations detected in Mtu and Zma are not natural variants. Additionally, we confirmed the presence of the mutations in Rps7 transcripts using Mtu/+ and Zma/+ cDNA for sequencing and real-time PCR, respectively. We also demonstrated a lack of complementation between the Mtu and Zma Rps7 alleles by performing an intercross of heterozygous mice from the two lines. Genotyping of newborn offspring revealed that no animals carried both the Mtu and Zma mutations (N = 27) (Figure S1D). The lack of viable double heterozygotes was consistent with homozygote lethality observed in each line, thus showing non-complementation of the two alleles. Collectively, the similar phenotypes, sequencing data, and genetic analyses provide strong evidence that the mutations identified in Rps7 are responsible for the observed Mtu and Zma phenotypes, hereafter referred to as Rps7Mtu and Rps7Zma.
Functional analysis of the RPS7 mutations
The high degree of evolutionary conservation of both mutated amino acids (p.V156G and p.Y177S) suggests that these alterations may disrupt normal function. The consequences of the RPS7Mtu and RPS7Zma mutant proteins were first assessed using the computational analyses PANTHER and SIFT. Results from the PANTHER coding SNP analysis tool [18] suggested that both mutations are likely to be deleterious (PANTHER subPSEC score of −5.00 and −5.06 for RPS7Mtu and RPS7Zma, respectively). Similarly, the Sorting Tolerant From Intolerant (SIFT) algorithm [19] predicted both alleles to affect protein function, however, the high degree of conservation in the 72 database sequences at each position resulted in a low confidence level for the prediction (SIFT prediction score 0.00, median conservation 3.59 for both RPS7Mtu and RPS7Zma). Collectively, the computational analysis was consistent with the Rps7Mtu and Rps7Zma alleles both having functional consequences.
The potential effects of p.V156G and p.Y177S on RPS7 secondary structure (Figure 1D, Figure S2) were also examined. There is no three-dimensional protein structure currently available for metazoan RPS7, therefore we used the available structures from Tetrahymena thermophila (PDB ID 2XZM) and Saccharomyces cerevisiae (PDB ID 3U5C), whose RPS7 ortholog sequences share 37% and 55% residue identity with mouse RPS7, respectively [20], [21]. RPS7 structural elements appear to be highly conserved across species: the S. cerevisiae and T. thermophila proteins themselves share only 34% amino acid identity, yet their solved three-dimensional structures superimpose well, and are consistent with secondary structural predictions of mouse RPS7 generated using PSSpred (http://zhanglab.ccmb.med.umich.edu/PSSpred) and PROFsec [22] (Figure S2B). Furthermore, the S. cerevisiae and T. thermophila sequences each contain a Tyr residue homologous to mouse p.Y177 (Figure S2A), and a Val residue homologous to mouse p.V156 is present in S. cerevisiae and substituted conservatively with Ile in T. thermophila. The introduction of either p.V156G or p.Y177S into the mouse sequence did not alter predictions of the beta strand and alpha helix at the respective locations of the substitutions (Figure S2B), suggesting that grossly altered secondary structure may not be responsible for the functional consequences of RPS7Mtu or RPS7Zma.
We next used biochemical analyses to examine the effects of p.V156G and p.Y177S on stability and subcellular localization of the 22 kiloDalton (kDa) protein as well as ribosome assembly and biogenesis. Previous studies have shown that nonsense mutations in RPS19 can result in decreased protein levels, and missense mutations can alter the capacity of RPS19 to localize to the nucleolus [23]. C - and N-terminal FLAG-tagged wild-type, RPS7Mtu and RPS7Zma proteins were transiently expressed in human embryonic kidney (HEK)-293 cells. Western blot analysis revealed no differences in protein levels for any of the RPS7 mutant proteins (Figure 2A, Figure S3A). The remaining in vitro studies were focused on the potentially more severe RPS7Mtu mutant protein. Analysis of the FLAG-tagged proteins revealed no notable differences in subcellular localization of the RPS7Mtu protein (Figure 2B). To verify the capacity of RPS7Mtu to be incorporated into the ribosome, cytoplasmic extracts from transiently transfected HEK-293 cells were fractionated to separate ribosomes and ribosomal subunits (in the pellet) from free cytoplasmic proteins (in the supernatant). A similar fraction of all transfected RPS7 proteins was observed in the ribosomal pellet (Figure S3B) indicating that the mutation does not alter RPS7 assembly into the ribosome. To confirm this finding, cytoplasmic extracts from Rps7Mtu/+ liver were analyzed by ultracentrifugation through sucrose gradients, and the ratio between the peaks of the ribosomal subunits in the absorbance profile was determined. This ratio can reveal defects in the synthesis of one of the two subunits, such as the net increase of the 60S/40S ratio seen in cultured human cells following depletion of single RPs [24], [25]. However, the observed 60S/40S ratio in Rps7Mtu/+ liver was similar to control (Figure S3C), indicating that RPS7Mtu does not drastically alter assembly of the ribosomal subunits.
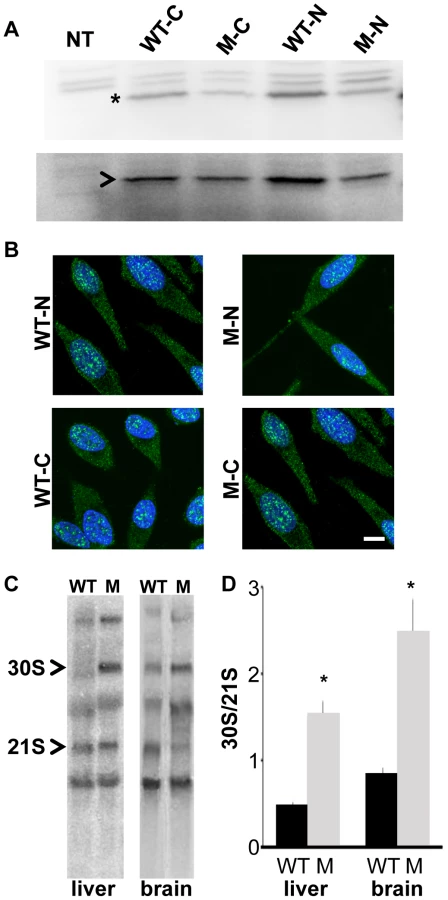
We further assessed whether RPS7Mtu affects ribosomal biogenesis by analyzing the pre-rRNA maturation pattern. Alterations are visualized by the accumulation of specific rRNA precursors: a defect in large subunit RPs affects cleavage of 28S precursors, whereas a defect in small subunit RPs alters that of 18S precursors. Northern blot analysis of total RNA from wild-type and Rps7Mtu/+ tissue, using a probe for the internal transcribed spacer (ITS) 1 of pre-rRNA that anneals to 18S rRNA precursors [26], [27], indicated that 18S rRNA pre-rRNA processing was altered both in liver and brain from Rps7Mtu/+ mutant mice (Figure 2C). Quantitative measurement of the hybridization signals showed a significant accumulation of the 30S precursor in Rps7Mtu/+ (indicated by an increased 30S/21S ratio; Figure 2D), confirming altered rRNA precursor processing and demonstrating that the RPS7Mtu mutation has functional consequences on ribosomal biogenesis.
Characterization of the Rps7 phenotype
Skeletal and eye malformation
X-ray examination of adult Rps7Zma/+ mice showed that vertebral fusion is responsible for the tail kinks (Figure 3A, 3B). Similar results were observed for Rps7Mtu/+ adult mice, indicating that in both mutants, defective somitogenesis occurs as a result of RPS7 alterations. Decreased vertebrae numbers were also observed in both mutants; a reduction in total vertebrae number in Rps7Mtu/+ (52–55 versus 55–57 in wild-type) and in caudal vertebrae number in Rps7Zma/+ (16–19 versus 20–22 in wild-type). The decreased vertebrae numbers indicate defective tail bud function associated with defects in RPS7. Alcian blue and alizarin red staining of late gestation Rps7+/+ and Rps7Zma/+ embryos (N = 3 for each genotype) revealed further Rps7Zma/+-associated skeletal abnormalities, including general disorganization of vertebral processes and arches in the cervical vertebrae (Figure 3C, 3D). The anterior tuberculum was appropriately located on C6, however a very small partial rib was observed on C7 (in 2 Rps7Zma/+ embryos), indicating an incompletely penetrant transformation of C7 to T1. Within the thoracic region, we observed a shortened first sternebrae and asymmetric attachment of 8 ribs to the sternum rather than the usual 7 (in all 3 Rps7Zma/+ embryos) (Figure 3E, 3F). Within the lumbar and sacral regions, a severe developmental delay was observed, and disorganization of the ossification centers was evident with some ossification centers being duplicated (Figure 3G, 3H). Additionally, we observed a transformation of L1 to T13 (in all 3 Rps7Zma/+ embryos) and also asymmetric fusion of the transverse processes of the first 5–6 sacral vertebrae (in 2 Rps7Zma/+ embryos) rather than the 3–4 observed in Rps7+/+ (Figure 3H). Collectively the skeletal phenotypes observed in Rps7 mutants are consistent with an anterior transformation of the axial skeleton.

Frequent microphthalmia and uveal coloboma were also observed as features of the Rps7 mutant phenotype (Figure 4). Rps7Zma/+ embryos showed eye malformations ranging from minor delay in optic fissure closure to severe microphthalmia and internalization of the entire eye structure. Whole mount microscopic imaging of E12.5 embryos revealed that 100% of Rps7Zma/+ embryos exhibited a unilateral or bilateral delay in closure of the optic fissure, as compared to Rps7+/+ embryos which all showed normal optic fissures (N = 5 Rps7Zma/+, N = 7 Rps7+/+) (Figure 4A–4C). By E14.5, a time when wild-type embryos have complete fissure, 100% of Rps7Zma/+ embryos still displayed optic fissure closure defects either unilaterally (N = 2) or bilaterally (N = 3) (Figure 4D–4F). Additionally, more severe microphthalmia and internalization of the eye was observed in 20% of Rps7Zma/+ embryos (Figure 4F, 4I; 1 out of 5 at both E12.5 and E14.5).

Circulating blood chemistry and erythroid differentiation
Because human RPS7 mutations are associated with DBA [11], a disease characterized by red cell aplasia, we undertook an initial characterization of circulating blood in adult Rps7Mtu/+ mice (on a C3H/He background) and Rps7Zma/+ mice (on a mixed C57BL/6J; BALB/cJ background). Neither Rps7 mutant allele caused any significant differences from Rps7+/+ littermates in total hemoglobin, red blood cell (RBC) counts, white blood cell (WBC) counts, or hematocrit (Figure 5A, 5B, Table S1). Mean corpuscular volume (MCV) was slightly elevated in Rps7Mtu/+ mice (elevated 1.026 fold over wild-type, p<0.05) (Figure 5A), however no significant difference in MCV was observed in Rps7Zma/+ mice (Figure 5B). Additional analyses of plasma biochemistry revealed no significant differences between Rps7Mtu/+ and Rps7+/+ animals in major electrolytes and metabolic indicators (Na+, K+, Cl−, urea, creatinine, ALP, AST, ALT, total cholesterol, triglycerides, iron, amylase, and free fatty acids) (Table S2).

To further assess erythroid development of Rps7Zma/+ mice, which exhibit perinatal lethality on a C57BL/6J background, E13.5 Rps7Zma/+ fetal liver samples were characterized by flow cytometry. We simultaneously used monoclonal antibodies against the erythroid cell lineage markers CD71, expressed at higher levels during early stages of erythroid differentiation, and Ter119, expressed at higher levels as erythrocytes mature, to profile 5 characteristic populations of erythroid precursors (R1 to R5) [28], [29] (Figure 5C). These cell populations correspond to increasingly differentiated developmental stages, with R1 marking erythroid progenitors and proerythroblasts, R2 marking proerythroblasts and early basophilic erythroblasts, R3 marking all basophilic erythroblasts, R4 marking chromatophilic and orthochromatophilic erythroblasts, and R5 marking late orthochromatophilic erythroblasts and reticulocytes. Compared to their Rps7+/+ littermates, Rps7Zma/+ E13.5 embryos showed a significant increase in the percentage of R3 cells (p<0.001) suggesting that erythroid precursors could be blocked in their maturation (Figure 5D). However, analysis of erythroid cell populations of E14.5 fetal livers showed that the percentages of cells in the R2–R5 populations were indistinguishable between Rps7+/+ and Rps7Zma/+ genotypes (Table S3). The presence of the more mature R4 and R5 cells at E14.5 indicates erythroid differentiation progresses, rather than being blocked at R3. Alternatively, the increase in R3 cells at E13.5 may result from a general developmental delay in Rps7Zma/+ embryos. This was confirmed by inspection of gross anatomical features of crown-rump length, along with eye, limb, and craniofacial development at E11.5–13.5, which showed that Rps7Zma/+ embryonic development is delayed by approximately one day relative to that of Rps7+/+ littermates (Figure S4). While we cannot rule out subtle alterations in erythroid maturation that may cause the differences observed in the precursor populations, these analyses collectively suggest that these two mutant alleles of Rps7 do not significantly impair red blood cell production or differentiation.
Early effect on melanoblast development
To assess the effects of RPS7 mutation on melanocyte development, we characterized Rps7Zma/+ mutant embryos from E10.5 to 14.5. To mark developing melanoblasts, we generated transgenic Rps7Zma/+ embryos carrying the melanoblast reporter Tg(Dct-LacZ) [30]. In these Rps7Zma/+; Tg(Dct-LacZ) embryos, Dct-positive melanoblasts were notably reduced by E10.5 relative to Rps7+/+; Tg(Dct-LacZ) littermates (Figure 6A, 6B). At E14.5, a significant reduction in melanoblast number was still observed in both the head and trunk of Rps7Zma/+; Tg(Dct-LacZ) mice compared to Rps7+/+; Tg(Dct-LacZ) littermates (Figure 6C–6F). Mutation of Rps7 did not cause a complete loss of melanoblasts, however, as Dct-positive cells could still be observed at E14.5 in regions where melanoblast density is normally highest, particularly around the developing pinna and eye (Figure 6C, 6D). This severe melanoblast reduction in Rps7Zma/+ embryos was independently confirmed by in situ hybridization with a probe targeting mRNA for the melanoblast/melanosome-specific protein Pmel17 (Figure S5). Qualitative observation of Pmel17-expressing melanoblasts showed that E12.5 Rps7Zma/+ mutant embryos displayed a marked reduction in melanoblasts relative to E12.5 Rps7+/+ littermates. The reduction in melanoblasts is more severe than can be attributed to the developmental delay observed in Rps7Zma/+ mutants, as E12.5 Rps7Zma/+ embryos showed a reduced melanoblast number even in comparison to E11.5 Rps7+/+ embryos (Figure S5). A combination of Rps7 and Sox10 haploinsufficiency (Rps7Zma/+; Sox10LacZ/+ double heterozygotes) further reduced melanoblast number at E12.5 (Figure S6), consistent with the increased spotting observed in rare Rps7Zma/+; Sox10LacZ/+ pups that survived postnatally (Figure S7A, S7B).

Postnatally, we observed that melanocytes were present in the tail skin of Rps7Zma/+ mice (Figure S7E, S7F), yet no dark skin phenotype occurred in the tails or footpads of adult Rps7Zma/+ or Rps7Mtu/+mice (Figure S7C–S7F). This indicates that these Rps7 mutations do not cause epidermal melanocytosis, as was previously observed for mutation of Rps19 and Rps20 [5]. Collectively, these data show that mutation of Rps7 causes a severe, early reduction in melanoblasts that is sufficient to account for the white belly spotting observed in viable heterozygotes.
Enlarged ventricles, cortical thinning, and deficits in working memory
We examined the neuroanatomical features of the Rps7 mutants, employing histological techniques and magnetic resonance microscopy (MRM). Gross observations and histological staining revealed marked cortical thinning accompanied by enlarged ventricles in adult Rps7Mtu/+ (Figure 7A–7B) and perinatal Rps7Zma/+ mice (Figure 7C, and data not shown). To investigate this phenotype further, we undertook MRM imaging of Rps7Zma/+ mice at E18.5 (N = 3) (Figure 7D–7I). Following manual segmentation of 10 major anatomic structures, the volume of each brain region was calculated as a percentage of the total brain volume (Figure 7J) and 3-dimensional (3D) rendering with smoothing was performed to generate 3D representations (Video S1 and Video S2). This quantitative analysis showed that the Rps7Zma/+ CNS phenotypes originate during development and confirmed a significant reduction in cortical (P = 0.0002) and hippocampal (P = 0.0044) size in Rps7Zma/+ mutants. Although lateral ventricle volume was not significantly different between these small groups, qualitative observations of ventricle size at dissection, in MRM images, and in histological sections consistently indicated Rps7Zma/+ mutants had enlarged ventricles, leading us to conclude that enlarged ventricles are a consistent feature of the Rps7 phenotype.

We investigated whether these gross neuroanatomical phenotypes affected the behavior of the Rps7Mtu/+ mice, assessing locomotor activity, anxiety and working memory. The results showed that while Rps7Mtu/+ mutants exhibited normal locomotor activity and anxiety-related phenotypes, they displayed deficits in working memory. Rps7Mtu/+ mutants displayed similar behavior to Rps7+/+ controls in an open-field test, with no significant differences in total distance travelled in the open field or time spent in the center (Figure S8A). Similarly, there were no significant differences between Rps7+/+ controls and Rps7Mtu/+littermates when assessed on the elevated plus-maze (Figure S8B–S8E), nor did Rps7Mtu/+ mutant animals display any ataxia that would be indicative of motor dysfunction. We also investigated whether Rps7Mtu/+ mice displayed any defects in working memory using a spontaneous alternation test, which can indicate hippocampal dysfunction [31]. The mouse is placed in the start arm of an enclosed T-maze (the base of the “T”), and allowed to enter a goal arm (the two arms of the top of the “T”) of its own choice. Upon repeating this test, the mouse can demonstrate memory of its previous choice by entering the arm not visited before, a behavior called spontaneous alternation. Here we observed a significant difference between Rps7+/+ controls, which exhibited an average alternation frequency of 83%, and Rps7Mtu/+ mutants, which exhibited an average alternation frequency of only 68% (N = 9, P = 0.01) (Figure 7K). Collectively, the MRM and behavioral data indicate that Rps7 is required for normal development of the central nervous system, particularly the telencephalon, and that mutations in this gene have the capacity to affect hippocampal-dependent behavior.
Increased neuronal apoptosis in Rps7 mutants
Our histological studies also revealed that late gestation Rps7Zma/+ embryos exhibited pyknotic nuclei in the white matter tracts of the spinal cord (Figure S9), consistent with CNS degeneration. As mutations in other RPs have been associated with increased cellular apoptosis [32]–[37], we used immunohistochemistry to detect cleaved caspase-3 (CC3) within the CNS. When compared to normal littermates, Rps7Zma/+ embryos showed increased apoptosis in the cortex and neural tube at both E11.5 (Figure 8A, 8B, 8D, 8E) and E12.5 (data not shown). Although mitotic, phospho-histone H3-positive (PH3+) cells surrounding the lumen of the neural tube appeared slightly disorganized in E11.5 and E12.5 Rps7Zma/+embryos (Figure 8G, 8H and data not shown), cell counting revealed no significant difference in PH3+ cell number per section compared to Rps7+/+ littermates (Figure 8J). In contrast, CC3 counts confirmed a significant increase in apoptosis in the neural tube of E11.5 Rps7Zma/+ embryos compared to Rps7+/+ (N = 3, p<0.001) (Figure 8K). Consistent with the location of apoptosis and with a putative role for RPS7 in CNS development, in situ hybridization studies on Rps7+/+ embryos revealed that Rps7 is highly expressed in proliferative regions, including the ventricular zone (Figure S10). As TRP53 levels are altered in other ribosomal mutants, we also investigated the expression of TRP53 in the developing CNS of Rps7 mutants. Using immunohistochemistry, we observed an increase in TRP53 staining in the cortex (E11.5) and neural tube (E11.5 and E12.5) of Rps7 mutants consistent with increased TRP53-mediated apoptosis (Figure S11). Collectively, the histology and immunohistochemistry data confirm that massive cell death in the Rps7 mutant CNS occurs at the onset of neurogenesis.

Phenotypic suppression of Rps7 mutant phenotypes byTrp53 haploinsufficiency
In an attempt to restore heterozygote viability by suppressing apoptosis, we crossed the Rps7Zma mutation onto a Trp53 mutant background. Trp53 mutations have been used to successfully suppress phenotypes in other RP mutants [5], [35]. Rps7Zma mice were originally identified on a mixed genetic background, and heterozygote lethality in Rps7Zma/+ mice increased as the mutation was outcrossed onto a C57BL/6J background. Thus we used early generation, mixed background Rps7Zma/+ mice to establish a cross with Trp53 mutant mice on a C57Bl/6J background (B6.129S2-Trp53tm1Tyj/J, hereafter Trp53KO). Within a few generations of outcrossing, no viable Rps7Zma/+ mice were observed after birth, as expected. However, Rps7Zma/+; Trp53 KO/+ double heterozygotes were recovered at the expected 50% frequency, suggesting restoration of Rps7Zma/+ viability in the context of Trp53 haploinsufficiency (Table S4). In addition to restoring viability, Trp53 mutation fully suppressed the Rps7Zma-associated vertebral fusion phenotype that was observed in earlier generations of Rps7Zma/+ mice (no tail kinks observed in >25 Rps7Zma/+; Trp53KO/+ mice). In most cases Rps7Zma/+; Trp53KO/+ double heterozygotes were indistinguishable from their wild-type littermates after weaning, with the exception of a very small belly spot observed in 47% of Rps7Zma/+; Trp53KO/+ mice. Trp53 haploinsufficiency also suppressed the phenotypes of gross embryonic developmental delay (Figure S4), the subtle delay in erythroid maturation (Table S3), and CNS apoptosis (Figure 8C, 8F, 8K). In summary, the pleiotropic phenotypes observed in Rps7Zma/+ mice all appeared to be suppressed by Trp53 haploinsufficiency.
Discussion
In this paper we present genetic, functional, and phenotypic evidence that the montu (Mtu) and zuma (Zma) mouse lines isolated from independent ENU screens harbor distinct point mutations in Rps7. These first-reported alleles of Rps7 in mice cause similar phenotypes including small body size, tail abnormalities, mid-ventral white spotting, eye defects, and an underdeveloped cerebral cortex.
We provide functional evidence that the Rps7Mtu mutation (p.V156G) affects ribosome biogenesis. The altered Rps7Mtu 30S/21S ratio is consistent with the altered pre-rRNA maturation reported for a DBA patient harboring an RPS7 donor splice-site mutation (c.147+1G>A) that results in accumulation of 30S and 45S precursors [11]. A requirement for RPS7 during rRNA maturation is supported by other studies showing a role for human RPS7 in early stages of 45S rRNA processing [24] or the nuclear stages of 40S maturation [38]. In yeast, depletion of the two RPS7 paralogs (S7a and S7b) results in a severe growth defect [39], [40] and S7 has been hypothesized to play an early role as a component of the small subunit processome [41] as well as a later role in ribosome biogenesis during assembly of polysomes [42]. However, the mechanism underlying the altered rRNA maturation in these murine Rps7 mutants is not clear, as neither the Rps7Mtu nor the Rps7Zma mutation is predicted to grossly disrupt protein secondary structure, and RPS7Mtu protein is correctly localized and incorporated into ribosomes in cultured cells. Presumably, the mutant RPS7 proteins could be altered in their interactions with rRNA, other ribosomal proteins, or non-ribosomal proteins. The possibility of interaction with non-ribosomal proteins is intriguing given the position of RPS7 at the surface of the ribosome (Figure S2C, S2D) in close proximity to the binding site for eIF4G [21], a eukaryotic initiation factor important for assembling the pre-initiation complex. Interestingly, most other known, viable mouse RP mutations also occur in eukaryotic-specific RPs located at the surface of the ribosome. Additional work is needed to determine the precise role RPS7 plays in mammalian ribosome biogenesis and how the Rps7Mtu and Rps7Zma mutations disrupt this function.
While modeling the pre-rRNA processing defect of an RPS7 DBA patient, these Rps7 mutant mice do not replicate the characteristic DBA phenotype of severe anemia. This lack of an anemia phenotype is not entirely unexpected, since the analysis of many mouse mutants suggests that mice as a species are generally less sensitive than humans to any gene haploinsufficiency [43], [44]. This is exemplified by the fact that, despite frequent mutation of RPS19 in DBA, Rps19 heterozygote knockout mice have been described with normal hematopoiesis [34], [45] and an ENU allele of Rps19 displays only a mild erythrocyte phenotype, along with an elevated MCV that parallels the slightly elevated MCV in Rps7Mtu [5]. It is possible that further studies of Rps7 alleles on different genetic backgrounds or in mice with more severe alleles may reveal additional red blood cell phenotypes. In the future, comparison among mouse DBA models may provide insight into why the murine and human hematological phenotypes do not fully overlap, and may also reveal why mutation in RPS19 has a uniquely high frequency in DBA compared to other RPs that cause the disease.
Both Rps7 mutants display a variety of skeletal phenotypes that collectively suggest that normal ribosome biogenesis is required for three distinct stages of somite development, which governs subsequent axial skeleton formation. First, the decreased number of tail vertebrae indicates an insufficient production of somite progenitors. Second, the mutant vertebral fusion leading to tail kinks is indicative of incorrect somite border formation. Third, the mutants exhibit an array of axial skeleton defects consistent with an anterior transformation, indicating that somite identity is perturbed. Widespread disturbances of the skeletal system encompassing each of these three defects are also found in Rpl38 (tail short, Ts) [46], [47] and Rpl24 (belly spot and tail, Bst) mutants [6], [48]. Indeed, there are striking similarities in the defects observed in Rpl38 and Rps7 mutants, including specific transformations within the cervical (C7 to T1), thoracic (T8 to T7), lumbar (L1 to T13) and sacral (fusion of additional transverse processes) regions. Mutations in Rpl38 and Rpl24 cause additional disturbances of the appendicular skeleton not observed in Rps7, however, given the similarities of these mutants to Rps7 it will be interesting to see if appendicular skeletal defects are observed with different Rps7 alleles and/or genetic backgrounds. Interestingly, severe skeletal malformations have not been reported for five other murine RP mutants (Rps19, Rps20, Rpl22, Rpl27a, Rpl29) [5], . This suggests that either mutation of RP proteins do not universally affect skeletal development, or skeletal defects remain to be identified in these other RP mutants. In the case of Rps7, the severity of skeletal defects was modified by both genetic background and by allele, suggesting that detailed characterization in existing and additional RP mutants is necessary to further explore the role of RP proteins in skeletal development.
We show that Rps7Zma-mediated white spotting results from a severe developmental reduction in melanoblasts. A developmental reduction in melanoblasts has also been reported in Rps19 and Rps20 heterozygous mutants, however, these mutants subsequently develop epidermal melanocytosis [5], a phenotype not seen in either Rps7 mutant. This difference is unlikely to be due to genetic background, as both Rps7 mutants and Rps19 and Rps20 mutants were examined on a predominantly C3H background. Alternatively, the difference could be a more severe developmental reduction in melanoblast numbers in Rps7 mutants, resulting in fewer melanoblasts available at later time points to generate epidermal melanocytosis. Another possibility is that loss of Rps7 may not have the same effect in keratinocytes as loss of Rps6, Rps19 and Rps20, which are thought to act in keratinocytes to produce TRP53-mediated dark skin [5]. Our in vitro experiments with Rps7 (Figure 2 and Figure S3) suggest that these mutations are hypomorphic alleles, so a third possibility is that allele severity, rather than gene-specific function, could account for pigmentation phenotype differences among RP mutants.
The mouse Rps7 mutants display additional developmental defects of reduced cortical and hippocampal size. We hypothesize that cell death within the developing CNS is sufficient to account for the neocortical thinning we observe in both Rps7 mutant lines. We also hypothesize that apoptosis within the developing telencephalon might account for the deficit in working memory observed in Rps7Mtu/+ heterozygotes. To our knowledge, this is a novel developmental brain abnormality in mouse arising from RP haploinsufficiency, differing substantially from the only other report of neuronal defects in RP mutant mice, that of cerebellar abnormalities and ataxia in Rpl27a sooty foot mutants [7]. Interestingly, the neuronal phenotypes in mouse Rps7 mutants correlate with the report of microcephaly as one of 11 congenital craniofacial anomalies that can be associated with DBA [51], and are consistent with RP mutant phenotypes observed in different model organisms. In zebrafish, gene-specific neuronal phenotypes were seen in a subset of knocked-down RPs [4] and further studies of RPL11 revealed brain deformities and reduced neuronal progenitor cells along with apoptosis in the affected regions [32]. In the zebra finch, RPL17 and RPL37 both regulate the sexually dimorphic formation of the song control regions of the brain within the VZ [52]. Thus, the murine Rps7 mutant phenotypes add to a growing body of literature linking individual RPs to specific features of neural development and suggesting that other RP mouse mutants may have previously uncharacterized defects in brain development.
Important questions remain regarding what underlying cellular mechanisms explain the similarities and differences among RP-associated phenotypes. A growing body of literature suggests a common mechanism where alterations in RP expression result in activation of TRP53, suggesting ribosomal stress is one of the many cellular anomalies detected by TRP53. For example, in their study of RP-mediated dark skin, McGowan and colleagues show that TRP53 acts as a sensor of ribosomal integrity, and that mutations in Rps19 and Rps20 can be phenotypically suppressed by Trp53 deficiency [5]. Trp53 haploinsufficiency also relieves the abnormal phenotypes of mice heterozygous for the sooty foot ataxia allele of Rpl27a [7] or the Bst allele of Rpl24 [6]. Similarly, our results show that loss of Rps7 increases TRP53 levels and that Trp53 haploinsufficiency suppresses all morphological aspects of the Rps7 phenotype. Our in vivo studies are supported by recent studies showing that Rps7 depletion in vitro as well as in vivo in zebrafish induces TRP53 expression [53], [54], and collectively suggest the mechanism underlying the Rps7 phenotype is TRP53 activation followed by TRP53-mediated apoptosis.
While these data suggest that RP mutant phenotypes may all be attributed to alterations in RP—TRP53 interactions, the variability among RP phenotypes suggests more complexity, potentially as a result of RP gene-specific or cell-specific functions [55]. Tissue-specific effects of mutations in different RPs have been observed in zebrafish [4], mouse [5], [6] and human [11]. These apparently tissue-specific phenotypes could simply be due to incomplete phenotypic characterization, differing severity of hypomorphic alleles, and/or differing genetic backgrounds. However, if they truly reflect tissue-specific RP functions, one explanation could be differences in the relative expression levels of individual RPs, with a consequent differential sensitivity to RP insufficiency that could vary with tissue type, developmental stage or differentiation state. Indeed, remarkable heterogeneity in RP mRNA levels has been observed in a variety of tissues [56] including specifically during mouse embryonic development [47] and human neuronal differentiation [57].
A cell type-specific sensitivity to RP insufficiency is strongly supported by the observation that keratinocyte-specific Rps6 hemizygosity causes hyperpigmentation while melanocyte-specific Rps6 hemizygosity instead causes hypopigmentation [5]. Similarly, the severe melanoblast reduction in Rps7 mutants could suggest a melanocyte-specific sensitivity to RPS7 loss. Given that Sox10 and Rps7 pathways interact in melanocytes in vivo (Figure S6, Figure S7), it will be interesting to determine if a melanocyte-specific transcriptional response to RPS7 deficiency is mediated by Sox10, which is expressed in melanocytes but absent in keratinocytes. TRP53 activation in melanocytes has already been associated with downregulation of another white spotting gene, Kit, which is proposed as a transcriptional target of TRP53 [7], [58]. Further work is needed in melanocytes and other cell types that display RP gene-specific phenotypes, to clarify if direct transcriptional regulation of lineage-specific genes plays a role in tissue-specific sensitivity to individual RP deficiencies.
Another explanation of tissue-specific RP phenotypes could be a function for individual RPs in the translation of specific mRNAs. This is supported by studies of RPL13 [59] and by a recent study reporting patterning defects in Rpl38 mutant mouse embryos due to RPL38 translational regulation of a subset of Hox transcripts [47]. The tissue-specific phenotypes observed in Rps7 mutants would be consistent with a role for RPS7 in facilitating translational regulation of transcripts critical for development of the affected tissues. Alternatively, tissue-specific phenotypes observed in Rps7 mutants could be a consequence of a direct interaction between RPS7 and tissue-specific developmental regulators at the protein level. Interestingly, support for protein-protein interactions between RPS7 and SOX10 comes from recent evidence that RPS7 interacts directly with SRY, an HMG box protein related to SOX10 [60]. This mechanism of direct interaction between RPs and tissue-specific, non-ribosomal proteins could explain tissue-specific phenotypes in RP mutants, and is supported by a number of studies reporting extra-ribosomal functions of specific RPs in regulation of ribosome biosynthesis and binding of transcription factor complexes (reviewed in ref. [55]).
The relative contributions of these various mechanistic explanations for tissue-specific and gene-specific RP mutant phenotypes will be clarified with further detailed comparison of mutants on consistent genetic backgrounds and with additional information from tissue-specific conditional alleles. In summary, these novel alleles of Rps7 add to the growing collection of mammalian ribosomal mutants and provide two new mouse models of a DBA-associated gene. Importantly, the unique CNS apoptosis and behavioral phenotypes reported here suggest that RPs need to be considered as candidate genes for not only DBA but also a broad spectrum of neurodevelopmental human diseases.
Materials and Methods
Ethics statement
All procedures performed in the UK were in accordance with the UK Animals (Scientific Procedures) Act 1986, and those performed in the United States were approved by the IACUC in accordance with NIH guidelines.
Mice
Montu mice were identified in a screen where BALB/c male mice were mutagenized using ENU and crossed to C3H/He for screening. Zuma mice were identified in an ENU screen sensitized by Sox10 haploinsufficiency (Sox10LacZ/+), where BALB/cJ mice were mutagenized using ENU and crossed to C57Bl/6J mice for further pedigree analysis. Trp53 null mice (B6.129S2-Trp53tm1Tyj/J, stock #002101) were purchased from JAX Mice. For behavioral studies, mice were housed in facilities with a 12∶12 light∶dark cycle at a temperature of 22±1°C with a 60–70% humidity. Upon weaning, mice were separated into single-sex littermate groups and food was available ad libitum.
For body weight analysis, each gender/genotype group represented in Figure 1B was weighed over 10 weeks (average N = 9 for each Figure 1B data point, range N = 2–19). A Student's t-test was used to compare wild-type to mutant within gender-matched groups at each age. Significance was assessed using the Bonferonni method to correct for multiple testing, with p<0.001 deemed significant. In females, all time points after 1 week were significantly different and in males all time points after 2 weeks were significantly different, with the exception of 8.5 weeks where only 2 mutant mice were weighed (p<0.003).
Alizarin red and alcian blue staining of skeletons was conducted using standard techniques. Briefly, embryos were skinned and eviscerated, fixed 24 hours in ethanol, then fixed 24 hours in acetone. Staining was performed for 3–4 hours at 37°C and then 3–4 days at room temperature. After staining, embryos were rinsed in water and cleared in 1% KOH for 3 hours at room temperature and moved to fresh 1% KOH overnight. After clearing, embryos were serially transferred through 20%/1% KOH, 50% glycerol/1% KOH, and 80% glycerol/1% KOH.
FACS analysis was completed using standard techniques. Briefly, fetal liver samples were collected in ice cold PBS and dispersed into a single cell suspension with passage through a 21 gauge needle. Samples were incubated for 20 minutes with the addition of 10 µl of each antibody (BD Parmingen Ter119-APC and CD71-FITC), then washed once with PBS before FACS analysis. At each age ANOVA analysis was performed with a post-hoc test to compare selected pairs: (Rps7+/+ versus Rps7Zma/+) and (Rps7Zma/+ versus Rps7Zma/+; Trp53KO/+).
Genetic mapping and sequencing
DNA was extracted from mice using standard techniques. For Mtu, the mutation was mapped using 11 affected animals and 51 polymorphic MIT makers that were amplified by PCR and visualized on a 4% agarose gel stained with ethidium bromide. Fine mapping was performed with 13 additional SNPs that distinguished between BALB/c and 101. Seventeen known and predicted genes were identified in the Mtu critical interval from the Ensembl database (ENSMUSG00000020633, ENSMUSG00000066544, ENSMUSG00000020636, ENSMUSG00000036655, ENSMUSG00000061477, ENSMUSG00000020630, ENSMUSG00000020629, ENSMUSG00000020628, ENSMUSG00000036613, ENSMUSG00000061911, ENSMUSG00000020674, ENSMUSG00000020673, ENSMUSG00000020672, ENSMUSG00000043061, ENSMUSG00000044573, ENSMUSG00000020669, ENSMUSG00000036136). All exons and adjacent splice sites for these 17 genes were sequenced using a BigDye dideoxy-terminator system and analyzed on an ABI3700 sequencer (Applied Biosystems). For Zma, DNA from 8 offspring was analyzed using the Illumina GoldenGate assay medium density linkage panel (Illumina, San Diego, CA). The addition of 24 microsatellite markers, from 4 regions with suggestive linkage (Chr 4, 6, 12, 17) in 8 additional affected mice, allowed localization to chromosome 12 (D12Mit182 -D12Mit60). Rps7 was sequenced as a candidate gene using the same methods as for Mtu.
Cellular studies and Western blots
mRNA was extracted from wild-type and Rps7Mtu/+ brains, and cDNA was prepared using reverse transcriptase (Invitrogen). A high fidelity PCR polymerase (KOD, Novagen) was then employed to amplify the Rps7 transcript encoding a fusion protein tagged at either the C - or N-terminus with a FLAG epitope, prior to cloning into the pcDNA3.1(+) vector. Constructs were transfected into HeLa cells grown on glass coverslips, before fixation and staining 48 hours later. The primary antibody employed was a rabbit anti-FLAG (Abcam, ab21536, 1∶1000), followed by a fluorescent secondary (1∶200, Molecular Probes). Five independent transfections were performed. Images were captured on a Zeiss LSM 510 confocal microscope.
For Western blot experiments, DNA constructs encoding wild-type RPS7, RPS7 p.V156G, and RPS7 p.Y177S fused to the FLAG epitope at the N - or C-terminus were used in transient transfection experiments in human embryonic kidney (HEK) 293 cells. Neomycin phosphotransferase II (NPT2) expressed by the cloning vector pcDNA3.1 was used as a control for transfection efficiency. After transfection, proteins were separated on a 12% SDS-PAGE, transferred onto a nitrocellulose Protran membrane (Schleicher and Schuell) and incubated with a rabbit anti-FLAG or anti-neomycin phosphotransferase II (NPT2) antibody for 1 hr. Following three consecutive washes in PBS/Tween, the membrane was incubated with horseradish peroxidase-conjugated goat anti-rabbit Ab (Jackson Immunoresearch) and visualized using a chemiluminescence detection kit (Pierce) and a LAS3000 Image System (Fuji). Equal loading was checked by Ponceau-S red staining of the membranes before Western blot analysis.
Ribosomal analysis
For Northern blots for rRNA processing, RNA was extracted from mouse tissues with Trizol (Invitrogen) according to the manufacturer's protocol. Total RNA was fractionated on formaldehyde-agarose gels and transferred to Gene Screen Plus membrane (NEN). To detect rRNA precursor transcripts, ITS1 probe was prepared by 5′ end-labeling of a 28-mer oligonucleotide (GCTCCTCCACAGTCTCCCGTTAATGATC) with 25 µCi of γ [32P]-ATP and T4 polynucleotide kinase according to standard protocols. Hybridization was carried out overnight at 42°C in 6× SSPE, 1% SDS, 0.25 mg/ml ssDNA and 5× Denhardt's solution. After hybridization, the blot was washed twice with 1% SDS in 2× SSPE for 30 min at 37°C. Quantitation of signals was obtained by phosphor screen scanning with a STORM PhosphorImager and ImageQuant software analysis (Molecular Dynamics). The experiments were performed on tissues from Rps7+/+ and Rps7Mtu/+ littermate mice (2 Rps7+/+ and 2 Rps7Mtu/+ for liver, 3 Rps7+/+ and 3 Rps7Mtu/+ for brain, each in triplicate). Quantitation is reported as the ratio between 30S and 21S rRNA precursors and was significantly different between genotypes in both brain (p<0.01) and liver (p<0.01) (two-way ANOVA with post hoc t-test).
For ribosome isolation, cytoplasmic extracts from RPS7 p.V156G transiently transfected HEK-293 cells were fractionated through ultracentrifugation on a sucrose cushion. After 2 hours of centrifugation at 100,000 g, ribosomes and ribosomal subunits were recovered in the pellet and free cytoplasmic proteins were recovered from the supernatant.
For sucrose gradients separating polysomes and ribosomal subunits, cytoplasmic extracts from Mtu/+ liver were loaded onto a 10%–30% linear sucrose gradient containing 30 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 10 mM MgCl2. Gradients were centrifuged in a Beckman SW 41 rotor for 5 hr at 37,000 rpm and then the absorbance profile of the gradient was used to evaluate the ratio between the peaks of ribosomal subunits (60S and 40S).
In situ hybridization
For whole mount, Pmel17-containing plasmid (Riken cDNA clone G370069C13; GenBank Acc: BB766987) was digested with Kpn1 and transcribed with T3 polymerase to generate DIG-labeled in situ probes. Hybridizations were performed using published protocols [61] with the following modifications. After probe hybridization, Ribonuclease A digestion was omitted, and Tris-buffered saline was used in place of PBS. BM-purple substrate (Roche, Molecular Biochemicals) was used in place of 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium. Embryos were photographed using a Zeiss SteREO Discovery V12 microscope with a Zeiss AxioCam camera. Probes to detect Rps7 by in situ hybridization were amplified by PCR and cloned into the pCRII - TOPO vector (Invitrogen) using the following primer pairs: R7_IN_F AAGGAAATCGAAGTTGGTG and R7_IN_ R AATTAACATCCTTGCCTGTG. Mouse embryos were fixed in 2–4% (w/v) paraformaldehyde, cryoprotected with 30% (w/v) sucrose in phosphate buffer before sectioning (10–20 µm) on a cryostat. In situ hybridization was carried out as previously described [62]. Briefly, hybridization of sections with 35S-UTP-labeled RNA probes was carried out in a 50% (v/v) formamide solution at 60°C. Sections were washed in 50% (v/v) formamide, an RNAse A treatment was performed for 30 min at 37°C, and then successively stringent SSC solution washes were performed, with a final wash at 0.1× SSC at 60°C.
Histology and immunohistochemistry
For Nissl staining, adult mice were perfused with 0.9% NaCl and 4% (w/v) paraformaldehyde before the brain was dehydrated in 30% sucrose, sectioned (40 µm) on a freezing microtome, and stained.. For embryonic histology, embryos were fixed in either Bouin's fixative or 4% paraformaldehyde overnight, washed extensively in PBS, and dehydrated in 70% ethanol before sectioning (5–7 µm paraffin) and hematoxylin and eosin (H and E) staining. For immunohistochemistry, embryos were fixed in 4% paraformaldehyde overnight, washed with PBS, dehydrated in 10% sucrose followed by 20% sucrose, then embedded in Neg-50 (Thermo Scientific) for cryosectioning (14 µm). Antibodies included anti-TRP53 (Leica NCL-p53-CM5p, 1∶200), anti-CC3 (Cell Signaling #9661, 1∶200), and anti-PH3 (Millipore #06-570, 1∶200) and resulting stains were imaged using a Zeiss Observer.D1 microscope. For apoptosis and proliferation experiments, transverse cryo-sections collected at the level of the forelimb from 3 embryos of each genotype, 6 sections per embryo, were counted for CC3+ and PH3+ cells. InStat software (GraphPad Software, Inc.) was used for one-way ANOVA statistical analysis. No significant difference among the three genotypes was observed in PH3+ cell counts (p = 0.902). CC3 counts were significantly different among genotypes (p = 0.0005) and a Tukey-Kramer multiple comparison post test was used for pairwise comparison among all genotypes with the following results: Rps7+/+ versus Rps7Zma/+ (p<0.001), Rps7+/+ versus Rps7Zma/+; Trp53KO/+ (P>0.05, no significant difference), Rps7Zma/+ versus Rps7Zma/+; Trp53KO/+ (p<0.01).
Behavioral phenotyping
Behavioral phenotyping was performed on mutant and unaffected sex-matched littermates between 8 and 12 weeks of age. Open-field tests were conducted for 5 minutes in a brightly lit, 60 cm diameter, enclosed white arena (field), and monitored by an automated tracking system to measure the total distance travelled and the amount of time spent in the center versus the border of the field. The elevated plus maze test was conducted with an elevated platform consisting of two enclosed arms and two open arms. An automated tracking system was employed to measure the number of entries into each arm and distance traveled in open and closed arms of the elevated plus maze over 5 minutes. Spontaneous alternation was performed in an enclosed T-maze built from gray plastic (arm dimensions 30 cm×10 cm×30 cm). Each mouse underwent 10 trials with an inter-trial interval of at least 20 minutes. For each trial the mouse was placed in the start arm facing the end wall and allowed to enter a goal arm of its own choice. The mouse was confined in the goal arm for 30 seconds and then put back in the start arm and allowed a free choice of either arm [31].
MRM imaging
Late gestation (E18.5) embryos were collected, fixed in 4% paraformaldehyde overnight, washed extensively in PBS, and stored in 70% ethanol. Prior to imaging, embryos were rehydrated in a solution of PBS and 0.5% (v/v) Magnevist (Bayer Healthcare Pharmaceuticals, Wayne, NJ), a contrast agent. MRM was performed on specimens using a Bruker 14.1T MR imaging spectrometer (Bruker Biospin, Billerica, MA) using a multi-echo RARE technique [63] with TR/TE = 200/6.9 ms, 8-echoes, and 4 signal averages. The resulting 3D images were acquired in 53 min with an acquisition resolution of 50 microns isotropic. Manual segmentation of the whole brain and 10 major anatomical regions was performed using Amira visualization software (v5.2.2, Visage Imaging Inc., Andover MA, USA) with guidance from an anatomical reference atlas [64]. The volume of these regions was estimated and 3D rendering with smoothing was performed to generate 3D representations. Student's t-tests were used to compare volumes of each Rps7+/+ and Rps7Zma/+ brain region. Significance was assessed using the Bonferonni method to correct for multiple testing, with p<0.005 deemed significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. UechiT, TanakaT, KenmochiN (2001) A complete map of the human ribosomal protein genes: assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics 72 : 223–230 doi:10.1006/geno.2000.6470.
2. BoisvertF-M, van KoningsbruggenS, NavascuésJ, LamondAI (2007) The multifunctional nucleolus. Nat Rev Mol Cell Biol 8 : 574–585 doi:10.1038/nrm2184.
3. MarygoldSJ, RooteJ, ReuterG, LambertssonA, AshburnerM, et al. (2007) The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol 8: R216 doi:10.1186/gb-2007-8-10-r216.
4. UechiT, NakajimaY, NakaoA, ToriharaH, ChakrabortyA, et al. (2006) Ribosomal protein gene knockdown causes developmental defects in zebrafish. PLoS ONE 1: e37 doi:10.1371/journal.pone.0000037.
5. McgowanKA, LiJZ, ParkCY, BeaudryV, TaborHK, et al. (2008) Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet 40 : 963–970 doi:10.1038/ng.188.
6. OliverER, SaundersTL, TarléSA, GlaserT (2004) Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 131 : 3907–3920 doi:10.1242/dev.01268.
7. TerzianT, DumbleM, ArbabF, ThallerC, DonehowerLA, et al. (2011) Rpl27a mutation in the sooty foot ataxia mouse phenocopies high p53 mouse models. J Pathol 224 : 540–552 doi:10.1002/path.2891.
8. FarrarJE, NaterM, CaywoodE, McDevittMA, KowalskiJ, et al. (2008) Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112 : 1582–1592 doi:10.1182/blood-2008-02-140012.
9. GazdaHT, GrabowskaA, Merida-LongLB, LatawiecE, SchneiderHE, et al. (2006) Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet 79 : 1110–1118 doi:10.1086/510020.
10. DohertyL, SheenMR, VlachosA, ChoesmelV, O'DonohueM-F, et al. (2010) Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet 86 : 222–228 doi:10.1016/j.ajhg.2009.12.015.
11. GazdaHT, SheenMR, VlachosA, ChoesmelV, O'DonohueM-F, et al. (2008) Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet 83 : 769–780 doi:10.1016/j.ajhg.2008.11.004.
12. GazdaHT, PretiM, SheenMR, O'DonohueM-F, VlachosA, et al. (2012) Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum Mutat doi:10.1002/humu.22081.
13. CmejlaR, CmejlovaJ, HandrkovaH, PetrakJ, PospisilovaD (2007) Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat 28 : 1178–1182 doi:10.1002/humu.20608.
14. DraptchinskaiaN, GustavssonP, AnderssonB, PetterssonM, WilligTN, et al. (1999) The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet 21 : 169–175 doi:10.1038/5951.
15. NolanPM, PetersJ, StrivensM, RogersD, HaganJ, et al. (2000) A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet 25 : 440–443 doi:10.1038/78140.
16. BoganiD, WarrN, ElmsP, DaviesJ, Tymowska-LalanneZ, et al. (2004) New semidominant mutations that affect mouse development. Genesis 40 : 109–117 doi:10.1002/gene.20071.
17. MateraI, Watkins-ChowDE, LoftusSK, HouL, IncaoA, et al. (2008) A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy. Hum Mol Genet 17 : 2118–2131 doi:10.1093/hmg/ddn110.
18. ThomasPD, KejariwalA, GuoN, MiH, CampbellMJ, et al. (2006) Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 34: W645–W650.
19. KumarP, HenikoffS, NgPC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4 : 1073–1081 doi:10.1038/nprot.2009.86.
20. RablJ, LeibundgutM, AtaideSF, HaagA, BanN (2011) Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331 : 730–736 doi:10.1126/science.1198308.
21. Ben-ShemA, Garreau de LoubresseN, MelnikovS, JennerL, YusupovaG, et al. (2011) The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334 : 1524–1529 doi:10.1126/science.1212642.
22. RostB, YachdavG, LiuJ (2004) The PredictProtein server. Nucleic Acids Res 32: W321–W326 doi:10.1093/nar/gkh377.
23. AngeliniM, CannataS, MercaldoV, GibelloL, SantoroC, et al. (2007) Missense mutations associated with Diamond-Blackfan anemia affect the assembly of ribosomal protein S19 into the ribosome. Hum Mol Genet 16 : 1720–1727 doi:10.1093/hmg/ddm120.
24. RobledoS, IdolRA, CrimminsDL, LadensonJH, MasonPJ, et al. (2008) The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 14 : 1918–1929 doi:10.1261/rna.1132008.
25. O'DonohueM-F, ChoesmelV, FaubladierM, FichantG, GleizesP-E (2010) Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol 190 : 853–866 doi:10.1083/jcb.201005117.
26. BowmanLH, RabinB, SchlessingerD (1981) Multiple ribosomal RNA cleavage pathways in mammalian cells. Nucleic Acids Res 9 : 4951–4966.
27. DianzaniI, LoreniF (2008) Diamond-Blackfan anemia: a ribosomal puzzle. Haematologica 93 : 1601–1604 doi:10.3324/haematol.2008.000513.
28. SocolovskyM, NamH, FlemingMD, HaaseVH, BrugnaraC, et al. (2001) Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood 98 : 3261–3273.
29. ZhangJ, SocolovskyM, GrossAW, LodishHF (2003) Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood 102 : 3938–3946 doi:10.1182/blood-2003-05-1479.
30. PotterfSB, MollaaghababaR, HouL, Southard-SmithEM, HornyakTJ, et al. (2001) Analysis of SOX10 function in neural crest-derived melanocyte development: SOX10-dependent transcriptional control of dopachrome tautomerase. Dev Biol 237 : 245–257 doi:10.1006/dbio.2001.0372.
31. DeaconRMJ, RawlinsJNP (2006) T-maze alternation in the rodent. Nat Protoc 1 : 7–12 doi:10.1038/nprot.2006.2.
32. ChakrabortyA, UechiT, HigaS, ToriharaH, KenmochiN (2009) Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS ONE 4: e4152 doi:10.1371/journal.pone.0004152.
33. GazdaHT, KhoAT, SanoudouD, ZauchaJM, KohaneIS, et al. (2006) Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells 24 : 2034–2044 doi:10.1634/stemcells.2005-0554.
34. MatssonH, DaveyEJ, DraptchinskaiaN, HamaguchiI, OokaA, et al. (2004) Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol Cell Biol 24 : 4032–4037.
35. BarkićM, CrnomarkovićS, GrabusićK, BogetićI, PanićL, et al. (2009) The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol 29 : 2489–2504 doi:10.1128/MCB.01588-08.
36. ToriharaH, UechiT, ChakrabortyA, ShinyaM, SakaiN, et al. (2011) Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br J Haematol 152 : 648–654 doi:10.1111/j.1365-2141.2010.08535.x.
37. SulicS, PanićL, BarkićM, MercepM, UzelacM, et al. (2005) Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev 19 : 3070–3082 doi:10.1101/gad.359305.
38. WildT, HorvathP, WylerE, WidmannB, BadertscherL, et al. (2010) A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8: e1000522 doi:10.1371/journal.pbio.1000522.
39. SynetosD, DabevaMD, WarnerJR (1992) The yeast ribosomal protein S7 and its genes. J Biol Chem 267 : 3008–3013.
40. SteffenKK, McCormickMA, PhamKM, MacKayVL, DelaneyJR, et al. (2012) Ribosome Deficiency Protects Against ER Stress in Saccharomyces cerevisiae. Genetics 191 : 107–118 doi:10.1534/genetics.111.136549.
41. BernsteinKA, GallagherJEG, MitchellBM, GrannemanS, BasergaSJ (2004) The Small-Subunit Processome Is a Ribosome Assembly Intermediate. Eukaryotic Cell 3 : 1619–1626 doi:10.1128/EC.3.6.1619-1626.2004.
42. Ferreira-CercaS, PöllG, GleizesP-E, TschochnerH, MilkereitP (2005) Roles of Eukaryotic Ribosomal Proteins in Maturation and Transport of Pre-18S rRNA and Ribosome Function. Molecular Cell 20 : 263–275 doi:10.1016/j.molcel.2005.09.005.
43. GoodmanFR (2003) Congenital abnormalities of body patterning: embryology revisited. Lancet 362 : 651–662 doi:10.1016/S0140-6736(03)14187-6.
44. BoganiD, WilloughbyC, DaviesJ, KaurK, MirzaG, et al. (2005) Dissecting the genetic complexity of human 6p deletion syndromes by using a region-specific, phenotype-driven mouse screen. Proc Natl Acad Sci USA 102 : 12477–12482 doi:10.1073/pnas.0500584102.
45. MatssonH, DaveyEJ, FröjmarkAS, MiyakeK, UtsugisawaT, et al. (2006) Erythropoiesis in the Rps19 disrupted mouse: Analysis of erythropoietin response and biochemical markers for Diamond-Blackfan anemia. Blood Cells Mol Dis 36 : 259–264 doi:10.1016/j.bcmd.2005.12.002.
46. MorganWC (1950) A new tail-short mutation in the mouse whose lethal effects are conditioned by the residual genotypes. J Hered 41 : 208–215.
47. KondrashovN, PusicA, StumpfCR, ShimizuK, HsiehAC, et al. (2011) Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145 : 383–397 doi:10.1016/j.cell.2011.03.028.
48. SouthardJ, EischerE (1977) Belly spot and tail (Bst). Mouse News Letter 56 : 40.
49. AndersonSJ, LauritsenJPH, HartmanMG, FousheeAMD, LefebvreJM, et al. (2007) Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity 26 : 759–772 doi:10.1016/j.immuni.2007.04.012.
50. StadanlickJE, ZhangZ, LeeS-Y, HemannM, BieryM, et al. (2011) Developmental arrest of T cells in Rpl22-deficient mice is dependent upon multiple p53 effectors. J Immunol 187 : 664–675 doi:10.4049/jimmunol.1100029.
51. Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, et al.. (2008) Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Vol. 142. pp. 859–876. doi:10.1111/j.1365-2141.2008.07269.x.
52. TangYP, WadeJ (2010) Sex - and age-related differences in ribosomal proteins L17 and L37, as well as androgen receptor protein, in the song control system of zebra finches. Neuroscience 171 : 1131–1140 doi:10.1016/j.neuroscience.2010.10.014.
53. FumagalliS, IvanenkovVV, TengT, ThomasG (2012) Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev 26 : 1028–1040 doi:10.1101/gad.189951.112.
54. DuanJ, BaQ, WangZ, HaoM, LiX, et al. (2011) Knockdown of ribosomal protein S7 causes developmental abnormalities via p53 dependent and independent pathways in zebrafish. The International Journal of Biochemistry & Cell Biology 43 : 1218–1227 doi:10.1016/j.biocel.2011.04.015.
55. WarnerJR, McIntoshKB (2009) How common are extraribosomal functions of ribosomal proteins? Molecular Cell 34 : 3–11 doi:10.1016/j.molcel.2009.03.006.
56. BortoluzziS, d'AlessiF, RomualdiC, DanieliGA (2001) Differential expression of genes coding for ribosomal proteins in different human tissues. Bioinformatics 17 : 1152–1157.
57. BévortM, LeffersH (2000) Down regulation of ribosomal protein mRNAs during neuronal differentiation of human NTERA2 cells. Differentiation 66 : 81–92 doi:10.1046/j.1432-0436.2000.660203.x.
58. TerzianT, TorchiaEC, DaiD, RobinsonSE, MuraoK, et al. (2010) p53 prevents progression of nevi to melanoma predominantly through cell cycle regulation. Pigment Cell & Melanoma Research 23 : 781–794 doi:10.1111/j.1755-148X.2010.00773.x.
59. KapasiP, ChaudhuriS, VyasK, BausD, KomarAA, et al. (2007) L13a Blocks 48S Assembly: Role of a General Initiation Factor in mRNA-Specific Translational Control. Molecular Cell 25 : 113–126 doi:10.1016/j.molcel.2006.11.028.
60. SatoY, YanoS, EwisAA, NakahoriY (2011) SRY interacts with ribosomal proteins S7 and L13a in nuclear speckles. Cell Biology International 35 : 449–452 doi:10.1042/CBI20090201.
61. WilkinsonDG, NietoMA (1993) Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Meth Enzymol 225 : 361–373.
62. Phan Dinh TuyF, SaillourY, KappelerC, ChellyJ, FrancisF (2008) Alternative transcripts of Dclk1 and Dclk2 and their expression in doublecortin knockout mice. Dev Neurosci 30 : 171–186 doi:10.1159/000109861.
63. HennigJ, NauerthA, FriedburgH (1986) RARE imaging: a fast imaging method for clinical MR. Magn Reson Med 3 : 823–833.
64. Paxinos G, Halliday GM, Watson C, Koutcherov Y, Wang H (2006) Atlas of the Developing Mouse Brain at E17.5, P0 and P6. Academic Press.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy