
A Role for the Malignant Brain Tumour (MBT) Domain Protein LIN-61 in DNA Double-Strand Break Repair by Homologous Recombination
Malignant brain tumour (MBT) domain proteins are transcriptional repressors that function within Polycomb complexes. Some MBT genes are tumour suppressors, but how they prevent tumourigenesis is unknown. The Caenorhabditis elegans MBT protein LIN-61 is a member of the synMuvB chromatin-remodelling proteins that control vulval development. Here we report a new role for LIN-61: it protects the genome by promoting homologous recombination (HR) for the repair of DNA double-strand breaks (DSBs). lin-61 mutants manifest numerous problems associated with defective HR in germ and somatic cells but remain proficient in meiotic recombination. They are hypersensitive to ionizing radiation and interstrand crosslinks but not UV light. Using a novel reporter system that monitors repair of a defined DSB in C. elegans somatic cells, we show that LIN-61 contributes to HR. The involvement of this MBT protein in HR raises the possibility that MBT–deficient tumours may also have defective DSB repair.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003339
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003339
Summary
Malignant brain tumour (MBT) domain proteins are transcriptional repressors that function within Polycomb complexes. Some MBT genes are tumour suppressors, but how they prevent tumourigenesis is unknown. The Caenorhabditis elegans MBT protein LIN-61 is a member of the synMuvB chromatin-remodelling proteins that control vulval development. Here we report a new role for LIN-61: it protects the genome by promoting homologous recombination (HR) for the repair of DNA double-strand breaks (DSBs). lin-61 mutants manifest numerous problems associated with defective HR in germ and somatic cells but remain proficient in meiotic recombination. They are hypersensitive to ionizing radiation and interstrand crosslinks but not UV light. Using a novel reporter system that monitors repair of a defined DSB in C. elegans somatic cells, we show that LIN-61 contributes to HR. The involvement of this MBT protein in HR raises the possibility that MBT–deficient tumours may also have defective DSB repair.
Introduction
DNA is maintained in the cell as chromatin: double-stranded DNA wrapped around core histone octomers to form nucleosome subunits. Chromatin folds into higher order structures depending on how tightly DNA is wrapped around the histones and how closely the nucleosomes interact [1]. Condensed chromatin acts as a physical barrier that restricts DNA access and therefore must be remodelled to enable various cellular processes such as gene transcription, DNA replication and DNA repair [2]. This is principally achieved by post-translational modification to the N-terminal tails of histones. One example of this is the methylation of lysine residues, which alters the degree of chromatin compaction and provides a binding site for the recruitment of non-histone proteins such as malignant brain tumour (MBT) domain proteins [2]. Once bound to histones, MBT domain proteins condense chromatin and repress transcription of target genes [3]. The MBT domain is a highly conserved motif of approximately 100 amino acids in length found throughout metazoans from C. elegans to humans [4].
Some MBT domain proteins act together with Polycomb group (PcG) repressor complexes that are best known for establishing and maintaining gene expression patterns during development [4]. The C. elegans MBT protein LIN-61 is also implicated in transcriptional regulation. It is a member of the synthetic multivulva (synMuv) class B group of proteins that act redundantly with synMuvA proteins to repress transcription of lin-3 EGF and lin-60 Ras [5]–[7]. Separate to its role within the synMuvB pathway, we found lin-61 is also involved in maintaining genome stability. Worms depleted of lin-61 have elevated rates of germline and somatic mutation, including small DNA insertions and deletions, but how LIN-61 maintains the genome fidelity was unknown [8]. Intriguingly, other MBT proteins have been shown to act as tumour suppressors: lethal(3)malignant brain tumour [l(3)mbt)] mutants of Drosophila develop malignant transformations of the adult optic neuroblast and ganglion mother cells of the larval brain [9]; furthermore, the human MBT domain genes L3MBTL2, L3MBTL3 and SCML2 are mutated in rare cases of medulloblastoma [10]. Also, depletion of L3MBTL1 (another LIN-61-related protein) causes genome instability [11]. Therefore it appears MBT proteins may have a general role in genome stability. It is not known how these proteins prevent tumourigenesis or protect the genome, but their ability to repress transcription likely plays a central role considering that the l(3)mbt malignancies of Drosophila ectopically express germline genes, the expression of which is required for tumour growth [12]. Preventing the expression of germline genes in somatic tissues may be a conserved function of MBT proteins because lin-61 mutants also express germline genes in the soma in a temperature-dependent manner [13].
As well as regulating transcription, an increasing number of chromatin-remodelling proteins (including PcG proteins) have been found to act within the DNA damage response (DDR). These proteins accumulate at sites of DNA damage where they locally modify chromatin to allow the recruitment of DNA repair proteins [14]. In the present study we investigate the cause of genomic instability in lin-61 mutants. We show that LIN-61 acts within the DDR where it is needed for efficient double-strand break (DSB) repair in both the germline and somatic cells of C. elegans. LIN-61 promotes DSB repair by homologous recombination (HR), but not the competing pathways, non-homologous end joining (NHEJ) or single-strand annealing (SSA). Despite the requirement for LIN-61 in HR, it is dispensable for meiotic recombination and the DNA damage checkpoints (cell cycle arrest and apoptosis) in the germline. We also use a novel GFP-based HR reporter assay that confirms LIN-61 is needed for HR. This reporter monitors the repair of a single defined DSB and is a new tool for measuring HR in C. elegans somatic cells. This is the first report demonstrating that an MBT protein promotes DNA repair and provides an explanation for why MBT-deficient cells have genomic instability.
Results
Genomic instability in lin-61 mutants
To investigate how LIN-61 contributes to genomic stability, we obtained three independently generated null alleles of lin-61 (n3809, pk2225 and tm2649; Figure 1A and Text S1). The fourth MBT domain [essential for binding H3K9me2/3; [15]] is truncated or deleted in each of the mutant LIN-61 proteins. Moreover, lin-61 mRNA is reduced approximately four-fold in n3809 and pk2225, likely due to nonsense-mediated decay (Figure 1B). Each of the three mutants produced small broods (17–24% fewer progeny than wild types; Figure 1C). This can be symptomatic of genomic instability as DNA repair mutants such as brc-1, rfs-1, blm-1 and smc-5/-6 also have small broods [16]–[19]. In accordance with their reduced fecundity, lin-61 mutants had considerably smaller germlines than wild types and contained fewer nuclei in the mitotic compartment (Figure 1D–1E). What is more, there were signs of DNA damage in these cells: their mitotic nuclei contained considerably more spontaneous RAD-51 foci than those of wild types (Figure 1F). RAD-51 is the DNA strand exchange protein, which accumulates at DSBs and blocked replication forks, and therefore is a marker for DNA damage [20]–[22].

LIN-61 is required for resistance to ionizing radiation but dispensable for meiotic recombination
Since lin-61 mutant germ cells displayed genomic instability and signs of persistent spontaneous DSBs, we wondered whether lin-61 mutants were sensitive to ectopically induced DSBs. We found that the germ cells of lin-61 mutants were hypersensitive to ionizing radiation (IR), which is a potent inducer of DSBs (Figure 2A). Also primordial germ cells that are arrested in the G2 stage of the cell cycle in L1 stage larvae, are hypersensitive to IR in lin-61 mutants animals (Figure S1).

The LIN-61 paralog, called MBTR-1 (Malignant Brain Tumour Repeat containing protein 1), shares a high degree of sequence conservation with LIN-61 and both proteins are comprised almost entirely of four MBT domains (Figure S2A). We wondered whether MBTR-1 too might be needed for resistance to IR-induced DSBs. To test this, we challenged mbtr-1(n4775) mutants with IR but found that they were not more sensitive than wild type controls (Figure S2B). Therefore LIN-61, but not the closely related MBT domain protein MBTR-1, is required for resistance to IR-induced DSBs in germ cells.
The IR-hypersensitivity of lin-61 mutant germlines suggested that LIN-61 might be required for DSB repair during gametogenesis. We therefore investigated if LIN-61 also had a role in the repair of programmed DSBs that arise during meiosis. Meiotic DSB repair is required for the proper segregation of chromosomes to gametes and involves the repair of programmed DSBs introduced by the topoisomerase-like protein SPO-11 [23]. These DSBs are repaired by HR using the homologous chromosome as the repair template (interhomolog HR). The progression of DSB repair can be monitored in meiosis by following RAD-51 foci, which first appear at prophase, peak at early/mid-pachytene, and are resolved by late pachytene once DSB repair is completed [24]. The distribution of RAD-51 foci in lin-61 meiotic cells was indistinguishable from those of wild types (Figure S3). This indicated that repair of SPO-11-introduced DSBs was unperturbed in lin-61 mutants. Interhomolog HR enables crossover (CO) formation, which establishes the physical connection (chiasmata) that holds homologs together until their separation at the first meiotic cell division. Diakinesis stage oocytes of lin-61 mutants contained the correct complement of six bivalents (paired homologs), which indicated that CO formation was competent in these mutants. Furthermore, lin-61 mutants produced mostly viable progeny and did not display an increased incidence of males (Him) phenotype (Figure 1C). Failed meiotic recombination causes nondisjunction and aneuploidy due to the uncontrolled segregation of chromosomes to gametes, which manifests as embryonic lethality and the Him phenotype [25]. We conclude that LIN-61 is necessary for the repair of IR-induced DSBs but dispensable for CO formation and meiotic recombination. This phenotype is paralleled by the HR mutant brc-1 and the cohesin-like mutants smc-5/-6. These mutants are IR hypersensitive due to defective DSB repair by HR that uses the sister chromatid (intersister HR) [19], [26], [27]. Our observation that lin-61 mutants were hypersensitivity to IR suggested that LIN-61 might also contribute to intersister HR.
lin-61 mutants are hypersensitive to interstrand crosslinks but not UV lesions
In addition to repairing IR-induced DSBs, intersister HR is needed for repair of interstrand crosslinks (ICLs). ICLs are particularly cytotoxic lesions that block the replication fork by covalently linking opposing strands of double-stranded DNA [28]. During ICL repair, the crosslinked lesion is excised, thus producing a DSB substrate for intersister HR [29]. HR-deficient mutants like brc-1, or the rad-51 paralog rfs-1 are therefore hypersensitive to ICLs [21]. Consistent with LIN-61 having a possible role in intersister HR, we found that lin-61 mutants were hypersensitive to nitrogen mustard (HN2), which is a potent inducer of ICLs (Figure 2B).
Other DNA lesions that block replication forks (such as bulky photoadducts made by UV light) do not cause a DSB and do not require HR for repair. Instead, translesion synthesis (TLS) DNA polymerases such as POLH-1 bypass these lesions to allow replication to proceed [30]. polh-1 mutants are therefore hypersensitive to UV-C [31] but HR-deficient mutants such as rfs-1 are not [21]. We found that lin-61 mutants were not hypersensitive to UV-C (Figure 2C). The sensitivity of lin-61 mutants to IR and HN2, but not UV-C, suggested that LIN-61 may promote DNA repair through HR, but is not required for the repair of other replication-blocking lesions such as photoadducts.
LIN-61 has a role in HR, but not NHEJ, in somatic cells
LIN-61 is broadly expressed in somatic and germ cells throughout development [6]. To determine if LIN-61 contributes to DSB repair in somatic cells, as it does in germ cells, we used established assays that test the proficiency of HR, as well as the other major DSB repair route, NHEJ [32]. Somatic cells use either HR or NHEJ depending on developmental context and phase of the cell cycle. HR is active during S and G2 phases (when sister chromatids are closely aligned), whereas NHEJ can be performed throughout the duration of the cell cycle, but is especially important during G1 when HR is unavailable [33]. Early stage embryonic cells (<6 hours post fertilisation) rapidly transition between S phase and M phase, without G1 and G2 gap phases [34], [35] and are particularly reliant on HR for DSB repair [32] (Figure 3A). Accordingly, early stage embryos of HR-deficient mutants are very sensitive to IR, while those of NHEJ-deficient mutants are not [32]. To test whether lin-61 promotes HR in somatic cells, we scored the viability of γ-irradiated early stage lin-61 embryos. These embryos were indeed hypersensitive to IR, which was indicative of an HR defect (Figure 3B). Their degree of IR sensitivity was similar to that of HR-deficient brc-1 embryos. While HR is the dominant DSB repair route in early embryos, NHEJ is the major repair pathway in late stage embryos and arrested L1 larvae because most of their cells are arrested in G1 [32] (Figure 3C). NHEJ-deficient L1 larvae have delayed or arrested growth in response to IR [32]. We found that wild type, lin-61(n3809) and lin-61(pk2225) L1 larvae did not display substantial growth delay following IR, whereas most NHEJ-deficient cku-80 mutants failed to develop to the L4 stage 48 hours after irradiation (Figure 3D). L1 larvae of the HR-deficient mutant, brc-1, were also not hypersensitive to IR (Figure S4). Taken together, these results suggest that LIN-61 has a role in repairing DSBs by HR, but not NHEJ, in somatic cells.

LIN-61 is not required for intersister HR in meiotic nuclei
Although lin-61 mutants phenocopy brc-1 mutants in many aspects of genome stability, they also differ in some important aspects. For example, brc-1 mutants display the Him phenotype, while lin-61 mutants do not. Him is an indication of problems with chromosome segregation at meiosis. Like brc-1 mutants, lin-61 mutants are able to successfully complete meiosis, indicating that their interhomolog HR is proficient. However, by genetically disrupting the synaptonemal complex (SC), and thereby preventing interhomolog HR, it has been possible to demonstrate that BRC-1 contributes to meiotic intersister HR [27]. Adamo and colleagues observed that chromosomal fragments appear in the diakinesis stage nuclei of brc-1 mutants that were depleted of key SC components [27]. Using this approach we tested whether LIN-61 also has a role in meiotic intersister HR. In contrast to brc-1 mutants, neither the oocytes of lin-61(pk2225) or lin-61(n3809) contained chromosomal fragmentation after depletion of the core SC component, SYP-2 (Figure 4A). These data, together with those showing normal RAD-51 kinetics and successful chiasmata formation in lin-61 mutants (Figure S3 and Figure 4A), indicate that LIN-61 is dispensable for HR in meiotic cells.

LIN-61 contributes to DSB repair in mitotic germ cells but not meiotic germ cells
lin-61 mutants are proficient in the repair, at meiosis, of SPO-11-introduced DSBs (using both intersister and interhomolog repair) but are hypersensitive to IR. To confirm that LIN-61 is required for DSB repair specifically in mitotic germ cells we used an assay that directly tests whether DSBs are adequately repaired in irradiated germ cells. Completion of DSB repair can be determined in germ cells by observing chromosomes at diakinesis because chromosome fragments are present if DSBs are unrepaired [36]. In the absence of exogenous damage, the diakinesis stage oocytes of lin-61 mutants contained six bivalents and were not fragmented (Figure 4B). This demonstrated that DSBs induced by SPO-11 were efficiently repaired in lin-61 mutants, as discussed earlier. Strikingly however, both lin-61 mutants and the HR-deficient mutant brc-1 had severely fragmented chromosomes 48 hours after γ-irradiation (Figure 4B–4C). We anticipated that these nuclei could have been located within the mitotic zone at the time of irradiation, having subsequently migrated to the diakinesis stage 48 hours later. Failure to repair the introduced DSBs could therefore be due to defective HR whilst in the mitotic zone, or later whilst in the meiotic zone, or both. To distinguish between these possibilities we analysed earlier time points following irradiation (7 h and 24 h). For these time points, the nuclei being analysed were in meiosis when DSBs were introduced. We found that brc-1 mutants had fragmented chromosomes at these earlier time points (7 h and 24 h) (Figure 4B–4C), which is consistent with BRC-1 acting in meiotic DSB repair [27]. In contrast, lin-61 mutants, like wild types, rarely had fragmented chromosomes at early time points following irradiation (Figure 4B–4C). Thus while BRC-1 contributes to DSB repair in both mitotic and meiotic cells, LIN-61 seems to promote DSB repair only in mitotic cells. In accordance with that notion, we found that brc-1 mutants were more sensitive to IR than lin-61 mutants (Figure 4D). Moreover, lin-61 brc-1 double mutants were no more sensitive to IR than brc-1 single mutants suggesting that lin-61 acts within the brc-1 genetic pathway (Figure 4D).
LIN-61 is dispensable for RAD-51 focus formation
Having established that LIN-61 promotes DSB repair via HR, we looked to address which step of HR fails in lin-61 mutants. The first stages of HR involve the nucleolytic processing at the DSB to expose single stranded 3′ overhangs (DNA end resection) and subsequent coating of these overhangs with RAD-51. RAD-51 foci rapidly formed in the γ-irradiated mitotic germ cells of both wild types and lin-61 mutants (Figure 5A). Foci were detected at a very early time point after γ-irradiation (10 minutes), which showed that DNA end resection was unperturbed in these cells (Figure 5A). The loading of RAD-51 at SPO-11-induced DSBs was also normal in lin-61 meiotic cells, as discussed earlier (Figure S3). Together this showed that DNA end resection at IR-induced and SPO-11-induced DSBs, as well as the loading of RAD-51 on resected DNA, was normal in lin-61 mutants. The number of RAD-51 foci that formed in γ-irradiated germ cells was similar between wild types and lin-61 mutants (4–5 foci per nucleus) (Figure 5B). Since the DNA in wild type and lin-61 nuclei were equally susceptible to IR, the hypersensitivity of these mutants was not due to an elevated damage load.

A novel GFP-based HR reporter system confirms that LIN-61 is required for HR in somatic cells
While IR is a potent source of DSBs, it also causes oxidative damage to proteins and cell membranes [37]. To confirm that the hypersensitivity displayed by lin-61 mutants was due to defective DSB repair (and not other types of damage), we developed an assay that specifically measures HR-mediated repair of a defined DSB. This assay was based on the DR-GFP reporter system, which has been used extensively to measure HR proficiency in cultured human cells [38]. Such an assay was previously unavailable to the C. elegans researcher. The new C. elegans reporter consisted of a gfp gene in which part of the open reading frame had been deleted and replaced by an I-SceI endonuclease recognition site, which rendered the GFP non-functional, and provided the defined location where the DSB could be introduced (Figure 6A). A fragment of gfp containing the sequences disrupted by the I-SceI site (but by itself non-functional) was located downstream of the reporter and served as a template for synthesis-dependent strand annealing (SDSA) (Figure 6A). SDSA is a sub-pathway of HR that results in gene conversion rather than a CO and is the most common HR pathway used to repair two-sided DSBs [39]. The reporter was designed such that repair of the DSB by SDSA (but not a CO pathway) would be able to restore expression to the corrupted gfp gene. Non-HR pathways such as NHEJ or SSA are unable to produce functional GFP (Figure 6B).
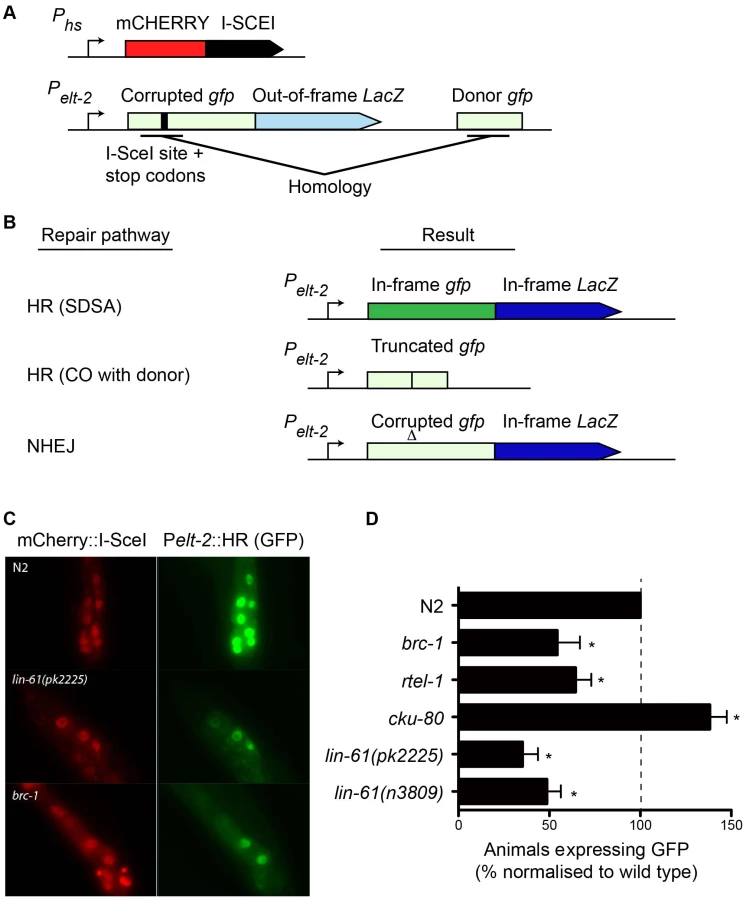
We created a transgenic strain that carried both the HR reporter and heat-shock inducible I-SceI endonuclease. I-SceI was fused to mCherry so that its expression could be easily monitored by epifluorescence. Since it is thought HR does not occur in postmitotic cells (i.e. G1/G0 stage cells), we chose to express the reporter in intestinal cells using the elt-2 promoter as their nuclei undergo endoreplication (S phase without mitosis) at several points during post-embryonic development [40]. We first confirmed that induction of mCherry::I-SceI resulted in GFP expression. 60–80% of wild type worms expressed GFP in intestinal nuclei 24 hours after mCherry::I-SceI expression. Importantly, reporter activation was dependent upon DSB induction because non-heat shocked worms did not express GFP (data not shown). Also, GFP expression was dependent upon the donor gfp sequences since a disabled version of the HR reporter, which lacked these sequences, was not able to express GFP (Figure S5). To confirm that GFP expression depended on HR, we tested the effect brc-1 mutation had on the reporter. BRC-1 promotes intersister HR in meiotic cells [27], and likely in somatic cells as well [41]. Indeed, brc-1 mutants had significantly reduced frequency of HR reporter activation (Figure 6C–6D). This confirmed that the assay provided a measure of HR proficiency. We also used an rtel-1 mutation to test whether reporter activation was dependent on the SDSA pathway. RTEL-1 is thought to influence HR pathway choice by removing the invaded DNA strand from its homologous template, which has the effect of promoting SDSA at the expense of CO outcomes [42]. The role of rtel-1 in somatic cells was previously untested but we found that rtel-1 mutants also had significantly reduced rates of HR reporter activation (Figure 6D). Therefore RTEL-1 likely promotes SDSA in somatic cells as it does in meiotic cells. A previous study showed that DSB repair pathways are dynamic and are in competition in C. elegans somatic cells such that the inhibition of one pathway caused increased activity in the others [41]. We therefore reasoned that inhibiting NHEJ should increase the frequency of HR reporter activation. As predicted, blocking NHEJ by cku-80 mutation resulted in substantial elevation of HR activity. More cku-80 animals expressed GFP than wild types (Figure 6D). This increase was likely an underestimation of HR activity as the GFP was also expressed much more brightly in cku-80 mutants than wild types. Brighter GFP likely results from multiple HR reporter genes being activated within a single cell. These experiments demonstrated that the HR reporter is able to measure relative changes in HR activity, in both HR-deficient and HR-hyperactive mutants.
Importantly, we found that both lin-61(n3809) and lin-61(pk2225) mutants showed a substantial reduction in the frequency of HR reporter activation compared with wild types (Figure 6D). In fact HR activation in lin-61 mutants was reduced to brc-1 levels. This confirmed LIN-61 is needed for DSB repair by the HR pathway. Further, it indicated that IR hypersensitivity of lin-61 mutants was likely due to defective DSB repair rather than other types of IR-induced cellular damage. While HR repairs DSBs in an error-free way, other DSB repair pathways such as NHEJ and SSA are error-prone processes. To test whether LIN-61 contributes to mutagenic DSB repair routes, we constructed a second reporter gene that specifically monitored SSA. This SSA reporter was similar to the HR reporter as both were expressed in intestinal nuclei and both received a single DSB from the mCherry::I-SceI enzyme, however the SSA reporter could only become active following an SSA event, and not an HR event (Figure S6A). We found that lin-61 mutants did not have reduced SSA activity but actually had increased SSA reporter activation compared to wild types (Figure S6B–S6C), in line with lin-61 mutants being HR-defective. A similar shift towards SSA has previously been found for DSB repair in brc-1 mutant animals [41]. We conclude that LIN-61 is necessary for efficient HR in somatic cells but is dispensable for SSA in somatic intestinal cells. Assays that measure sensitivity to DNA-damaging agents revealed that embryonic and germline cells of lin-61 mutants are defective for DSB repair (Figure 2 and Figure 3). The data generated using the HR and SSA reporters demonstrated that cell types other than those of the germline and embryo are defective for DSB repair in lin-61 mutants. Together, these complementary experiments suggested that lin-61 mutants have a systemic defect in DSB repair.
DNA damage checkpoints are proficient in lin-61 mutants
Sensitivity to DNA damage can be caused by failure to activate DNA damage checkpoints [43]. The G2/M checkpoint is triggered in response to DNA damage and keeps mitotic germ cells in G2 phase to provide sufficient time for DNA repair (Figure 7A) [44]. Arrested cells do not divide, but continue to grow, making them readily identifiable by their enlarged size [43]. Following exposure to IR, all three lin-61 mutants displayed proficient cell cycle arrest. Like wild type worms (and mbtr-1 mutants that are not IR sensitive), the lin-61 mutants had enlarged mitotic nuclei and a reduced number of germ cells 24 hours after γ-irradiation (Figure 7B–7C).

In addition to the G2/M checkpoint, DNA damage also triggers apoptosis in pachytene stage meiotic cells via a process dependent upon the p53 homologue, CEP-1 [43], [45]. Upon challenge with IR, apoptotic corpses accumulated in the germlines of wild type, lin-61(n3809) and lin-61(pk2225) animals, while cep-1 mutants failed to undergo DNA damage-dependent apoptosis (Figure 6D–6E). CEP-1 drives the apoptotic programme by up-regulating egl-1/BH3-only transcription [43], [45], [46]. In response to IR, egl-1 expression was increased in wild type and lin-61 worms, but not cep-1 mutants, as determined by qRT-PCR (Figure 7F). Together these results indicated that the activation of DNA damage checkpoints (cell cycle arrest and apoptosis) was normal in lin-61 mutants. The hypersensitivity of lin-61 mutants to IR could therefore not be attributed to defective checkpoint activation.
DNA repair genes are expressed at normal levels in lin-61 mutants
Since LIN-61 is a transcriptional repressor, we checked whether DDR genes were appropriately expressed in lin-61 mutants, as this could be the underlying cause of their HR defect. Using microarrays, we compared the expression profiles of wild types and lin-61 animals. Young adult worms (24 hours post L4) were analysed in order to increase the proportion of germ cells present in the samples, considering LIN-61 is needed for repair of DSBs in both somatic and germ cells. Microarrays were performed on two different lin-61 alleles (n3809 and pk2225) in order to control for changes in gene expression that were due by background mutations present within only one of the single strains. 58 genes were identified that, in both mutants, had a 1.5-fold or greater change in expression level (p-value<0.01) (Table S1). Most of these alternatively expressed genes were upregulated in lin-61 mutants (52 genes, 90%), with only 6 genes (10%) downregulated. This is consistent with LIN-61 acting as a transcriptional repressor. Importantly, none of the genes alternatively expressed in lin-61 mutants were implicated in DNA repair. The lin-61 transcript served as a positive control in the microarray analysis as we had previously shown, using qRT-PCR, that this transcript was reduced approximately 4-fold in lin-61 mutants, likely due to nonsense-mediated decay (Figure 1B). According to the microarray data, lin-61 mRNA was reduced 3.25-fold, which in good agreement with the qRT-PCR data. The expression analysis showed that while LIN-61 does indeed act as a transcriptional repressor, lin-61 mutation by itself (in the absence of an additional synMuvA mutation) has only a minor effect on global gene transcription. Finally, since these experiments indicated that DNA repair genes are expressed at normal levels in lin-61 mutants, it is likely that LIN-61 influences DSB repair directly and not by ensuring that other DDR genes are appropriately expressed.
Discussion
In this study we have identified the underlying cause of genomic instability in lin-61 mutants: DSBs are not adequately repaired due to defective HR. Accordingly, these animals are hypersensitive to IR and nitrogen mustard and DSBs remain unrepaired in diakinesis oocytes of γ-irradiated lin-61 mutants. LIN-61 contributes to HR in mitotic cells but it is dispensable for DSB repair during meiosis. Sensitivity of lin-61 germ cells to DSBs is not due to faulty DNA damage checkpoints as both cell cycle arrest and apoptosis are functional. Moreover, DNA repair genes are not inappropriately expressed in lin-61 mutants. The role of LIN-61 in HR is not restricted to germ cells because the somatic cells of early stage embryo are also very sensitive to IR. Also, later in development, intestinal cells are HR defective, as determined by the GFP-based HR reporter system. HR is essential for genome stability, as it is the principal DSB repair route in germ cells. It is also an error-free repair pathway. Blocking HR enables mutagenic and toxic repair routes to become active, which likely contributes to genomic instability in lin-61 mutants.
The role of LIN-61 in HR is restricted to mitotic cells
LIN-61 is expressed in all nuclei, both in the germline and somatic tissues [6]. Despite this, several observations suggests that LIN-61 contributes to HR only in mitotic cells and is dispensable for both meiotic interhomolog and intersister HR. Meiotic cells rely on interhomolog HR to repair at least one programmed DSBs per chromosome pair so that the obligate CO will be established [47]. Meiotic recombination is not defective in lin-61 mutants as they form chiasmata normally and produce nearly completely viable broods. What is more, RAD-51 foci that appear in prophase are resolved by late pachytene in both wild type and lin-61 mutants, indicative of the successful repair of programmed DSBs. The proficiency of intersister HR can be tested in meiotic cells by disrupting the SC in order to prevent interhomolog HR. In this situation, DSBs remain unrepaired if intersister HR too is defective, which manifests as chromosomal fragmentation at diakinesis. Unlike brc-1 and smc-5/-6 mutants [19], [27,27], lin-61 mutants depleted of the SC component SYP-2 do not have fragmented diakinesis chromosomes, indicating that intersister HR is proficient in the meiotic cells of these mutants. Moreover, DSBs introduced by IR into lin-61 meiotic cells, but not brc-1 meiotic cells, are efficiently repaired.
While lin-61 mutants are proficient in meiotic HR, their mitotic cells are defective in HR. These cells display signs of persistent and spontaneous DNA damage. Further, γ-irradiation of mitotic germ cells causes severe chromosome fragmentation in lin-61 mutants. Finally, lin-61 mutants are also hypersensitive to ICLs and the repair of these lesions occurs in S/G2 phase using the newly synthesised sister chromatid as the HR repair template [29]. The somatic (mitotic) cells of lin-61 are also hypersensitive to IR and mitotic cells exclusively use the sister chromatid for HR [39]. Together, these observations indicate that LIN-61 contributes to DSB repair via intersister HR in mitotic cells but does not participate in meiotic HR.
How does LIN-61 promote DSB repair?
Considering that the transcriptional profile of lin-61 mutants cannot explain their HR defect, LIN-61 likely acts directly at sites of DNA damage to promote DSB repair. This is an attractive hypothesis considering that chromatin can act as a physical barrier that must be remodelled to allow access of DDR factors to sites of damage. In addition, many proteins that alter chromatin structure have recently been implicated in the DDR including NuRD components MTA1, MTA2, CHD4, HDAC1 and HDAC2 [48]–[50]; and PcG proteins BMI1, RING1, RING2 and HP1 [51]–[55]. Each of these proteins is rapidly recruited to DNA damage and is necessary for DNA repair. The C. elegans counterparts of these proteins are also synMuvB proteins like LIN-61. Intriguingly, L3MBTL2, the putative human orthologue of LIN-61, is part of a PcG-like complex (PRC1L4) that shares RING1, RING2 and HP1γ as partner members [56]. Moreover, human cells depleted of RING2 [55], and C. elegans hlp-2 HP1 mutants [53], are radiosensitive like lin-61 mutants. PRC1L4, or a related L3MBTL2-containing PcG complex, may therefore act in DSB repair like LIN-61. Using immunofluorescence, we were not able to detect a change in LIN-61 intracellular localisation upon IR (data not shown). However LIN-61 is abundantly present and localised at chromatin in all cells, which may conceal its relocalisation around sites of DNA damage. Recruitment to sites of DNA damage has also not been observed for any other C. elegans synMuvB proteins, likely for similar reasons.
It is unknown how PcG activity promotes DSB repair but it is argued that inhibiting transcription locally at the DSB may be important as the transcriptional machinery could interfere with repair proteins or with DNA repair intermediates [50], [57]. PRC1L4 represses transcription of target genes by monoubiquitinating lysine 119 of histone H2A via its E3 ubiquitin ligase activity [56]. This histone mark is also implicated in the DDR as it was recently shown to rapidly accumulate at DSBs [52], [58]. It will be of interest to determine whether L3MBTL2 and the other members of PRC1L4 are involved in DSB repair in human cells.
One possible explanation we considered for why lin-61 mutants were HR-defective was that they might have altered expression of DDR genes. But contrary to this, microarray expression analysis did not reveal any alternatively expressed DDR genes in these mutants. Some alternatively expressed genes were identified but none are implicated in DNA repair. The vast majority of the alternatively expressed genes were upregulated rather than downregulated, which is in accordance with LIN-61 being a transcriptional repressor. A previous study found that germline genes were ectopically expressed in the somatic tissues of lin-61 mutants, but only when maintained at the relatively high temperature of 26°C [13]. In line with this, we found that lin-61 mutants grown at the normal laboratory temperature of 20°C had only minor changes in gene expression and did not overexpress germline genes. Importantly, lin-61 mutants grown at 20°C displayed a profound HR defect, which further indicated that altered gene expression was not the cause of defective DNA repair. The microarrays were performed using RNA from a mixed population of germ and somatic cells. We cannot strictly exclude the possibility that a distinct population of cells had altered DDR gene expression that went undetected. This is unlikely though, as the defect in DSB repair was systemic, occurring in multiple tissues and at various stages of development, and not isolated to a small number of cells.
A novel GFP-based HR reporter system for C. elegans
In this study we introduce a novel reporter system for monitoring HR in C. elegans somatic cells. The reporter confirmed that LIN-61 is needed for HR. This tool was previously unavailable for C. elegans researchers. We propose it as a method for testing candidate HR genes, for example it confirmed that both BRC-1 and RTEL-1 have roles in HR in somatic cells, analogous to their functions previously only described in meiotic germ cells. Our experiments with the HR reporter also supported previous findings that suggested DSB repair pathways are dynamic and are in competition in somatic cells [41] since mutations that blocked NHEJ, increased HR reporter activity.
Though this system is a new tool that provides for the readout of repair, probably by an SDSA mechanism, of a defined DSB, it does have limitations. For example, the HR reporter does not easily allow for dissection of the biochemical processes that underpin HR pathways. These approaches are not well suited to C. elegans. Also, in its current form the HR reporter is expressed only in intestinal cells, which in contrast to most C. elegans somatic cells still cycle postembryoniccally. This choice of cell type was largely motivated by the likely need for S - and G2 phase dependent DNA end resection at DSBs for HR type of repair to occur. However, when interpreting the data it must be considered that these cells are atypical because they progress and grow through cycles of endoreduplication and not via canonical cell cycle stages including mitosis. It is thus possible that the response to the HR reporter is cell type-dependent. Finally, since formation of the DSB relies on expression of the I-SceI transgene using the heatshock promoter, any possible differences in heatshock response must be carefully controlled for as these differences may affect the level of DSB induction.
Implications for HR deficiency in MBT mutants
This is the first report showing that an MBT protein is needed for DSB repair. Genes encoding MBT proteins have previously been linked with tumourigenesis and can act as tumour suppressor genes. However, their contribution to DNA repair and genome stability is unknown. Our finding that LIN-61 is required for efficient HR may have implications for the treatment of MBT-deficient tumours, which may also be HR defective. HR-deficient tumours, such as those with BRCA1 or 2 hypomorphic mutations, are very susceptible to poly(ADP ribose) polymerase (PARP) inhibitors [39]. It will be important to determine whether the role of LIN-61 in DSB repair is conserved in human MBT proteins and whether MBT mutated tumours, such as medulloblastomas with mutations in L3MBTL2, L3MBTL3 or SCML2 [10], are HR deficient as they too may prove responsive to treatment with PARP inhibitors.
Materials and Methods
Genetics
The Bristol N2 strain was used as the wild type strain and maintained at 20°C according to standard protocols [59]. Alleles used in the study include LG I: lin-61(n3809) [6], lin-61(pk2225) (this study), lin-61(tm2649) [15], mbtr-1(n4775) [6], cep-1(gk138) [60] and rtel-1(tm1866) [61]; LG III: brc-1(tm1145) [62], cku-80(ok861) [63], polh-1(lf31) [31], lfIs129 [elt-2::HR-reporter; hsp16-41::mCherry::I-SceI] (this study); and LG X: lfIs82 [elt-2::SSA-reporter; hsp16-41::mCherry::I-SceI] (this study). To determine brood sizes, L4 larvae were singled on 6 cm plates with OP50 E. coli and transferred each day for three days. The number of viable progeny and unhatched eggs were counted, as well as the number of males in the brood.
DNA damage sensitivity, checkpoint activation, and chromosome fragmentation assays
All γ-irradiation was performed with a dose rate of 15 Gy/minute using an electronic X-ray generator set to 200 kV 12 mA (XYLON International). For L4 larval IR sensitivity, three L4 animals per plate (three plates per condition) were treated with various doses of γ-irradiation. For UV-C sensitivity, young adult (24 post L4 stage) worms were exposed to UV (254 nm lamp, Philips). HN2 sensitivity assays were performed as described [64]. γ-irradiation of embryos and L1 larvae was preformed as described [32]. Apoptosis assays were performed in as [45]. Cell cycle arrest and fragmentation assays were as in [36]. syp-2 RNAi was performed as in [19]. For cell cycle arrest, 4–5 germlines were analysed per condition, except for irradiated lin-61(tm649) for which a single germ line was scored.
Germline dissections and RAD-51 immunofluorescence
Germlines were dissected in egg salts, Tween, levamisole and fixed in 2% paraformaldehyde for 5 minutes at room temperature, and snap frozen on dry ice, then placed in methanol at −20°C for 10 minutes, washed three times for 10 minutes in PBS with 1% Triton X-100 and blocked in PBST (PBS with 0.1% Tween 20) and 1% BSA for 30 minutes at room temperature. Samples were incubated overnight at 4°C with rabbit anti-RAD-51 antibodies (Novus Biologicals) diluted 1∶200 in PBST 1% BSA and detected with Alexa488 goat anti-rabbit antibodies (Invitrogen) diluted 1∶1000. DNA was counterstained with 0.5 µg/ml DAPI and samples were mounted with VectaShield. RAD-51 foci were imaged with a Leica DM6000 deconvolution microscope collecting 0.5 µm Z-sections. The number of foci per nucleus was counted for each of the seven zones of the germline as described [64]. Three to five germlines were quantified per condition.
Microarray and qRT–PCR
Worms were synchronised as L1 larvae by bleaching and grown to the L4 stage. Total RNA was isolated with Trizol reagent (Invitrogen), and cleaned with RNeasy kit (Qiagen). Service XS (Leiden, NL) performed the Affymetrix expression analysis according to standard protocols. Data was analysed with the MAS 5.0 algorithm using Tukey's biweight estimator. Significance (p-value) was determined using Wilcoxon's rank test. Sequence of qRT-PCR primers is available in Text S1.
Pelt-2::HR and Pelt-2::SSA reporter
Details on construction of the Pelt-2::HR and Pelt-2::SSA reporter strains are provided in Text S1. For HR reporter assays, expression of mCherry::ISce-I was induced in L4 larvae by heatshock twice at 34°C for 1 hour (with 30 min rest at 20°C). 24 hours after induction, worms were mounted on agarose pads and their intestinal nuclei were scored for GFP expression using a Leica DM6000 microscope with 63× objective. Experiments were performed in triplicate with 50–100 animals tested for each condition. Statistical significance was tested using the Cochran-Mantel-Haenszel test.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. TremethickDJ (2007) Higher-order structures of chromatin: the elusive 30 nm fiber. Cell 128 : 651–654 doi:10.1016/j.cell.2007.02.008.
2. KouzaridesT (2007) Chromatin modifications and their function. Cell 128 : 693–705 doi:10.1016/j.cell.2007.02.005.
3. TrojerP, LiG, SimsRJ, VaqueroA, KalakondaN, et al. (2007) L3MBTL1, a histone-methylation-dependent chromatin lock. Cell 129 : 915–928 doi:10.1016/j.cell.2007.03.048.
4. BonasioR, LeconaE, ReinbergD (2010) MBT domain proteins in development and disease. Semin Cell Dev Biol 21 : 221–230 doi:10.1016/j.semcdb.2009.09.010.
5. SafferAM, KimDH, van OudenaardenA, HorvitzHR (2011) The Caenorhabditis elegans Synthetic Multivulva Genes Prevent Ras Pathway Activation by Tightly Repressing Global Ectopic Expression of lin-3 EGF. PLoS Genet 7: e1002418 doi:10.1371/journal.pgen.1002418.
6. HarrisonMM, LuX, HorvitzHR (2007) LIN-61, one of two Caenorhabditis elegans malignant-brain-tumor-repeat-containing proteins, acts with the DRM and NuRD-like protein complexes in vulval development but not in certain other biological processes. Genetics 176 : 255–271 doi:10.1534/genetics.106.069633.
7. PoulinG, DongY, FraserAG, HopperNA, AhringerJ (2005) Chromatin regulation and sumoylation in the inhibition of Ras-induced vulval development in Caenorhabditis elegans. EMBO J 24 : 2613–2623 doi:10.1038/sj.emboj.7600726.
8. PothofJ, van HaaftenG, ThijssenK, KamathRS, FraserAG, et al. (2003) Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev 17 : 443–448 doi:10.1101/gad.1060703.
9. WismarJ, LöfflerT, HabtemichaelN, VefO, GeissenM, et al. (1995) The Drosophila melanogaster tumor suppressor gene lethal(3)malignant brain tumor encodes a proline-rich protein with a novel zinc finger. Mech Dev 53 : 141–154.
10. NorthcottPA, NakaharaY, WuX, FeukL, EllisonDW, et al. (2009) Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet 41 : 465–472 doi:10.1038/ng.336.
11. GurvichN, PernaF, FarinaA, VozaF, MenendezS, et al. (2010) L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci USA 107 : 22552–22557 doi:10.1073/pnas.1017092108.
12. JanicA, MendizabalL, LlamazaresS, RossellD, GonzalezC (2010) Ectopic Expression of Germline Genes Drives Malignant Brain Tumor Growth in Drosophila. Science 330 : 1824–1827 doi:10.1126/science.1195481.
13. PetrellaLN, WangW, SpikeCA, RechtsteinerA, ReinkeV, et al. (2011) synMuv B proteins antagonize germline fate in the intestine and ensure C. elegans survival. Development 138 : 1069–1079 doi:10.1242/dev.059501.
14. LuijsterburgMS, van AttikumH (2011) Chromatin and the DNA damage response: The cancer connection. Mol Oncol doi:10.1016/j.molonc.2011.06.001.
15. Koester-EiserfunkeN, FischleW (2011) H3K9me2/3 binding of the MBT domain protein LIN-61 is essential for Caenorhabditis elegans vulva development. PLoS Genet 7: e1002017 doi:10.1371/journal.pgen.1002017.
16. YoudsJL, O'NeilNJ, RoseAM (2006) Homologous recombination is required for genome stability in the absence of DOG-1 in Caenorhabditis elegans. Genetics 173 : 697–708 doi:10.1534/genetics.106.056879.
17. GrabowskiMM, SvrzikapaN, TissenbaumHA (2005) Bloom syndrome ortholog HIM-6 maintains genomic stability in C. elegans. Mech Ageing Dev 126 : 1314–1321 doi:10.1016/j.mad.2005.08.005.
18. YanowitzJL (2008) Genome integrity is regulated by the Caenorhabditis elegans Rad51D homolog rfs-1. Genetics 179 : 249–262 doi:10.1534/genetics.107.076877.
19. BickelJS, ChenL, HaywardJ, YeapSL, AlkersAE, et al. (2010) Structural maintenance of chromosomes (SMC) proteins promote homolog-independent recombination repair in meiosis crucial for germ cell genomic stability. PLoS Genet 6: e1001028 doi:10.1371/journal.pgen.1001028.
20. AlpiA, PasierbekP, GartnerA, LoidlJ (2003) Genetic and cytological characterization of the recombination protein RAD-51 in Caenorhabditis elegans. Chromosoma 112 : 6–16 doi:10.1007/s00412-003-0237-5.
21. WardJD, BarberLJ, PetalcorinMI, YanowitzJ, BoultonSJ (2007) Replication blocking lesions present a unique substrate for homologous recombination. EMBO J 26 : 3384–3396 doi:10.1038/sj.emboj.7601766.
22. HayashiM, ChinGM, VilleneuveAM (2007) C. elegans germ cells switch between distinct modes of double-strand break repair during meiotic prophase progression. PLoS Genet 3: e191 doi:10.1371/journal.pgen.0030191.
23. DernburgAF, McDonaldK, MoulderG, BarsteadR, DresserM, et al. (1998) Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell 94 : 387–398.
24. ColaiácovoMP, MacQueenAJ, Martinez-PerezE, McDonaldK, AdamoA, et al. (2003) Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev Cell 5 : 463–474.
25. HodgkinJ, HorvitzHR, BrennerS (1979) Nondisjunction Mutants of the Nematode CAENORHABDITIS ELEGANS. Genetics 91 : 67–94.
26. BoultonSJ, MartinJS, PolanowskaJ, HillDE, GartnerA, et al. (2004) BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Current Biology 14 : 33–39.
27. AdamoA, MontemauriP, SilvaN, WardJD, BoultonSJ, et al. (2008) BRC-1 acts in the inter-sister pathway of meiotic double-strand break repair. EMBO Rep 9 : 287–292 doi:10.1038/sj.embor.7401167.
28. DeansAJ, WestSC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11 : 467–480 doi:10.1038/nrc3088.
29. LongDT, RäschleM, JoukovV, WalterJC (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333 : 84–87 doi:10.1126/science.1204258.
30. CicciaA, ElledgeSJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40 : 179–204 doi:10.1016/j.molcel.2010.09.019.
31. RoerinkSF, KooleW, StapelLC, RomeijnRJ, TijstermanM (2012) A Broad Requirement for TLS Polymerases η and κ, and Interacting Sumoylation and Nuclear Pore Proteins, in Lesion Bypass during C. elegans Embryogenesis. PLoS Genet 8: e1002800 doi:10.1371/journal.pgen.1002800.
32. ClejanI, BoerckelJ, AhmedS (2006) Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics 173 : 1301–1317 doi:10.1534/genetics.106.058628.
33. JacksonSP, BartekJ (2009) The DNA-damage response in human biology and disease. Nature 461 : 1071–1078 doi:10.1038/nature08467.
34. KipreosET (2005) C. elegans cell cycles: invariance and stem cell divisions. Nat Rev Mol Cell Biol 6 : 766–776 doi:10.1038/nrm1738.
35. EdgarLG, McGheeJD (1988) DNA synthesis and the control of embryonic gene expression in C. elegans. Cell 53 : 589–599.
36. BaillyAP, FreemanA, HallJ, DéclaisA-C, AlpiA, et al. (2010) The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet 6: e1001025 doi:10.1371/journal.pgen.1001025.
37. KriskoA, RadmanM (2010) Protein damage and death by radiation in Escherichia coli and Deinococcus radiodurans. Proc Natl Acad Sci USA 107 : 14373–14377 doi:10.1073/pnas.1009312107.
38. PierceAJ, JohnsonRD, ThompsonLH, JasinM (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13 : 2633–2638.
39. HelledayT, LoJ, van GentDC, EngelwardBP (2007) DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair 6 : 923–935 doi:10.1016/j.dnarep.2007.02.006.
40. HedgecockEM, WhiteJG (1985) Polyploid tissues in the nematode Caenorhabditis elegans. Dev Biol 107 : 128–133.
41. PontierDB, TijstermanM (2009) A robust network of double-strand break repair pathways governs genome integrity during C. elegans development. Current biology: CB 19 : 1384–1388 doi:10.1016/j.cub.2009.06.045.
42. YoudsJL, MetsDG, McIlwraithMJ, MartinJS, WardJD, et al. (2010) RTEL-1 enforces meiotic crossover interference and homeostasis. Science 327 : 1254–1258 doi:10.1126/science.1183112.
43. GartnerA, MilsteinS, AhmedS, HodgkinJ, HengartnerMO (2000) A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5 : 435–443.
44. MoserSC, Elsner vonS, BüssingI, AlpiA, SchnabelR, et al. (2009) Functional dissection of Caenorhabditis elegans CLK-2/TEL2 cell cycle defects during embryogenesis and germline development. PLoS Genet 5: e1000451 doi:10.1371/journal.pgen.1000451.
45. SchumacherB, HofmannK, BoultonS, GartnerA (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Current biology: CB 11 : 1722–1727.
46. ConradtB, HorvitzHR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93 : 519–529.
47. HillersKJ, VilleneuveAM (2003) Chromosome-wide control of meiotic crossing over in C. elegans. Current biology: CB 13 : 1641–1647 doi:10.1016/j.cub.2003.08.026.
48. MillerKM, TjeertesJV, CoatesJ, LegubeG, PoloSE, et al. (2010) Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol doi:10.1038/nsmb.1899.
49. SmeenkG, WiegantWW, VrolijkH, SolariAP, PastinkA, et al. (2010) The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol 190 : 741–749 doi:10.1083/jcb.201001048.
50. ChouDM, AdamsonB, DephoureNE, TanX, NottkeAC, et al. (2010) A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA 107 : 18475–18480 doi:10.1073/pnas.1012946107.
51. FacchinoS, AbdouhM, ChatooW, BernierG (2010) BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci 30 : 10096–10111 doi:10.1523/JNEUROSCI.1634-10.2010.
52. GinjalaV, NacerddineK, KulkarniA, OzaJ, HillSJ, et al. (2011) BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol 31 : 1972–1982 doi:10.1128/MCB.00981-10.
53. LuijsterburgMS, DinantC, LansH, StapJ, WiernaszE, et al. (2009) Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol 185 : 577–586 doi:10.1083/jcb.200810035.
54. IsmailIH, AndrinC, McDonaldD, HendzelMJ (2010) BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 191 : 45–60 doi:10.1083/jcb.201003034.
55. PanM-R, PengG, HungW-C, LinS-Y (2011) Monoubiquitination of H2AX Protein Regulates DNA Damage Response Signaling. J Biol Chem 286 : 28599–28607 doi:10.1074/jbc.M111.256297.
56. TrojerP, CaoAR, GaoZ, LiY, ZhangJ, et al. (2011) L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol Cell 42 : 438–450 doi:10.1016/j.molcel.2011.04.004.
57. IacovoniJS, CaronP, LassadiI, NicolasE, MassipL, et al. (2010) High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29 : 1446–1457 doi:10.1038/emboj.2010.38.
58. ShanbhagNM, Rafalska-MetcalfIU, Balane-BolivarC, JanickiSM, GreenbergRA (2010) ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141 : 970–981 doi:10.1016/j.cell.2010.04.038.
59. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
60. HofmannER, MilsteinS, BoultonSJ, YeM, HofmannJJ, et al. (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Current Biology 12 : 1908–1918.
61. BarberLJ, YoudsJL, WardJD, McIlwraithMJ, O'NeilNJ, et al. (2008) RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135 : 261–271 doi:10.1016/j.cell.2008.08.016.
62. PolanowskaJ, MartinJS, Garcia-MuseT, PetalcorinMIR, BoultonSJ (2006) A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J 25 : 2178–2188 doi:10.1038/sj.emboj.7601102.
63. DmitrievaNI, CelesteA, NussenzweigA, BurgMB (2005) Ku86 preserves chromatin integrity in cells adapted to high NaCl. Proc Natl Acad Sci USA 102 : 10730–10735 doi:10.1073/pnas.0504870102.
64. SaitoTT, YoudsJL, BoultonSJ, ColaiácovoMP (2009) Caenorhabditis elegans HIM-18/SLX-4 interacts with SLX-1 and XPF-1 and maintains genomic integrity in the germline by processing recombination intermediates. PLoS Genet 5: e1000735 doi:10.1371/journal.pgen.1000735.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy