
The Conserved SKN-1/Nrf2 Stress Response Pathway Regulates Synaptic Function in
The Nrf family of transcription factors plays a critical role in mediating adaptive responses to cellular stress and defends against neurodegeneration, aging, and cancer. Here, we report a novel role for the Caenorhabditis elegans Nrf homolog SKN-1 in regulating synaptic transmission at neuromuscular junctions (NMJs). Activation of SKN-1, either by acute pharmacological treatment with the mitochondrial toxin sodium arsenite or by mutations that cause constitutive SKN-1 activation, results in defects in neuromuscular function. Additionally, elimination of the conserved WD40 repeat protein WDR-23, a principal negative regulator of SKN-1, results in impaired locomotion and synaptic vesicle and neuropeptide release from cholinergic motor axons. Mutations that abolish skn-1 activity restore normal neuromuscular function to wdr-23 mutants and animals treated with toxin. We show that negative regulation of SKN-1 by WDR-23 in the intestine, but not at neuromuscular junctions, is necessary and sufficient for proper neuromuscular function. WDR-23 isoforms differentially localize to the outer membranes of mitochondria and to nuclei, and the effects of WDR-23 on neuromuscular function are dependent on its interaction with cullin E3 ubiquitin ligase. Finally, whole-transcriptome RNA sequencing of wdr-23 mutants reveals an increase in the expression of known SKN-1/Nrf2-regulated stress-response genes, as well as neurotransmission genes not previously implicated in SKN-1/Nrf2 responses. Together, our results indicate that SKN-1/Nrf2 activation may be a mechanism through which cellular stress, detected in one tissue, affects cellular function of a distal tissue through endocrine signaling. These results provide insight into how SKN-1/Nrf2 might protect the nervous system from damage in response to oxidative stress.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003354
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003354
Summary
The Nrf family of transcription factors plays a critical role in mediating adaptive responses to cellular stress and defends against neurodegeneration, aging, and cancer. Here, we report a novel role for the Caenorhabditis elegans Nrf homolog SKN-1 in regulating synaptic transmission at neuromuscular junctions (NMJs). Activation of SKN-1, either by acute pharmacological treatment with the mitochondrial toxin sodium arsenite or by mutations that cause constitutive SKN-1 activation, results in defects in neuromuscular function. Additionally, elimination of the conserved WD40 repeat protein WDR-23, a principal negative regulator of SKN-1, results in impaired locomotion and synaptic vesicle and neuropeptide release from cholinergic motor axons. Mutations that abolish skn-1 activity restore normal neuromuscular function to wdr-23 mutants and animals treated with toxin. We show that negative regulation of SKN-1 by WDR-23 in the intestine, but not at neuromuscular junctions, is necessary and sufficient for proper neuromuscular function. WDR-23 isoforms differentially localize to the outer membranes of mitochondria and to nuclei, and the effects of WDR-23 on neuromuscular function are dependent on its interaction with cullin E3 ubiquitin ligase. Finally, whole-transcriptome RNA sequencing of wdr-23 mutants reveals an increase in the expression of known SKN-1/Nrf2-regulated stress-response genes, as well as neurotransmission genes not previously implicated in SKN-1/Nrf2 responses. Together, our results indicate that SKN-1/Nrf2 activation may be a mechanism through which cellular stress, detected in one tissue, affects cellular function of a distal tissue through endocrine signaling. These results provide insight into how SKN-1/Nrf2 might protect the nervous system from damage in response to oxidative stress.
Introduction
To prevent cellular damage caused by environmental stresses, organisms have developed defense mechanisms at the cellular level which ultimately result in system-wide protective changes. Oxidative stress is generated in part by an increase in reactive oxygen species and has been associated with increased protein and lipid damage and reduced mitochondrial function. Elevated levels of reactive oxygen species and a concomitant decrease in antioxidants have been linked to aging, neurodegeneration and diabetes [1], [2]. How organisms are able to coordinate an appropriate, systemic response to prevent damage caused by oxidative stress is not well understood.
The Nrf (nuclear factor E2-related factor) family of transcription factors, homologous to CNC [3] and SKN-1 [4] in Drosophila melanogaster and Caenorhabditis elegans, respectively, is activated in response to heightened oxidative stress or drugs (xenobiotics) [1], [5]. When activated, Nrf proteins move into the nucleus where they bind the antioxidant response element, or ARE, in enhancer regions of target genes [1], [5]. Nrf nuclear translocation results in the expression of a host of phase II detoxificants and antioxidants, including glutathione-synthesizing enzymes, thioredoxins, and proteasomal subunits; together, these enzymes are proposed to collectively mitigate the harmful effects of reactive oxygen species [6]–[9].
Nrf family members promote systemic changes in many tissues which together enhance organismal survival and longevity [1], [5]. For example, in mice cortical cultures, activation of neuronal Nrf2 results in ARE-mediated transcription of many genes, including the excitatory amino acid transporter 3 (EAAT3), which allows neurons to import the antioxidant glutathione [10]; at the same time, Nrf2 activation in astrocytes is proposed to confer protection to neurons by increasing glutathione release [6], [8], [11]. In another example, disruption of Nrf-1 in mice results in embryonic lethality due to a cell non-autonomous defect in erythropoiesis [12]. Similarly, in C. elegans, SKN-1 functions in a pair of neurons to promote longevity through endocrine signaling that affects distal tissues [13], [14]. Thus, SKN-1/Nrf signaling acts cell-autonomously, as well as non-autonomously, to coordinate adaptive responses in response to stress.
Due in part to the high metabolic demands required for neuronal function, the nervous system is particularly susceptible to oxidative damage [1], [2]. Reactive oxygen species cause protein aggregation, mitochondrial dysfunction, altered synaptic transmission, and are associated with neurodegeneration [2], [15]. In addition to promoting neuron survival, Nrf2 activation improves cognitive performance following traumatic brain injury [16], raising the possibility that Nrf2 might regulate aspects of neuronal function. Gene expression analyses across multiple species indicate that, in addition to antioxidants, activation of SKN-1/Nrf2 also increases the expression of genes involved in calcium homeostasis, synaptic function, and signaling [8], [17], [18]; however, whether SKN-1/Nrf2 transcriptional programs directly regulate neuronal function is not known.
In C. elegans, SKN-1 activity is negatively regulated by the WD40 repeat protein WDR-23. WDR-23 contains seven WD repeats and is proposed to negatively regulate SKN-1 activity by targeting SKN-1 for degradation [19], [20]. wdr-23 mutants are resistant to oxidative stress and display increased expression of SKN-1 transcriptional targets [19], [20]. This has led to the model that oxidative stress stabilizes nuclear SKN-1 by disrupting WDR-23-mediated SKN-1 degradation. WDR-23 is highly conserved, sharing 41% amino acid similarity with its human ortholog, WDR23. WDR23 is a component of the DDB1-CUL4 ubiquitin ligase complex and is proposed to act as a substrate specificity subunit that targets proteins for degradation [21]–[23].
Here, we demonstrate that WDR-23 promotes synaptic transmission at neuromuscular junctions in C. elegans by negatively regulating skn-1. We show that mutants lacking wdr-23 have defects in locomotion and neurotransmitter secretion. We provide functional evidence that the effects of wdr-23 on synaptic function are entirely mediated by skn-1, and activation of SKN-1 by mitochondrial stress can mimic the effects of wdr-23 on synaptic function. WDR-23 acts cell non-autonomously in the intestine, localizes to the nucleus as well as to mitochondria, and loss of wdr-23 results in altered expression of over 2000 genes. Our results support a model whereby presynaptic function is negatively regulated by a SKN-1 dependent transcriptional pathway in the intestine in response to stress.
Results
wdr-23 Mutants Have Altered Locomotion Rates
We previously identified wdr-23 in an RNA interference (RNAi) screen for genes that regulate synaptic transmission at the C. elegans neuromuscular junction [24]. To further characterize the effects of wdr-23 function on the nervous system, we examined putative null wdr-23(tm1817) mutants, which have a 635 bp deletion, generating a premature stop in the third WD repeat (Figure 1A). In addition to their small size and delayed growth rates [19], wdr-23 mutants had reduced locomotion rates and spent significantly less time moving compared to wild type animals (Figure S1A–S1C, Figure 1B and 1C, and Table S1). Transgenes expressing wdr-23 cDNA driven by a 1.3 kb wdr-23 promoter fragment fully rescued these phenotypes of wdr-23 mutants, including the locomotion defects (Figure S1C, Figure 1B and 1C, and data not shown). Impaired locomotion could be due to presynaptic defects in neurotransmitter release or postsynaptic defects in muscle response to acetylcholine at NMJs. To distinguish between these possibilities, we examined the response of wdr-23 mutants to drugs that alter neuromuscular function. Aldicarb is an acetylcholine esterase inhibitor, and treatment with aldicarb causes acetylcholine to accumulate in synaptic clefts, resulting in muscle contraction and eventual paralysis. Mutations that decrease the rate of acetylcholine release from motor neurons reduce the rate of aldicarb-induced paralysis, whereas mutations that increase acetylcholine release increase the rate of paralysis [25]. wdr-23 mutants were highly resistant to the paralytic effects of aldicarb, and expression of a genomic wdr-23 fragment in wdr-23 mutants completely rescued this defect (Figure 1D). These results indicate that wdr-23 mutants have impaired acetylcholine release from motor neurons. Consistent with these results, wdr-23 mutants were hypersensitive to the paralytic effects of the muscle agonist levamisole (Figure 1E), indicating that muscle responsiveness was not reduced in these mutants.

SKN-1 Mediates the Effects of wdr-23 on Neuromuscular Function
Mammalian WDR23 binds to the DDB1-CUL4 ubiquitin ligase complex via an interaction between the cullin adaptor DDB1 and a conserved ‘DxR’ box within the WDR40 repeat domains [21], [22]. WDR-23 contains two conserved DxR boxes within WD repeats four and five (Figure 1A). To determine whether interaction with DDB1 is necessary for the function of WDR-23, we mutated the final arginines in the DxR boxes required for DDB binding to make WDR-23(DxR), which is predicted to be deficient in binding the DDB1 ortholog, DDB-1 [21], [22]. The WDR-23(DxR) mutation did not appear to alter the expression or localization of WDR-23 (Figure S2). However, expression of WDR-23(DxR) driven by the endogenous wdr-23 promoter fragment failed to restore wild type aldicarb responsiveness to wdr-23 mutants (Figure 2A). This suggests that the interaction between WDR-23 and DDB-1/CUL-4 is essential for regulating neuromuscular function.
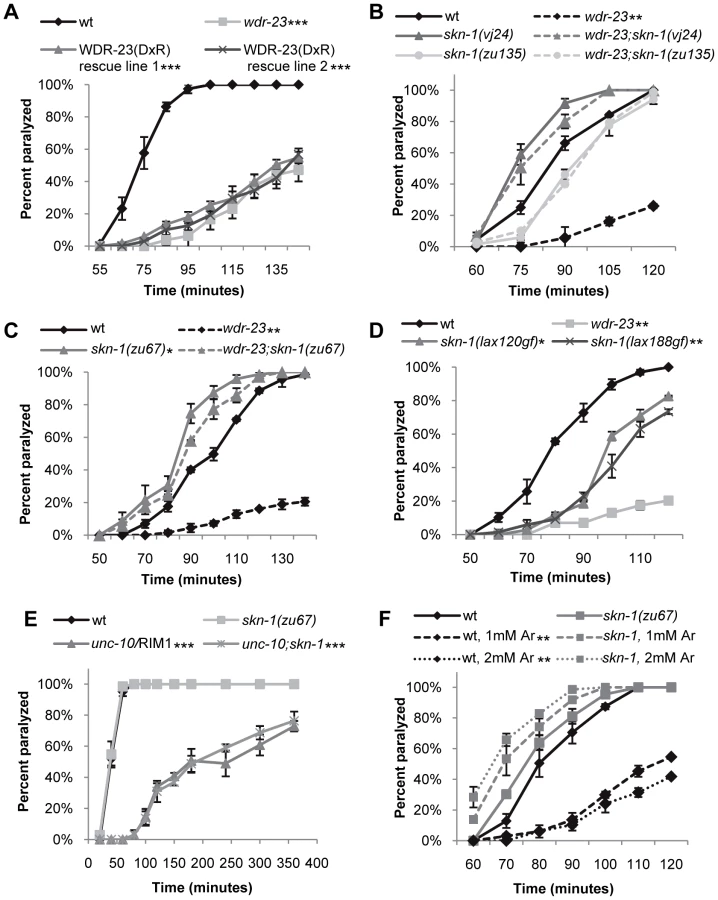
To identify targets of WDR-23 that promote presynaptic function, we performed a forward genetic screen for suppressors of the wdr-23 aldicarb resistant phenotype. We identified an allele of skn-1, vj24, which encodes a nonsense mutation in the DNA binding domain of SKN-1 (R519STOP of the skn-1a isoform); skn-1(vj24) completely restored the small size, delayed growth and aldicarb resistance of wdr-23 mutants to wild type levels (Figure 2B and data not shown). An independently isolated skn-1 allele, zu135, which also encodes a premature stop in the SKN-1 DNA binding domain, also completely restored the size, delayed growth and aldicarb response of wdr-23 mutants (Figure 2B and data not shown). SKN-1 encodes three isoforms that share the DNA binding domain (www.wormbase.org). Two SKN-1 isoforms, SKN-1a and SKN-1c, function in the intestine to mediate stress response [4], [26], whereas the third isoform, SKN-1b, functions in two sensory neurons for dietary restriction-induced longevity [13]. skn-1(zu67) mutants, which specifically lack the skn-1a and skn-1c isoforms, restored wild type locomotion rates, aldicarb response, size and growth rates to wdr-23 mutants (Figure 1C, Figure 2C, Figure S1B and S1C). None of the skn-1 mutants examined displayed significant size, growth, locomotion or aldicarb response defects alone (Figure 2B and 2C, Figure S1B, and data not shown). These results suggest that the primary function of WDR-23 is to negatively regulate SKN-1a and/or SKN-1c, and increased SKN-1a/c activity leads to defects in synaptic function. Consistent with this idea, skn-1 gain-of-function (gf) mutants, which are predicted to enhance the activity of SKN-1a and SKN-1c isoforms [27], displayed aldicarb resistance reminiscent of wdr-23 mutants (Figure 2D). Thus, wdr-23 is likely to promote neuromuscular function by negatively regulating SKN-1 activity.
Because SKN-1 induces transcriptional programs that detoxify xenobiotics, the aldicarb resistance of wdr-23 mutants could be due to increased aldicarb detoxification by SKN-1 in these mutants rather than a specific effect of SKN-1 activity on synaptic function. If this were the case, we would predict that the aldicarb resistance of mutants that directly reduce acetylcholine secretion should also be suppressed by skn-1 mutations. unc-10/RIM1 is a presynaptic protein that is required for acetylcholine release from synaptic vesicles, and unc-10 mutants are aldicarb resistant [25]. However, skn-1(zu67) mutations failed to suppress the aldicarb resistant phenotype of unc-10/RIM1 mutants (Figure 2E). Thus, the aldicarb and locomotion defects observed in wdr-23 mutants likely reflect SKN-1-dependent changes in neuromuscular function, rather than alterations in drug detoxification.
Environmental Stress Alters NMJ Function
Several environmental stressors have been identified that promote SKN-1 nuclear translocation, induce SKN-1-dependent transcriptional programs, and have SKN-1-dependent effects on stress response and aging [4], [19], [28], [29]. In order to identify stressors that might cause SKN-1-dependent changes in neuronal function, we tested a variety of conditions that activate SKN-1 for their effects on aldicarb-induced paralysis. Interestingly, treatment of wild type animals with paraquat, peroxide or heat shock did not cause aldicarb resistance in our assays (data not shown). In contrast, we found that prolonged exposure to the toxin sodium arsenite resulted in robust aldicarb resistance (Figure 2F). Arsenite stimulates reactive oxygen species production and protein misfolding and has previously been shown to cause SKN-1 nuclear translocation in intestinal cells [7], [30]. Importantly, arsenite-induced aldicarb resistance was completely blocked by skn-1 mutations (Figure 2F). In contrast, no change in aldicarb responsiveness was observed following acute exposure to arsenite, consistent with previous findings (data not shown, [31]). Together, these results suggest arsenite may reduce NMJ function by inducing SKN-1 dependent transcriptional programs.
wdr-23 Mutants Have Defective Neurotransmitter and Neuropeptide Release
We next examined the nature of the neuronal defects in wdr-23 mutants in more detail by analyzing synaptic structure and function. Examination of cholinergic neurons labeled with GFP (using the acr-2 promoter [32]) revealed no gross morphological changes in axonal projections or in the number of cholinergic neurons in wdr-23 mutants (wild type = 31.0±0.6; wdr-23 = 31.9±0.4, p = 0.2 Student's t-test, and Figure S1A). In addition, quantification of the active zone protein UNC-10/RIM1 tagged to GFP [33] showed no significant changes in the number of synapses (measured by interpunctal interval, Figure 3B and Table S2) or synaptic size (measured by punctal width, wild type = 0.75±0.03 µm; wdr-23 = 0.72±0.02 µm, p = 0.3) in wdr-23 mutants compared to wild type controls. These results indicate that wdr-23 mutants are likely to have normal neuronal and synaptic development. In support of this idea, we found that RNAi knockdown of wdr-23 after hatching (when development is largely complete) resulted in aldicarb resistance similar to that seen in wdr-23 mutants (Figure 4D).


To investigate defects in neurotransmitter release, we visualized the cycling of synaptic vesicles at cholinergic synapses in vivo. Synaptic vesicle-associated proteins labeled with GFP can be used to assess synaptic vesicle cycling [33], [34]; in axons, the fluorescent intensity of individual puncta labeled with synaptic vesicle markers correlates with the number of vesicles at presynaptic sites [34]. Along these lines, we utilized two synaptic vesicle markers, GFP-SNB-1 and GFP-RAB-3, to visualize steady-state vesicle abundance at release sites in wdr-23 mutants. We found that mutants lacking wdr-23 had significantly increased GFP-SNB-1 and GFP-RAB-3 punctal fluorescence, but not axonal fluorescence, relative to wild type controls. The increase in punctal fluorescence of GFP-RAB-3 in wdr-23 mutants was fully rescued by WDR-23 cDNA (Figure 3A, 3B and Table S2). These results are consistent with a defect in synaptic vesicle exocytosis from cholinergic motor neurons in wdr-23 mutants.
C. elegans cholinergic axons also secrete the neuropeptides NLP-21 and INS-22, which are FMRF amide-related peptide (FaRP) and an insulin-like growth factor, respectively, and are packaged in dense core vesicles [35]. Fluorescently tagged versions of these proteins adopt a punctate pattern in axons of cholinergic motor neurons, and defects in dense core vesicle secretion release result in increased punctal fluorescence within axons. We found that wdr-23 mutants had significant increases in INS-22-YFP and NLP-21-YFP punctal fluorescence, but no change in axonal fluorescence, compared to wild type controls (Figure 3A, 3B and Table S2), consistent with a defect in dense core vesicle secretion. Expression of WDR-23 cDNA fully rescued the increases in punctal fluorescence of INS-22-YFP in wdr-23 mutants to wild type levels. Next, we examined neuropeptide accumulation in specialized scavenger cells called coelomocytes which endocytose neuropeptides after they are secreted from neurons [36]. Mutants with defective neuropeptide secretion accumulate less fluorescence in coelomocytes [35]. We found that coelomocyte fluorescence in wdr-23 mutants secreting NLP-21-YFP from motor neurons was significantly dimmer than in wild type controls (Figure 3C and Table S2). In contrast, coelomocyte fluorescence in wdr-23 mutants secreting GFP fused to a signal sequence (ssGFP) from motor neurons [37] was not significantly different than wild type controls, indicating that constitutive vesicle release and coelomocyte uptake are normal in wdr-23 mutants (Figure 3C and Table S2). Together, our results suggest that wdr-23 specifically promotes the regulated exocytosis of synaptic vesicles and dense core vesicles from motor neurons.
WDR-23 Functions in the Intestine to Regulate Synaptic Transmission
To determine in which tissues WDR-23 functions to regulate neurotransmitter release, we first examined the expression pattern of wdr-23. wdr-23 encodes two isoforms, WDR-23a and WDR-23b, distinguishable by their unique amino termini (Figure 1A; www.wormbase.org). wdr-23a is the predominant isoform (Figure 7C), and the endogenous 1.3 kb wdr-23a promoter fragment driving wdr-23a fully rescued wdr-23 locomotion phenotypes (Figure 1B and 1C). A reporter consisting of the 1.3 kb wdr-23a promoter tagged to gfp was detected in the hypodermis (skin), intestine, muscle and neurons, consistent with previous findings (Figure 4A, [19]). A 2.0 kb wdr-23b promoter fragment tagged to gfp was detected in muscle, hypodermis, and intestine, but not in neurons (Figure S3). Next, we selectively expressed wdr-23 in different tissues using tissue-specific promoters and assayed its ability to rescue the neuromuscular defects of wdr-23 mutants. Expression of functional WDR-23-GFP in the intestine (using either the ges-1 or nlp-40 promoter) fully restored wild type aldicarb responses and locomotion to wdr-23 mutants (Figure 1C, Figure 4B and data not shown). In contrast, expression of WDR-23-GFP in muscle (myo-3 promoter) or neurons (rab-3 or snb-1 promoter) was not sufficient to restore normal aldicarb responsiveness to wdr-23 mutants (Figure 4B).
To confirm the intestinal site of action of wdr-23, we knocked down wdr-23 by tissue-specific RNA interference. Neurons of wild type animals are resistant to gene knockdown by feeding double stranded RNA (dsRNA), whereas neurons of nre-1 lin-15b mutants are less resistant [38], [39]. unc-17 encodes the vesicular acetylcholine transporter that functions specifically in neurons, and unc-17/VAChT loss-of-function mutants are aldicarb resistant [25]. As expected, feeding knockdown of unc-17/VAChT was only effective in eliciting aldicarb resistance in nre-1 lin-15b mutants, but not in wild type animals (Figure 4D), confirming that unc-17/VACht acts in neurons. In contrast, wdr-23 knockdown elicited robust aldicarb resistance in both wild type animals and nre-1 lin-15b mutants (Figure 4D), suggesting that wdr-23 activity is likely to be required outside of the nervous system for its effects on aldicarb response. Next, we selectively knocked down wdr-23 activity in specific tissues by expressing inverted repeat hairpin transgenes directed toward wdr-23 using tissue specific promoters (in a sid-1 mutant background to prevent systemic spreading of RNAi) [40], [41]. wdr-23 hairpin expression in the intestine (using ges-1 and nlp-40 promoters) but not in neurons or muscles (using the rab-3 and myo-3 promoters, respectively) resulted in aldicarb resistance comparable to that seen with the endogenous wdr-23 promoter fragment (Figure 4C and Table S1). Together, these results indicate that wdr-23 in the intestine is necessary and sufficient to positively regulate synaptic function and suggest that activation of SKN-1 in the intestine is responsible for the reduced neuronal function of wdr-23 mutants.
WDR-23 Localization and Function
Next, to determine the sub-cellular localization of WDR-23, we expressed functional GFP tagged versions of WDR-23a or WDR-23b in the intestine, and we found that the two isoforms localized to distinct sub-cellular compartments. WDR-23b-GFP was entirely nuclear, whereas WDR-23a-GFP adopted a ring-like pattern of fluorescence in the intestinal cytoplasm (Figure 5B). The identity of the ring-like structures could not be determined unambiguously by co-localization studies, since fluorescent markers were difficult to resolve in the intestine (Figure S4). Thus, we examined WDR-23a-GFP localization in muscles—which also express wdr-23 but are more amenable to fluorescent imaging studies [42].

In muscles, WDR-23a-GFP fluorescence was observed at dense bodies, which are actin rich-sites of muscle attachment to the hypodermis, and on ring-like structures similar to those seen in the intestine (Figure 5C). The ring-like structures were regularly spaced along muscle fibers and co-localized with an outer mitochondrial membrane marker tagged with RFP (INVOM-RFP). WDR-23a-GFP fluorescence appeared to be distributed on the outer surface of the rings occupied by the mitochondrial marker, suggesting that it may be associated with outer mitochondrial membranes (Figure 5C). To test this idea, we examined the distribution of WDR-23a-GFP in drp-1/Drp1 loss of function mutants. Drp1 is required for outer mitochondria membrane fission; C. elegans mutants lacking drp-1 activity have normal inner membrane fission, but form continuous elongated tubes of outer membrane [43]. The WDR-23a-GFP fluorescence pattern was dramatically altered in drp-1 mutants; fluorescence was seen in elongated tubes that co-localized with INVOM-RFP, consistent with WDR-23a-GFP being associated with the outer mitochondrial membrane (Figure 5C′). In contrast, WDR-23b-GFP was detected exclusively in nuclei (Figure 5D).
To test whether mitochondrial or nuclear localization of WDR-23 is critical for its function, we introduced wdr-23 isoforms into wdr-23 mutants using Mos-mediated low copy insertion [44] to avoid artifacts due to high copy expression of wdr-23. We found that both WDR-23a and WDR-23b transgenes restored normal aldicarb responsiveness, synaptic vesicle and dense core vesicle release to wdr-23 mutants (Figure 3A and Figure 5A). Last, a truncated version of WDR-23 that contains only the common WD40 repeat region, referred to as WDR-23(repeats), also fully restored aldicarb response of wdr-23 mutants to wild type (Figure 6A and 6B). This variant was not concentrated at mitochondria or nuclei but was distributed uniformly throughout the muscle. Taken together, these results indicate that the functional domain of WDR-23 is contained within the WD40 repeats, and although WDR-23 isoforms are concentrated at distinct cellular compartments, the precise sub-cellular localization of WDR-23 may not be critical for its function.

Transcriptional Targets of SKN-1 Are Increased in wdr-23 Mutants
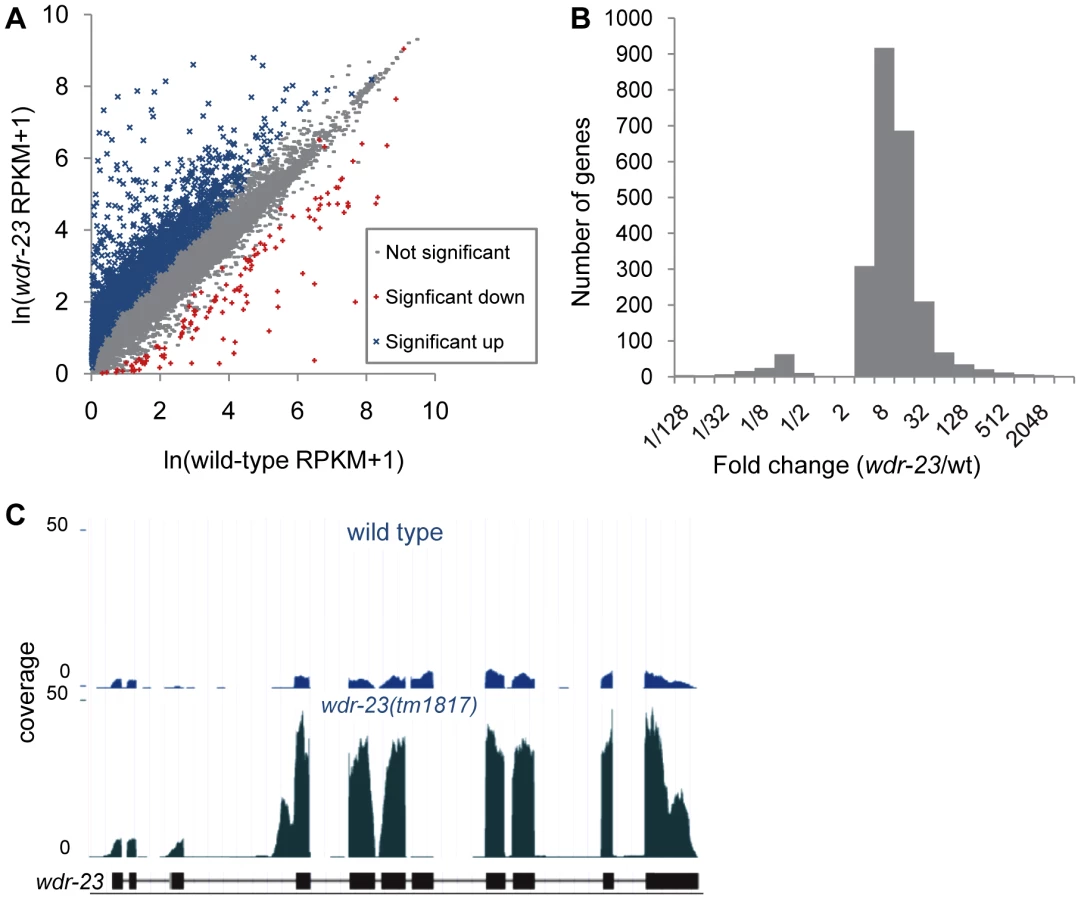
What might be the critical transcriptional targets of SKN-1 that inhibit synaptic transmission? To begin to address this question, we compared gene expression profiles of wdr-23 mutants to wild type controls using whole transcriptome RNA sequencing. Approximately 29.2 and 29.4 million reads were mapped to the genome for wild type and wdr-23 mutants, respectively, corresponding to an average coverage of 698.9 reads per gene for wild type and 728.5 reads per gene for wdr-23 mutants. Using the Cuffdiff module of the statistical Cufflinks package [45], we identified 2285 genes that were significantly up-regulated and 134 genes that were significantly down-regulated in wdr-23 mutants (Figure 7A, Tables S4 and S6). The amplitude of the up-regulated genes ranged from 1.04 (rpl-3/ribosomal protein L3, p = 0.001) to 3517.69 fold (clec-74/c-type lectin, p<0.001), with an average fold change of 22.9 (Figure 7B). Significantly down-regulated genes ranged from 0.001 (ocr-1/capsaicin receptor related, p<0.001) to 0.96 fold (eft-3/elongation factor 1-alpha, p<0.001) and had a mean fold change of 0.16 (Figure 7B). Microarray analysis has previously identified 118 genes that are induced by arsenite in a SKN-1 dependent manner [7], [17]. We identified 80 genes among these that were significantly up-regulated in wdr-23 mutants (Table S4), supporting the idea that SKN-1-dependent transcriptional programs are increased in wdr-23 mutants.
We classified all significantly up-regulated genes in wdr-23 mutants using the Database for Annotation, Visualization and Integrated Discovery (DAVID) by GO terms and InterPro protein domains (Figure 8A and Table 1) [46]. Approximately twenty percent of genes with known functions were classified as being involved in metabolism or stress resistance and detoxification. Among these are genes involved in SKN-1/Nrf2 dependent oxidative and xenobiotic stress resistance, including glutathione S-transferases, UDP-glucuronosyl transferases and cytochrome P450s [7], [17]. Two glutathione S-transferases, gst-4 and gst-30, whose expression is increased by SKN-1 activation [7], [47], increased by 255.5 and 1921.1 fold, respectively in wdr-23 mutants. We validated these increases and confirmed that their expression was SKN-1 dependent by qRT-PCR (Table 2).



In addition to stress response and metabolic genes, several other categories of genes were up-regulated in wdr-23 mutants, including those involved in signaling (27%), protein degradation (6%) and the extracellular matrix (7%). In addition, 165 genes (13%) likely involved in neuronal function or behavior were up-regulated (Table S5). Genes directly involved in synaptic transmission and calcium sensing at neuromuscular junctions, such as unc-25/glutamic acid decarboxylase, snt-1/synaptotagmin, unc-13/Munc13, and unc-10/RIM1, all had statistically increased expression in wdr-23 mutants (Table S5). qRT-PCR of unc-25/glutamic acid decarboxylase and snt-1/synaptotagmin (but not the negative control unc-17/VAChT) verified a roughly three-fold increase in expression in wdr-23 mutants, in agreement with the RNA sequencing results (Table 2). Finally, approximately 50 neuropeptide genes expressed in diverse tissues were up-regulated in wdr-23 mutants, suggesting that endocrine signaling is extensively altered in these mutants.
To determine the number of up-regulated genes in wdr-23 mutants likely to be direct binding targets of SKN-1, we used Regulatory Sequence Analysis Tools (RSAT) oligo analysis [48] to detect and quantify potential SKN-1 binding motifs. Previous studies have established the SKN-1 consensus to be WWTDTCAT or the related sequence TTDTCATC (W = A/T, D = G/A/T), where the underlined sequence is thought to be directly bound by SKN-1 [4], [7], [49]. Consistent with these studies, we found the sequence WWTDTCAT to be enriched in the promoter regions within 1 kb upstream of the transcriptional start of the most up-regulated genes. This motif was found in 90.3% of the top 300 up-regulated genes, compared to 78.6% of promoters from a randomized list of genes not significantly different in wdr-23 mutants relative to wild type controls (Table S3). Further analysis by RSAT identified the more restrictive sequence TTDTCATC to be over-represented among these promoter fragments (Figure 8B). This restricted element was enriched in the stress related genes (found in 52% of the stress and detoxification gene promoters, compared to 25% of the randomized gene list; Table S3), consistent with these genes being direct SKN-1 targets. In contrast, this restrictive motif was found in only 24.6% of the promoters of the synaptic or behavioral genes, implying that a majority of these genes may not be direct SKN-1 targets. Thus, changes in gene expression are likely to reflect both direct and indirect regulation by SKN-1. Together, these results indicate that extensive changes in gene expression profiles of functionally diverse genes in multiple tissues takes place in wdr-23 mutants.
wdr-23(tm1817) mutants have a deletion that spans exon seven (of wdr-23a), and examination of wdr-23(tm1817) mutants confirmed the presence of a truncated wdr-23 transcript in which exons six and eight were spliced together (Figure 7C), resulting in a frame shift and premature stop. Surprisingly, wdr-23 transcripts were 7.6 fold more abundant in wdr-23 mutants compared to wild type controls (Figure 7C). The wdr-23a promoter contains three restrictive and five canonical SKN-1 binding sites within 1 kb upstream of the start site. Together, these results indicate that wdr-23 expression itself may be under direct control of SKN-1 (Table S4).
Finally, using DAVID, we analyzed the functional classifications of the 134 genes down-regulated in wdr-23 mutants (Figure 8C and Table 3). Grouping by GO terms revealed that genes involved in aging were enriched this group. Interestingly, previous microarray studies identified 63 genes to be transcriptionally down-regulated under basal conditions by SKN-1 but did not detect any genes to be down-regulated in response to stress [7]. RSAT promoter analysis identified the motif TCTTATCAG to be over-represented in the genes down-regulated in wdr-23 mutants (Figure 8D). This sequence is different than the sequence TGAGTCAC, which is enriched in genes down-regulated by skn-1 RNAi [7]. This suggests that genes constitutively down-regulated by SKN-1 might utilize a different binding site than genes down-regulated by SKN-1 during stress.

Discussion
Inducible transcriptional programs are critical cellular mechanisms which allow organisms to respond systemically to varying environmental conditions such as caloric restriction, hypoxia or inflammation in order to promote survival. The conserved transcription factor SKN-1/Nrf2 has well known roles in preventing cellular damage caused by oxidative and xenobiotic stress. In this study, we establish that WDR-23 is the principle negative regulator of SKN-1 in vivo. We show that SKN-1/Nrf2 negatively regulates function of the C. elegans NMJ by reducing neurotransmitter secretion. We find that SKN-1/Nrf2 pathway activation is cell non-autonomous and results in global transcriptional changes, some of which may regulate synaptic transmission (Figure 6C). Thus, Nrf family activation may be a conserved mechanism by which stress alters neuronal function during adverse conditions.
Cell Non-Autonomous Role of SKN-1 on NMJ Function
Our genetic and phenotypic analysis of wdr-23 mutants supports the idea that the negative regulation of SKN-1 by WDR-23 is required for the normal release of neurotransmitter at NMJs. Interestingly, we found that although WDR-23 was nearly ubiquitously expressed in multiple tissues, WDR-23 acts cell non-autonomously in the intestine to regulate neuronal function, suggesting that increased SKN-1 activity in the intestine negatively regulates synaptic transmission. High copy skn-1 transgenes expressed in the intestine are toxic [26], and even low copy transgenes make animals sick (data not shown), precluding direct behavioral characterization of SKN-1 over-expressing animals. The intestine is a first line of defense for C. elegans in their natural environment. Similarly, analysis of human disease-associated Nrf2 polymorphisms indicate that the organ systems which come in contact with environmental stressors, such as skin, lungs and intestines, are often highly compromised [1]. Thus, detection of environmental stress by the intestine could result in increased intercellular communication between the intestine and nervous system to promote neuronal survival.
Our results raise the possibility that a diffusible factor released from the intestine might mediate the effects of SKN-1 activation on the nervous system. One possible model is that increased SKN-1 activity alters the expression and/or release of neuropeptides from the intestine. Indeed, RNA sequencing indicates that genes involved in neuropeptide signaling in the intestine are up-regulated in wdr-23 mutants, including neuropeptides, insulins and FMRFamides. Furthermore, our observation that WDR-23 promotes neuropeptide secretion from neurons suggests that neuropeptide secretion from the intestine might also be under WDR-23/SKN-1 regulation. Interestingly, recent work has established signaling from the nervous system to the intestine during activation of mitochondrial unfolded protein response [41], and protons released from the intestine directly affect muscular activity [50]. Together, these studies highlight the importance of endocrine signaling from the intestine in regulating survival and behavior.
Mitochondrial Function of WDR-23
We show that WDR-23a is primarily localized to mitochondria whereas WDR-23b is primarily nuclear, yet both wdr-23a and wdr-23b transgenes rescue wdr-23 mutant phenotypes. Thus specific sub-cellular localization of WDR-23 may not be critical for its function. Alternatively, it is possible that localization of WDR-23 is critical for function, but localized fluorescence of WDR-23-GFP transgenes may be below our limits of detection by fluorescence microscopy.
Nrf2 is proposed to be dynamically associated with the cytosol, nucleus, actin filaments and mitochondria, and its nuclear abundance might be influenced by factors regulating its association with these compartments during stress [1]. Nrf2 is tethered to mitochondria via its interaction with Keap1 and PGAM5 [1], [51], [52], and Keap1 directly detects increased levels of reactive oxygen species released by mitochondria [5], [53], [54]. C. elegans do not encode a clear Keap1 ortholog. Thus, WDR-23 might play a homologous role in C. elegans to detect increased levels of oxidative stress or mitochondrial damage. We speculate that WDR-23a may associate with mitochondria dynamically and may be released from mitochondria during stress, where it can regulate SKN-1 translocation or degradation. In support of this hypothesis, a recent study has established that SKN-1 interacts with the mitochondrial protein PGAM-5, and inhibition of this interaction results in constitutively activated SKN-1 and a perceived state of starvation [27].
Why might increased levels of SKN-1 result in decreased neuromuscular activity? During stress, organisms might reallocate energy for critical cellular processes by diverting energy consumption away from non-essential processes, such as energy demanding synaptic transmission. Consistent with this prediction, previous work has established that changes in reactive oxygen species can result in altered evoked and spontaneous neurotransmitter release [15]. Furthermore, reduced energy results in the activation of AMP-activated protein kinase, AMPK, which results in increased mitophagy [55] and SKN-1-dependent changes in longevity [28]. Thus, it is interesting to speculate that reduced energy, possibly caused by damaged mitochondria, activates SKN-1, resulting in broad transcriptional changes meant to scavenge oxidants and conserve energy across multiple tissues.
Stress Regulation of Synaptic Transmission
Our results indicate that not all environmental stressors that activate SKN-1 impact synaptic transmission. We found that arsenite, which has previously been shown to strongly activate skn-1 [7], [30] robustly induced skn-1-dependent aldicarb resistance, whereas other classical skn-1 activators did not. Thus, arsenite may be a more potent activator of SKN-1. Alternatively, it is possible that different environmental stressors elicit differential SKN-1 dependent transcriptional programs. SKN-1/Nrf2 is regulated by diverse signaling pathways, and Nrf2 differentially associates with multiple cofactors, providing potential mechanisms for stress-specific and cell-type-specific SKN-1/Nrf2 responses [1]. Indeed, microarray analyses of C. elegans indicate that different stressors activate distinct but overlapping SKN-1-dependent gene expression profiles [7], [17].
While previous microarray studies have identified approximately 200 genes to be transcriptionally regulated by SKN-1 [7], [17], [27], [47], our RNA sequencing indicates that the expression of over 2000 genes is altered in wdr-23 mutants. We do not think that the increased number of genes in our study is due to SKN-1 independent functions of wdr-23, since skn-1 mutations fully block all wdr-23 defects. Instead, this increase is likely due to greater sensitivity of RNA sequencing compared to microarrays, allowing detection of genes with low abundance or restricted tissue distribution [56], [57]. For example, we found the expression of the GPCR signaling genes odr-3 and odr-10, which are both expressed in a few sensory neurons, to be up regulated approximately 6 fold in wdr-23 mutants. Although our behavioral and phenotypic analyses indicate that the sole function of wdr-23 in adults is to negatively regulate SKN-1, we cannot exclude the possibility that some of the transcriptional changes detected in wdr-23 mutants may be SKN-1-independent.
Increases in expression of synaptic transmission genes could explain the reduction in NMJ function in wdr-23 mutants. rab-3/RAB3 expression was increased 3.3 fold in wdr-23 mutants, for example, and over-expression of Rab3 in adrenal chromaffin cells causes reduced exocytosis [58], [59]. However, given that our results show intestinal SKN-1 regulates synaptic function, it seems likely that these synaptic transmission genes are indirectly regulated by SKN-1, possibly by an endocrine mechanism. Identifying the functionally relevant transcriptional targets that mediate the effects of wdr-23 on synaptic transmission will be important in understanding regulation of neuronal function by SKN-1.
Methods
C. elegans Strains
Strains were cultured at 20° using standard methods. All experiments were performed on young adult hermaphrodites unless otherwise indicated. Strains skn-1(zu67), skn-1(zu135) and glo-1(zu391) were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). Strain wdr-23(tm1817) was provided by National BioResource Project (Japan). The wild type reference strain was N2 Bristol. The following strains were also used: drp-1(tm1108), unc-104(e1265), unc-10(e102), nre-1(hd20) lin-15b(hd126), sid-1(qt2), skn-1(lax120), skn-1(lax188), EG4322 ttTi5605;unc-119(ed9), PS6268[Pmyo-3-INVOM-YFP, Pmyo-3-MatrixCFP, rol-6], nuIs122[Pmyo-2-GFP, Pacr-2-synaptophluorin], nuIs152[Pttx-1-RFP, Punc-129-GFP-SNB-1]II, nuIs321[Pmyo-2-GFP, Punc-17-mCherry]. All strains were outcrossed at least 6 times. Transgenic lines expressing integrated fluorescence markers in a skn-1 mutant background resulted in larval arrest, making fluorescence imaging of skn-1 mutant adults impossible.
Molecular Biology
The genomic wdr-23 fragment, wdr-23 promoter, ges-1 promoter, and col-12 promoter were amplified by PCR from mixed-stage genomic DNA. C. elegans cDNA was used to clone all genes and truncations into pPD49.26 or pCFJ151 using standard molecular biology techniques. The WDR-23(DxR) mutation was made by QuikChange PCR (Stratagene) targeting the two DxR motifs (R342H, R389H). For a detailed list of all primers used, refer to Text S1.
Transgenic Lines
Transgenic strains were generated by injecting N2 or wdr-23(tm1817) with expression constructs (2.5–25 ng/µL) and the co-injection marker KP#708 (Pttx-3-rfp, 40 ng/µL) or KP#1106 (Pmyo-2-gfp, 10 ng/µL). Microinjection was performed using standard techniques as previously described [60]. At least three lines for each transgene were examined, and a representative transgene is shown. For a detailed list of all transgenes used, refer to Text S1.
Locomotion Assays
The body bends assay was completed using age matched young adult animals. Animals were individually moved onto a NGM plate without food and allowed to acclimate for 30 seconds. After 30 seconds, body bends were counted per worm for 60 seconds for at least 30 animals per genotype.
Quantification of time spent in locomotion was completed using age matched young adult animals. Single L4 animals were moved onto freshly seeded OP50 NGM plates and incubated overnight at 20°. The next day, at least 20 animals per genotype were evaluated for time spent in forward locomotion. To induce locomotion, the lid of the plate was removed and quickly replaced. This generally resulted in a quick bout of backward locomotion followed by forward locomotion. Quantification began once the animal resumed forward locomotion. Time was scored using Ethotimer software (Thomas Lab, University of Washington).
Pharmacology
For analysis of sensitivity to the drugs aldicarb and levamisole, paralysis of adult worms was scored every 10 or 15 minutes, starting at 40 minutes, using 1.0 mM aldicarb (Bayer) or 200 µM levamisole (Sigma) unless otherwise specified. For experiments, three replicates of at least 20 worms per genotype were placed on NGM plates supplemented with paralytic, and the number of worms paralyzed on each plate was counted to calculate the percent paralyzed at each time point per genotype. The percentages were averaged at each time point per genotype and plotted graphically. For each experiment, the genotype was blind to the scorer, and the analysis was repeated at least two times. Results were analyzed using GraphPad Prism 5 software, and significances were calculated by one-way repeated measures ANOVA with Tukey's test for multiple comparisons.
For pretreatment with sodium arsenite (RICCA Chemicals), age matched L4 animals were placed on NGM plates supplemented with 1.0 or 2.0 mM sodium arsenite. Animals were incubated at 20° for 14 hours, then moved to NGM without arsenite for recovery for 3 hours prior to aldicarb assay.
Microscopy and Analysis
Fluorescence microscopy experiments were performed as previously described [33]. Our preliminary studies indicated expression of unc-129, the DA motor neuron promoter used by Ch'ng et al., 2008, increased modestly in a skn-1 dependent manner in wdr-23 mutants (data not shown). Therefore, we used the unc-17 promoter for all of our quantitative imaging. Expression of unc-17 does not change in wdr-23 mutants according to an unc-17 reporter construct, qRT-PCR or RNAseq (Figure 3B, Table 3 and Table S4). To image animals, L4 or adult worms were paralyzed using 2,3-butanedione monoxime (BDM, 30 µg/µL; Sigma) and mounted on 2% agarose pads for imaging. Images of synapses were captured from dorsal axons of DA neurons near the posterior gonadal bend of the worm. At least 30 worms were imaged for all quantitative experiments. For all fluorescence microscopy experiments, images were captured with a Nikon eclipse 90i microscope equipped with a Nikon PlanApo 60× or 100× objective (NA = 1.4) and a Photometrics Coolsnap ES2 camera. Metamorph 7.0 software (Universal Imaging/Molecular Devices) was used to capture serial image stacks, and the maximum intensity projection was used for analysis of the dorsal cords. Line scans of the maximum intensity projection image were also recorded using Metamorph. The fluorescence intensity values were then quantified using Puncta 6.0 software written with Igor Pro (Wavemetrics), as previously described [33], [34]. For all experiments, fluorescence values were normalized to the values of 0.5 µm FluoSphere beads (Invitrogen) captured during each imaging session. This was performed to provide a standard for comparing absolute fluorescence levels between animals from different sessions.
RNA Interference
Feeding RNAi: Feeding RNAi was completed as described [61]. Briefly, animals were synchronized by a hypochloride/sodium hydroxide egg preparation and placed on RNAi plates containing HT1115(DE3) bacteria specific for unc-17, wdr-23, or the empty vector L4440 from the Ahringer library [61]. Aldicarb analysis was completed three or four days after the egg preparation.
Hairpin RNAi: The inverted repeat hairpin targeting wdr-23 was constructed as described [62] and injected directly into sid-1(qt2) mutants. Multiple lines were analyzed for each injection.
Whole-Genome Sequencing
From an ethyl methanesulfonate (EMS) screen to identify genes that suppress the small size and aldicarb response of wdr-23 mutants, we isolated the allele vj24. DNA was extracted using Qiagen Gentra Purgene system from mixed staged wdr-23;vj24 heterozygous animals outcrossed three times. Sequencing was completed in single reads of 101 bp using an Illumnia HiSeq2000 with HiSeqv3 flow cell at an average sequencing depth of 38. Data was analyzed using MAQGene [63].
RNA Sequencing
Total RNA was isolated from approximately 10,000 mixed stage animals for wild type and wdr-23 mutants using Stat60 (Tel-test B, Texas). Transcriptome libraries were prepared using TruSeq RNA sample preparation kit (Illumina) according to manufacturer's TruSeq protocol rev. A. In brief, polyadenylated mRNA was isolated from 0.1–4 ug of total RNA using oligo(dT) beads, fragmented, first strand cDNA synthesized using random primers and SuperScript II reverse transcriptase (Invitrogen) followed by synthesis of a second strand. Ends of resulting double-stranded fragments were repaired, 3′ ends were adenylated and adapters were ligated to the fragment, completing the library preparation. Library then was amplified by PCR and quality and quantity of libraries were evaluated on BioAnalyzer 2100 (Agilent). Sequencing was performed on HiSeq2000 (Illumina). Sequencing reads were aligned to the C. elegans genome (release WS210) using TopHat [45]. Libraries were sequenced to a depth of 36.1 and 34.9 million reads for wild type and wdr-23 mutants, respectively. Gene models were downloaded from ENSEMBL and quantified using Cufflinks. Differentially expressed genes at false discovery rate (FDR) of 0.05 were identified using the Cuffdiff module of the Cufflinks package.
qRT–PCR
cDNA was synthesized from total RNA using ProtoScript M-MuLV RT-PCR (NEB). SYBR Premix Ex Taq (Clontech) was used in an BioRad Chromo4 Real-Time PCR Detector in triplicate. Data was analyzed using the comparative Ct method. Relative mRNA levels were normalized to rpl-2, and calculated from at least two biological replicates. Primers were designed to be intron-spanning.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SykiotisGP, BohmannD (2010) Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal 3: re3.
2. HalliwellB (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97 : 1634–1658.
3. SykiotisGP, BohmannD (2008) Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell 14 : 76–85.
4. AnJH, BlackwellTK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17 : 1882–1893.
5. KobayashiM, YamamotoM (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul 46 : 113–140.
6. ShihAY, JohnsonDA, WongG, KraftAD, JiangL, et al. (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23 : 3394–3406.
7. OliveiraRP, Porter AbateJ, DilksK, LandisJ, AshrafJ, et al. (2009) Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8 : 524–541.
8. LeeJM, ShihAY, MurphyTH, JohnsonJA (2003) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278 : 37948–37956.
9. LeeJM, CalkinsMJ, ChanK, KanYW, JohnsonJA (2003) Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 278 : 12029–12038.
10. EscartinC, WonSJ, MalgornC, AureganG, BermanAE, et al. (2011) Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J Neurosci 31 : 7392–7401.
11. KraftAD, JohnsonDA, JohnsonJA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24 : 1101–1112.
12. ChanJY, KwongM, LuR, ChangJ, WangB, et al. (1998) Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J 17 : 1779–1787.
13. BishopNA, GuarenteL (2007) Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447 : 545–549.
14. ParkSK, LinkCD, JohnsonTE (2010) Life-span extension by dietary restriction is mediated by NLP-7 signaling and coelomocyte endocytosis in C. elegans. FASEB J 24 : 383–392.
15. KeatingDJ (2008) Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 104 : 298–305.
16. SaykallyJN, RachmanyL, HaticH, ShaerA, RubovitchV, et al. (2012) The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience
17. ParkSK, TedescoPM, JohnsonTE (2009) Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8 : 258–269.
18. LandisGN, AbduevaD, SkvortsovD, YangJ, RabinBE, et al. (2004) Similar gene expression patterns characterize aging and oxidative stress in Drosophila melanogaster. Proc Natl Acad Sci U S A 101 : 7663–7668.
19. ChoeKP, PrzybyszAJ, StrangeK (2009) The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol Cell Biol 29 : 2704–2715.
20. HasegawaK, MiwaJ (2010) Genetic and cellular characterization of Caenorhabditis elegans mutants abnormal in the regulation of many phase II enzymes. PLoS One 5: e11194.
21. AngersS, LiT, YiX, MacCossMJ, MoonRT, et al. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443 : 590–593.
22. HeYJ, McCallCM, HuJ, ZengY, XiongY (2006) DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev 20 : 2949–2954.
23. HigaLA, WuM, YeT, KobayashiR, SunH, et al. (2006) CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol 8 : 1277–1283.
24. SieburthD, Ch'ngQ, DybbsM, TavazoieM, KennedyS, et al. (2005) Systematic analysis of genes required for synapse structure and function. Nature 436 : 510–517.
25. MillerKG, AlfonsoA, NguyenM, CrowellJA, JohnsonCD, et al. (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A 93 : 12593–12598.
26. TulletJM, HertweckM, AnJH, BakerJ, HwangJY, et al. (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132 : 1025–1038.
27. PaekJ, LoJY, NarasimhanSD, NguyenTN, Glover-CutterK, et al. (2012) Mitochondrial SKN-1/Nrf Mediates a Conserved Starvation Response. Cell Metab 16 : 526–537.
28. OnkenB, DriscollM (2010) Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One 5: e8758.
29. LiX, MatilainenO, JinC, Glover-CutterKM, HolmbergCI, et al. (2011) Specific SKN-1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS Genet 7: e1002119.
30. InoueH, HisamotoN, AnJH, OliveiraRP, NishidaE, et al. (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev 19 : 2278–2283.
31. LiaoVH, LiuJT, LiWH, YuCW, HsiehYC (2010) Caenorhabditis elegans bicarbonate transporter ABTS-1 is involved in arsenite toxicity and cholinergic signaling. Chem Res Toxicol 23 : 926–932.
32. JospinM, QiYB, StawickiTM, BoulinT, SchuskeKR, et al. (2009) A neuronal acetylcholine receptor regulates the balance of muscle excitation and inhibition in Caenorhabditis elegans. PLoS Biol 7: e1000265.
33. Ch'ngQ, SieburthD, KaplanJM (2008) Profiling synaptic proteins identifies regulators of insulin secretion and lifespan. PLoS Genet 4: e1000283.
34. DittmanJS, KaplanJM (2006) Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc Natl Acad Sci U S A 103 : 11399–11404.
35. SieburthD, MadisonJM, KaplanJM (2007) PKC-1 regulates secretion of neuropeptides. Nat Neurosci 10 : 49–57.
36. LiC, KimK (2008) Neuropeptides. Worm Book 1–36.
37. PerryMD, LiW, TrentC, RobertsonB, FireA, et al. (1993) Molecular characterization of the her-1 gene suggests a direct role in cell signaling during Caenorhabditis elegans sex determination. Genes Dev 7 : 216–228.
38. FireA (1999) RNA-triggered gene silencing. Trends Genet 15 : 358–363.
39. SchmitzC, KingeP, HutterH (2007) Axon guidance genes identified in a large-scale RNAi screen using the RNAi-hypersensitive Caenorhabditis elegans strain nre-1(hd20) lin-15b(hd126). Proc Natl Acad Sci U S A 104 : 834–839.
40. WinstonWM, MolodowitchC, HunterCP (2002) Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 295 : 2456–2459.
41. DurieuxJ, WolffS, DillinA (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 : 79–91.
42. MeissnerB, RogalskiT, ViveirosR, WarnerA, PlastinoL, et al. (2011) Determining the sub-cellular localization of proteins within Caenorhabditis elegans body wall muscle. PLoS One 6: e19937.
43. LabrousseAM, ZappaterraMD, RubeDA, van der BliekAM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4 : 815–826.
44. Frokjaer-JensenC, DavisMW, HopkinsCE, NewmanBJ, ThummelJM, et al. (2008) Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40 : 1375–1383.
45. TrapnellC, WilliamsBA, PerteaG, MortazaviA, KwanG, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28 : 511–515.
46. DennisGJr, ShermanBT, HosackDA, YangJ, GaoW, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: P3.
47. PrzybyszAJ, ChoeKP, RobertsLJ, StrangeK (2009) Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech Ageing Dev 130 : 357–369.
48. Thomas-ChollierM, SandO, TuratsinzeJV, JankyR, DefranceM, et al. (2008) RSAT: regulatory sequence analysis tools. Nucleic Acids Res 36: W119–127.
49. BlackwellTK, BowermanB, PriessJR, WeintraubH (1994) Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science 266 : 621–628.
50. BegAA, ErnstromGG, NixP, DavisMW, JorgensenEM (2008) Protons act as a transmitter for muscle contraction in C. elegans. Cell 132 : 149–160.
51. LoSC, HanninkM (2008) PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res 314 : 1789–1803.
52. LoSC, HanninkM (2006) PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem 281 : 37893–37903.
53. KobayashiA, KangMI, OkawaH, OhtsujiM, ZenkeY, et al. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24 : 7130–7139.
54. ItohK, WakabayashiN, KatohY, IshiiT, IgarashiK, et al. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13 : 76–86.
55. EganDF, ShackelfordDB, MihaylovaMM, GelinoS, KohnzRA, et al. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331 : 456–461.
56. WangZ, GersteinM, SnyderM (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 : 57–63.
57. TariqMA, KimHJ, JejelowoO, PourmandN (2011) Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Res 39: e120.
58. JohannesL, LledoPM, RoaM, VincentJD, HenryJP, et al. (1994) The GTPase Rab3a negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J 13 : 2029–2037.
59. ThiagarajanR, TewoldeT, LiY, BeckerPL, RichMM, et al. (2004) Rab3A negatively regulates activity-dependent modulation of exocytosis in bovine adrenal chromaffin cells. J Physiol 555 : 439–457.
60. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10 : 3959–3970.
61. KamathRS, AhringerJ (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30 : 313–321.
62. Strange K (2006) C. elegans Methods and Applications. Totowa, New Jersey: Humana Press.
63. BigelowH, DoitsidouM, SarinS, HobertO (2009) MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods 6 : 549.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy