
Extensive Copy-Number Variation of Young Genes across Stickleback Populations
After a locus is duplicated in a genome, individuals from a population instantaneously differ in the number of copies of this locus producing a copy-number variation (CNV). Over time, the joint effects of selection and other evolutionary forces will act to either eliminate the extra genetic copy or retain it. Depending on this evolutionary interplay, young duplications, including newly duplicated genes, can persist for millions of years as CNVs. CNVs may especially be prevalent between populations that have colonized and adapted to disparate environments in which selective pressures differ. Using whole genome sequences from several populations of three-spined sticklebacks that inhabit different environments, we find that a third of young duplicated genes are CNVs. These young CNV genes are enriched with environmental response functions and evolving rapidly at the molecular level, making them promising candidates for a role in the rapid ecological adaptation to novel environments.
Published in the journal:
. PLoS Genet 10(12): e32767. doi:10.1371/journal.pgen.1004830
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004830
Summary
After a locus is duplicated in a genome, individuals from a population instantaneously differ in the number of copies of this locus producing a copy-number variation (CNV). Over time, the joint effects of selection and other evolutionary forces will act to either eliminate the extra genetic copy or retain it. Depending on this evolutionary interplay, young duplications, including newly duplicated genes, can persist for millions of years as CNVs. CNVs may especially be prevalent between populations that have colonized and adapted to disparate environments in which selective pressures differ. Using whole genome sequences from several populations of three-spined sticklebacks that inhabit different environments, we find that a third of young duplicated genes are CNVs. These young CNV genes are enriched with environmental response functions and evolving rapidly at the molecular level, making them promising candidates for a role in the rapid ecological adaptation to novel environments.
Introduction
Structural polymorphisms such as copy-number variations (CNVs) epitomize the dynamic nature of genomes. Inter - and intra-specific comparisons of whole genomes have revealed large genomic portions deleted and duplicated between individuals [1]–[7]. The substantial contribution of these CNVs to genetic diversity is fuelled by their high mutation rates, which have been estimated to be orders of magnitude greater than that of single nucleotide polymorphisms in mutation accumulation lines [8]–[12]. Although most deletions and duplications are thought to be under purifying selection and eventually eliminated from genomes [11], [12], high gene duplication rates provide ample opportunities for functional diversification and adaptation given the right ecological circumstances [13]–[16]. In this study, we report genomic CNVs across natural populations inhabiting distinct ecological niches, their evolutionary dynamics, and their putative role in local adaptation.
When a gene is initially duplicated, it appears as a CNV at the population level. That is, the duplication event occurs in one individual genome within the population, producing a locus that varies in quantity (copy-number) amongst individuals. Under neutrality, this early copy-number polymorphic stage of a new duplicate gene can persist for millions of years before fixation or loss in a population [17]. But the ultimate probability of (and time to) fixation depends on numerous factors including mutation rates, effective population size, and natural selection. As a small subset of CNVs may eventually give rise to new genes [18]–[20], their evolutionary dynamics can give insights into the earliest life stages of genes. New genes provide a platform for the evolution of novel functions [21]–[24], and their persistence may be associated with environmental adaptations [25]–[27]. Exposure to distinct environments thus sets the stage for differential gene loss and fixation, potentially reflecting local adaptation, and culminating in varying gene content between populations. Here we use population genomics to access the proportion of young genes that are CNVs across populations from different habitats.
The three-spined stickleback (Gasterosteus aculeatus) provides an excellent opportunity to evaluate the frequency, distribution and functional impact of CNVs and young genes as a response to local environmental conditions. Sticklebacks have repeatedly colonized and adapted to various freshwater habitats since the last glaciation period (approximately 12,000 years ago), making this fish an important evolutionary and ecological vertebrate model [28]–[30]. The ability of sticklebacks to thrive in several distinct environments and the associated recurrence of ecotypes and rapid adaptations may to some extent rely on ancestral variation and the differential sorting of CNVs and young genes among populations. Using 66 whole genomes, we have extended earlier scans of polymorphisms in sticklebacks [7], [30]–[33] to evaluate CNVs across populations. Our sampling design of replicated lake-river population pairs that have diversified post-glacially allows us to estimate CNV frequencies as well as gene gain and loss across individuals from the same population, from neighboring populations in distinct habitats, and from populations that have diverged at various time scales. The combination of population-level approaches with comparative genomics enables us to evaluate the dynamic evolution of young lineage-specific genes in stickleback genomes.
Results
Genome-wide copy-number variation in sticklebacks
Three-spined sticklebacks were sampled from eleven populations across Europe and North America (herein referred to as “Atlantic” and “Pacific”) including lake, river/stream and marine populations (Fig. 1A). This sampling design allows for both an assessment of large-scale distribution of CNVs as a function of geographic proximity, and of the putative adaptive role of CNVs in two recently derived ecotypes from lakes and rivers. Whole genomes of 66 fish, six individuals from each population, were sequenced using two paired-end libraries (100 bp reads with 140 bp and 300 bp insert sizes) and a mate-pair library (50 bp reads with a 3 kb insert size), achieving an average depth of 26 fold and covering over 99% of the reference genome (Table 1 and S1 Table, study accession number ERP004574).


CNVs were inferred based on three complementary approaches: depth of coverage, discordant paired-end mapping, and split-reads in comparison with the reference genome (see Methods). We found that the combined 66 stickleback genomes contained 758 duplications (mean length of 20,373 bp) and 3,550 deletions (mean length of 7,189 bp). After merging overlapping duplications and deletions we delineated a total of 3,898 CNV regions covering 36.3 Mbp (∼8% of the genome, mean length of 9,310 bp). The pattern of CNV sharing across individuals follows the geographic and phylogenomic distributions (Fig. 1B-C and S1 Figure). Based on independent PCR validation of CNVs, we confirmed the presence of 96% of the tested CNV loci (7/7 deletions and 15/16 duplications) and recovered 88% (284/321) concordant genotypes among these CNV loci (S13 Table and S2 Figure).
Most CNVs are at low frequencies
We first address the question of how CNVs are distributed among individuals and across populations using both the allele frequency spectrum and CNV presence/absence per individual. Allelic frequencies were analyzed using genotypes inferred at bi-allelic sites, constituting 38% of all CNVs (S2 Table). CNVs generally occur at very low frequencies across all individuals (Fig. 2A) and are rarely “fixed” (all homozygous) within populations (Fig. 2B); 72% of bi-allelic CNVs are found at overall frequencies below 0.05, wherein about half of all bi-allelic duplications are singletons found in a single individual (S3A Figure). Overall, CNVs are maintained at lower frequencies than intergenic SNPs (S3B Figure) and although some CNVs are shared among multiple populations (Fig. 2C), they are often heterozygous with a high abundance of singleton alleles. We also found that CNVs fully overlapping genes have higher allele frequencies and are found in more individuals than other CNVs (S4 and S5 Figures). An excess of low frequency variants can be caused by selection, but demographic processes (bottlenecks and population expansions) and population structure can also elicit similar patterns [34]. Given our limited sample size per population, we used our whole dataset to get more reliable variant frequency estimates, accepting the existence of an underlying population structure. However, since we compare CNVs with intergenic SNPs called form the same dataset, both types of variants have experienced the same demographic history. To evaluate the potential influence of selection on the frequencies of CNVs versus SNPs, we estimated the scaled coefficient of natural selection γ and 95% confidence intervals (CI) using the Poisson Random Field approach implemented in the program prfreq [35]. Whereas intergenic SNPs appear to be near neutral (γ = 0.2, CI 0.1 to 0.3), purifying selection may be shaping the distribution of both deletions (γ = −2.2, CI −2.0 to −2.5) and duplications (γ = −5.4 CI −4.6 to −6.8). Taken together, CNVs appear to be for the most part deleterious compared to intergenic SNPs based on the allele frequency spectrum.

Although most CNVs are at low frequencies, a few bi-allelic CNVs (1%) are fixed within populations (Fig. 2B), and a total of 370 CNVs (9%) are present at least in a heterozygous state in all six individuals from at least one population (Fig. 2D). Most of these CNVs are shared across multiple populations suggesting they are of ancestral origin, although five are specific to a population and ten are specific to a group of neighboring populations. The aforementioned private CNVs intersect ten genes in total, including a G-protein signaling modulator (SGSM3), calmegin (CLGN) - a gene associated with spermatogenesis in mammals, an enzymatic gene (B4GALNT2) that is duplicated in marine individuals, and transposable element (POGK) members that are deleted in Atlantic individuals (S21 Figure). Other private CNVs intersect non-coding regions such as a fixed (homozygous) deletion upstream of an opsin gene (TMTOPSA) in a German river (G1_R) population (S3 Table). In general though, CNVs are copy-number polymorphic within populations (Fig. 2D).
Given our sampling design, we were able to evaluate the extent of CNV sharing between populations with different divergence times. This revealed that 50% of CNVs are population specific in that we only detect them in a single population, and another 25% are shared across continents (Fig. 2E). Despite generally low frequency estimates, many CNVs are shared across populations either due to gene flow, incomplete lineage sorting from ancestral polymorphisms pre-dating population divergence, or recurrent mutations. The extent of CNV sharing between individuals follows patterns of common ancestry that decreases with geographic distance (Fig. 2F and S6 Figure).
Substantial differences in gene content between individuals
To evaluate the effect of CNVs on the emergence, duplication and loss of genes, we focus the rest of our analyses on the genes encompassed in CNV regions, herein referred to as “CNV genes”. Both deletion CNVs and duplication CNVs harbor genes, but despite being more abundant and covering more nucleotides, deletion CNVs were preferentially found in non-coding regions compared to duplications (χ2 with Yates correction, p<0.0001) even after correcting for CNV length (S1 Text); only 7% of deletion CNVs overlap entire genes compared with 33% of duplication CNVs (Fig. 3A). In total, we found 1,016 protein-coding genes (5%) and 174 RNA genes (11%) that are autosomal CNV genes (S4 Table and S7 Figure). CNV genes mirror the distribution patterns of total CNVs; CNV genes are found at low frequencies, are generally population specific, and are more often shared between individuals from adjacent populations (S8–S12 Figures).

A subset of CNV genes (86 protein-coding, 7 RNA genes and 2 pseudogenes) was identified as “gene losses”, genes that were present in some genomes but completely missing from others due to homozygous deletions (Fig. 3B and S5 Table). A detailed analysis of unmapped reads suggests that the identification of CNV genes, including gene losses, is minimally affected by a reference genome bias (S1 Text). Based on the assembly of unmapped reads we detected over 100 putative genes that are not currently assembled in the reference genome [30], however these are generally short contigs and could represent partial genes or pseudogenes (S6 Table). Based on mapped data, each individual was found to have on average 142 CNV genes (ranging from 68 to 236) and 22 gene losses (ranging from 5 to 39, S7 Table), leading to two individuals differing on average by a combined 242 CNV genes (1.1% of genes).
CNV genes are predominantly young genes
As newly duplicated genes undergo an early evolutionary stage that consists of copy-number polymorphism at the population level, we rationalized that young genes that have not reached copy-number fixation would be well represented among CNV genes. To evaluate the proportion of young genes that are copy-number fixed versus copy-number polymorphic, we first identified young genes using a comparative genomics approach based on orthology and paralogy relationships from Ensembl (Fig. 4A). Young genes included lineage-specific genes (LSGs) that are only found in sticklebacks (putative orphan genes) and lineage-specific duplications (LSDs) that have recently duplicated in sticklebacks, both of which show hallmarks of new genes (S1 Text). Concordant with our expectations, a substantial overrepresentation of young stickleback genes overlaps with CNVs (χ2 with Yates correction, p<0.0001, Fig. 4B) including gene losses (homozygous deletions; χ2 with Yates correction, p<0.0001). About a third of LSDs and 10% of LSG singletons are CNV genes (S4 Table), indicating that many young genes have not been fixed across populations since they emerged, or that recurrent mutations causing CNVs preferentially involve young genes. The CNVs in LSGs and LSDs have fewer singletons and higher allele frequencies than those in non-LSGs (Mann-Whitney test W = 4631, p = 4.672e-06), demonstrating that CNVs in young genes are not simply single events.

LSGs and LSDs are on average shorter in length than other genes (S1 Text). After correcting for gene length (see Methods), we found that protein-coding LSGs and LSDs remain overrepresented among both deletions and duplications (p<0.001) whereas non-LSGs are underrepresented (p<0.001). Similar results are also found for RNA genes (S1 Text). A comparative molecular analysis (see Methods) revealed that, like young genes, CNV genes are evolving rapidly at the sequence level, with higher dN (nonsynonymous substitutions per nonsynonymous site) compared to non-CNV genes (Mann-Whitney test W = 505097.5, p = 1.482e-06) and higher dN/dS (the ratio of nonsynonymous substitutions per nonsynonymous site versus synonymous substitutions per synonymous site; Mann-Whitney test W = 477084.5, p = 0.0004). Elevated dN and dN/dS are found for both deletion CNV genes (p = 2.742e-06 and p = 0.0003 respectively) and duplication CNV genes (p = 0.0011 and p = 0.0172 respectively) when analyzed separately. These results suggest that the early evolution of young genes involves a period of relaxed purifying selection or positive selection, which may promote their genomic persistence. Surprisingly, almost half of the autosomal genes (9/20) found to be under positive selection from the Selectome database [36], [37] are CNV genes, and all are LSDs (Table 2).

Segmental duplications, stretches greater than 1kb that are similar in sequence (>90%), are known hotspots for structural variations such as CNVs in humans [3], [4], [38], [39]. Not surprisingly, the great majority of genes in stickleback segmental duplications are young genes including 64% of all protein-coding LSDs, therefore most CNV genes in segmental duplications are also young genes (Fig. 4C and S13 Figure). As for annotated RNA genes, most (59%) reside in segmental duplications, including almost all (88%) of the 174 RNA CNV genes. CNV regions that are outside of segmental duplications are generally gene-poor and are found in fewer individuals (Mann-Whitney test W = 201332, p = 7.592e-06, S14 Figure). However, CNVs outside of segmental duplications remain enriched with LSGs and LSDs (χ2 with Yates correction, p<0.0001; S15 Figure and S4 Table). In other words, the relationship between young genes and CNVs is not solely determined by segmental duplications.
Lineage-specific genes and duplications identified by our methods may vary in age and could be as old as the most recent common ancestor of sticklebacks and other fishes. To evaluate whether CNV genes are primarily younger versus older lineage-specific genes, we calculated the number of synonymous substitutions per synonymous sites (dS) between LSD gene pairs as a proxy for their age (see Methods). We found that LSD pairs in which both are CNV genes have significantly smaller dS than LSD pairs in which only one is a CNV gene (Mann-Whitney test W = 832, p = 0.007), which in turn have significantly smaller dS than LSD pairs in which neither is a CNV gene (Mann-Whitney test W = 5155, p = 0.004). From these results we infer that younger paralogs are more likely to be CNV genes than older paralogs (Fig. 5A). Alternatively, gene conversion between paralogs (see Methods) would reduce dS making them appear younger than they are [40], [41]. However, we did not find a positive correlation between CNV genes and gene conversion, but rather a non-significant negative relationship (Fisher's exact test, p = 0.070).
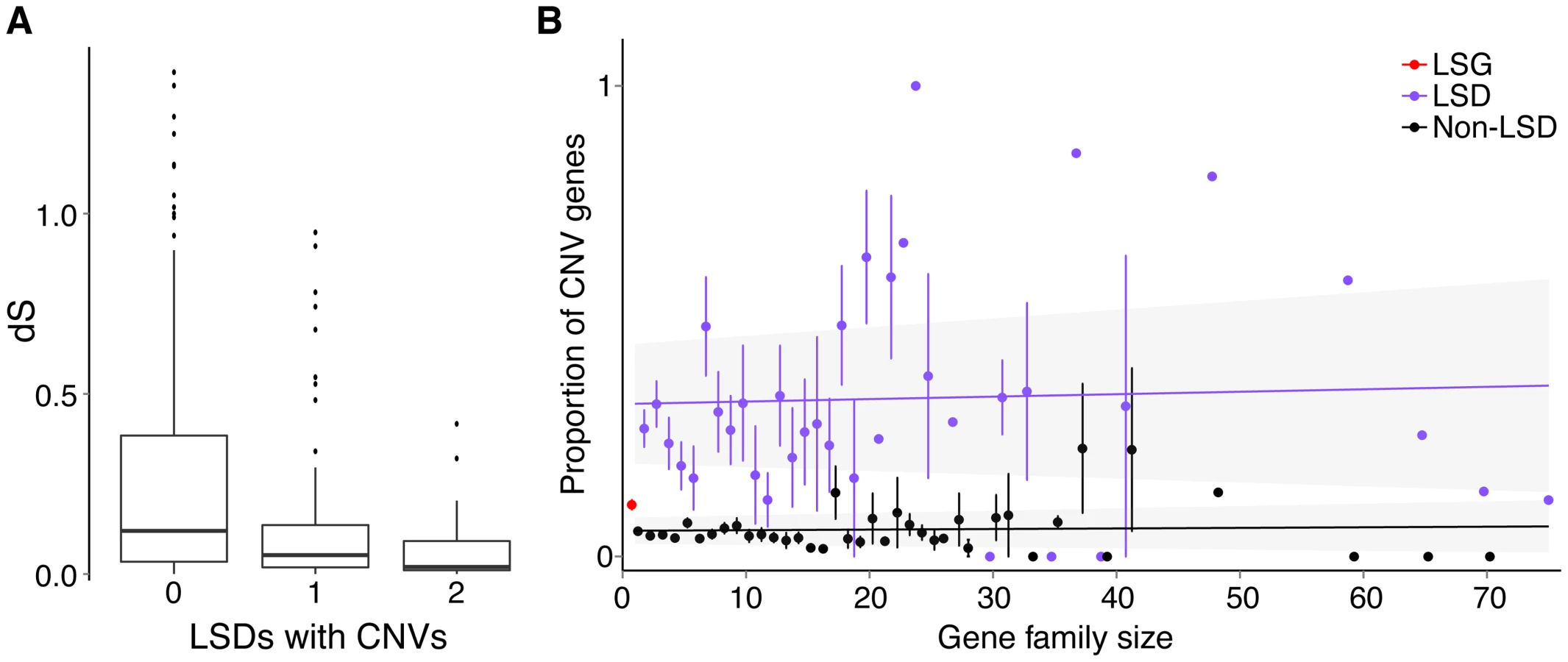
Since CNV genes are enriched with young duplicated genes, we tested for an association between the proportion of CNV genes in a gene family and family size. Here the idea was to examine whether larger gene families with many paralogs might be more volatile in terms of copy-number changes through duplications and deletions. We found that CNV genes are not significantly associated with gene family size for both LSDs (ANCOVA, p = 0.225) and non-LSDs (ANCOVA, p = 0.835), whether or not we include low-frequency (<0.05) CNVs. However, LSDs consistently make up a large proportion of CNV genes across families of all sizes (Fig. 5B). We also found that LSG singletons are significantly more often CNV genes than non-LSG singletons (Fisher's exact test, p<0.0001), emphasizing the dynamic evolution of very young duplicate genes and potentially novel genes that have arisen by mechanisms other than duplication such as de novo gene birth [20].
Ecological response of CNVs and young genes
The comparison of gene copy-numbers between populations can help identify candidate genes under positive selection. We investigated the effects of population differentiation between lake and river ecotypes using the VST statistic [4], a measure analogous to Wright's FST on allele frequency differentiation, to identify the genes and exons that have the most differentiated copy-numbers between parapatric lake-river populations. The use of VST allowed us to incorporate copy-number information from each individual and each locus, including multi-allelic sites, in the evaluation of copy-number variance between populations inhabiting different environments. High VST indicates larger variance between populations relative to the variance within each population, a pattern consistent with positive selection on copy-number in one or both populations. Although high differentiation can reveal loci under selection, this signal can also be caused by drift (especially from founder effects). Young duplicated genes (LSDs) make up half of the 14 genes with VST values above the 99.9% percentile of 0.89 (Table 3), and have higher average VST compared to older genes (0.17 versus 0.12, Mann-Whitney test p<0.0001). Similar results were found when VST was calculated across genic exons to evaluate the impact of CNVs on partially duplicated and deleted genes (S10 Table). The gene with the highest VST is a multi-allelic gene similar to the lysosomal protective protein cathepsin A (CTSA) that is duplicated in almost every German river individual compared to only one heterozygous German lake individual (Fig. 6). Cathepsins are proteases with both stabilizing and activating properties, and are involved in immune response within the MHC class II antigen presentation pathway [42], [43]. Other genes with immune functions also have extremely high VST including CMKLR1 between the G1 populations, and PYCARD between the Ca populations.


Over half of the genes in certain ecologically relevant families are CNVs, including Major Histocompatibility Complex (MHC) immune genes (18 out of 32), and groups of G-protein coupled receptor genes including olfactory receptor (OR) genes (121 out of 216) and trace amine associated receptors (TAAR – 49 out of 64). The average VST values of immune response genes (0.17) and G-protein coupled receptor genes (0.16) were both significantly higher than the average of all genes (0.14, Mann-Whitney test p = 0.0301 and p = 0.0037, respectively). TAARs had particularly high average VST (0.24). Consistent with the low amount of CNV sharing across populations, VST values across the genome were mostly not correlated across the different pairs of lake-river populations (S16 Figure), with the highest correlation being between the German groups G1 and G2 with a Pearson's r of 0.18.
Discussion
Like other mutations, most duplications and deletions are likely neutral or detrimental to fitness and the majority of duplicate genes are expected to be lost over time by purifying selection or by drift alone [17], [21]. Assuming that the limited sample sizes per population and the underlying population structure does not adversely skew our allele frequency estimates, most CNVs are present at low frequencies across several diverged populations and habitats, even lower than intergenic SNPs, suggesting that CNVs are generally experiencing purifying selection in nature. Although the majority of CNVs occur at low frequencies, a subset of CNVs was found to reach high frequencies within specific populations, some of which are highly differentiated in copy-numbers between parapatric lake-river populations. CNV genes may reach different frequencies between populations due to selection, but also drift (for example from bottlenecks and founder effects). The observation that CNVs have higher average dN and dN/dS compared to non-CNV genes, coupled with the high incidence of CNV genes in the Selectome database, suggests that a subset of CNV genes may be preserved by positive selection. The wealth of genomic CNVs as standing genetic variation [7] possibly facilitates the rapid adaptation of sticklebacks to various distinct habitats and changing ecology, e.g. infectious diseases. Examples of CNVs associated with environmental change include an expansion of an amylase gene associated with high-starch diets in humans [13], and a recurrent deletion of a gene enhancer associated with an adaptive trait in sticklebacks [44]. It is therefore possible that some genes remain copy-number polymorphic across populations due to selection after habitat or lifestyle diversification.
A quarter of CNVs are shared among distantly related populations, either due to the presence of ancient polymorphisms spanning across continents or to more recent gene flow, or due to recurrent mutations affecting the same genetic region. Recurrent CNVs appear to be common [4] and can be caused by non-allelic homologous recombination hotspots and maintained by gene conversion [45], leading to high turnover rates and the same genes appearing as CNVs across species [46], [47]. This implies that the underlying genomic architecture will provide differential opportunities across the genome for the formation and subsequent preservation of CNVs; CNVs can have higher frequencies due to recurrent mutations in recombination hotspots even if they are under strong purifying selection. However, here we report that CNV sharing is much greater between individuals from the same population or adjacent populations, suggesting that shared ancestry may explain much of the distribution patterns observed, including ancestral CNVs that are shared across continents. Given that Atlantic and Pacific individuals separated at most a few million years ago [48], some ancestral CNV genes may have been maintained even when they are slightly deleterious; genes originating via duplication can take several million years before becoming fixed or pseudogenized [17]. Taken together, we surmise that CNVs occurring across continents are probably a combination of ancestral and recurrent CNVs. In either case, CNVs may have greater opportunities for further persistence when encountering different environments, especially those affecting genes involved in environmental response, e.g. olfactory genes and MHC immune genes associated with adaptation to infectious diseases.
Focusing on the impact of CNVs on genes, we found that around 5% of protein-coding genes were completely encompassed in CNVs. There were proportionally twice as many RNA genes as protein-coding genes among CNVs, which may reflect rapid RNA gene turnover through duplications and deletions. We estimate that two individuals differ by over 200 CNV genes, double that which has been predicted in humans [49]. Whereas many of these CNV genes may eventually be lost, this finding is in line with the differing genic content observed between species due to new genes [20]. The substantial amount of genetic structural variation affecting genes reported here has consequences on the interpretations from comparative approaches that use only one reference genome from a species rather than population-wide data, for example to calculate the number of orphan genes in a species. Our results demonstrate the highly dynamic landscape of stickleback genomes and the impressive contribution of genic CNVs to biological diversity across environments in addition to sequence divergence (see companion paper submitted by Feulner et al.), supporting the idea that structural changes play an important role during population differentiation.
The earliest stage of gene evolution involves a period of presence-absence polymorphism that can be detected as a CNV. Consistent with this notion, young genes in sticklebacks were enriched with CNVs. This was also independent of gene family size, in which younger gene family members were often CNVs across a range of family sizes. Within LSD pairs, more recently duplicated genes were also more frequently associated with CNVs, similar to recent findings in humans using different methods [19]. These are strong indications that CNVs offer a way of capturing young genes during an early evolutionary stage; as much as 30% of lineage-specific paralogs in genomes may be copy-number polymorphic since their emergence. Alternatively, recurrent duplication and deletion events of genomic regions containing young genes may predispose them to be CNVs. Consistent with this latter scenario, we found that LSGs and LSDs are also enriched in segmental duplications, genomic regions that have strong relationships with structural variations in other animals [1], [3], [39], [46], [50], and that can produce raw material potentiating new genes [51], [52]. Segmental duplications may especially affect the dynamics of RNA genes, since we found a strikingly large overlap between RNA CNV genes and segmental duplications. Together these observations point to a strong association between segmental duplications, CNVs and young genes, confirmed here in natural populations spanning a range of divergence times. Although segmental duplications have been conceptualized as fixed CNVs [53], [54], these are normally annotated based on a single genome and thus a subset of segmental duplications may alternatively represent duplicated regions that are actually segregating CNVs [39]. While there is a noticeable impact of segmental duplications on CNVs, we also found a significant enrichment of CNVs among young genes outside of segmental duplications, suggesting a more complex interplay between young genes and CNVs.
Gene ontology enrichment analysis of CNV genes returned several overrepresented categories with functions involved in environmental response, similar to results in other species [50], [55], [56]. These functions include protein ubiquitination and glycosylation, G-protein coupled receptor signaling pathway and immune activity (S8 Table). The same is true after removing low frequency CNVs occurring in only one individual, as well as for genes that show large copy-number differences among individuals (genes with both deletions and duplications in different individuals). This overrepresentation is driven by LSDs, which make up a significant portion of the gene families in these categories (S9 Table). Although the overrepresentation of these functional categories may hint at an ecological role of many CNVs, it could also indicate that these gene categories are simply under relaxed purifying selection and can better tolerate copy-number fluctuations compared to other functions.
Over half of the genes in some immune and olfaction gene families are CNV genes, and on average they show high population differentiation. The differentiation and population specificity of some of these genes (S19–S22 Figures and S1 Text) may reflect important differences across habitats such as parasite resistance [57]. In sticklebacks, both MHC and olfactory genes are involved in mate choice and ecological diversification among lake and river ecotypes [58], [59]. Immune genes were among the most differentiated CNV genes between parapatric populations, including a gene resembling a lysosomal protease in the two German population pairs (Fig. 6). These same populations are known to experience different parasite communities and parasite loads [60], perhaps contributing to differentiating lysosomal proteases that play critical roles in antigen presentation by MHC genes [43]. As for olfactory receptor gene families, TAARs show particularly high population differentiation on average, and have recently expanded and diversified in teleosts while displaying evolutionary signatures of strong positive selection [61]. It is thus tempting to speculate that some TAARs have perhaps emerged and persisted through adaptations to lineage-specific odours [62], [63]. Although we initially hypothesized similar ecological selection gradients among the habitat pairs, we found that differentiated CNV genes were mostly unique to populations rather than ecotypes. Interestingly then, recurrent CNVs originating due to biased mutational mechanisms and the underlying genome architecture generally have different evolutionary trajectories in different populations, possibly due to influences such as local adaptation. However, further experimental and functional studies would be required to formally test the role of candidate CNVs, especially considering that gene ontology assignment, which may be particularly biased against young genes [64], is based on sequence similarity to genes in other vertebrates that may not reflect functions in sticklebacks.
We have combined comparative and population genomics approaches to characterize the evolution of CNVs and of young genes across several distinct environments. Our results demonstrate that most CNV genes in the genome constitute recently emerged genes – as much as a third of all lineage-specific duplications – that are not fixed after millions of years. These young genes often have annotated functions associated with environmental response, some of which potentially play a role in adaptation soon after the colonization of a new habitat. The high prevalence of genes with copy-number differences across populations highlights their contribution in shaping the diversity of stickleback genomes.
Methods
Sampling and data processing
Three-spined stickleback fish were caught from five pairs of lakes and rivers in North America and Northern Europe (more details can be found in companion paper submitted by Feulner et al.), as well as from a marine population in the North Sea [7] (S1 Table and Fig. 1A). Muscle tissue from six sampled individuals from each location (aiming for an equal sex ratio) was used for DNA extraction (using a Qiagen DNA Midi Kit following the manufacturer's protocol for high molecular weight DNA) and Illumina sequencing following our previous methods [7]. To capture natural variation present in the wild, we randomly picked individual fish for sequencing, thus without pre-selection of any particular morphological or parasitological characteristics. For each individual, two paired-end libraries (100 bp reads, average insert size of 140 bp and 300 bp) and a mate-pair library (50 bp reads, average insert gap of 3 kb) was produced, achieving an average depth of coverage of 26x (Table 1 and S1 Table). Raw sequence data was processed and filtered following previous procedures [7] and mapped against the three-spined stickleback reference genome [30] from Ensembl version 68 [65]. The use of six individuals per population was chosen to have a balance between a wide geographical range of samples, an adequate number of individuals represented per population, and sufficient depth of sequence coverage per genome to reliably call SNPs and CNVs. Raw sequence reads from the 66 genomes are accessible under the European Nucleotide Archive study accession ERP004574.
We called CNVs for each individual separately based on the combination of signals from a read depth approach (CNVnator), paired-end approach (Breakdancer and delly) and split-reads approach (Pindel). Deletions and duplications were defined as the decrease or increase of copy-number relative to the reference genome, wherein duplications in sequenced genomes could actually be deletions in the reference genome, but this occurrence may be low [6], [49]. This also means that we considered large insertions and deletions to be CNVs, even though the mechanism of formation is different from duplications. The read depth software CNVnator [66] was used to evaluate CNVs along the genome in 500 bp windows. The paired-end mapping approach implemented in Breakdancer [67] made use of paired-end data (with the longer insert size of ∼300 bp) and mate-pair libraries that were processed separately, and only using uniquely mapped reads. We kept the default cut-off for the insert size deviation, but increased the required read pairs to establish a connection to 4. Haploid sequence coverage was adjusted when calling variants on all 66 libraries combined. We also used delly and duppy [68] to infer CNVs with the default parameters. The split-reads approach implemented in Pindel [69] was used with the default settings on all 66 paired-end libraries together.
For each individual, deletions and duplications that overlapped (more than 50% of their length) within the same CNV calling approach were merged together and then compared across approaches; we only kept deletion and duplication calls from CNVnator that overlapped (more than 50% of their length) with calls from at least one other approach. In an effort to reduce the impact of the reference genome when calling CNVs, deletions and duplications found in all individuals were excluded as well as calls that overlapped (more than 50% of their length) with repeat-masked regions from the Ensembl annotations (version 68). We also removed short CNVs (<500 bp) and those found on the sex chromosome that are influenced by male hemizygosity. Deletions and duplications were then compared across individuals to evaluate CNV regions; contiguous chromosomal stretches encompassed by CNVs. All copy-number variations are reported in S11 Table. The visualization of CNV clustering based on shared variants was performed using principal components analysis as implemented by prcomp() in R [70].
Complete overlap of genes and genic regions was given a 5% leeway due to imperfect breakpoint calling in CNVnator (>95% of the gene length covered by a CNV was considered a CNV gene). Gene information was acquired using EnsemblCompara version 68 [65], [71]. Read depth averages were used to infer CNV genotypes for each gene and were retrieved via CNVnator using the default genotyping output. For each individual, gene read depth was normalized by the median and then centered around two (to represent diploids). Differences between individuals were evaluated based on this normalized read depth. Gene loss was inferred when the normalized read depth average fell below 0.25, effectively 8-fold lower coverage than expected. For each gene (or exon) loss, at least one individual had a normalized read depth average above 2 to curtail regional mapping biases. These inferences are made using the average read depth across a region, and thus ignore heterogeneity within the region. Shell and perl scripts were used with BEDTools [72] to evaluate the distribution of CNVs, overlap of gene regions, and variance in read depth between populations. We evaluated VST between parapatric lake-river population pairs for each gene following the definition in [4].
To identify CNV sharing across individuals, we required a reciprocal overlap of CNVs (more than 50% of their lengths) between individuals. For the evaluation of CNV occurrence across individuals, we tabulated presence and absence of each CNV for each individual. In addition, CNV allele frequencies (determined based on the copy-number on each chromosome) were calculated (and polarized) compared to the reference genome, such that each reference locus was considered to be in a diploid state with two single-copy alleles. Copy-number was inferred for each CNV and gene based on normalized read depth using CNVnator, in which read depth at each locus was normalized by the median across all individuals, centered around two (diploid), and rounded to the closest integer as a proxy for copy-number. We then retained all putative bi-allelic CNV loci – those loci with either two or three different genotypes that could be explained by a combination of zero deletion alleles, one deletion allele and two deletion alleles, or by a combination of zero duplication alleles, one duplication allele and two duplication alleles. In other words, genotypes were assumed to only differ by one copy per allele. Allele frequencies and genotypes were inferred from 1492 putative bi-allelic CNVs (38% of CNVs) and used to determine allele frequency spectra.
A phylogeographic analysis on the genomic data was performed using the Neighbor-net method [73] as implemented in SPLITSTREE4 [74] and using default settings, except that ambiguous states were averaged over all possible resolutions. A network is preferable over a tree because of the potential gene flow and shared ancestral polymorphisms across populations. The phylogenomic network (Fig. 1A) was created using 50,000 randomly selected high-quality single nucleotide polymorphisms (SNPs) that were outside of repeat-masked and CNV regions, as well as at least 10 bp from any indels. These same SNPs were also used to compare with the allele frequency spectrum of CNVs. After the initial processing and filtering of the raw sequencing data following the procedures stated above in [7], SNPs and indels were called with GATKv1.6 [75] using concordant SNP calls from SAMtools v0.1.18 [76] for variant recalibration. Phasing and imputation was performed with BEAGLE v3.1 [77]. VCFtools v0.1.11 [78] was utilized for processing genotypes and allele frequency spectra.
To test for the influence of selection on the allele frequency spectrum of CNVs, we used a Poisson Random Field approach implemented in prfreq [35], which evaluates the expected allele frequency distributions given different demographic and selection models. We compared the estimates of the scaled selection coefficient γ of CNVs (both deletions and duplications) with intergenic SNPs, by fitting the “single point mass” model over a range of γ (between −20 and 10). Estimates were taken modeling a stationary population, under the assumption that demographics affect intergenic SNPs and CNVs in a similar fashion.
Assembly of unmapped reads
We extracted all unmapped reads for each individual and performed a de novo assembly with Velvet [79], using a k-mer of 21 bp, a minimum contig length of 99 bp and coverage cutoff of 4 reads. The resulting contigs for each individual were then clustered and assembled using CAP3 [80] with a 97 percent identity. This returned 161,780 contigs that were made up of sequences from at least two individuals, and had an average length of 456 bp (max = 12,780 bp). BLASTX [81] was then performed on the assembly output versus the nr protein database, and hits below 1e-05 were kept as putative orthologs. BLASTX was also run on the three-spined stickleback protein database (Ensembl version 68) and BLAT was run on the three-spined stickleback reference genome (S17 Figure). Blast2GO [82] was used to annotate the hits. In total we found over 100 putative genes that were not found in the reference genome (S6 Table and S1 Text), in addition to another 100 genes absent from the Blast2GO results but annotated through BLASTX.
Gene annotations and lineage-specific genes and duplications
Gene annotations were taken from Ensembl, including protein-coding genes and RNA genes: ribosomal RNA (rRNA), micro RNA (miRNA), small nucleolar RNA (snoRNA) and small nuclear RNA (snRNA). The annotations of olfactory receptor (OR) genes were taken from multiple sources, including 122 general odorant receptors [63], six V1R genes [83], 24 V2R genes [63] and 64 TAAR genes [62]. Gene ontology (GO) terms were inferred using the Ensembl annotations (version 68), and significant enrichment of GO terms was acquired using the topGO weight algorithm and determined by FDR adjusted p-values to help correct for multiple testing [84].
We categorized genes as LSGs and LSDs using orthology and paralogy relationships from EnsemblCompara version 68 [65], [71]. BLASTX searches versus the nr database returns some hits between some of our identified LSG singletons (34%) and most LSG LSDs (74%), suggesting that these may not have strictly originated in sticklebacks but alternatively have substantially diverged from orthologs while retaining enough similarities to be recognized as potential homologs (S12 Table). Nevertheless, LSGs and LSDs have properties associated with young genes (S1 Text). It is also possible that some of these genes are on their way to pseudogenization, although we found that 48% have expression information in EST databases. Expression data was collected from the EST database on NCBI, mapped against the stickleback genome, and expression profiles were examined across gene categories. Segmental duplications were downloaded from http://humanparalogy.gs.washington.edu/stickleback/data/.
We used permutations to test for overrepresentation or underrepresentation of genes among CNVs. We permuted (n = 1000) the CNV data to randomly select across each chromosome the same number (and length) of regions as deletions and duplications. We then evaluated the overlap of genes based on our observed data versus the distribution of this random sampling. See S1 Text for detailed results.
Validation of CNV calls
We validated some CNV calls by PCR and quantitative PCR. We designed primers in and surrounding CNV calls to (1) genotype genic deletions in individuals using PCR and gel assays, and to (2) measure relative copy-numbers between individuals using qPCR. This allowed us to evaluate concordance for CNV loci, genotypes and read depth. Overall, we validated 96% (22/23) of the CNV loci, in terms of CNV presence, with lower concordance (88%) for genotypes, most of which were called heterozygotes by PCR. For the same individuals for which genome sequencing was performed, DNA was re-extracted using the DNeasy 96 Qiagen extraction kit (Hilden Germany) and adjusted the concentration to 10 ng/uL. For six deletions affecting between 3 to 42 individuals and showing clear genotype distinctions between a homozygote deletion, heterozygote deletion and no deletion in our sequencing data, we performed a gel assay in all 66 individuals scoring presence or absence based on different primer pairs; one spanning the complete CNV deletion and the other falling within the deletion. In this way, primer pairs were designed such that a positive signal in both pairs would return a heterozygote (primer sequences can be found in S13 Table). Some combinations in certain individuals gave inconclusive signals (in case one of the primer pairs failed), but in total we confirmed the 6 deletions with 88% (284/321) concordant presence/absence genotypes across individuals. Secondly, a qPCR assay was performed on 17 loci, selected to have a range of low to high CNV allele frequencies, and a range of low to high copy-numbers, to evaluate concordance of relative read depth. Using a standard housekeeping gene (ribosomal protein L13) as an internal control reference, we followed a modified version of the comparative CT method [85]–[87], in which the internal control was also used as the calibrator and the ΔCT values were used to directly compare relative copy-number between individuals. We estimated absolute copy-number assuming that the reference gene is diploid in all individuals (the CNVnator read depth across the 66 individuals ranged from 1.78 to 2.35). Concordance of read depth signal was evaluated using the Pearson correlation statistic (as implemented in R) on the relative copy-number between individuals evaluated from the read depth analysis in CNVnator versus the qPCR analysis. This returned 94% (16/17) concordant relative read depth calls after Bonferroni correction (100% with FDR q-values <0.05) and an average correlation of 0.78, including a high concordance for the gene with the highest VST (S2 Figure).
Molecular evolution
We evaluated the relationship between the age of paralog pairs and CNVs. A proxy for relative age of paralogs was calculated by first aligning LSD pairs (n = 350) using MACSE [88], and then by estimating pairwise synonymous substitutions per synonymous site (dS) using a maximum likelihood approach implemented in PAML 4.5 [89]. Genes with dS greater than 1.5 (n = 123) were excluded from analysis due to poor alignment or substitution saturation. 89% of these pairs (dS >1.5) encountered no CNVs. We performed comparative genomic analyses using sequences from nine-spined sticklebacks to evaluate molecular rates of evolution (dN, dS and dN/dS) among genes. Raw nine-spined stickleback 454 sequences from Guo et al. [90] were assembled with iAssembler v1.3.0 [91] using a strict 100% identity threshold and resulting contigs were mapped to three-spined stickleback gene transcripts from Ensembl version 68 using BLAST. Top hits with an e-value threshold of 1e-4, score above 90 and >90% percent identity were used in a sequence alignment with MACSE. The longest transcript for each gene was used as the representative gene alignment. Pairwise rates of molecular evolution were conducted using PAML 4.5, and threshold values for dN were set to a maximum of 10 and dS to a maximum of 1.0.
Evidence for gene conversion was identified using default settings in GENECONV v.1.81 [92], with the exception of listing pairwise p-values (-ListPair) and to include monomorphic sites (-Include_monosites). After excluding gene pairs with dS <0.05 due to difficulties of detecting gene conversion events with highly similar sequences [93], LSD pairs with CNVs still did not show a significant association with gene conversion (two-sided Fisher's exact test, p = 1), and still have significantly smaller dS than LSD pairs without CNV regions (Mann-Whitney, W = 2388, p = 0.0105).
Ethics statement
This study was performed according to the requirements of the German Protection of Animals Act (Tierschutzgesetz) and was approved by the 'Ministry of Energy, Agriculture, the Environment and Rural Areas' of the state of Schleswig-Holstein, Germany (reference number: V 312-72241.123-34). Wild sticklebacks were caught using minnow traps or hand nets. Before dissection the fish were anesthetized with MS222 and sacrificed by an incision into the brain followed by immediate decapitation, and every effort was made to minimize suffering. No further animal ethics committee approval was needed. The species used in this study are not endangered or protected.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. IafrateAJ, FeukL, RiveraMN, ListewnikML, DonahoePK, et al. (2004) Detection of large-scale variation in the human genome. Nature Genet 36 : 949–951 doi:10.1038/ng1416
2. WaszakSM, HasinY, ZichnerT, OlenderT, KeydarI, et al. (2010) Systematic inference of copy-number genotypes from personal genome sequencing data reveals extensive olfactory receptor gene content diversity. PLoS Comput Biol 6: e1000988 doi:10.1371/journal.pcbi.1000988
3. SebatJ, LakshmiB, TrogeJ, AlexanderJ, YoungJ, et al. (2004) Large-scale copy number polymorphism in the human genome. Science 305 : 525–528 doi:10.1126/science.1098918
4. RedonR, IshikawaS, FitchKR, FeukL, PerryGH, et al. (2006) Global variation in copy number in the human genome. Nature 444 : 444–454 doi:10.1038/nature05329
5. KorbelJO, UrbanAE, AffourtitJP, GodwinB, GrubertF, et al. (2007) Paired-end mapping reveals extensive structural variation in the human genome. Science 318 : 420–426 doi:10.1126/science.1149504
6. EmersonJJ, Cardoso-MoreiraM, BorevitzJO, LongM (2008) Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science 320 : 1629–1631 doi:10.1126/science.1158078
7. FeulnerPGD, ChainFJJ, PanchalM, EizaguirreC, KalbeM, et al. (2013) Genome-wide patterns of standing genetic variation in a marine population of three-spined sticklebacks. Mol Ecol 22 : 635–649 doi:10.1111/j.1365-294X.2012.05680.x
8. LynchM, SungW, MorrisK, CoffeyN, LandryCR, et al. (2008) A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci USA 105 : 9272–9277 doi:10.1073/pnas.0803466105
9. TurnerDJ, MirettiM, RajanD, FieglerH, CarterNP, et al. (2008) Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nature Genet 40 : 90–95 doi:10.1038/ng.2007.40
10. LipinskiKJ, FarslowJC, FitzpatrickKA, LynchM, KatjuV, et al. (2011) High spontaneous rate of gene duplication in Caenorhabditis elegans. Curr Biol 21 : 306–310 doi:10.1016/j.cub.2011.01.026
11. SchriderDR, HouleD, LynchM, HahnMW (2013) Rates and genomic consequences of spontaneous mutational events in Drosophila melanogaster. Genetics 194 : 937–954 doi:10.1534/genetics.113.151670
12. KatjuV, BergthorssonU (2013) Copy-number changes in evolution: rates, fitness effects and adaptive significance. Front Genet 4 : 273 doi:10.3389/fgene.2013.00273
13. PerryGH, DominyNJ, ClawKG, LeeAS, FieglerH, et al. (2007) Diet and the evolution of human amylase gene copy number variation. Nature Genet 39 : 1256–1260 doi:10.1038/ng2123
14. CookDE, LeeTG, GuoX, MelitoS, WangK, et al. (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338 : 1206–1209 doi:10.1126/science.1228746
15. IskowRC, GokcumenO, LeeC (2012) Exploring the role of copy number variants in human adaptation. TIG 28 : 245–257 doi:10.1016/j.tig.2012.03.002
16. KondrashovFA (2012) Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc R Soc Lond [Biol] 279 : 5048–5057 doi:10.1098/rspb.2012.1108
17. LynchM, ConeryJS (2000) The evolutionary fate and consequences of duplicate genes. Science 290 : 1151–1155.
18. KorbelJO, KimPM, ChenX, UrbanAE, WeissmanS, et al. (2008) The current excitement about copy-number variation: how it relates to gene duplications and protein families. Current Opinion in Structural Biology 18 : 366–374 doi:10.1016/j.sbi.2008.02.005
19. JuanD, RicoD, Marques-BonetT, Fernandez-CapetilloO, ValenciaA (2013) Late-replicating CNVs as a source of new genes. Biology Open. doi:10.1242/bio.20136924
20. LongM, VanKurenNW, ChenS, VibranovskiMD (2013) New gene evolution: little did we know. Annu Rev Genet 47 : 307–333 doi:10.1146/annurev-genet-111212-133301
21. Ohno S (1970) Evolution by gene duplication. Springer-Verlag. 1 pp.
22. Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press.1 pp.
23. LongM, BetránE, ThorntonK, WangW (2003) The origin of new genes: glimpses from the young and old. Nat Rev Genet 4 : 865–875 doi:10.1038/nrg1204
24. ConantGC, WolfeKH (2008) Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet 9 : 938–950 doi:10.1038/nrg2482
25. KhalturinK, HemmrichG, FrauneS, AugustinR, BoschTCG (2009) More than just orphans: are taxonomically-restricted genes important in evolution? TIG 25 : 404–413 doi:10.1016/j.tig.2009.07.006
26. ColbourneJK, PfrenderME, GilbertD, ThomasWK, TuckerA, et al. (2011) The ecoresponsive genome of Daphnia pulex. Science 331 : 555–561 doi:10.1126/science.1197761
27. TautzD, Domazet-LošoT (2011) The evolutionary origin of orphan genes. Nat Rev Genet 12 : 692–702 doi:10.1038/nrg3053
28. PeichelCL, NerengKS, OhgiKA, ColeBL, ColosimoPF, et al. (2001) The genetic architecture of divergence between threespine stickleback species. Nature 414 : 901–905 doi:10.1038/414901a
29. GibsonG (2005) Evolution. The synthesis and evolution of a supermodel. Science 307 : 1890–1891 doi:10.1126/science.1109835
30. JonesFC, GrabherrMG, ChanYF, RussellP, MauceliE, et al. (2012) The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484 : 55–61 doi:10.1038/nature10944
31. HohenlohePA, BasshamS, EtterPD, StifflerN, JohnsonEA, et al. (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet 6: e1000862 doi:10.1371/journal.pgen.1000862
32. RoestiM, HendryAP, SalzburgerW, BernerD (2012) Genome divergence during evolutionary diversification as revealed in replicate lake-stream stickleback population pairs. Mol Ecol 21 : 2852–2862 doi:10.1111/j.1365-294X.2012.05509.x
33. DeagleBE, JonesFC, AbsherDM, KingsleyDM, ReimchenTE (2013) Phylogeography and adaptation genetics of stickleback from the Haida Gwaii archipelago revealed using genome-wide single nucleotide polymorphism genotyping. Mol Ecol 22 : 1917–1932 doi:10.1111/mec.12215
34. WilliamsonSH, HernandezR, Fledel-AlonA, ZhuL, NielsenR, et al. (2005) Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc Natl Acad Sci USA 102 : 7882–7887 doi:10.1073/pnas.0502300102
35. BoykoAR, WilliamsonSH, IndapAR, DegenhardtJD, HernandezRD, et al. (2008) Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4: e1000083 doi:10.1371/journal.pgen.1000083
36. ProuxE, StuderRA, MorettiS, Robinson-RechaviM (2009) Selectome: a database of positive selection. Nucleic Acids Res 37: D404–D407 doi:10.1093/nar/gkn768
37. MorettiS, LaurenczyB, GharibWH, CastellaB, KuzniarA, et al. (2013) Selectome update: quality control and computational improvements to a database of positive selection. Nucleic Acids Res 42: D917–21 doi:10.1093/nar/gkt1065
38. SharpAJ, LockeDP, McGrathSD, ChengZ, BaileyJA, et al. (2005) Segmental duplications and copy-number variation in the human genome. Am J Hum Genet 77 : 78–88 doi:10.1086/431652
39. CooperGM, NickersonDA, EichlerEE (2007) Mutational and selective effects on copy-number variants in the human genome. Nature Genet 39: S22–S29 doi:10.1038/ng2054
40. TeshimaKM, InnanH (2004) The effect of gene conversion on the divergence between duplicated genes. Genetics 166 : 1553–1560.
41. KatjuV, BergthorssonU (2010) Genomic and population-level effects of gene conversion in Caenorhabditis paralogs. Genes (Basel) 1 : 452–468 doi:10.3390/genes1030452
42. HiraiwaM (1999) Cathepsin A/protective protein: an unusual lysosomal multifunctional protein. Cell Mol Life Sci 56 : 894–907.
43. HsingLC, RudenskyAY (2005) The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev 207 : 229–241 doi:10.1111/j.0105-2896.2005.00310.x
44. ChanYF, MarksME, JonesFC, VillarrealG, ShapiroMD, et al. (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327 : 302–305 doi:10.1126/science.1182213
45. FawcettJA, InnanH (2013) The role of gene conversion in preserving rearrangement hotspots in the human genome. TIG 29 : 561–568 doi:10.1016/j.tig.2013.07.002
46. PerryGH, TchindaJ, McGrathSD, ZhangJ, PickerSR, et al. (2006) Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci USA 103 : 8006–8011 doi:10.1073/pnas.0602318103
47. Kehrer-SawatzkiH, CooperDN (2008) Comparative analysis of copy number variation in primate genomes. Cytogenet Genome Res 123 : 288–296 doi:10.1159/000184720
48. OrtiG, BellMA, ReimchenTE, MeyerA (1994) Global survey of mitochondrial DNA sequences in the threespine stickleback: evidence for recent migrations. Evolution 48 : 608–622.
49. SchriderDR, HahnMW (2010) Gene copy-number polymorphism in nature. Proc R Soc Lond [Biol] 277 : 3213–3221 doi:10.1073/pnas.0710524104
50. GuryevV, SaarK, AdamovicT, VerheulM, van HeeschSAAC, et al. (2008) Distribution and functional impact of DNA copy number variation in the rat. Nature Genet 40 : 538–545 doi:10.1038/ng.141
51. Marques-BonetT, GirirajanS, EichlerEE (2009) The origins and impact of primate segmental duplications. TIG 25 : 443–454 doi:10.1016/j.tig.2009.08.002
52. SudmantPH, KitzmanJO, AntonacciF, AlkanC, MaligM, et al. (2010) Diversity of human copy number variation and multicopy genes. Science 330 : 641–646 doi:10.1126/science.1197005
53. KimPM, LamHYK, UrbanAE, KorbelJO, AffourtitJ, et al. (2008) Analysis of copy number variants and segmental duplications in the human genome: Evidence for a change in the process of formation in recent evolutionary history. Genome Res 18 : 1865–1874 doi:10.1101/gr.081422.108
54. GazaveE, DarreF, Morcillo-SuarezC, Petit-MartyN, CarrenoA, et al. (2011) Copy number variation analysis in the great apes reveals species-specific patterns of structural variation. Genome Res 21 : 1626–1639 doi:10.1101/gr.117242.110
55. GhanemN, Uring-LambertB, AbbalM, HauptmannG, LefrancMP, et al. (1988) Polymorphism of MHC class III genes: definition of restriction fragment linkage groups and evidence for frequent deletions and duplications. Hum Genet 79 : 209–218.
56. SjödinP, JakobssonM (2012) Population genetic nature of copy number variation. Methods Mol Biol 838 : 209–223 doi:_10.1007/978-1-61779-507-7_10
57. XuL, HouY, BickhartDM, SongJ, Van TassellCP, et al. (2014) A genome-wide survey reveals a deletion polymorphism associated with resistance to gastrointestinal nematodes in Angus cattle. Funct Integr Genomics 14 : 333–9 doi:10.1007/s10142-014-0371-6
58. MilinskiM, GriffithsS, WegnerKM, ReuschTBH, Haas-AssenbaumA, et al. (2005) Mate choice decisions of stickleback females predictably modified by MHC peptide ligands. Proc Natl Acad Sci USA 102 : 4414–4418 doi:10.1073/pnas.0408264102
59. EizaguirreC, LenzTL, TraulsenA, MilinskiM (2009) Speciation accelerated and stabilized by pleiotropic major histocompatibility complex immunogenes. Ecology Letters 12 : 5–12 doi:10.1111/j.1461-0248.2008.01247.x
60. EizaguirreC, LenzTL, SommerfeldRD, HarrodC, KalbeM, et al. (2011) Parasite diversity, patterns of MHC II variation and olfactory based mate choice in diverging three-spined stickleback ecotypes. Evol Ecol 25 : 605–622 doi:10.1007/s10682-010-9424-z
61. HussainA, SaraivaLR, KorschingSI (2009) Positive Darwinian selection and the birth of an olfactory receptor clade in teleosts. Proc Natl Acad Sci USA 106 : 4313–4318 doi:10.1073/pnas.0803229106
62. HashiguchiY, NishidaM (2007) Evolution of trace amine associated receptor (TAAR) gene family in vertebrates: lineage-specific expansions and degradations of a second class of vertebrate chemosensory receptors expressed in the olfactory epithelium. Mol Biol Evol 24 : 2099–2107 doi:10.1093/molbev/msm140
63. HashiguchiY, FurutaY, NishidaM (2008) Evolutionary patterns and selective pressures of odorant/pheromone receptor gene families in teleost fishes. PLoS ONE 3: e4083 doi:10.1371/journal.pone.0004083
64. ZhangYE, LandbackP, VibranovskiM, LongM (2012) New genes expressed in human brains: Implications for annotating evolving genomes. Bioessays 34 : 982–991 doi:10.1002/bies.201200008
65. FlicekP, AmodeMR, BarrellD, BealK, BrentS, et al. (2012) Ensembl 2012. Nucleic Acids Res 40: D84–D90 doi:10.1093/nar/gkr991
66. AbyzovA, UrbanAE, SnyderM, GersteinM (2011) CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21 : 974–984 doi:10.1101/gr.114876.110
67. ChenK, WallisJW, McLellanMD, LarsonDE, KalickiJM, et al. (2009) BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods 6 : 677–681 doi:10.1038/nmeth.1363
68. RauschT, ZichnerT, SchlattlA, StützAM, BenesV, et al. (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339 doi:10.1093/bioinformatics/bts378
69. YeK, SchulzMH, LongQ, ApweilerR, NingZ (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25 : 2865–2871 doi:10.1093/bioinformatics/btp394
70. Team RC (2013) R: a language and environment for statistical computing.
71. VilellaAJ, SeverinJ, Ureta-VidalA, HengL, DurbinR, et al. (2008) EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19 : 327–335 doi:10.1101/gr.073585.107
72. QuinlanAR, HallIM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 : 841–842 doi:10.1093/bioinformatics/btq033
73. BryantD, MoultonV (2004) Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21 : 255–265 doi:10.1093/molbev/msh018
74. HusonDH, BryantD (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23 : 254–267 doi:10.1093/molbev/msj030
75. DePristoMA, BanksE, PoplinR, GarimellaKV, MaguireJR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet 43 : 491–498 doi:10.1038/ng.806
76. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079 doi:10.1093/bioinformatics/btp352
77. BrowningSR, BrowningBL (2007) Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81 : 1084–1097 doi:10.1086/521987
78. DanecekP, AutonA, AbecasisG, AlbersCA, BanksE, et al. (2011) The variant call format and VCFtools. Bioinformatics 27 : 2156–2158 doi:10.1093/bioinformatics/btr330
79. ZerbinoDR, BirneyE (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 : 821–829 doi:10.1101/gr.074492.107
80. HuangX, MadanA (1999) CAP3: A DNA sequence assembly program. Genome Res 9 : 868–877.
81. AltschulSF, GishW, MillerW, MyersEW, LipmanDJ (1990) Basic local alignment search tool. J Mol Biol 215 : 403–410 doi:10.1016/S0022-2836(05)80360-2
82. ConesaA, GötzS, García-GómezJM, TerolJ, TalónM, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 : 3674–3676 doi:10.1093/bioinformatics/bti610
83. SaraivaLR, KorschingSI (2007) A novel olfactory receptor gene family in teleost fish. Genome Res 17 : 1448–1457 doi:10.1101/gr.6553207
84. AlexaA, RahnenführerJ, LengauerT (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22 : 1600–1607 doi:10.1093/bioinformatics/btl140
85. LivakKJ, SchmittgenTD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25 : 402–408 doi:10.1006/meth.2001.1262
86. WegnerKM, KalbeM, RauchG, KurtzJ, SchaschlH, et al. (2006) Genetic variation in MHC class II expression and interactions with MHC sequence polymorphism in three-spined sticklebacks. Mol Ecol 15 : 1153–1164 doi:10.1111/j.1365-294X.2006.02855.x
87. SchmittgenTD, LivakKJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3 : 1101–1108 doi:10.1038/nprot.2008.73
88. RanwezV, HarispeS, DelsucF, DouzeryEJP (2011) MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE 6: e22594 doi:10.1371/journal.pone.0022594
89. YangZ (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24 : 1586–1591 doi:10.1093/molbev/msm088
90. GuoB, ChainFJ, Bornberg-BauerE, LederEH, MeriläJ (2013) Genomic divergence between nine - and three-spined sticklebacks. BMC Genomics 14 : 756 doi:10.1186/1471-2164-14-756
91. ZhengY, ZhaoL, GaoJ, FeiZ (2011) iAssembler: a package for de novo assembly of Roche-454/Sanger transcriptome sequences. BMC Bioinformatics 12 : 453 doi:10.1186/1471-2105-12-453
92. SawyerS (1989) Statistical tests for detecting gene conversion. Mol Biol Evol 6 : 526–538.
93. McGrathCL, CasolaC, HahnMW (2009) Minimal effect of ectopic gene conversion among recent duplicates in four mammalian genomes. Genetics 182 : 615–622 doi:10.1534/genetics.109.101428
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Tetraspanin (TSP-17) Protects Dopaminergic Neurons against 6-OHDA-Induced Neurodegeneration in
- Maf1 Is a Novel Target of PTEN and PI3K Signaling That Negatively Regulates Oncogenesis and Lipid Metabolism
- The IKAROS Interaction with a Complex Including Chromatin Remodeling and Transcription Elongation Activities Is Required for Hematopoiesis
- Echoes of the Past: Hereditarianism and
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy