
Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
Genome rearrangements are associated with eukaryotic evolutionary processes ranging from tumorigenesis to speciation. Rearrangements are especially common following interspecific hybridization, and some of these could be expected to have strong selective value. To test this expectation we created de novo interspecific yeast hybrids between two diverged but largely syntenic Saccharomyces species, S. cerevisiae and S. uvarum, then experimentally evolved them under continuous ammonium limitation. We discovered that a characteristic interspecific genome rearrangement arose multiple times in independently evolved populations. We uncovered nine different breakpoints, all occurring in a narrow ∼1-kb region of chromosome 14, and all producing an “interspecific fusion junction” within the MEP2 gene coding sequence, such that the 5′ portion derives from S. cerevisiae and the 3′ portion derives from S. uvarum. In most cases the rearrangements altered both chromosomes, resulting in what can be considered to be an introgression of a several-kb region of S. uvarum into an otherwise intact S. cerevisiae chromosome 14, while the homeologous S. uvarum chromosome 14 experienced an interspecific reciprocal translocation at the same breakpoint within MEP2, yielding a chimaeric chromosome; these events result in the presence in the cell of two MEP2 fusion genes having identical breakpoints. Given that MEP2 encodes for a high-affinity ammonium permease, that MEP2 fusion genes arise repeatedly under ammonium-limitation, and that three independent evolved isolates carrying MEP2 fusion genes are each more fit than their common ancestor, the novel MEP2 fusion genes are very likely adaptive under ammonium limitation. Our results suggest that, when homoploid hybrids form, the admixture of two genomes enables swift and otherwise unavailable evolutionary innovations. Furthermore, the architecture of the MEP2 rearrangement suggests a model for rapid introgression, a phenomenon seen in numerous eukaryotic phyla, that does not require repeated backcrossing to one of the parental species.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003366
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003366
Summary
Genome rearrangements are associated with eukaryotic evolutionary processes ranging from tumorigenesis to speciation. Rearrangements are especially common following interspecific hybridization, and some of these could be expected to have strong selective value. To test this expectation we created de novo interspecific yeast hybrids between two diverged but largely syntenic Saccharomyces species, S. cerevisiae and S. uvarum, then experimentally evolved them under continuous ammonium limitation. We discovered that a characteristic interspecific genome rearrangement arose multiple times in independently evolved populations. We uncovered nine different breakpoints, all occurring in a narrow ∼1-kb region of chromosome 14, and all producing an “interspecific fusion junction” within the MEP2 gene coding sequence, such that the 5′ portion derives from S. cerevisiae and the 3′ portion derives from S. uvarum. In most cases the rearrangements altered both chromosomes, resulting in what can be considered to be an introgression of a several-kb region of S. uvarum into an otherwise intact S. cerevisiae chromosome 14, while the homeologous S. uvarum chromosome 14 experienced an interspecific reciprocal translocation at the same breakpoint within MEP2, yielding a chimaeric chromosome; these events result in the presence in the cell of two MEP2 fusion genes having identical breakpoints. Given that MEP2 encodes for a high-affinity ammonium permease, that MEP2 fusion genes arise repeatedly under ammonium-limitation, and that three independent evolved isolates carrying MEP2 fusion genes are each more fit than their common ancestor, the novel MEP2 fusion genes are very likely adaptive under ammonium limitation. Our results suggest that, when homoploid hybrids form, the admixture of two genomes enables swift and otherwise unavailable evolutionary innovations. Furthermore, the architecture of the MEP2 rearrangement suggests a model for rapid introgression, a phenomenon seen in numerous eukaryotic phyla, that does not require repeated backcrossing to one of the parental species.
Introduction
Eukaryotic genome content and architecture can vary dramatically as populations of organisms evolve, or as populations of cells evolve during disease processes like cancer [1], [2]. Chromosome number may change, resulting in polyploidy and/or aneuploidy, or chromosomes may be restructured by translocations, inversions, deletions and amplifications. A striking example of genomic change, homoploid hybrid speciation, occurs when gametes of closely related species fuse to form viable hybrids. If both parental species have the same number of chromosomes, the homoploid hybrid will contain a “diploid” genome that has the same chromosome number as its ancestors; such hybrids can also be called “F1 hybrids,” as they arise in the first filial generation following hybridization. By contrast, allopolyploid hybrid speciation typically results in a doubling (or more) of the ancestral chromosome number. Although homoploid hybrid speciation has been most commonly observed in plants [3], it has been documented in every eukaryotic Kingdom (e.g., [4]–[7]). In the wild, as well as in brewing and wine-making, both homoploid and allopolyploid hybrid yeast have been isolated whose genomes are wholly or partly derived from two or more different members of the Saccharomyces “sensu stricto” group [8]–[10]. These Saccharomyces species can also be mated in the lab to create de novo interspecific hybrids [11]–[13].
In addition to homoploid hybrids that bear one copy of each of their parental species' chromosomes, “introgressive hybridization,” also known as introgression, has been observed among the sensu stricto group of Saccharomyces. This term was first coined by Anderson and Hubricht in 1938 [14] to denote the infiltration of the “germplasm” of one species into that of another following hybridization and repeated backcrossing. If this region is not selected against, in time it can become established as an “island” of the minor species' genome encompassed within the major species' genome. Introgressive hybridization is thought to be a long-term process, requiring an initial interspecific hybridization event, followed by the repeated backcrossing with only one of its parent species [15]. Since its first description, introgressive hybridization has been identified in numerous eukaryotic phyla (see [16], [17] for reviews). Introgression events have been documented among many of the Saccharomyces sensu stricto species, in yeasts isolated from natural environments [11], [18], clinical and animal sources [19], [20], and from wine, beer, and other industrial environments ([8], [21]–[28]; also see [29] for review). Like homoploid and polyploid hybridization, introgression is also considered to be important as a mechanism leading to speciation [30], [31]. Indeed, hybridization and introgression have been suggested as sources of unexpected, extreme ‘transgressive’ phenotypic traits upon which natural selection can act [32], facilitating rapid within-lineage evolution [30].
Although the evolutionary implications of interspecific hybridization in general, and introgressive hybridization in particular, have been appreciated for some time, their molecular bases and relative importance as evolutionary mechanisms among various Kingdoms are incompletely understood [33]. Moreover, while genomic technologies have greatly expanded our understanding of genome content and stability during adaptive evolution, we have limited knowledge of how genomes stabilize following the initial ‘shock’ of interspecific hybridization [34]. Significantly, the actual process of introgressive hybridization has never been captured in action. Budding yeasts of the genus Saccharomyces provide an ideal eukaryotic system in which to close these knowledge gaps. Not only do Saccharomyces yeasts readily form interspecific hybrids, they also have a relatively simple life cycle, reproduce quickly, tolerate aneuploidy [35] and can be propagated as stable haploids or diploids. Environmental variables and the size and structure of yeast populations can also be controlled experimentally, and because yeasts can be preserved cryogenically, it is possible to compare evolved to ancestral strains or to replicate any stage of an experiment [36].
Saccharomyces cerevisiae and S. uvarum (previously called S. bayanus) are distantly related members of the Saccharomyces sensu stricto group, having diverged ∼20 million years ago [37]. Despite having only 80% sequence identity in coding regions and 62% in intergenic regions, the S. cerevisiae and S. uvarum genomes are largely syntenic, with the exception of 3 large reciprocal translocations and within some regions of their telomeres, where rapid structural evolution has occurred [37], [38]. Because of their synteny and their sequence divergence, which allows their genomes to be distinguished, we experimentally investigated the evolution of an F1 homoploid interspecific hybrid formed between S. cerevisiae and S. uvarum. We evolved three independent replicate populations of this hybrid under continuous nitrogen limitation, a selective pressure often encountered in wine-making as well as in other ecological settings where S. uvarum and S. cerevisiae likely occur [39]–[41]. We determined each parental species' contribution to the evolving genomes by array Comparative Genomic Hybridization (aCGH) as well as by whole genome sequencing of select ancestral and evolved hybrid clones. We discovered a recurrent genomic rearrangement in all three independently evolved hybrid populations. This rearrangement ultimately produces two copies of an interspecific MEP2 fusion gene, which in both S. cerevisiae and S. uvarum encodes for a high-affinity ammonium permease. In all cases the fusion gene is structured such that the 5′ end of the gene is derived from S. cerevisiae sequences and the 3′ end is derived from S. uvarum sequences, an evolutionary innovation that could only arise in a hybrid genome. Repeated evolution of this novel fusion gene in independent populations suggests that it is adaptive under nitrogen-poor environments where ammonium is the sole nitrogen source. The architecture of the rearrangement suggests a model for rapid introgression without the need for repeated backcrossing to one of the parental species.
Results
We created an S. cerevisiae-S. uvarum interspecific F1 homoploid hybrid, strain GSY86 (Table 1), by mass-mating a haploid S. cerevisiae strain (S288c background) to haploid spores of S. uvarum (CBS7001 strain background), as described in Materials and Methods and shown schematically in Figure S1. Experimental populations were founded by GSY86 in three independent vessels, which hereafter we call vessel A, B, or C. Each independent population was evolved for >200 generations in continuous, aerobic culture, limiting on ammonium (NH4+, supplied as (NH4)2SO4) as described in Materials and Methods. The hybrid strain grew robustly, achieving steady state within 10 culture generations at the target dilution rate, D = 0.16 h−1. Because a previous study [42] had demonstrated that S. cerevisiae×S. uvarum interspecific hybrids evolving under stress can shed one of their ancestral genomes, we performed flow cytometry on hybrids isolated at the beginning and end of our experiments. In all cases genomes were diploid, indicating that there was no large-scale loss of genome content during the experiments (data not shown); we also microscopically observed the cultures periodically during the course of the evolution and saw no evidence of asci or spores.

Populations of newly formed interspecific hybrids show “hybrid vigor” but limited scope for physiological improvement under nitrogen limitation
At steady state, the interspecific hybrid performed better than either of its ancestral species under aerobic ammonium limitation at 25°C. At the beginning of steady state growth (t-initial), residual ammonium was near or below detection limit (<0.01 ppm or 0.01 µg L−1) for both parental species and for the interspecific hybrid. Residual glucose at t-initial in diploid S. cerevisiae, diploid S. uvarum and in the interspecific hybrid was 5.0±0.34, 5.7±0.23 and 2.3±1.20, respectively (mean ± Std. Error, g L−1; P = 0.04, one-way ANOVA followed by Student-Newman-Kuels (SNK) test), while optical density at A600 was 0.90±0.04, 1.11±0.02 and 1.55±0.17, (mean ± Std. Error, P<0.01 one-way ANOVA followed by SNK test); thus for both parameters the hybrid showed superior growth performance compared to either of its parents. In three independent hybrid populations evolved for 200 generations (t-final) we detected no significant change in either residual glucose or optical density relative to the ancestral unevolved hybrid. The extent to which uptake of the limiting nutrient was enhanced could not be assessed, as ammonium concentration in the experimental populations was close to our assay detection limit at t-initial and below this limit at t-final. Based on these observations, we concluded that in a nitrogen-limited, glucose-sufficient environment, S. cerevisiae×S. uvarum interspecific hybrids had limited scope for measurable improvement in the physiological parameters we measured.
Independently evolved hybrid clones are more fit than their common ancestor
To directly test whether individual clones from the evolved populations were more fit than their common ancestor, we performed short-term (15 generation) competitive chemostat experiments. We competed the founder S. cerevisiae×S. uvarum hybrid (GSY86) and each of three individual 200-generation evolved clones (GSY2532, GSY2533 and GSY2535, representing one isolate from each vessel; Table 1) against a fluorescently marked unevolved S. cerevisiae×S. uvarum interspecific hybrid strain (GSY2590; Table 1), under the same ammonium-limited conditions used for our long-term evolution experiments. GSY2590 is identical to the ancestral founder strain except for the presence of an integrated Green Fluorescent Protein (GFP) gene, as described in Materials and Methods. Because we observed only modest fitness differences between the founder hybrid (GSY86) and GSY2590 (competition coefficient = 1.04±0.009), we concluded that the latter could serve as a surrogate “founder” in the competitive chemostat experiments, with a 0.04 correction to the competition coefficient. We found that each of the three evolved F1 hybrids consistently outcompeted GSY2590 under continuous ammonium limitation: corrected selection coefficients for GSY2532, GSY2533 and GSY2535 were 1.15 (±0.005), 1.14 (±0.003), and 1.11 (±0.008), respectively (mean ± Std. Error). These fitness gains are statistically significant (P<0.001) and similar in magnitude to values reported for S. cerevisiae evolving under ammonium limitation (1.09 in [43]), but less than fitness gains reported for S. cerevisiae evolved under aerobic glucose limitation (1.16 to 1.60 in [44]). The scope for fitness improvement in yeast evolving at low growth rates is likely greater under aerobic glucose limitation because cells can switch from respiro-fermentative to respiratory metabolism, which greatly increases the efficiency of converting substrate to biomass [45]. Furthermore, as fungi in nature face chronic nitrogen limitation [39], [40], natural selection has likely fine-tuned mechanisms to scavenge inorganic nitrogen.
Karyotypic evolution is evident in independent hybrid populations
For time points corresponding to generations ∼50, ∼100, ∼150, and ∼200, archived population samples from vessels A, B and C were revived from cryogenic storage and plated on YPD; for each time point two clones were selected at random for karyotype analysis using CHEF (Clamped Homogeneous Electric Fields) gel-electrophoresis; one clone from the founding (t-initial) population was also included (Figure 1). Although most isolates exhibited the parental karyotype, several variants exhibited size changes in one or two chromosomes. For example, both isolates from generation 200 of vessel A exhibited an increase in size of one of the chromosomes corresponding to the S. cerevisiae chromosome 7+15 doublet at 1200 Kb (Figure 1, yellow arrow). Interestingly, in both vessels B and C (inoculated independently with different starter cultures), clones isolated at generation 100 and generation 200, respectively, demonstrated absence of the ∼650 Kb band, apparently corresponding to chromosome 11 of S. uvarum (Figure 1, red ovals). Other karyotypes transiently appeared in the populations, such as that observed in vessel C at 100 generations involving a size increase in S. uvarum chromosome 2–4 at 1500 Kb (Figure 1, blue arrow), as well as multiple instances of size variation in S. cerevisiae and S. uvarum chromosome 12 (1640/1900 Kb; topmost chromosomal band seen in Figure 1). This last observation may reflect variation in copy number of tandemly-arrayed ribosomal DNA repeats on chromosome 12, as this region of the yeast genome is known to be labile [46].

Array-Comparative Genomic Hybridization (aCGH) reveals additional changes in genome content
CHEF analyses clearly demonstrated genome malleability in interspecific hybrids evolving under continuous nitrogen limitation. However, while CHEF analysis reveals the phenomenon of malleability, and serves as a screen to identify interesting karyotypes, it tell us little about the underlying architectural changes, and nothing at all about the molecular mechanisms that might be at play. To further investigate the evolved clones, we used aCGH to assay whole genome copy number variation arising from non-copy-neutral changes such as deletions, amplifications and non-reciprocal translocations. aCGH profiles of evolved hybrids revealed that a small number of chromosomes had undergone rearrangement; however, the rearrangements detected using aCGH were not those detected by CHEF, indicating that the rearrangements detected by CHEF were most likely copy-neutral events.
aCGH analysis showed that clones isolated from each of two independent t-initial founding populations had the expected, non-rearranged F1 hybrid genome configuration, i.e., they contained one complete non-rearranged chromosomal set from each of the input genomes, within the limits of detection of aCGH (Figure S2). Strikingly, however, aCGH revealed that 9 out of the 10 evolved hybrid clones we examined—four of four 150-generation clones (one from Vessel A, two from Vessel B, and one from Vessel C), and five of six 200-generation clones (2 each from Vessels A, B, and C)—contained a distinctive and apparently identical, or extremely similar, rearrangement on chromosome 14, whereby fully half of the S. uvarum chromosome 14 (much of the distal portion of the left arm) was replaced with the corresponding region of the S. cerevisiae chromosome 14 (Figure S2). This appeared in all cases to be a “non-reciprocal” translocation event, resulting in increased copy number of the distal left portion of the S. cerevisiae chromosome 14, with the concomitant deletion of the corresponding S. uvarum chromosome 14 region (detailed aCGH results for the chromosome 14 region are shown for three 200-generation clones, one from each vessel, in Figure 2). Because the lengths of the translocated regions of the two chromosome 14 s are roughly equivalent between these species, we would not expect to see in these clones any change in chromosome 14 mobility by CHEF, and in fact, none was seen (see Figure 1).
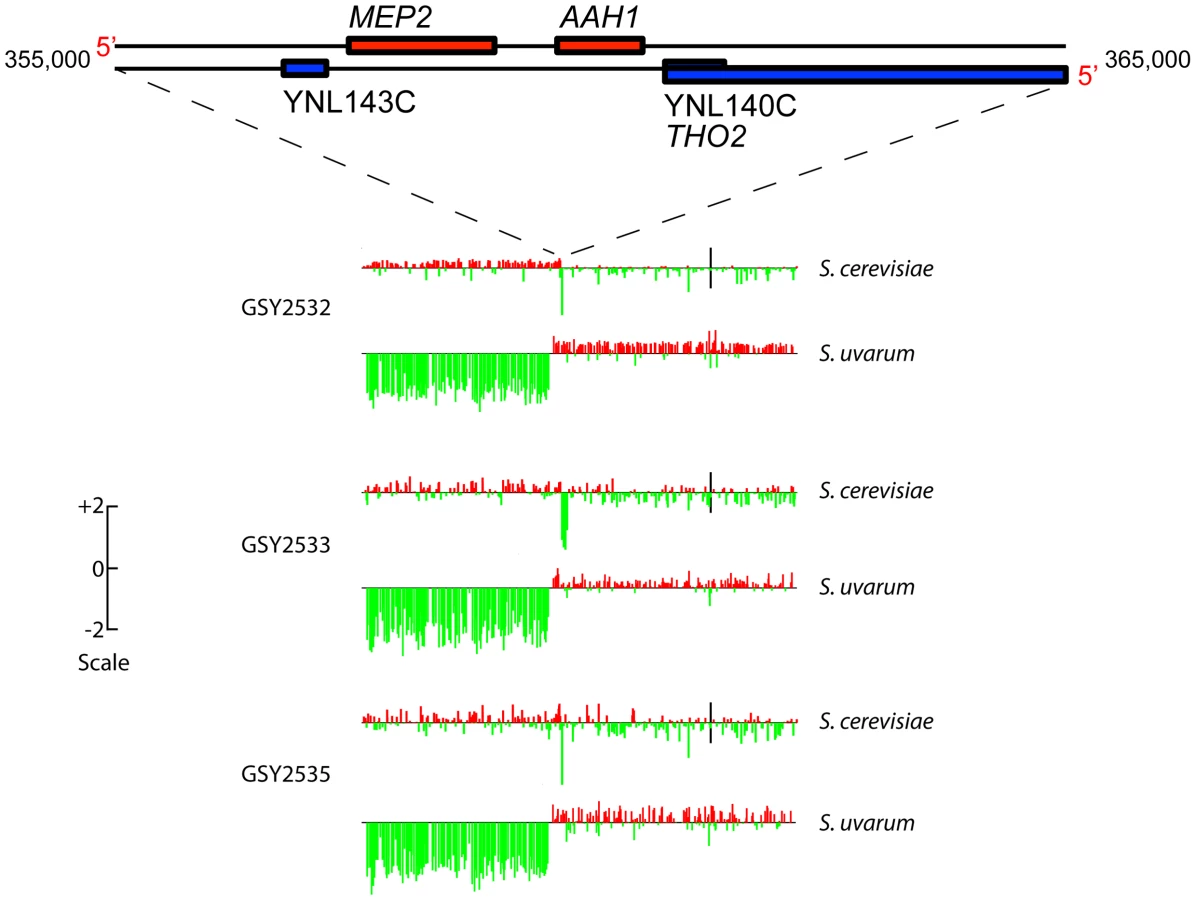
Because this rearrangement is seen in clones from all three vessels, it must have arisen independently. The fact that the chromosome 14 rearrangement occurred independently and is seen in a large majority of evolved clones examined suggests that it is adaptive under inorganic nitrogen limitation; indeed, as shown below, the rearrangement always occurs precisely within the MEP2 gene (YNL142W), which encodes the high-affinity, low capacity ammonium permease in Saccharomyces [47], [48]. The MEP2 gene is found in both the S. uvarum and S. cerevisiae genomes, in the same (syntenic) position on each genome's chromosome 14, sharing 85% DNA sequence identity. In addition to the MEP2 rearrangement, a few additional rearrangements resulting in copy number variation—including deletions of ∼15 to ∼50 kb occurring on chromosomes 5, 12, and 15 of S. cerevisiae and chromosome 9 of S. uvarum, plus a probable extra copy of S. cerevisiae chromosome 12 in Vessel B clones—were evident among some of the evolved clones, but none of these were shared across vessels (Figure S2).
Sequencing of evolved clones' chromosome 14 junction regions
We designed primers well outside the chromosome 14 fusion junctions detected by aCGH in the evolved clones (Table S1) to PCR-amplify the junction-containing regions of the three 200-generation evolved clones whose aCGH results are shown in Figure 2; these are clones GSY2532, GSY2533, and GSY2535, coming from Vessels A, B, and C, respectively (Table 1). Sanger sequencing of these PCR products revealed that the junction breakpoints of the rearrangement differed among clones (Figure S3A), indicating that despite appearing almost identical by aCGH, the rearrangements were indeed independent, as expected since the clones arose in three separate vessels. The junction sites for these three clones were all located within the coding sequence of the MEP2 gene and in all three cases the gene remained in-frame. For GSY2532 and GSY2535 the junctions result in a predicted fusion protein with the N-terminal one-third (approximately) of the protein coming from S. cerevisiae and the C-terminal two-thirds from S. uvarum; for GSY2533, these proportions are swapped (Figure S3B). The S. cerevisiae and S. uvarum Mep2 proteins are each 499 amino acids long, with 17 amino acid differences between them; each of the three predicted Mep2 fusion proteins has a novel predicted protein sequence derived from the combination of the S. cerevisiae and S. uvarum MEP2 genes.
Whole-genome sequencing of ancestral and evolved clones
To further elucidate genomic changes that occurred during evolution of the interspecific F1 hybrids, we performed Illumina whole genome sequencing on the three independent 200-generation evolved clones containing MEP2 fusion genes, whose junctions we had sequenced as described above (GSY2532, 2533, 2535), and also on the ancestral clone used to found the three replicate vessels (GSY86). Read depths across the MEP2 regions indicated that as expected for the ancestral GSY86 clone, there were no rearrangements resulting in copy number changes in either genome's MEP2 region (Figure 3A).
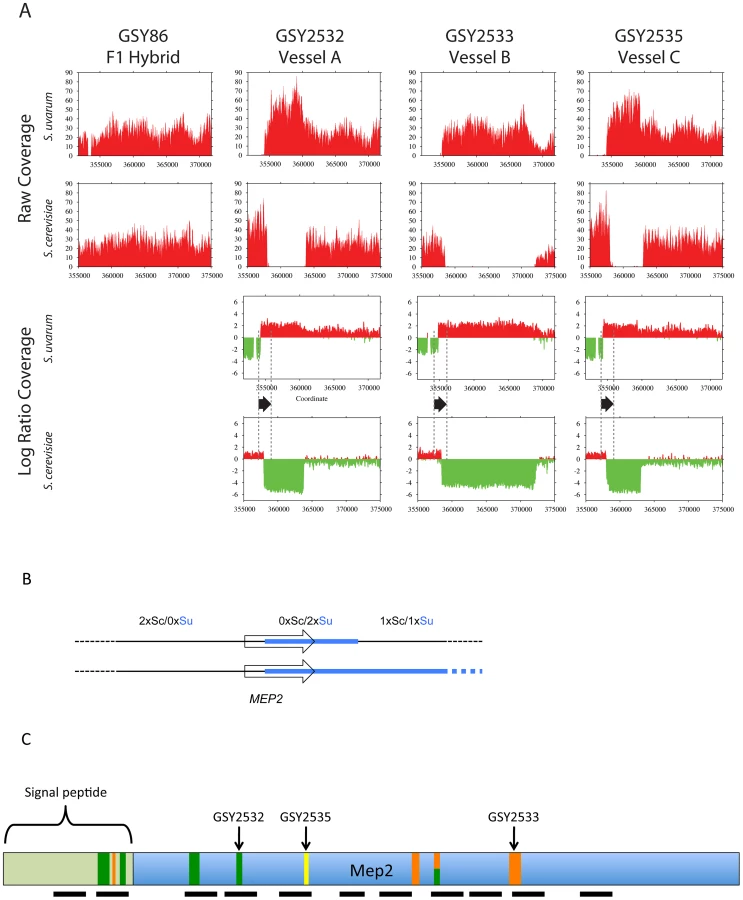
In contrast, and confirming our aCGH results, we detected large-scale copy number changes in the MEP2 region for each of the three evolved clones (Figure 3A). Surprisingly, however, we observed from our whole genome sequencing that the architecture of the genome rearrangement was more complex than we had predicted by aCGH. Instead of a simple translocation (and/or “breakage-induced replication” event) to yield one S. cerevisiae – S. uvarum chimaeric chromosome (with the junction located within the MEP2 gene) and one intact S. cerevisiae chromosome, in all three cases the expected S. cerevisiae – S. uvarum chimaeric chromosome was present, but there was an additional rearrangement on the S. cerevisiae chromosome (Figure 3A,B). This additional event resulted in a complete deletion of 5 to 15 kb of the S. cerevisiae chromosome, the region instead being precisely replaced with the corresponding S. uvarum chromosomal region within an otherwise intact S. cerevisiae chromosome; this is an event that can be considered to be the equivalent of an “introgression” of the S. uvarum genome into the S. cerevisiae chromosome (Figure 3B). In each case the distal junction on the S. cerevisiae chromosome occurred within the MEP2 gene, with exactly the same junction as that found in the partner S. cerevisiae – S. uvarum chimaeric chromosome (note: this is why our Sanger sequencing of PCR products described above gave readable sequences). In all cases the junction found by whole genome sequencing matched exactly the junction we had found by Sanger sequencing. The proximal junction was always well “downstream” from the MEP2 gene and varied for each clone, occurring anywhere from ∼5 Kb (GSY2532 and GSY2533; within THO2) to 15 kb (GSY2535; near FPR2) toward the centromere (Figure 3A). The most interesting outcome of this additional rearrangement within the S. cerevisiae chromosome is that each of the evolved clones contains two copies of identical MEP2 fusion genes (with junctions as shown in Figure S3A and S3B), and no copies of either the S. cerevisiae or the S. uvarum endogenous (“wild-type”) MEP2 genes. Analysis of the whole genome sequences for shared SNPs (and/or shared SNP-containing genes) revealed that no such shared mutations existed among the evolved clones (Table S2).
Multiple MEP2 gene fusion alleles coexist in the same population, and MEP2 fusions coexist with S. uvarum MEP2 in the same genome
Because we saw MEP2 rearrangement events occurring on both the S. uvarum and S. cerevisiae chromosomes of the evolved clones, we wished to know if the rearrangements occurred in a single concerted step, or whether a sequential multi-step process led to the final configuration. We therefore performed diagnostic PCRs on 12 single colony isolates from evolved populations corresponding to 0, ∼50, ∼100, ∼150 and ∼200 generations, from Vessels A and B, for a total of 120 isolates (12 per time point, 60 per vessel). We used 4 PCR primer combinations for each clone, using primer combinations (Table S1) specific for the S. cerevisiae MEP2 gene, the S. uvarum MEP2 gene, the S. cerevisiae-S. uvarum fusion MEP2 gene (found in evolved clones), or the S. uvarum-S. cerevisiae “reverse-fusion” MEP2 gene (not found in the evolved clones described above that were examined by aCGH and/or sequencing). Almost all clones from generations 0 and 50, as expected for an un-rearranged (“ancestral”) hybrid, showed the coexistence of the S. cerevisiae MEP2 gene and the S. uvarum MEP2 gene, with no evidence of a MEP2 fusion gene (Figure S4A). We further found that the MEP2 S. cerevisiae-S. uvarum fusion gene appeared in both vessels starting at 100 generations and persisted through to the 200-generation time point (Figure S4A). At 100 generations, in both vessels, less than 20% of the clones contained the ancestral un-rearranged MEP2 genes; instead most clones contained the MEP2 fusion gene either alone (presumably in two copies as seen in GSY2532, 2533, and 2535), or the fusion gene in conjunction with the S. uvarum-only MEP2 gene. By the 150 and 200-generation time points, the MEP2 fusion gene alone was predominant. In these later time points, there also appeared clones containing only the S. cerevisiae MEP2 gene or only the S. uvarum MEP2 gene, without the presence of the MEP2 fusion gene (Figure S4A). Interestingly, although we observed the MEP2 fusion gene in conjunction with the S. uvarum MEP2 gene, we never observed the S. cerevisiae MEP2 gene occurring with the MEP2 fusion gene. Finally, the “reverse-fusion” MEP2 gene was not found in any of the 120 clones.
We Sanger-sequenced the PCR products corresponding to the MEP2 fusion gene from all clones yielding such PCR products. We found that there were several additional MEP2 fusion junctions present in the evolved clones of both vessels, with junctions differing from those found in the three clones (GSY2532, 2533, and 2535) we had previously characterized by Sanger and whole genome sequencing. As seen in Figure 3C and Figure S4B, in addition to the junctions found for GSY2532 (Vessel A) and GSY2533 (Vessel B), four additional distinct and separate novel MEP2 fusion junctions were found in Vessel A clones, and a further three distinct and separate novel MEP2 fusion junctions were found in Vessel B, for an observed total of nine different MEP2 gene fusion junctions (including that of GSY2535); in all cases, the junctions occurred within the MEP2 coding sequence and were in-frame.
qRT–PCR assays reveal only slight difference in transcription levels of MEP2 genes from each genome within a hybrid
Because the evolved fusion genes have the S. cerevisiae MEP2 promoter, we hypothesized that the MEP2 gene fusion events may have been selected because that promoter might result in higher transcript levels. We thus performed qRT-PCR reactions for each genome's version of the MEP2 gene on the founding ancestor GSY86, assaying (in triplicate) two independent biological replicates of GSY86 that had been grown to steady state in the same nitrogen-limited media and chemostats used for the original evolutions. We determined that the S. cerevisiae genome's copy of the MEP2 gene is indeed expressed at a somewhat higher level than the S. uvarum copy, by almost 2-fold, supporting our hypothesis (Figure S5; raw and normalized data given in Table S4). However, when we determined the expression of the fusion gene in an evolved clone (GSY2532), it appeared to produce less transcript per locus than either the S. cerevisiae or S. uvarum genes did in the founding hybrid (Figure S5, Table S4; note, in the evolved clone, the transcript quantified by qPCR is produced from 2 fusion loci, so the amount per locus is less). The mechanistic basis for MEP2 fusion genes' adaptive advantage is therefore more complex than increased expression, and may relate instead to changes in protein structure that increase the novel permeases' catalytic efficiency, decrease their Km for ammonium, and/or alter their activity as nutrient signaling molecules.
Discussion
Fungal genome architecture varies both in nature and in the laboratory
Laboratory strains of S. cerevisiae are the best-studied group of fungi in terms of their genome structure. However, even within this relatively homogeneous group, strains differ widely in rates of mitotic chromosome loss and levels of chromosome-length polymorphism [49]–[51]. Furthermore, mitotic genome instability in S. cerevisiae has been shown to be evolutionarily significant in the laboratory [52]–[54], in wine fermentation [24], [55] and in biomass conversion to fuel ethanol [56]. A large amount of standing genomic variation (e.g., ploidy differences, transposon copy number, and chromosome length polymorphism) is found among Saccharomyces isolates collected from natural and industrial settings (e.g. [57]–[59]), reinforcing the view that genomic plasticity may be evolutionarily important in diverse settings (see [10], [24] for reviews). Of special relevance to our study is the discovery that this variation very often takes the form of mosaic genomes that result from natural interspecific hybridization events [20], [28], [60]–[63]. Mosaic genomes arising from interspecific hybridization have been discovered in other yeasts. For example, Pichia sorbitophila appears to have arisen in recent centuries via allopolyploidization between two species affiliated with the genus Millerozyma [64]. Resolution of the initial hybridization event has produced 7 chromosome pairs that are either completely homozygous, completely heterozygous or mosaics. In mosaic chromosomes, breakpoints between homozygous and heterozygous regions can occur in protein coding genes [64], though with unknown phenotypic consequences. While the foregoing example provides an interesting snapshot of a recent hybridization event, no published study to date has explored genome dynamic changes that occur as experimentally-created interspecific hybrids evolve.
Interspecific hybrids evolved under limiting nitrogen exhibit recurrent independent rearrangements of the MEP2 ammonium permease gene
Using CHEF and aCGH analysis we were able to detect chromosomal loss and/or size changes, large indels, and non-reciprocal translocations in evolving interspecific F1 hybrids. Overall, however, the frequency with which we observed genomic rearrangements in hybrids evolving under nitrogen limitation was considerably less than that reported for S. cerevisiae evolving under glucose limitation [53]. Further, very few large-scale genomic changes were observed by CHEF and aCGH analysis when the diploid parental species themselves, S. cerevisiae and S. uvarum, were evolved under nitrogen limitation (Dunn, Piotrowski et al. in prep.). However, for a large number of evolved interspecific hybrid clones we observed a distinctive recurrent rearrangement, involving both parental genomes at the locus encoding high affinity ammonium permease, which we first observed by aCGH and then confirmed by Sanger and whole genome sequencing. These recurrent MEP2 rearrangements in S. cerevisiae×S. uvarum hybrids provide an interesting contrast with the results of experimentally evolving haploid S. cerevisiae under different types of limiting nitrogen. There, recurrent rearrangements were observed at the GAP1 locus, which encodes for the general amino acid permease [43]. A single homologous recombination event was seen to produce two different alleles: GAP1extrachromosomal circle or gap1Δ; the former being associated with higher fitness in clones adapted to L-glutamine and L-glutamate, the latter with higher fitness in clones adapted to urea, allantoin, and ammonium. Owing to differences in genome content, hybrid interspecific diploids are able to explore adaptive possibilities not open to haploid S. cerevisiae.
While we failed to detect any mutations or rearrangements that inactivate GAP1 in any of the ammonium-adapted clones that we sequenced, our observations are remarkably similar to what occurs when either haploid or diploid S. cerevisiae is evolved under sulfur limitation, where a recurrent rearrangement, resulting in gene amplification, has been observed at the SUL1 locus which encodes a high affinity sulfate permease of the SulP anion transporter family [65]. In our experiments, the recurrent event is a complex rearrangement at the MEP2 locus, which encodes a high affinity ammonium permease. This rearrangement yields a genome containing two copies of a fusion MEP2 gene with the 5′ portion derived from S. cerevisiae and the 3′ portion from S. uvarum. It is likely that this rearrangement is adaptive under N-limitation, due to both its high allele frequency and the fact that it was selected for independently multiple times. As expected, when tested in direct head-to-head competition experiments, each of three independently-evolved evolved clones having the characteristic MEP2 rearrangement showed significant fitness increases relative to an unevolved ancestral clone. We have not yet shown that the presence of only the two MEP2 fusion genes is necessary and sufficient to confer this selective advantage within the context of an otherwise unevolved hybrid. Nevertheless, whole genome sequencing of these three evolved clones revealed no shared SNPs or rearrangements in other genes, suggesting that the recurrent MEP2 rearrangement is a key shared adaptive innovation in these evolutions, an innovation unavailable to either parent alone.
Fusion genes as sources of evolutionary innovation
A number of recent studies suggest that gene fusions may contribute to the evolution of novel functions (reviewed in [66]). Because new folding structures could quickly produce traits unattainable by point mutation alone, fusion genes could be potent drivers of adaptive change [67], and indeed, in vitro generation of fusion genes has been directly shown to create novel enzymatic functions [68]. Fusion genes have been discovered in many organisms, and even play an important role in the initial steps of tumorigenesis [69]. Examining hybrid lager yeast, Usher and Bond recently described a fusion gene formed by recombination between homoeologous chromosomes of S. cerevisiae and S. eubayanus [21]. The result, a chimaeric gene for GPH1, which encodes for glycogen phosphorylase, fails to produce mature mRNA because of a frameshift in its coding sequence; loss-of-function at GPH1 leads to a glycogen phenotype typical of haploid cells. In contrast, the chimaeric genes that we discovered in the course of evolving yeast hybrids are always formed by in-frame fusions of the 5′ end of the S. cerevisiae MEP2 coding sequence to the 3′ end of the S. uvarum MEP2 coding sequence.
What is the nature of the fitness advantage in the evolved clones?
Chemostat theory [70], [71] predicts that when cells evolve under nutrient limitation, adaptive genotypes arise as a result of either increased efficiency of nutrient use or increased capacity for assimilating the limiting nutrient. Previous studies have shown that in S. cerevisiae evolving under glucose limitation both mechanisms come into play [45], [53], [72], resulting in increased yield biomass and diminished concentrations of residual substrate at steady state. To test whether this was also the case in our experiments, we grew to steady state under ammonium limitation single-colony isolates of the ancestral hybrid and the three evolved clones that we sequenced. We found that in all cases residual ammonium was near or below detection limits (0.01 ppm), which follows from the very low Km of ammonium permease (1–2 µmolar; [73]) and the high velocity of ammonium uptake (<0.25 µmolar per second at D = 0.1 h−1), under glucose-sufficient conditions. We found that culture density and residual glucose concentrations for ancestral and evolved strains were also not statistically different (though interestingly, GSY2532 and GSY2535 produced a small but significantly greater amount of dry weight biomass than their ancestor, see Table S3, P = 0.002). These biochemical results, are, as for the genomic results, again reminiscent of those obtained when S. cerevisiae is experimentally evolved under inorganic sulfur limitation [65]. Evolved sulfur-limited populations show very modest increases in cell biomass, compared to evolved glucose and phosphate-limited populations, even though SUL1, which encodes a high-affinity sulfate transporter, is amplified in multiple independent evolutions, and even though this mutation demonstrably increases fitness when crossed into an unevolved wild-type background. Gresham et al. [65] conclude that the scope for metabolic innovation in inorganic sulfur metabolism is constrained, in this case by the small contribution of sulfur to cell biomass, relative to that of glucose or phosphate, and that this constraint results in the repeated evolution of a rearrangement resulting in SUL1 amplification. Our nitrogen-limitation results suggest metabolic constraints of a similar nature, which may be driven in part by the fact that fungi as a group are chronically nitrogen limited and have likely been under strong selection to acquire the capacity to scavenge this element to extremely low levels.
Our qRT-PCR results from the unevolved hybrid show a relatively modest two-fold difference in expression of the MEP2 gene from each of the genomes present in the unevolved hybrid, with S. cerevisiae the higher expressed gene of the two. This would seem to indicate that the unidirectional nature of the fusion gene rearrangement, whereby we always observe the S. cerevisiae promoter and 5′ end of the gene fused to the 3′ end of the S. uvarum gene, arises simply to increase overall transcript levels of the MEP2 gene. However, in an evolved clone, we surprisingly discovered that the fusion gene produces slightly lower transcript levels per locus than does either the wild-type S. cerevisiae or S. uvarum locus in the progenitor hybrid. It is unclear exactly what this finding means – possibly that transcription from the MEP2 gene is governed by a feedback mechanism that reduces its transcription.
It may be that the actual fusion proteins themselves, despite the few amino acid differences they show relative to the two parental genes (Figure S3B), provide an adaptive advantage. Possibly the novel chimaeric ammonium permeases differ from their ancestors in having a lower Km, which would lead to lower residual ammonium levels, and/or a higher kcat, which would result in greater overall uptake velocity. Alternatively, because in S. cerevisiae the Mep2 protein forms multimeric complexes in the plasma membrane [74], it may be of adaptive benefit to hybrids to produce Mep2 proteins that contain only S. cerevisiae N-termini and/or only S. uvarum C-termini; this could possibly result in better oligomerization for improved transport function and/or prevent dominant negative interactions between the two species' proteins. Indeed, dominant negative interactions have previously been noted between different alleles of the closely-related Mep1 and Mep3 proteins in yeast [75]. In this regard it is also provocative that we did not observe the coexistence of the MEP2 fusion gene with the S. cerevisiae MEP2 gene in any of the 120 clones that we genotyped, although we did see coexistence of the MEP2 fusion gene with the S. uvarum MEP2 gene; this may be evidence for dominant negative interactions between the MEP2 fusion gene and the S. cerevisiae MEP2 gene.
A novel mechanism to generate introgressions rapidly
Based on our genotyping results shown in Figure S4A, we believe that a two-step recombination event such as that shown in Figure 4 occurred between S. cerevisiae and S. uvarum chromosomes to generate the rearrangements seen in the evolved clones. We presume the event began with a double-strand break (DSB) in or near the S. cerevisiae MEP2 gene, followed by some amount of resection of the sequences surrounding the break (as described in [76], [77]). Strand invasion into a homologous region of the S. uvarum chromosome would have then been followed by repair of the resected sequences using the S. uvarum chromosome as a template, creating a gene conversion event with resultant loss of heterozygosity (LOH) (top portion of Figure 4). At the end of the first event, the S. uvarum chromosome would have been intact, while the other resultant chromosome would still be almost completely composed of S. cerevisiae sequences, aside from several Kb of S. uvarum genome precisely substituted at the MEP2 region (the exact Kb of S. uvarum sequences would depend on the amount of resection on either side of the DSB). Subsequently, either a DSB in the S. uvarum chromosome within the shared MEP2 gene region, followed by break-induced replication (BIR), or alternatively, a mitotic crossover event in G2 (left and right lower portions, respectively, of Figure 4) would have led to the final evolved genome configuration of two fusion MEP2 genes, sharing an identical fusion junction. Such gene conversion and BIR mechanisms have been previously well-documented and described in detail for yeast and many other organisms (see [78], [79] for reviews). We believe that a two-step process brought about the final evolved clone configuration, because some of the isolates from the ∼100 generation time-points in two independent populations (Figure 3A) showed the coexistence of the fusion MEP2 gene with the S. uvarum MEP2 gene (as for the intermediate shown in the first step of Figure 4). We further believe that genetic information was always transferred unidirectionally by a gene conversion event from S. uvarum to S. cerevisiae, because we never observed coexistence of the fusion MEP2 gene with the intact S. cerevisiae MEP2 gene (as depicted in Figure S6A and S6B), and we never detected the S. uvarum - S. cerevisiae “reverse” fusion (as depicted in Figure S6B). These findings suggest that the alternative models shown in Figure S6A and S6B are unlikely. Interestingly, this same type of event, leading to a virtually identical rearrangement configuration, has been seen in chromosomes of mouse cell lines lacking the Bloom syndrome helicase [80]; other similar, but not identical, patterns of rearrangement have been seen in yeast using plasmid-based “chromosome fragmentation vectors” and are thought to arise from template switching [81].

Introgression—infiltration of the “germplasm” of one species into that of another—occurs widely [16] and may produce extreme transgressive traits [32], which can drive rapid evolution and even speciation [30], [31]. Horizontal gene transfer, long known to be an engine of biodiversity in prokaryotes, has also been observed between eukaryotes [82], and in yeasts has recently been shown to be a mechanism by which “germplasm infiltration” can rapidly occur. Galeote et al. reported variable integration of a 17 kb ARS-containing Zygosaccharomyces bailii genome segment into dozens of S. cerevisiae wine strains [83]; this Z. bailii insertion was first discovered by whole genome sequencing of a wine strain [84]. The organization of Z. bailii insertions and the conspicuous absence of sequence similarity at breakpoints suggest they replicate via an extrachromosomal circular intermediate and insert via nonhomologous recombination. By contrast, introgression that arises via interspecific hybridization is currently thought to occur slowly, requiring repeated backcrossing with one of the parental species [15]. However, the structure of the MEP2 rearrangements we have discovered suggests a mechanism by which introgressive hybridization can occur rapidly. In each case, one of the rearranged chromosomes consists almost exclusively of S. cerevisiae sequences except for a precise replacement of several Kb with S. uvarum sequences. Diploid MEP2 fusion hybrids that undergo meiosis may produce a small number of spores that contain a haploid complement of only S. cerevisiae chromosomes including the one S. cerevisiae chromosome with the S. uvarum MEP2 region. Alternatively, loss of the S. uvarum chromosomes from the MEP2 fusion hybrid, similar to what has been described before for interspecific hybrids undergoing selection [42], could result in an “S. cerevisiae” strain containing just the introgressed S. uvarum MEP2 region. These scenarios do not require repeated backcrosses to one of the parents, and open up the possibility for rapidly evolving adaptive innovations forbidden to either parental species.
Materials and Methods
Strains
The S. cerevisiae parental strain is a derivative of laboratory strain S288C (strain “CC230”; MATα; ura3-52; ho), while the S. uvarum parental strain is derived from strain CBS7001 (strain “CC6”; MATa/MATα; lys2-5/lys2-5; HO/HO; see Table 1 for complete list of strains used in this study). Their F1 interspecific hybrid, GSY86, was obtained by mass-mating CC230 with mass-sporulated CC6 and selecting for prototrophy. Individual evolved clones that were further studied are also shown in Table 1.
Chemostat media and culture conditions
The “Delft” nitrogen-limiting medium used for batch and chemostat cultures was based on that described by Boer et al. [85] as follows: the basal nitrogen-limiting medium (“basal salts”) consisted of the following components per liter: 0.15 g (NH4)2SO4, 5.3 g K2SO4, 3.0 g KH2PO4, and 0.5 g MgSO4.7H2O, to which was added 1× vitamins and 1× trace metals (both as in [86]), as well as 0.02 g uracil, 0.03 g lysine, 0.06 g leucine and 9 g glucose. Strains were grown at 25°C in 500 mL fermenters (INFORS AG) with a working volume of 300 mL. Impeller speed was set to 300 rpm; airflow to 10 L h−1; the target dilution rate was 0.16 h−1.
Founding and sampling of the experimental populations
The founder F1 hybrid was first grown overnight in 2 mL of YPD-1% glucose, whereupon 500 µL of this culture was transferred into 25 mL Delft nitrogen-limiting media and grown overnight at 25°C. Three mL of this culture were sterilely transferred into 27 mL of sterile glucose, vitamins, metals, uracil, leucine, and lysine at the prescribed concentrations, and the suspension added by positive pressure to an INFORS vessel containing 270 mL of autoclaved basal salts. Three separate fermenters (Vessels A, B, and C) were inoculated in this manner. Populations were sampled every 48 h (∼10 generations). Two and one-half mL of cell suspension were withdrawn from the chemostat vessels and apportioned as follows: (i) 500 µL were added to 500 µL sterile 30% glycerol, then archived in duplicate at −80°C; (ii) 1 mL of cells were sterile-filtered through a 0.45 µm in-line filter and retained for assay of extracellular metabolites, (iii) 100 µL were diluted 9∶1 in glass-distilled water, and optical density measured at 600 nm using a Spectronic Biomate 3 spectrophotometer. Every 50 generations, archived populations were streaked onto YPD agar, and a random subset karyotyped by CHEF gel electrophoresis, as described below. A subset of these clones was further analyzed for changes in genome architecture by aCGH and by Illumina whole genome sequencing, as described below.
Metabolite and biomass assays
Residual glucose was assayed spectrophotometrically on cell-free filtrate using R-Biopharm assay Kit #716251 (R-Biopharm, Darmstadt, Germany). Residual ammonium was determined by a modified version of the Berthelot reaction [87], scaled down for 96-well format. Biomass was estimated by filtering 50 mL of chemostat culture onto tared 0.45 µm nylon filters, and drying filters in a desiccator at 37°C for 48 hrs. To test for statistically significant differences in growth and residual metabolites between experimental populations from the first steady-state (t-initial, 10 generations) and the last (t-final at ∼200 generations) time points, and to test physiological data obtained by growing single clones to steady state, we used ANOVA followed by a Student-Newman-Kuels multiple comparison test. All statistics were calculated with Sigma Plot 11 (Systat Software, San Jose, CA).
Analysis of yeast karyotypes
CHEF analysis was performed on two randomly-chosen single colonies isolated on YPD agar from frozen glycerol stocks of ∼50, ∼100, ∼150, and ∼200 generation population samples, from each of the three independent experimental populations. Analyses were performed essentially as described [88], [89].
Array-based Comparative Genomic Hybridization (aCGH)
A complete description of the 2-species (S. cerevisiae and S. uvarum) array design is given in [62]; arrays were manufactured by Agilent and contain ∼5500 oligonucleotide species-specific probes per species, approximately evenly spaced across each genome. Genomic DNA from ancestral and evolved clones was prepared using Zymo Research YeaStar columns according to the manufacturer's recommendations, and then digested with HaeIII. We then labeled 350 ng of this DNA with Cy5 (red). We similarly labeled, but instead with Cy3 (green), the same amount of reference DNA, which consisted of an equimolar mix of sheared genomic DNA from S. uvarum (CBS7001) and from S. cerevisiae (S288c). The labeled experimental and reference DNAs were then mixed together and hybridized to the 2-species microarrays as described [62]. Microarray data have been deposited in the Gene Expression Omnibus (GEO) repository [http://www.ncbi.nlm.nih.gov/geo/] under accession GSE18060. The Caryoscope program [90] was used to view microarray data in a genomic context.
Colony–PCR assay of multiple clones from population samples
Single-colony isolates were obtained by plating onto YPD agar small aliquots of the populations corresponding to 0, ∼50, ∼100, ∼150 and ∼200 generations, from Vessels A and B. Twelve isolates per time point, per vessel (60 isolates total per vessel) were subjected to colony lysis and subsequent PCR using primer combinations (Table S1) specific for the: (1) S. cerevisiae MEP2 gene, (2) S. uvarum MEP2 gene, (3) S. cerevisiae-S. uvarum fusion MEP2 gene found in evolved clones, and (4) S. uvarum-S. cerevisiae “reverse-fusion” MEP2 gene. PCR products that arose from the S. cerevisiae-S. uvarum MEP2 fusion gene-specific PCRs were Sanger-sequenced using the sequencing primers shown in Table S1.
Whole-genome DNA sequencing and analysis
DNA isolation was performed, using Qiagen G-100 genomic-tip columns as described by the manufacturer, from strains GSY86, GSY2532, GSY2533, and GSY2535. The DNA was then used to prepare libraries for Illumina sequencing as described [91], using barcoded adaptors for multiplexed paired-end sequencing. Flow cells for the Illumina HiSeq 2000 platform were prepared according to manufacturer's instructions and sequencing was performed for 100 cycles for each of paired-end reads. Read data has been deposited at the NCBI under BioProject PRJNA172024. Reads were mapped using BWA [92] to a combined S. cerevisiae – S. uvarum reference genome (S. cerevisiae genome downloaded from http://www.yeastgenome.org on Feb 24, 2011, plus S. uvarum genome assembly downloaded from http://saccharomycessensustricto.org May 26, 2011). SNPs were identified using the GATK [93], [94]. No subsequent hard-filtering of identified SNPs was performed; instead, SNPs present in the unevolved GSY86 ancestor were discarded from the analysis, and the remaining SNPs present in the evolved clones were manually inspected using Samtools tview [95], with only those showing sufficient coverage and quality given further consideration; these are shown in Table S2.
Quantitative RT–PCR
The founder interspecific hybrid GSY86 and evolved strain GSY2532 were cultured in monoculture to steady state (∼15 generations) in two independent NH4+-limited chemostats each; cultures were harvested by fast-filtration on 0.45 µm Nylon filters, frozen in liquid nitrogen, then stored at −80°C until RNA purification. Hot phenol RNA preparation was performed as described previously [96], followed by treatment with Ambion TURBO-DNAfree DNAse using manufacturer's recommendations (Life Technologies). 2 µg of total RNA were reverse transcribed using oligo dT primer and Superscript III according to the manufacturer's instructions (Invitrogen). Real-time qPCR was performed on a Bio-rad CFX96 cycler using SsoFast EvaGreen Supermix (Bio-rad). For GSY86, separate PCR reactions for detecting the S. cerevisiae and the S. uvarum MEP2 transcripts were performed, using primers shown in Table S1, in technical triplicates for each of the two biological replicates. Note that the primer pairs for detecting the S. cerevisiae and the S. uvarum MEP2 transcripts were first determined by PCR (using genomic DNA) to be specific for each species. Control PCR reactions (in triplicate, for each biological duplicate) for the reference S. cerevisiae TFC1 and S. uvarum YDR458C genes [97] (primers shown in Table S1) were also performed. For GSY2532, qRT-PCR was performed similarly, except using primers to detect the S. cerevisiae - S. uvarum MEP2 fusion transcript (see Table S1).
Competition in chemostats
Competitive chemostat experiments were performed for the interspecific hybrid GSY86 and each of three independently-evolved strains from Vessels A, B, and C, respectively (GSY2532, GSY2533 and GSY2535). Each strain was competed pairwise (in triplicate) against a common GFP-marked F1 hybrid reference strain (GSY2590, see Table 1; it is similar to GSY86 but with the S. cerevisiae parent genome containing the fluorescent GFP marker inserted into the YBR209W locus [72]). Each strain was grown to steady state, as determined by constant optical density for ∼24 h; culture volume for the reference strain was set at 500 mL, and its competitors at 300 mL. 150 ml were removed from each competitor vessel and replaced with 150 mL of the reference strain. The competition was followed for ∼15 generations. Beginning at the time of mixing (t-zero), 1 mL samples of the mixed populations were collected every 8–12 h, spun at 11,000×g for 2 min, resuspended in 0.5 mL of 1× PBS, then stored at 4°C until FACS analysis. Flow cytometry was performed using a FACSCaliber flow cytometer (Becton Dickinson, San Jose, CA) using a 488 nm laser for excitation of GFP and signal collection using a 530-30 bypass filter. Analysis was performed using CellQuest 3.3. Selection coefficients were determined from the linear regression of ln [Test/Reference] against generations, using methods developed by [65]. We used ANOVA followed by Tukey's HSD to test for differences among competition coefficients.
Supporting Information
Zdroje
1. GreavesM, MaleyCC (2012) Clonal evolution in cancer. Nature 481 : 306–313 doi:10.1038/nature10762.
2. PodlahaO, RiesterM, DeS, MichorF (2012) Evolution of the cancer genome. Trends Genet 28 : 155–163 doi:10.1016/j.tig.2012.01.003.
3. GrossBL, RiesebergLH (2005) The ecological genetics of homoploid hybrid speciation. Journal of Heredity 96 : 241–252 doi:10.1093/jhered/esi026.
4. GreigD, LouisEJ, BortsRH, TravisanoM (2002) Hybrid speciation in experimental populations of yeast. Science (New York, NY) 298 : 1773–1775 doi:10.1126/science.1076374.
5. HiraiH, TaguchiT, SaitohY, KawanakaM, SugiyamaH, et al. (2000) Chromosomal differentiation of the Schistosoma japonicum complex. Int J Parasitol 30 : 441–452.
6. MavarezJ, LinaresM (2008) Homoploid hybrid speciation in animals. Mol Ecol 17 : 4181–4185 doi:10.1111/j.1365-294X.2008.03898.x.
7. MavarezJ, SalazarCA, BerminghamE, SalcedoC, JigginsCD, et al. (2006) Speciation by hybridization in Heliconius butterflies. Nat Cell Biol 441 : 868–871 doi:10.1038/nature04738.
8. de Barros LopesM, BellonJR, ShirleyNJ, GanterPF (2002) Evidence for multiple interspecific hybridization in Saccharomyces sensu stricto species. FEMS Yeast Research 1 : 323–331.
9. LitiG, PeruffoA, JamesSA, RobertsIN, LouisEJ (2005) Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex. Yeast 22 : 177–192 doi:10.1002/yea.1200.
10. QuerolA, BondU (2009) The complex and dynamic genomes of industrial yeasts. FEMS Microbiology Letters 293 : 1–10 doi:10.1111/j.1574-6968.2008.01480.x.
11. LitiG, BartonDBH, LouisEJ (2006) Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174 : 839–850 doi:10.1534/genetics.106.062166.
12. GreigD (2008) Reproductive isolation in Saccharomyces. Heredity (Edinb) 102 : 39–44 doi:10.1038/hdy.2008.73.
13. MacleanCJ, GreigD (2011) Reciprocal gene loss following experimental whole-genome duplication causes reproductive isolation in yeast. Evolution 65 : 932–945 doi:10.1111/j.1558-5646.2010.01171.x.
14. AndersonE, HubrichtL (1938) Hybridization in Tradescantia. III. The evidence for introgressive hybridization. American Journal of Botany 25 : 396–402.
15. DowlingTE, SecorCL (1997) The role of hybridization and introgression in the diversificataion of animals. Annu Rev Ecol Syst 28 : 593–619.
16. DowlingTE, DeMaraisBD (1993) Evolutionary significance of introgressive hybridization in cyprinid fishes. Nature 362 : 444–446.
17. MalletJ (2005) Hybridization as an invasion of the genome. Trends in Ecology & Evolution 20 : 229–237 doi:10.1016/j.tree.2005.02.010.
18. ZhangH, SkeltonA, GardnerRC, GoddardMR (2010) Saccharomyces paradoxus and Saccharomyces cerevisiae reside on oak trees in New Zealand: evidence for migration from Europe and interspecies hybrids. FEMS Yeast Research 10 : 941–947 doi:10.1111/j.1567-1364.2010.00681.x.
19. WeiW, McCuskerJH, HymanRW, JonesT, NingY, et al. (2007) Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci USA 104 : 12825–12830 doi:10.1073/pnas.0701291104.
20. MullerLAH, McCuskerJH (2009) A multispecies-based taxonomic microarray reveals interspecies hybridization and introgression in Saccharomyces cerevisiae. FEMS Yeast Research 9 : 143–152 doi:10.1111/j.1567-1364.2008.00464.x.
21. UsherJ, BondU (2009) Recombination between homoeologous chromosomes of lager yeasts leads to loss of function of the hybrid GPH1 gene. Appl Environ Microbiol 75 : 4573–4579 doi:10.1128/AEM.00351-09.
22. DonigerSW, KimHS, SwainD, CorcueraD, WilliamsM, et al. (2008) A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet 4: e1000183 doi:10.1371/journal.pgen.1000183.
23. EsbergA, MullerLAH, McCuskerJH (2011) Genomic structure of and genome-wide recombination in the Saccharomyces cerevisiae S288C progenitor isolate EM93. PLoS ONE 6: e25211 doi:10.1371/journal.pone.0025211.
24. DequinS, CasaregolaS (2011) The genomes of fermentative Saccharomyces. C R Biol 334 : 687–693 doi:10.1016/j.crvi.2011.05.019.
25. NaumovaES, NaumovGI, (null), (null), Masneuf-PomarèdeI (2011) Genetic diversity study of the yeast Saccharomyces bayanus var. uvarum reveals introgressed subtelomeric Saccharomyces cerevisiae genes. Research in Microbiology 162 : 204–213 doi:10.1016/j.resmic.2010.09.023.
26. RoncoroniM, SantiagoM, HooksDO, MoroneyS, HarschMJ, et al. (2011) The yeast IRC7 gene encodes a β-lyase responsible for production of the varietal thiol 4-mercapto-4-methylpentan-2-one in wine. Food Microbiology 28 : 926–935 doi:10.1016/j.fm.2011.01.002.
27. ErnyC, RaoultP, AlaisA, ButterlinG, DelobelP, et al. (2012) Ecological success of a group of Saccharomyces cerevisiae/Saccharomyces kudriavzevii hybrids in the Northern European wine making environment. Appl Environ Microbiol doi:10.1128/AEM.06752-11.
28. DunnB, RichterC, KvitekDJ, PughT, SherlockG (2012) Analysis of the Saccharomyces cerevisiae pan-genome reveals a pool of copy number variants distributed in diverse yeast strains from differing industrial environments. Genome Res 22 : 908–924 doi:10.1101/gr.130310.111.
29. DujonB (2010) Yeast evolutionary genomics. Nature Reviews Genetics 11 : 512–524 doi:10.1038/nrg2811.
30. SalzburgerW, BaricS, SturmbauerC (2002) Speciation via introgressive hybridization in East African cichlids? Mol Ecol 11 : 619–625.
31. VershininAV, AllnuttTR, KnoxMR, AmbroseMJ, EllisTHN (2003) Transposable elements reveal the impact of introgression, rather than transposition, in Pisum diversity, evolution, and domestication. Molecular Biology and Evolution 20 : 2067–2075 doi:10.1093/molbev/msg220.
32. RiesebergLH, ArcherMA, WayneRK (1999) Transgressive segregation, adaptation and speciation. Heredity (Edinb) 83 : 363–372.
33. TwyfordAD, EnnosRA (2012) Next-generation hybridization and introgression. Heredity (Edinb) 108 : 179–189 doi:10.1038/hdy.2011.68.
34. McClintockB (1983) The significance of responses of the genome to challenge. Nobel lecture 1–20.
35. WaghmareSK, BruschiCV (2005) Differential chromosome control of ploidy in the yeast Saccharomyces cerevisiae. Yeast 22 : 625–639 doi:10.1002/yea.1226.
36. ZeylC, VanderfordT, CarterM (2003) An evolutionary advantage of haploidy in large yeast populations. Science (New York, NY) 299 : 555–558 doi:10.1126/science.1078417.
37. KellisM, PattersonN, EndrizziM, BirrenB, LanderES (2003) Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423 : 241–254 doi:10.1038/nature01644.
38. BrownCA, MurrayAW, VerstrepenKJ (2010) Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20 : 895–903 doi:10.1016/j.cub.2010.04.027.
39. VitousekPM, HowarthRW (1991) Nitrogen limitation on land and in the sea: How can it occur? Biogeochemistry 13 : 87–115.
40. GoddardMR, BradfordMA (2003) The adaptive response of a natural microbial population to carbon - and nitrogen-limitation. Ecology Letters 6 : 594–598.
41. ColemanMC, FishR, BlockDE (2007) Temperature-dependent kinetic model for nitrogen-limited wine fermentations. Appl Environ Microbiol 73 : 5875–5884 doi:10.1128/AEM.00670-07.
42. PiotrowskiJS, NagarajanS, KrollE, StanberyA, ChiottiKE, et al. (2012) Different selective pressures lead to different genomic outcomes as newly-formed hybrid yeasts evolve. BMC Evolutionary Biology 12 : 46 doi:10.1186/1471-2148-12-46.
43. GreshamD, UsaiteR, GermannSM, LisbyM, BotsteinD, et al. (2010) Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. Proc Natl Acad Sci USA 107 : 18551–18556 doi:10.1073/pnas.1014023107.
44. WengerJW, PiotrowskiJ, NagarajanS, ChiottiK, SherlockG, et al. (2011) Hunger artists: yeast adapted to carbon limitation show trade-offs under carbon sufficiency. PLoS Genet 7: e1002202 doi:10.1371/journal.pgen.1002202.
45. FereaTL, BotsteinD, BrownPO, RosenzweigRF (1999) Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc Natl Acad Sci USA 96 : 9721–9726.
46. KobayashiT, HeckDJ, NomuraM, HoriuchiT (1998) Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev 12 : 3821–3830.
47. MariniAM, VissersS, UrrestarazuA, AndréB (1994) Cloning and expression of the MEP1 gene encoding an ammonium transporter in Saccharomyces cerevisiae. EMBO J 13 : 3456–3463.
48. BoeckstaensM, AndréB, MariniAM (2007) The yeast ammonium transport protein Mep2 and its positive regulator, the Npr1 kinase, play an important role in normal and pseudohyphal growth on various nitrogen media through retrieval of excreted ammonium. Mol Microbiol 64 : 534–546 doi:10.1111/j.1365-2958.2007.05681.x.
49. FritschES, SchachererJ, Bleykasten-GrosshansC, SoucietJ-L, PotierS, et al. (2009) Influence of genetic background on the occurrence of chromosomal rearrangements in Saccharomyces cerevisiae. BMC Genomics 10 : 99 doi:10.1186/1471-2164-10-99.
50. PutnamCD, PennaneachV, KolodnerRD (2005) Saccharomyces cerevisiae as a model system to define the chromosomal instability phenotype. Mol Cell Biol 25 : 7226–7238 doi:10.1128/MCB.25.16.7226-7238.2005.
51. VernonM, LobachevK, PetesTD (2008) High rates of “unselected” aneuploidy and chromosome rearrangements in tel1 mec1 haploid yeast strains. Genetics 179 : 237–247 doi:10.1534/genetics.107.086603.
52. AdamsJ, Puskas-RozsaS, SimlarJ, WilkeCM (1992) Adaptation and major chromosomal changes in populations of Saccharomyces cerevisiae. Curr Genet 22 : 13–19.
53. DunhamMJ, BadraneH, FereaT, AdamsJ, BrownPO, et al. (2002) Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 99 : 16144–16149 doi:10.1073/pnas.242624799.
54. CoyleS, KrollE (2008) Starvation induces genomic rearrangements and starvation-resilient phenotypes in yeast. Molecular Biology and Evolution 25 : 310–318 doi:10.1093/molbev/msm256.
55. QuerolA, Fernández-EspinarMT, del OlmoM-L, BarrioE (2003) Adaptive evolution of wine yeast. Int J Food Microbiol 86 : 3–10.
56. StambukBU, DunnB, AlvesSL, DuvalEH, SherlockG (2009) Industrial fuel ethanol yeasts contain adaptive copy number changes in genes involved in vitamin B1 and B6 biosynthesis. Genome Res 19 : 2271–2278 doi:10.1101/gr.094276.109.
57. MartínezC, GacS, LavinA, GangaM (2004) Genomic characterization of Saccharomyces cerevisiae strains isolated from wine-producing areas in South America. J Appl Microbiol 96 : 1161–1168 doi:10.1111/j.1365-2672.2004.02255.x.
58. LucenaBTL, Silva-FilhoEA, CoimbraMRM, MoraisJOF, SimõesDA, et al. (2007) Chromosome instability in industrial strains of Saccharomyces cerevisiae batch cultivated under laboratory conditions. Genet Mol Res 6 : 1072–1084.
59. LitiG, CarterDM, MosesAM, WarringerJ, PartsL, et al. (2009) Population genomics of domestic and wild yeasts. Nature 458 : 337–341 doi:10.1038/nature07743.
60. GonzálezSS, BarrioE, GafnerJ, QuerolA (2006) Natural hybrids from Saccharomyces cerevisiae, Saccharomyces bayanus and Saccharomyces kudriavzevii in wine fermentations. FEMS Yeast Research 6 : 1221–1234 doi:10.1111/j.1567-1364.2006.00126.x.
61. GonzálezSS, BarrioE, QuerolA (2008) Molecular characterization of new natural hybrids of Saccharomyces cerevisiae and S. kudriavzevii in brewing. Appl Environ Microbiol 74 : 2314–2320 doi:10.1128/AEM.01867-07.
62. DunnB, SherlockG (2008) Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res 18 : 1610–1623 doi:10.1101/gr.076075.108.
63. BellochC, Pérez-TorradoR, GonzálezSS, Pérez-OrtínJE, García-MartínezJ, et al. (2009) Chimeric genomes of natural hybrids of Saccharomyces cerevisiae and Saccharomyces kudriavzevii. Appl Environ Microbiol 75 : 2534–2544 doi:10.1128/AEM.02282-08.
64. LouisVL, DesponsL, FriedrichA, MartinT, DurrensP, et al. (2012) Pichia sorbitophila, an Interspecies Yeast Hybrid, Reveals Early Steps of Genome Resolution After Polyploidization. G3 (Bethesda) 2 : 299–311 doi:10.1534/g3.111.000745/-/DC1.
65. GreshamD, DesaiMM, TuckerCM, JenqHT, PaiDA, et al. (2008) The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet 4: e1000303 doi:10.1371/journal.pgen.1000303.
66. KaessmannH (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res 20 : 1313–1326 doi:10.1101/gr.101386.109.
67. RogersRL, BedfordT, LyonsAM, HartlDL (2010) Adaptive impact of the chimeric gene Quetzalcoatl in Drosophila melanogaster. Proc Natl Acad Sci USA 107 : 10943–10948 doi:10.1073/pnas.1006503107.
68. CrameriA, RaillardSA, BermudezE, StemmerWP (1998) DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391 : 288–291 doi:10.1038/34663.
69. MitelmanF, JohanssonB, MertensF (2007) The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7 : 233–245 doi:10.1038/nrc2091.
70. Kubitschek HE (1970) Introduction to research with continuous cultures. Englewood Cliffs, NJ: Prentice Hall. 1 pp.
71. Monod J (1958) Recherches sur la croissance des cultures bacteriennes. Paris: Hermann & cie. 1 pp.
72. KaoKC, SherlockG (2008) Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet 40 : 1499–1504 doi:10.1038/ng.280.
73. MariniA-M, Soussi-BoudekouS, VissersS, AndréB (1997) A Family of Ammonium Transporters in Saccharomyces cerevisiae. Mol Cell Biol 17 : 4282–4293.
74. RutherfordJC, ChuaG, HughesT, CardenasME, HeitmanJ (2008) A Mep2-dependent transcriptional profile links permease function to gene expression during pseudohyphal growth in Saccharomyces cerevisiae. Mol Biol Cell 19 : 3028–3039 doi:10.1091/mbc.E08-01-0033.
75. MariniAM, SpringaelJY, FrommerWB, AndréB (2000) Cross-talk between ammonium transporters in yeast and interference by the soybean SAT1 protein. Mol Microbiol 35 : 378–385.
76. SzostakJW, Orr-WeaverTL, RothsteinRJ, StahlFW (1983) The double-strand-break repair model for recombination. Cell 33 : 25–35.
77. BernsteinKA, RothsteinR (2009) At loose ends: Resecting a double-strand break. Cell 137 : 807–810 doi:10.1016/j.cell.2009.05.007.
78. LlorenteB, SmithCE, SymingtonLS (2008) Break-induced replication: what is it and what is it for? Cell cycle (Georgetown, Tex) 7 : 859–864.
79. HaberJE (1999) DNA recombination: the replication connection. Trends Biochem Sci 24 : 271–275.
80. LaRocqueJR, StarkJM, OhJ, BojilovaE, YusaK, et al. (2011) Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells. Proc Natl Acad Sci USA 108 : 11971–11976 doi:10.1073/pnas.1104421108.
81. SmithCE, LlorenteB, SymingtonLS (2007) Template switching during break-induced replication. Nature 447 : 102–105 doi:10.1038/nature05723.
82. KeelingPJ, PalmerJD (2008) Horizontal gene transfer in eukaryotic evolution. Nature Reviews Genetics 9 : 605–618 doi:10.1038/nrg2386.
83. GaleoteV, BigeyF, BeyneE, NovoM, LegrasJ-L, et al. (2011) Amplification of a Zygosaccharomyces bailii DNA Segment in Wine Yeast Genomes by Extrachromosomal Circular DNA Formation. PLoS ONE 6: e17872 doi:10.1371/journal.pone.0017872.g008.
84. NovoM, BigeyF, BeyneE, GaleoteV, GavoryF, et al. (2009) Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA 106 : 16333–16338 doi:10.1073/pnas.0904673106.
85. BoerVM, de WindeJH, PronkJT, PiperMDW (2003) The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem 278 : 3265–3274 doi:10.1074/jbc.M209759200.
86. VerduynC, PostmaE, ScheffersW, Van DijkenJ (1992) Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast 8 : 501–517.
87. WillisR, SchwabG, GentryC (1993) Elimination of interferences in the colorimetric analysis of ammonium in water and soil extracts. Communications in Soil Science and Plant Analysis 24 : 1009–1019.
88. CarleG, OlsonMV (1985) An electrophoretic karyotype for yeast. Proc Natl Acad Sci USA 82 : 3756–3760.
89. ChuG, VollrathD, DavisRW (986AD) Separation of large DNA molecules by contour-clamped homogeneous electric fields. Science (New York, NY) 234 : 1582–1585.
90. AwadIA, ReesCA, Hernandez-BoussardT, BallCA, SherlockG (2004) Caryoscope: An Open Source Java application for viewing microarray data in a genomic context. BMC Bioinformatics 5 : 151 doi:10.1186/1471-2105-5-151.
91. SchwartzK, WengerJW, DunnB, SherlockG (2012) APJ1 and GRE3 homologs work in concert to allow growth in xylose in a natural Saccharomyces sensu stricto hybrid yeast. Genetics 191 : 621–632 doi:10.1534/genetics.112.140053.
92. LiH, DurbinR (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 : 589–595 doi:10.1093/bioinformatics/btp698.
93. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303 doi:10.1101/gr.107524.110.
94. DePristoMA, BanksE, PoplinR, GarimellaKV, MaguireJR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43 : 491–498 doi:10.1038/ng.806.
95. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079 doi:10.1093/bioinformatics/btp352.
96. LeeA, HansenKD, BullardJ, DudoitS, SherlockG (2008) Novel low abundance and transient RNAs in yeast revealed by tiling microarrays and ultra high–throughput sequencing are not conserved across closely related yeast species. PLoS Genet 4: e1000299 doi:10.1371/journal.pgen.1000299.t002.
97. BullardJH, MostovoyY, DudoitS, BremRB (2010) Polygenic and directional regulatory evolution across pathways in Saccharomyces. Proc Natl Acad Sci USA 107 : 5058–5063 doi:10.1073/pnas.0912959107.
98. EdgarRC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 : 1792–1797 doi:10.1093/nar/gkh340.
99. KvitekDJ, SherlockG (2011) Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet 7: e1002056 doi:10.1371/journal.pgen.1002056.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy