
Formation of Linear Amplicons with Inverted Duplications in Requires the MRE11 Nuclease
Extrachromosomal DNA amplification is frequent in the human protozoan parasite Leishmania when challenged with drug or other stressful conditions. DNA amplicons, either circular or linear, are formed by recombination between direct or inverted repeats spread throughout the genome of the parasite. The recombinase RAD51 is involved in the formation of circular amplicons, but the mechanism by which linear amplicons are formed is still unknown in this parasite. Studies in other organisms have provided some evidence that a DNA break is required for linear amplifications, and that the DNA repair protein MRE11 can be involved in this process. In this work, we present our biochemical, cellular and molecular characterization of the Leishmania infantum MRE11 orthologue and provide evidence that this nuclease is involved in the formation of linear amplicons in Leishmania. Our results highlight a novel MRE11-dependent pathway used by Leishmania to amplify portions of its genome to respond to a changing environment.
Published in the journal:
. PLoS Genet 10(12): e32767. doi:10.1371/journal.pgen.1004805
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004805
Summary
Extrachromosomal DNA amplification is frequent in the human protozoan parasite Leishmania when challenged with drug or other stressful conditions. DNA amplicons, either circular or linear, are formed by recombination between direct or inverted repeats spread throughout the genome of the parasite. The recombinase RAD51 is involved in the formation of circular amplicons, but the mechanism by which linear amplicons are formed is still unknown in this parasite. Studies in other organisms have provided some evidence that a DNA break is required for linear amplifications, and that the DNA repair protein MRE11 can be involved in this process. In this work, we present our biochemical, cellular and molecular characterization of the Leishmania infantum MRE11 orthologue and provide evidence that this nuclease is involved in the formation of linear amplicons in Leishmania. Our results highlight a novel MRE11-dependent pathway used by Leishmania to amplify portions of its genome to respond to a changing environment.
Introduction
The protozoan parasite Leishmania is responsible for a group of diseases named leishmaniasis, affecting approximately 12 million people worldwide. No vaccine is currently available against Leishmania and treatments mainly rely on chemotherapy [1], [2]. Pentavalent antimony is the main anti-leishmanial drug although treatment failure due to resistance has been reported in most endemic regions [3]–[7]. Locus amplification is a frequent resistance mechanism allowing the parasite to modulate gene copy number and increased gene expression. Indeed, the parasite Leishmania is an early diverging eukaryotic parasite with no control of gene expression at the level of transcription initiation [8]–[10] and amplification of DNA loci is one strategy to increase the expression of resistance genes. Resistance genes can be amplified as part of extrachromosomal circular DNAs (circular amplicons) or as inverted duplications (linear amplicons) under drug pressure [11]–[16]. Gene rearrangements leading to locus amplification always occur at the level of either homologous direct or inverted repeated (IRs) sequences leading respectively to circular or linear extrachromosomal amplification [14], [15], [17]–[19]. A model for the generation of linear amplicons is shown in Figure 1.

A recent bioinformatics screen revealed that repeated sequences are widely distributed in the Leishmania genome, which is continuously being rearranged at the level of those repeated sequences. This process is adaptive as the copy number of advantageous extrachromosomal circular or linear elements increases upon selective pressure [19]. The whole genome of Leishmania is thus stochastically rearranged at the level of repeated sequences and the selection of parasite subpopulations with changes in the copy number of specific loci is used as one strategy to respond to drug pressure.
Circular or linear amplification has been observed when parasites were selected against a wide variety of drugs including the mainstay antimony [15], [20] but one drug that has proven highly useful in deciphering gene amplification mechanisms in Leishmania is the model antifolate drug methotrexate (MTX). Two loci are frequently amplified after MTX selection, one encoding the dihydrofolate reductase-thymidylate synthase (DHFR-TS) gene, the target of MTX, usually as part of circular elements [14], [21]–[23] the second encoding the pteridine reductase 1 (PTR1) gene, which is less sensitive to MTX but can reduce folates when DHFR-TS is blocked [24], [25]. The PTR1 gene is amplified as part of either circular amplicons [22], [26], [27] or linear amplicons [13], [14], [19], [28], [29].
We have recently provided mechanistic insights into the formation of circular amplicons mediated by homologous recombination between direct repeated sequences catalyzed by the RAD51 recombinase [19]. However rearrangements at IRs leading to linear amplicons were not RAD51-dependent. Studies in other organisms have provided some evidence that a DNA break is required for palindromic amplifications formed by annealing of IRs [30]–[33] and that an exonuclease activity must be recruited to perform DNA end resection after chromosomal breakage in order to allow annealing of IRs [34]–[36] (see also Figure 1). Based on these observations, we hypothesized the involvement of the nuclease MRE11 (Meiotic REcombination 11) in the generation of linear amplicons in Leishmania (Figure 1). MRE11 interacts with RAD50 and NBS1 to form the MRN complex [37], [38]. Indeed, the nuclease MRE11 is a sensor of DNA double-strand breaks in cells and is important for the DNA double-strand break repair pathway [39], [40] by homologous recombination (HR) or non-homologous end joining (NHEJ) [41]. Leishmania infantum encodes a putative MRE11 with conserved endo - and exonuclease domains as well as DNA-binding domains [41]. In this manuscript, we present our biochemical, cellular and molecular characterization of the L. infantum MRE11 orthologue and provide evidence that this nuclease is involved in the formation of linear amplicons in the parasite Leishmania.
Results
Purification and biochemical characterization of LiMRE11
Since the critical catalytic residues of MRE11 are conserved in Leishmania [41] and the replacement of the histidine (H) at position 217 by a tyrosine (Y) is known to abolish the nuclease activity of the human MRE11 but not its nucleic acid binding property [42], we scrutinized the amino acid alignment between the human and Leishmania sequences and found the equivalent of human H217 at position 210 of LiMRE11 (Figure 2A, upper panel and Figure S1). We therefore produced a LiMRE11 mutated at the corresponding amino acid (LiMRE11H210Y) and used a two-step affinity purification procedure to purify LiMRE11WT and LiMRE11H210Y as described in Material and Methods (Figure 2A, lower panel).

We used the electrophoretic mobility shift assay to study DNA interactions with MRE11 proteins (Figure 2B). We observed that the splayed arm (SA) and single-strand (SS) DNA structures were shifted in the presence the wild-type and mutated MRE11 protein in a dose-dependent manner while neither version of the protein were able to shift the double-strand (DS) structure in this competitive assay. The binding was quantitated and at 15 nM of either protein, 65% binding was observed with either SS and SA DNA structures while we observed only 10% binding for DS DNA (Figure 2C).
We next tested whether purified LiMRE11WT and LiMRE11H210Y displayed exonuclease activity (Figure 3A), in comparison with human MRE11WT and hMRE11H217Y proteins (Figure 3B). Our findings suggest that LiMRE11WT is enzymatically active and can perform exonucleolytic degradation with a 3′ to 5′ polarity but it is less effective than the human MRE11WT protein in cleaving DNA into smaller fragments (Figure 3A–B, lanes 1–4). As expected, LiMRE11H210Y was unable to perform DNA resection (Figure 3A, lanes 5–7), similar to its human mutated counterpart (Figure 3B, lanes 5–7). The substrate specificity was also monitored by using 25 nM of LiMRE11WT protein with DS DNA, either blunt or with 3′ or 5′ overhang DNA structures (Figure 3C). The same extensive degradation was observed with DS DNA and 5′-overhang ends (Figure 3C, lane 2 and 4) while the protein was blocked by 3′-overhang extremities (Figure 3C, lane 3). LiMRE11WT also exhibits endonuclease activity, as shown by the 13 bp band found at the bottom of the gel.

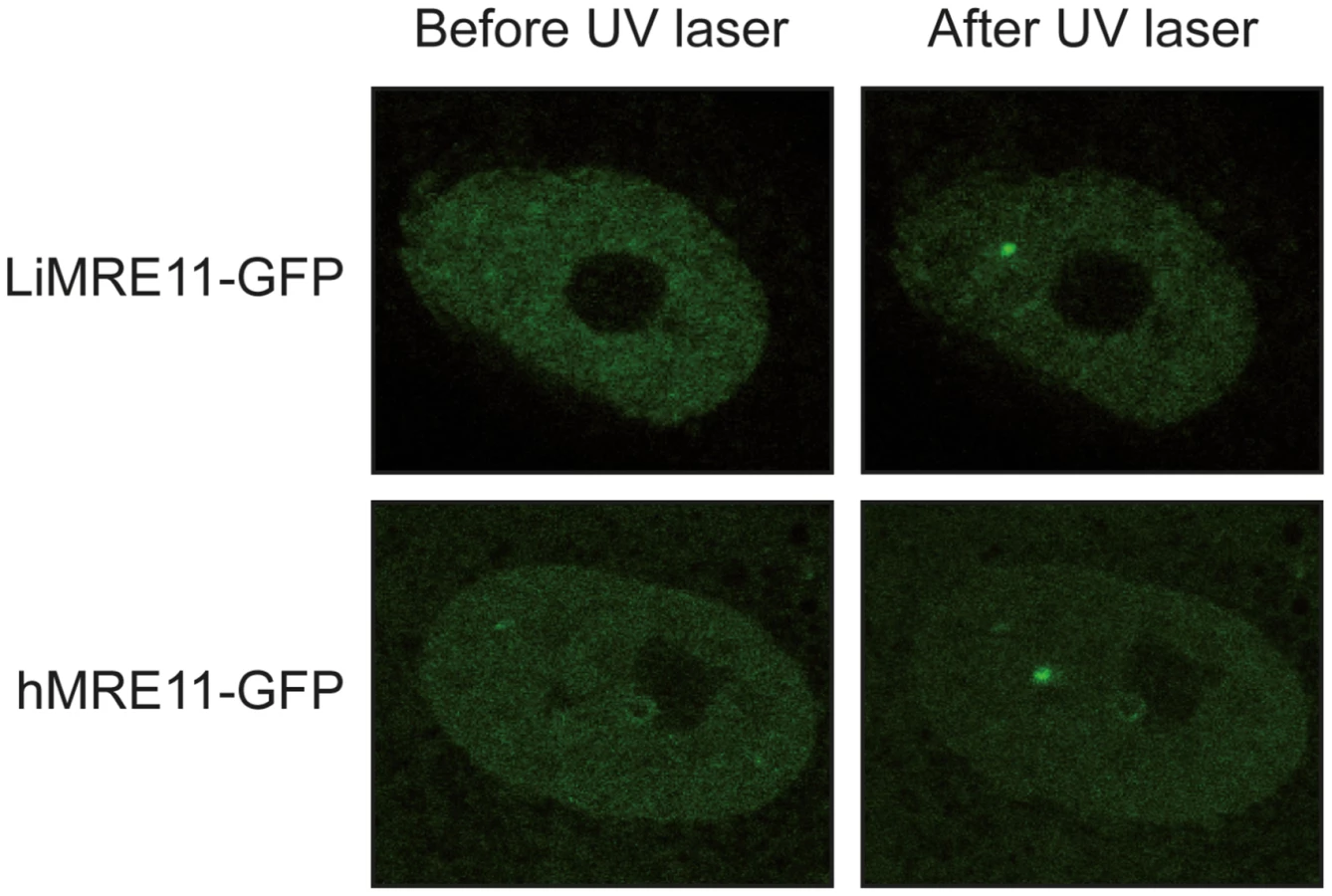
LiMRE11-GFP is recruited at DNA damage loci in human MRE11-deficient cells (ATLD)
We generated a LiMRE11-GFP fusion construct that was transfected in L. infantum cells. However, we could never achieve a high copy number of the plasmid (overexpression of MRE11 can be toxic to the cell, see below) and fluorescence levels were too low for analysis. We then turned to a heterologous system to study LiMRE11 in vivo. DNA constructs encoding the fusion protein LiMRE11WT-GFP and the human counterpart hMRE11WT-GFP were transfected in human ATLD cells, which are deficient for hMRE11 [43]. After laser-induced DNA damage in these cells, we detected a localized fluorescent foci representative of the recruitment of LiMRE11WT-GFP in micro-irradiated nuclear regions (Figure 4, upper panels), similar to what was observed for the human MRE11WT-GFP fusion protein (Figure 4, bottom panels). Among 24 ATLD cells micro-irradiated, we observed a recruitment of LiMRE11 to DNA damages sites in 75% of the cases, while the human homolog was recruited in 100% of the cells. No recruitment was observed for the control GFP alone. These observations confirmed the ability of the Leishmania MRE11 protein to be recruited at DNA damage sites, in a heterologous cellular model. Altogether, these results show that LiMRE11 display similar localization properties as the human enzyme.
Inactivation of the Leishmania infantum MRE11 gene
L. infantum MRE11 null mutant parasites were generated by replacing the entire ORF (LinJ27.1790) with genes coding for the neomycin (NEO) and hygromycin (HYG) phosphotransferases. The two resistant markers were cloned between the 5′ - and 3′-MRE11 flanking regions and targeting constructs were transfected independently in two rounds by electroporation. Southern blot analysis confirmed the homologous chromosomal integration of the two antibiotic markers in the MRE11 locus (Figures 5A and 5B). Genomic DNAs of the WT and the HYG/NEO MRE11−/− null mutant were digested with XhoI, transferred onto membranes and hybridized. Hybridization with a probe recognizing the 5′UTR region of MRE11 yielded a 3 kb band in WT cells (Figure 5A and 5B-lane 1) while hybridization with a 3′UTR probe generated a 3,4 kb band as expected (Figure 5A and 5B, lane 5). In the HYG/NEO MRE11−/− strain, replacement of both MRE11 wild-type alleles by NEO and HYG led, as expected, to 4,7 kb and 4,9 kb bands respectively, with either UTR probes (Figure 5A and 5B, lanes 2 and 6).

It is standard practice to introduce episomal copies of the corresponding wild-type gene in a null mutant background to reverse a potential phenotype. However, we noticed that episomal overexpression of MRE11 as Psp72-α-PUR-α-MRE11WT in WT cells led to a growth defect (Figure S2A). This prompted us to use an alternative to generate revertants. We replaced the NEO chromosomal integrated cassette in the MRE11 null mutant by a re-expressing cassette containing either a WT or a mutated allele (H210Y) of LiMRE11 along with the PUR gene in order to generate respectively the HYG/PUR-MRE11WT and HYG/PUR-MRE11H210Y re-expressing add back strains. Hybridization of the DNAs of the add-back strains with a 5′UTR probe led, as expected, to a 3 kb band in both strains corresponding to the restoration of a WT allele at the MRE11 locus, a 4,9 kb band corresponding to the HYG chromosomal integration, and a loss of the 4,7 kb-NEO containing band (Figure 5A and 5B-lanes 3 and 4) which was replaced by the MRE11-α-PUR re-expressing cassette as supported by the hybridization of a 2,7 kb band when using a 3′UTR probe (Figure 5A and 5B-lanes 7 and 8). MRE11 expression level was assessed by quantitative real-time RT-PCR in the various cell lines generated. As expected MRE11 expression level was not detectable in the MRE11−/− null mutant while it was approximately half the level of the WT in both add back strains, consistent with one new active allele (Figure S3).
A growth defect was observed in L. infantum HYG/NEO MRE11−/− parasites compared to the WT strain. Promastigotes of the WT strain had a calculated generation time of 12 hours while the MRE11 null mutant had a generation time of 26 hours. (Figure 5C). Reintroduction of one intact WT or mutated (H210Y) allele of MRE11 into the chromosomal locus in add back strains partially rescued the growth defect with respective generation time of 15 and 22 hours.(Figure 5C).
Since the MRE11 complex is known to promote repair of DNA double-strand breaks (DSBs) [44], [45], we tested the impact of the LiMRE11 inactivation using the alkylating damaging agent methyl methanesulphonate (MMS), a compound known to induce DSBs [46]. The HYG/NEO MRE11−/− cells were significantly more sensitive to MMS compared to both WT and MRE11 add back re-expressing cells (Figure 5D). As indicated above, intriguingly, overexpressing MRE11 in WT cells led to a growth defect (Figure S2A) but also to a significant increase in MMS sensitivity (Figure S2B). However, episomal overexpression of MRE11WT in HYG/NEO MRE11−/− cells restores the growth defect and MMS susceptibility of the mutant strain to WT levels (Figures S2A and S2B).
Gene amplification in cells with varying copy of MRE11
We compared the ability of the MRE11 null mutants and WT cells to generate extrachromosomal linear amplicons. We selected clones of wild-type cells and of HYG/NEO MRE11−/− for MTX resistance in a stepwise manner (up to 1600 nM, a 16-fold increase in resistance compared to starting parent cells), a drug known to select for PTR1 linear DNA amplifications [13], [18], [28]. Leishmania chromosomes extracted from ten MTX resistant clones derived from either WT or HYG/NEO MRE11−/− parasites were separated by pulse field gel electrophoresis (PFGE) and hybridized with a PTR1 probe. Ethidium bromide stained gels already indicated that some linear amplicons smaller than the smallest chromosome were present in some resistant clones derived from WT but not in the MTX resistant MRE11−/− mutants (Figure S4). Hybridization data revealed that all ten MTX resistant clones derived from WT cells displayedPTR1 linear amplicons of varying size of 125 kb, 250 kb, 450 kb and 565 kb (Figure 6A). Clones 6 and 7 also gave rise to PTR1 circular amplicons, as suggested from the hybridizing smears (Figure 6A). The situation was drastically different in the HYG/NEO MRE11−/− parasites selected for MTX resistance. We observed only one resistant clone from the MRE11 null mutant with a faint hybridization signal corresponding to a PTR1 linear amplification (Figure 6B, clone 1), while a PTR1 circular amplification was present in clones 4 and 5 derived from the MRE11−/− mutant (Figure 6B). Clone 3 displayed a hybridization signal at around 1150 kb (Figure 6B) which could correspond to either a very large linear amplicon or to a chromosomal translocation. The difference in formation of linear amplicons between WT and MRE11−/− null mutant was found to be significant (p<0,01). We also selected the add back strains HYG/PUR-MRE11WT and HYG/PUR-MRE11H210Y for MTX resistance for testing for the specificity of the phenotype and for assessing the role of the MRE11-exonuclease activity in the generation of linear amplicons. While the MRE11−/− mutants had a decreased capacity to generate linear amplicons after MTX selection (Figure 6B), nine out of ten MTX resistant clones derived from the HYG/PUR-MRE11WT add back strain had PTR1 linear amplicons (Figure 6C, clones 1, 2, 4–10). Similar to the mutants derived from the wild-type cells (Figure 6A), four different PTR1 linear amplicons of 125, 250, 450 and 565 kb (Figure 6C) were present and four clones derived from HYG/PUR-MRE11WT had additional PTR1 circular amplicons (Figure 6C, clones 1, 2, 6 and 7). This phenotype reversion was not observed when the MRE11−/− cells were complemented with MRE11WT as part of an episomal construct. Clones derived from the latter transfectants and selected for MTX resistance were similar to the MRE11−/− mutants with no PTR1 linear amplicons (Figure S5). The results were even more surprising with the MTX resistant clones derived from HYG/PUR-MRE11H210Y. Strikingly all mutants had circular amplifications and the PTR1 hybridization intensity was in general much higher suggesting a higher copy number of the circles. Four clones derived from this add-back revertant also had a PTR1 linear amplicon (Figure 6D, clones 2, 7, 9, 10).

Previous data has indicated that linear amplicons are constituted of inverted duplications rearranged at the level of IRs with the formation of a new junction that can be amplified by PCR (see Figure 1). The diversity in size of linear amplicons observed in Figure 6A and 6C would suggest that different IRs were used and we tested for the presence of IRs in the chromosome 23 that could lead to PTR1 amplicons with size (125, 250, 450 and 565 kb) consistent with what observed in the blots. We detected a potential of 5 such IRs with size ranging from 440 to 790 bp and with a minimum of 85% identity (Figure 7A), a finding consistent with our demonstration that low copy repeated sequences are widespread throughout the genomes [19]. We performed PCR assays using five different pairs of primers recognizing the five different pairs of IRs under the principle shown in Figure 1. Amplification of the GAPDH gene was also done as a control. In the ten MTX resistant clones derived from WT cells and the HYG/PUR-MRE11WT add back strain, we detected several junctions by PCR (Figure 7B and D). The junction formed following a rearrangement at the level of IRs AA′ was the most frequently observed rearrangement, but junctions BB′, EE′ were also detected frequently while the junctions DD′ and CC′ were detected once in clones derived respectively from the WT or add back strains (Figures 7B and 7D). In clones in which we detected circles by Southern blots (Figure 6), we also obtained a positive signal for junction FF′ where the repeats are in direct orientation (Figure 7). There was a general good agreement between the number of amplicons detected by southern blots and PCR although PCR was more sensitive. For example, we could not detect a linear amplicon in clone 2 in MRE11−/− (Figure 6B) but it had a positive PCR reaction for the junction AA′ (Figure 7C).

The MRE11−/− MTX resistant mutants do not have PTR1 amplification (Figure 6B) and these cells must resist MTX by other means. Several mechanisms of resistance have been described [47], [48] including transport defects and gene amplification. We have carried out transport experiments in the mutants and observed no difference in MTX uptake between the MRE11−/− and the MRE11−/− MTX resistant mutants. We also hybridized PFGE blots with a DHFR-TS probe and while we failed in detecting circular DHFR-TS amplification, we observed an extra high molecular weight band hybridizing to DHFR-TS (Figure S6). The exact mechanism leading to this rearrangement is not known but it leads to a 2-fold increase in DHFR-TS expression (Figure S7) and it may contribute to MTX resistance.
Discussion
Gene amplification as part of linear or circular extrachromosomal elements is frequently observed in the parasite Leishmania selected for drug resistance or subjected to nutritional stresses [11]. The circles or linear elements are formed at the level of homologous direct or inverted repeats, with more than 2000 repeats of more than 200 bp representing close to 5% of the Leishmania genome [19]. The genome of Leishmania is continuously being rearranged at the level of these repeats and while each cell has a core genome, they each differ by a complement of circular or linear amplicons. Upon selection the copy number of these elements increases and in the absence of selection the copy number of these elements decreases [19]. It was shown that circular elements are formed by homologous recombination between direct repeated sequences which is catalyzed by the RAD51 recombinase, known to be involved in the homologous recombination process in kinetoplastids [49], [50]. However, the rate of formation of linear amplicons was unchanged in a RAD51−/− mutant and linear amplicons must then be formed through another pathway [19]. We hypothesized that DNA repair proteins with nuclease activities may be involved since genomic DNA must be processed when inverted repeats are annealing for the formation of linear amplicons (Figure 1). We thus focused our efforts on the nuclease MRE11 that is part of the MRN complex [1], [51].
The biochemical characterization of the Leishmania MRE11 protein indicated that it has properties similar to other MRE11 orthologues. Indeed, as previously reported for MRE11 homologs in other organisms [42], [51]–[53], it binds preferentially SS and SA DNA structures in competitive assays (Figure 2); it is capable of DNA end resection and exhibited a 3′→5′ exonuclease activity on DS DNA structures, albeit with less effectiveness than the human enzyme (Figure 3). A H210Y mutant abolished its nuclease activity without impairing its DNA binding properties (Figures 2 and 3A). Finally, LiMRE11 recruitment at DNA damage loci was demonstrated in human ATLD cells, an indication that LiMRE11 detects and binds DNA breaks in these cells (Figure 4).
Having established that the Leishmania MRE11 protein bears all the hallmarks of the MRE11 family of DNA nucleases, we generated a recombinant parasite where the two alleles were inactivated (Figure 5). These parasites were viable but displayed a growth defect (Figure 5C). The parasites were also more sensitive to the DNA damaging agent MMS (Figure 5D) as also observed for Trypanosoma brucei [54]. The growth defect and susceptibility to MMS were reverted when we re-introduced one allele of MRE11 at its original chromosomal locus (Figures 5C and 5D) or as part of an episomal construct (Figure S2). Surprisingly, the expression of an episomal MRE11 gene in a WT strain severely impaired its growth rate (Figure S2A) and led to increased sensitivity to MMS (Figure 2). This result strongly suggests that in a wild-type background, the overproduction of MRE11 is somehow affecting the parasite cell growth, a phenomenon usually not observed in other cell types. The expression level of GFP-MRE11 in WT cells was very low, a phenotype also reported in the trypanosomatid parasite Trypanosoma brucei [55]. These early divergent eukaryotes may be more sensitive to exonuclease overexpression. Alternatively, overexpression of MRE11 in Leishmania may alter more acutely the stoichiometry of interactions with partner proteins such as RAD50 [56] and this could have an impact on cell growth. Interestingly and in support of the above hypothesis, we have shown in an independent study that RAD50 is essential in Leishmania WT cells but its gene can be inactivated in a MRE11−/− background (Laffitte et al., unpublished data). Possibly that recombination pathways are changed in MRE11−/− to compensate for the loss of MRE11, therefore altering the importance of the MRN complex and its components. Growth delay observed in WT cells overexpressing MRE11 may also relate to the known role of MRE11 in cell cycle regulation [37]. Indeed, MRE11 is involved in control of DNA replication initiation [57] and overexpression of MRE11 in Leishmania may have stronger effect on replication of Leishmania chromosomes.
Selection for MTX resistance often leads to linear amplifications of PTR1 in Leishmania [13], [14], [28]. We selected wild-type cells, MRE11−/− null mutants and reverted lines for MTX resistance. Amplified linear PTR1-containing amplicons were observed in all the clones derived from the WT strain but in only 1 out of 10 clones derived from the MRE11−/− null mutant (Figure 6). The capacity to generate circular amplicons was similar in the two different lines (Figure 6A and 6B). This strong phenotype was specific to MRE11, as reintroduction of MRE11 at the original chromosomal locus restored the ability of the parasites to generate PTR1 linear amplicons upon MTX selection (Figure 6C). Leishmania differs from yeast in this process since the MRN complex can prevent palindrome amplification in yeast. This process requires the interaction with the CtIP protein [36], [58] which is absent in Leishmania [41], possibly explaining the difference between the two organisms. Two others important parameters to consider are the length of the inverted repeats, which are very long in Leishmania, and the length of the sequences between these repeats. Indeed, it was previously suggested in yeast that hairpins with large loops are handled differently than hairpin with smaller loops [59]. This explanation is consistent with our study where IRs are few kb apart (Figure 7A), creating large loops in the hairpin structure, while most of the experiments done in yeast presents IRs closer to each other [58]–[61]. Further experiments could be interesting to determine whether the hairpin strength and structure influences DNA processing by the MRN complex.
The reversion of linear amplicons phenotype is dependent on MRE11 nuclease activity since reintegration of the mutated version MRE11H210Y led to parasites generating more efficiently circular PTR1 amplicons but not linear ones (Figure 6D). The mutated MRE11 therefore appears to favor homologous recombination in rescued parasites at the level of direct repeated sequences leading to circular amplicons. It is known that MRE11 is involved in initial events of homologous recombination in many organisms [37], [38], [62] and can interact with a number of nucleases and helicases, several of which are encoded in the L. infantum genome [41]. We suggest that the Leishmania MRE11H210Y is still capable of binding DNA and therefore MRN complex formation is intact, as it was previously suggested in yeast [63]. However, MRE11 lack of nuclease activity possibly makes it a better bait for recruiting HR proteins including RAD51. This facilitated recruitment could be due for example to putative longer association kinetics of the mutated MRE11 to DNA. Alternatively, the inability of LiMRE11H210Y to perform DNA resection may alter the first steps of DNA repair and possibly increase HR proteins recruitment, hence facilitating the formation of circular amplicons. This phenotype is observed in a MRE11−/− background in which we believe that recombination pathways have changed, possibly for compensating loss of MRE11. Thus, a combination of alterations in recombination pathways along with the mutated MRE11 may be responsible for the phenotype observed. It is salient to reiterate that while the episomal expression of MRE11 in the MRE11−/− null mutants reverted the growth phenotype and sensitivity to MMS (Figure S2), it did not revert the phenotype of generating linear amplicons upon MTX selection (Figure S5). This is a further demonstration of the importance of a suitable level of expression to recover proper MRE11 functions.
We have shown that gene rearrangements are continuously taking place at the level or repeated sequences, and that these rearrangements can be highlighted by PCR assays. Using PCR, we have shown that the PTR1 linear amplicons are generated at the level of 5 different inverted repeats, indicating that different rearrangements led to the linear amplicons (Figure 7). The IRs most frequently used are AA′, BB′ and EE′ and these are relatively close to one another (10 kb between A and A′ as well as between E and E′, 3 kb between B and B′) while IRs CC′ and DD′ are further apart (respectively 38 and 53 kb) and used only once. This suggests that the length of the intervening sequences between the IRs may impact the rate of annealing of the IRs and the rearrangements leading to linear amplicons. Few amplicons detected by southern blot were not observed by PCR, suggesting that either smaller inverted repeats were used or secondary rearrangements occurred. Our bioinformatics screen has revealed only one direct repeat that could entertain PTR1 circular amplification and indeed the PCR assay has revealed that in every cell in which a circular amplicon was observed in Figure 6, we observed a positive PCR signal indicative of recombination between direct repeat sequences FF′ (Figure 7).
Because of its lack of control at the level of transcription initiation, Leishmania is likely to use several mechanisms to regulate its expression. We have suggested that gene rearrangements leading to copy number variation is one such mechanism. Indeed the whole Leishmania genome is continuously and stochastically rearranged at the level of repeated sequences. We have shown that there are at least two pathways of rearrangement. One leads to circles after recombination between two direct repeated sequences and this requires RAD51. Here we have shown that linear amplicons, formed at the annealing of two IRs, is facilitated by the presence of an active MRE11. We proposed that double-strand breaks (see Figure 1) would be necessary, although this will require experimental validation, which may be challenging in Leishmania as they are no suitable inducible systems. Gene rearrangement is one main mechanism of resistance in Leishmania and a further understanding of the proteins involved in gene rearrangements may provide a strategy to circumvent the emergence of drug resistance.
Materials and Methods
Strains, culture conditions
Promastigotes of Leishmania infantum (MHOM/MA/67/ITMAP-263) and all recombinants were grown in SDM-79 medium at 25°C supplemented with 10% fetal bovine serum, 5 µg/ml of hemin at pH 7.0. Independent clones of all cells generated in this study were selected for methotrexate (MTX) resistance, using a stepwise selection starting from an EC50 of 100 nM up to 1600 nM of MTX. All chemical reagents were purchased from Sigma-Aldrich unless specified and were of the highest grades.
DNA constructs and protein purification
The L. infantum MRE11 gene (LinJ.27.1790) was amplified by PCR using primers 1 and 2 (Table S1) on genomic DNA template and cloned in a modified pFASTBAC1 plasmid (Invitrogen) [64] encoding the glutathione-S-transferase tag (GST) at the N-terminus of MRE11 and a 10-histidine tag at its C-terminus for protein purification. Site-directed mutagenesis (Stratagene, Quickchange) was used to generate the LiMRE11 mutant H210Y using primers 15 and 16 (Table S1). The LiMRE11WT protein and the mutated version LiMRE11H210Y were purified from baculovirus-infected SF9 cells and the GST tag was removed by PreScission cleavage as described in [64]. Full-length human MRE11 cDNAs cloned in pFASTBAC were generously provided by Tanya Paull (University of Texas, Austin). Primers 22 and 23 (Table S1) were used for site-directed mutagenesis (Stratagene, Quickchange) to generate the human MRE11 mutant H217Y. Proteins hMRE11WT and hMRE11H217Y were purified as described [65]. Full-length human MRE11 cDNAs cloned in pEYFP-C1 (Clontech) was generously provided by John Petrini (University of California, San Francisco). The fluorescence observed with pEYFP-C1 is equivalent to that from pEGFP-C1. The L. infantum gene LiMRE11WT was cloned in pEGFP-C1 plasmid (Clontech, encoding a GFP tag located at the N-terminus) for FRAP analysis.
DNA binding assays
DNA substrates were made by the annealing of the 32P-labelled primer 21 with either primer 17 for double-stranded DNA substrate (DS) or primer 20 for splayed arm (SA) (Table S1). Reactions (10 µL) contained 25 nM of 32P-labeled DNA oligonucleotides with the indicated concentration of proteins (see Figure 2) in MOPS buffer (25 mM MOPS (morpholine-propanesulfonic acid) pH 7.0, 0,2% tween-20, 2 mM CaCl2 and 2 mM DTT). After 15 minutes of incubation at 37°C, reactions were fixed at 37°C during 15 minutes with 0.2% glutaraldehyde. Samples were loaded onto a 8% TBE 1× acrylamide gel, run at 150 V for 1h30, followed by autoradiography.
Exonuclease assays
Exonuclease assays were performed in MOPS/EXO buffer (25 mM MOPS (morpholine-propanesulfonic acid) pH 7.0, 60 mM KCl, 0.2% tween-20, 2 mM DTT, 2 mM ATP, 5 mM MnCl2). Double-stranded DNA substrate (DS) was generated as stated above. The indicated concentration of proteins (see Figure 3) were incubated in MOPS/EXO buffer with 200 nM of 32P-labeled DNA for 30 minutes at 37°C, followed by deproteinization in one-fifth volume of stop buffer (20 mM Tris-Cl pH 7.5 and 2 mg/mL proteinase K) for 30 minutes at 37°C. The reactions were boiled during 5 minutes at 95°C after the addition of formamide blue (50% final) then loaded on 8% acrylamide/urea gels. Gels were run at 75W for 60 minutes, dried onto DE81 filter paper, followed by autoradiography. For exonuclease assay on different DNA substrates, 32P-labeled oligonucleotide 21 (Table S1) was labeled at the 5′-end (using the terminal transferase and the New England Biolabs protocol) and hybridized to primers 17, 18 and 19 (Table S1).
FRAP analysis
ATLD human cells (kindly obtained from Yossi Shiloh, University of Tel Aviv, Israël) were maintained in DMEM medium supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin (Life Technologies). ATLD cells were transfected by electroporation with 50 µg of LiMRE11-GFP or hMRE11-GFP DNA constructs. After 16 hours, we performed Fluorescence recovery after photobleaching (FRAP) analysis. Briefly, fluorescence was monitored on a Leica TCS SP5 II confocal microscope and laser-induced DNA damage was created using a 405-nm UV laser. Visualization of GFP fluorescence within the micro-irradiated nuclear region was achieved using a 488 nm excitation filter and a 63× objective. Background and photo-bleaching corrections were applied to each dataset using the Volocity-software.
Generation of LiMRE11 (LinJ27.1790) null mutant cells
The L. infantum MRE11 null mutant (MRE11−/−) cells were obtained by targeted gene replacement. MRE11 flanking regions were amplified from L. infantum wild-type genomic DNA and fused to both neomycin phosphotransferase (NEO) and hygromycin phosphotransferase (HYG) genes using a PCR fusion based-method as described previously [66]. Briefly, 5′UTR of MRE11 was amplified using primers 3 and 4 for the NEO cassette and primers 3 and 5 for the HYG cassette. The NEO gene was amplified with primers 7 and 10 and the HYG gene with primers 8 and 11. 3′UTR of MRE11 was amplified using primers 13 and 14 for both inactivation cassettes (see primer sequences in Table S1). At least 3 µg of the 5′UTR-NEO-3′UTR and 5′UTR-HYG-3′UTR linear fragments were successively transfected by electroporation (as described in [67]) into L. infantum WT to replace both MRE11 alleles. Recombinants were selected in the presence of 300 µg/ml of hygromycin B (New England Biolabs, Beverly, MA, USA) and 40 µg/ml of G418 (Geneticin; Sigma-Aldrich). After 4–5 passages, cells resistant to the drug selection were cloned in SDM-Agar plates (1%) in the presence of antibiotics at the same concentrations. Ten clones of each recombinant were picked up after 10 days and used for further analysis.
Re-expression of MRE11 in null mutant cells
A re-expression cassette, 5′UTR-MRE11-α-PUR-3′UTR was designed to reintroduce MRE11 into its original chromosomal locus in the HYG/NEO MRE11−/− null mutant. Briefly, this cassette was obtained by PCR of the PUR gene using primers 9 and 12 on the plasmid template Psp72-α-PUR-α [68] encoding the puromycin acetyltransferase marker. This fragment was fused to the 5′UTR and coding sequences of MRE11 (amplified using primers 3 and 6) and 3′UTR fragments (amplified using primers 13 and 14 in Table S1). The cassette was then transfected by electroporation in the L. infantum HYG/NEO MRE11−/− parasites to replace the NEO allele and recombinants were selected with 100 µg/ml of puromycin (Sigma–Aldrich) and 300 µg/ml of hygromycin B (New England Biolabs). The same strategy was used to introduce MRE11 containing the mutation H210Y in the HYG/NEO MRE11−/− strain. The MRE11 ORF was also cloned in the episomal plasmid Psp72-α-puro-α, transfected in L. infantum WT and HYG/NEO MRE11−/− parasites, and stable transfectants were selected with 100 µg/ml of puromycin.
Southern blot analyses
MRE11 allele replacement was confirmed by Southern blot analyses. Genomic DNAs from clones were isolated using DNAzol as recommended by the manufacturer (Invitrogen). Digested genomic DNAs or separated chromosomes were subjected to Southern blot hybridization with [α-32P]dCTP-labeled DNA probes according to standard protocols [69]. All probes were obtained by PCR (see primers in Table S1) from L. infantum genomic DNAs.
Quantitative real-time RT-PCR
RNAs were extracted using RNeasy plus mini kit (Sigma) according to the manufacturer recommendations. The cDNA was synthesized using Oligo dT12–18 and SuperScript II RNase H-Reverse Transcriptase (Invitrogen) and amplified in SYBR Green Supermix (Bio-Rad) using a rotator thermocycler Rotor Gene (RG 3000, Corbett Research). The expression level was derived from three technical and three biological replicates and was normalized to constitutively expressed mRNA encoding glyceraldehyde-3-phosphate dehygrogenase (GAPDH, LinJ.36.2480). The sequences of the primers used in this assay are listed in Table S1.
Methylmethane sulfonate (MMS) assays
L. infantum WT, HYG/NEO MRE11−/−, MRE11 re-expressing cells (HYG/PUR-MRE11WT and HYG/PUR-MRE11H210Y), WT Psp72-α-puro-α-MRE11 and HYG/NEO MRE11−/− Psp72-α-puro-α-MRE11 were resuspended at a concentration of 5×106 cells/ml and exposed to increasing doses of MMS (Sigma–Aldrich). Cells were counted after 72 h and reported in survival rate.
Pulsed-field gel electrophoresis
Intact chromosomes were prepared from L. infantum promastigotes harvested from log phase cultures, washed once in 1× Hepes-NaCl buffer then lysed in situ in 1% low melting agarose plugs. Briefly, cells were resuspended in HEPES-NaCl buffer at a density of 5×107 cells/ml and mixed with an equal volume of low melting-point agarose (Invitrogen). Cells were then lysed overnight at 50°C in lysis buffer (0.5M ethylenediaminetetraacetic acid (EDTA) pH 9.5, 1% sodium dodecyl sulfate (SDS), 350 ug/ml proteinase K). Leishmania intact chromosomes were separated in 1× TBE buffer (from 10× TBE: 1M Tris, 1M Acid boric, 0,02M EDTA) by Pulsed-Field Gel Electrophoresis (PFGE) using a Bio-Rad CHEF-DR III apparatus at 5 V/cm and a 120°C separation angle as described previously [70]. The range of chromosome separation was between 150 and 1500 kb.
Bioinformatics analyses and primer design
Repeated intergenic sequences were already characterized [19]. Primers (see Table S1) used to detect new junctions created by amplicon formation (Figure 1) were designed for all putative recombination/annealing events between repeated sequences. Primers were chosen within 150 nucleotides from the repeated sequences with their orientation shown in Figure 1. Optimal primer length was 20 nucleotides and optimal melting temperature (Tm) was 64°C.
DNA preparation for semi quantitative qPCR assays
Late log phase promastigotes (30 ml) were pelleted at 3000 rpm for 5 minutes and pellets were washed once with 1× HEPES-NaCl, resuspended in suspension buffer (100 mM EDTA, 100 mM NaCl, 10 mM Tris pH 8.0), then lysed in 1% SDS and 50 µg/ml proteinase K at 37°C for 2 hours. Genomic DNAs were extracted with 1 volume phenol, precipitated with 2 volume 99% ethanol, washed with 70% ethanol twice and dissolved in 1 ml 1× TE buffer. RNAse A (Qiagen) was added at 20 µg/ml and DNAs were incubated at 37°C for 30 minutes, followed by the addition of 50 µg/ml of proteinase K and 0.1% SDS at 37°C for 30 minutes. DNAs were extracted with 1 volume of phenol, precipitated and washed in ethanol, and dissolved in DNase free-water (Millipore) at 37°C overnight.
PCR reaction mixtures consisted of 100 ng of genomic DNA isolated as described above, 1 µl of forward and reverse primers at 100 µM (Table S1), 0.5 µl dNTP mix at 10 mM, 1.25 U of FastStart Taq DNA polymerase (Roche), 1× PCR buffer+MgCl2 and 1.25 µl BSA at 66 mg/ml. The total reaction mixture was made up to 25 µl by addition of the genomic DNA. For each PCR reaction, the number of cycles was optimized to prevent saturation of the amplification. Saturation of band intensities of the amplified PCR products was determined using the AlphaImager 2000 software. The housekeeping chromosomal gene GAPDH (LinJ36.2480) was used as an internal control (primers pair gg' in Table S1) to normalize the amount of DNA loaded in each reaction.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HandmanE (2001) Leishmaniasis: current status of vaccine development. Clin Microbiol Rev 14 : 229–243.
2. MurrayHW, BermanJD, DaviesCR, SaraviaNG (2005) Advances in leishmaniasis. Lancet 366 : 1561–1577.
3. LiraR, SundarS, MakhariaA, KenneyR, GamA, et al. (1999) Evidence that the high incidence of treatment failures in Indian kala-azar is due to the emergence of antimony-resistant strains of Leishmania donovani. J Infect Dis 180 : 564–567.
4. SundarS, MoreDK, SinghMK, SinghVP, SharmaS, et al. (2000) Failure of pentavalent antimony in visceral leishmaniasis in India: report from the center of the Indian epidemic. Clin Infect Dis 31 : 1104–1107.
5. HadighiR, MohebaliM, BoucherP, HajjaranH, KhamesipourA, et al. (2006) Unresponsiveness to Glucantime treatment in Iranian cutaneous leishmaniasis due to drug-resistant Leishmania tropica parasites. PLoS Med 3: e162.
6. RojasR, ValderramaL, ValderramaM, VaronaMX, OuelletteM, et al. (2006) Resistance to antimony and treatment failure in human Leishmania (Viannia) infection. J Infect Dis 193 : 1375–1383.
7. TorresDC, AdauiV, Ribeiro-AlvesM, RomeroGA, ArevaloJ, et al. (2010) Targeted gene expression profiling in Leishmania braziliensis and Leishmania guyanensis parasites isolated from Brazilian patients with different antimonial treatment outcomes. Infect Genet Evol 10 : 727–733.
8. CampbellDA, ThomasS, SturmNR (2003) Transcription in kinetoplastid protozoa: why be normal? Microbes Infect 5 : 1231–1240.
9. HaileS, PapadopoulouB (2007) Developmental regulation of gene expression in trypanosomatid parasitic protozoa. Curr Opin Microbiol 10 : 569–577.
10. Martinez-CalvilloS, Vizuet-de-RuedaJC, Florencio-MartinezLE, Manning-CelaRG, Figueroa-AnguloEE (2010) Gene expression in trypanosomatid parasites. J Biomed Biotechnol 2010 : 525241.
11. BeverleySM (1991) Gene amplification in Leishmania. Annu Rev Microbiol 45 : 417–444.
12. BorstP, OuelletteM (1995) New mechanisms of drug resistance in parasitic protozoa. Annu Rev Microbiol 49 : 427–460.
13. GrondinK, KundigC, RoyG, OuelletteM (1998) Linear amplicons as precursors of amplified circles in methotrexate-resistant Leishmania tarentolae. Nucleic Acids Res 26 : 3372–3378.
14. UbedaJM, LegareD, RaymondF, OuameurAA, BoisvertS, et al. (2008) Modulation of gene expression in drug resistant Leishmania is associated with gene amplification, gene deletion and chromosome aneuploidy. Genome Biol 9: R115.
15. LeprohonP, LegareD, RaymondF, MadoreE, HardimanG, et al. (2009) Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res 37 : 1387–1399.
16. DowningT, ImamuraH, DecuypereS, ClarkTG, CoombsGH, et al. (2011) Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res 21 : 2143–2156.
17. OuelletteM, HettemaE, WustD, Fase-FowlerF, BorstP (1991) Direct and inverted DNA repeats associated with P-glycoprotein gene amplification in drug resistant Leishmania. EMBO J 10 : 1009–1016.
18. GrondinK, RoyG, OuelletteM (1996) Formation of extrachromosomal circular amplicons with direct or inverted duplications in drug-resistant Leishmania tarentolae. Mol Cell Biol 16 : 3587–3595.
19. UbedaJM, RaymondF, MukherjeeA, PlourdeM, GingrasH, et al. (2014) Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite leishmania. PLoS Biol 12: e1001868.
20. HaimeurA, BrochuC, GenestP, PapadopoulouB, OuelletteM (2000) Amplification of the ABC transporter gene PGPA and increased trypanothione levels in potassium antimonyl tartrate (SbIII) resistant Leishmania tarentolae. Mol Biochem Parasitol 108 : 131–135.
21. CoderreJA, BeverleySM, SchimkeRT, SantiDV (1983) Overproduction of a bifunctional thymidylate synthetase-dihydrofolate reductase and DNA amplification in methotrexate-resistant Leishmania tropica. Proc Natl Acad Sci U S A 80 : 2132–2136.
22. BeverleySM, CoderreJA, SantiDV, SchimkeRT (1984) Unstable DNA amplifications in methotrexate-resistant Leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38 : 431–439.
23. KundigC, LeblancE, PapadopoulouB, OuelletteM (1999) Role of the locus and of the resistance gene on gene amplification frequency in methotrexate resistant Leishmania tarentolae. Nucleic Acids Res 27 : 3653–3659.
24. BelloAR, NareB, FreedmanD, HardyL, BeverleySM (1994) PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc Natl Acad Sci U S A 91 : 11442–11446.
25. WangJ, LeblancE, ChangCF, PapadopoulouB, BrayT, et al. (1997) Pterin and folate reduction by the Leishmania tarentolae H locus short-chain dehydrogenase/reductase PTR1. Arch Biochem Biophys 342 : 197–202.
26. HightowerRC, Ruiz-PerezLM, WongML, SantiDV (1988) Extrachromosomal elements in the lower eukaryote Leishmania. J Biol Chem 263 : 16970–16976.
27. WhiteTC, Fase-FowlerF, van LuenenH, CalafatJ, BorstP (1988) The H circles of Leishmania tarentolae are a unique amplifiable system of oligomeric DNAs associated with drug resistance. J Biol Chem 263 : 16977–16983.
28. PapadopoulouB, RoyG, OuelletteM (1993) Frequent amplification of a short chain dehydrogenase gene as part of circular and linear amplicons in methotrexate resistant Leishmania. Nucleic Acids Res 21 : 4305–4312.
29. ChiqueroMJ, OlmoA, NavarroP, Ruiz-PerezLM, CastanysS, et al. (1994) Amplification of the H locus in Leishmania infantum. Biochim Biophys Acta 1227 : 188–194.
30. ButlerDK, YasudaLE, YaoMC (1995) An intramolecular recombination mechanism for the formation of the rRNA gene palindrome of Tetrahymena thermophila. Mol Cell Biol 15 : 7117–7126.
31. OkunoY, HahnPJ, GilbertDM (2004) Structure of a palindromic amplicon junction implicates microhomology-mediated end joining as a mechanism of sister chromatid fusion during gene amplification. Nucleic Acids Res 32 : 749–756.
32. VanHulleK, LemoineFJ, NarayananV, DowningB, HullK, et al. (2007) Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol 27 : 2601–2614.
33. RosenbergSM, SheeC, FrischRL, HastingsPJ (2012) Stress-induced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. Bioessays 34 : 885–892.
34. LinCT, LinWH, LyuYL, Whang-PengJ (2001) Inverted repeats as genetic elements for promoting DNA inverted duplication: implications in gene amplification. Nucleic Acids Res 29 : 3529–3538.
35. RattrayAJ, McGillCB, ShaferBK, StrathernJN (2001) Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics 158 : 109–122.
36. TanakaH, YaoMC (2009) Palindromic gene amplification–an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer 9 : 216–224.
37. AssenmacherN, HopfnerKP (2004) MRE11/RAD50/NBS1: complex activities. Chromosoma 113 : 157–166.
38. StrackerTH, PetriniJH (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12 : 90–103.
39. ShibataA, MoianiD, ArvaiAS, PerryJ, HardingSM, et al. (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53 : 7–18.
40. MimitouEP, SymingtonLS (2011) DNA end resection—unraveling the tail. DNA Repair (Amst) 10 : 344–348.
41. GenoisMM, PaquetER, LaffitteMC, MaityR, RodrigueA, et al. (2014) DNA repair pathways in trypanosomatids: from DNA repair to drug resistance. Microbiol Mol Biol Rev 78 : 40–73.
42. PaullTT, GellertM (2000) A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc Natl Acad Sci U S A 97 : 6409–6414.
43. StewartGS, MaserRS, StankovicT, BressanDA, KaplanMI, et al. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99 : 577–587.
44. DolganovGM, MaserRS, NovikovA, TostoL, ChongS, et al. (1996) Human Rad50 is physically associated with human Mre11: identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol Cell Biol 16 : 4832–4841.
45. BressanDA, OlivaresHA, NelmsBE, PetriniJH (1998) Alteration of N-terminal phosphoesterase signature motifs inactivates Saccharomyces cerevisiae Mre11. Genetics 150 : 591–600.
46. StraussB, WahlR (1964) The presence of breaks in the deoxyribonucleic acid of Bacillus subtilis treated in vivo with the alkylating agent, methylmethanesulfonate. Biochimica et Biophysica Acta (BBA)-Specialized Section on Nucleic Acids and Related Subjects 80 : 116–126.
47. NareB, LubaJ, HardyLW, BeverleyS (1997) New approaches to Leishmania chemotherapy: pteridine reductase 1 (PTR1) as a target and modulator of antifolate sensitivity. Parasitology 114 Suppl: S101–110.
48. OuelletteM, DrummelsmithJ, El-FadiliA, KundigC, RichardD, et al. (2002) Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int J Parasitol 32 : 385–398.
49. McCullochR, BarryJD (1999) A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev 13 : 2875–2888.
50. GenoisMM, MukherjeeA, UbedaJM, BuissonR, PaquetE, et al. (2012) Interactions between BRCA2 and RAD51 for promoting homologous recombination in Leishmania infantum. Nucleic Acids Res 40 : 6570–6584.
51. WenQ, ScorahJ, PhearG, RodgersG, RodgersS, et al. (2008) A mutant allele of MRE11 found in mismatch repair-deficient tumor cells suppresses the cellular response to DNA replication fork stress in a dominant negative manner. Mol Biol Cell 19 : 1693–1705.
52. LeeJH, GhirlandoR, BhaskaraV, HoffmeyerMR, GuJ, et al. (2003) Regulation of Mre11/Rad50 by Nbs1: effects on nucleotide-dependent DNA binding and association with ataxia-telangiectasia-like disorder mutant complexes. J Biol Chem 278 : 45171–45181.
53. YuZ, VogelG, CoulombeY, DubeauD, SpehalskiE, et al. (2012) The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res 22 : 305–320.
54. RobinsonNP, McCullochR, ConwayC, BrowittA, BarryJD (2002) Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J Biol Chem 277 : 26185–26193.
55. TanKS, LealST, CrossGA (2002) Trypanosoma brucei MRE11 is non-essential but influences growth, homologous recombination and DNA double-strand break repair. Mol Biochem Parasitol 125 : 11–21.
56. HopfnerKP, KarcherA, CraigL, WooTT, CarneyJP, et al. (2001) Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 105 : 473–485.
57. OlsonE, NieveraCJ, LiuE, LeeAY, ChenL, et al. (2007) The Mre11 complex mediates the S-phase checkpoint through an interaction with replication protein A. Mol Cell Biol 27 : 6053–6067.
58. LobachevKS, GordeninDA, ResnickMA (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108 : 183–193.
59. ZhangY, SainiN, ShengZ, LobachevKS (2013) Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genet 9: e1003979.
60. ButlerDK, YasudaLE, YaoMC (1996) Induction of large DNA palindrome formation in yeast: implications for gene amplification and genome stability in eukaryotes. Cell 87 : 1115–1122.
61. BrewerBJ, PayenC, RaghuramanMK, DunhamMJ (2011) Origin-dependent inverted-repeat amplification: a replication-based model for generating palindromic amplicons. PLoS Genet 7: e1002016.
62. LamarcheBJ, OrazioNI, WeitzmanMD (2010) The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 584 : 3682–3695.
63. KroghBO, LlorenteB, LamA, SymingtonLS (2005) Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics 171 : 1561–1570.
64. MaityR, PautyJ, KrietschJ, BuissonR, GenoisMM, et al. (2013) GST-His purification: a two-step affinity purification protocol yielding full-length purified proteins. J Vis Exp e50320.
65. DeryU, CoulombeY, RodrigueA, StasiakA, RichardS, et al. (2008) A glycine-arginine domain in control of the human MRE11 DNA repair protein. Mol Cell Biol 28 : 3058–3069.
66. MoreiraW, LeblancE, OuelletteM (2009) The role of reduced pterins in resistance to reactive oxygen and nitrogen intermediates in the protozoan parasite Leishmania. Free Radic Biol Med 46 : 367–375.
67. PapadopoulouB, RoyG, OuelletteM (1992) A novel antifolate resistance gene on the amplified H circle of Leishmania. EMBO J 11 : 3601–3608.
68. El FadiliA, KundigC, OuelletteM (2002) Characterization of the folylpolyglutamate synthetase gene and polyglutamylation of folates in the protozoan parasite Leishmania. Mol Biochem Parasitol 124 : 63–71.
69. SambrookJ, FritschEF, ManiatisT (1989) Molecular cloning: Cold spring harbor laboratory press New York.
70. DumasC, OuelletteM, TovarJ, CunninghamML, FairlambAH, et al. (1997) Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J 16 : 2590–2598.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Tetraspanin (TSP-17) Protects Dopaminergic Neurons against 6-OHDA-Induced Neurodegeneration in
- Maf1 Is a Novel Target of PTEN and PI3K Signaling That Negatively Regulates Oncogenesis and Lipid Metabolism
- The IKAROS Interaction with a Complex Including Chromatin Remodeling and Transcription Elongation Activities Is Required for Hematopoiesis
- Echoes of the Past: Hereditarianism and
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy