
Surveillance-Activated Defenses Block the ROS–Induced Mitochondrial Unfolded Protein Response
Disturbance of cellular functions results in the activation of stress-signaling pathways that aim at restoring homeostasis. We performed a genome-wide screen to identify components of the signal transduction of the mitochondrial unfolded protein response (UPRmt) to a nuclear chaperone promoter. We used the ROS generating complex I inhibitor paraquat to induce the UPRmt, and we employed RNAi exposure post-embryonically to allow testing genes whose knockdown results in embryonic lethality. We identified 54 novel regulators of the ROS–induced UPRmt. Activation of the UPRmt, but not of other stress-signaling pathways, failed when homeostasis of basic cellular mechanisms such as translation and protein transport were impaired. These mechanisms are monitored by a recently discovered surveillance system that interprets interruption of these processes as pathogen attack and depends on signaling through the JNK-like MAP-kinase KGB-1. Mutation of kgb-1 abrogated the inhibition of ROS–induced UPRmt, suggesting that surveillance-activated defenses specifically inhibit the UPRmt but do not compromise activation of the heat shock response, the UPR of the endoplasmic reticulum, or the SKN-1/Nrf2 mediated response to cytosolic stress. In addition, we identified PIFK-1, the orthologue of the Drosophila PI 4-kinase four wheel drive (FWD), and found that it is the only known factor so far that is essential for the unfolded protein responses of both mitochondria and endoplasmic reticulum. This suggests that both UPRs may share a common membrane associated mechanism.
Published in the journal:
. PLoS Genet 9(3): e32767. doi:10.1371/journal.pgen.1003346
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003346
Summary
Disturbance of cellular functions results in the activation of stress-signaling pathways that aim at restoring homeostasis. We performed a genome-wide screen to identify components of the signal transduction of the mitochondrial unfolded protein response (UPRmt) to a nuclear chaperone promoter. We used the ROS generating complex I inhibitor paraquat to induce the UPRmt, and we employed RNAi exposure post-embryonically to allow testing genes whose knockdown results in embryonic lethality. We identified 54 novel regulators of the ROS–induced UPRmt. Activation of the UPRmt, but not of other stress-signaling pathways, failed when homeostasis of basic cellular mechanisms such as translation and protein transport were impaired. These mechanisms are monitored by a recently discovered surveillance system that interprets interruption of these processes as pathogen attack and depends on signaling through the JNK-like MAP-kinase KGB-1. Mutation of kgb-1 abrogated the inhibition of ROS–induced UPRmt, suggesting that surveillance-activated defenses specifically inhibit the UPRmt but do not compromise activation of the heat shock response, the UPR of the endoplasmic reticulum, or the SKN-1/Nrf2 mediated response to cytosolic stress. In addition, we identified PIFK-1, the orthologue of the Drosophila PI 4-kinase four wheel drive (FWD), and found that it is the only known factor so far that is essential for the unfolded protein responses of both mitochondria and endoplasmic reticulum. This suggests that both UPRs may share a common membrane associated mechanism.
Introduction
In order to survive, organisms have to deal with an adverse environment either by avoiding unfavorable or toxic conditions, or by dealing with the consequences of such exposure. The nematode C. elegans for this purpose has developed a number of survival strategies. First, the sensory capabilities of this soil-dwelling animal enable the detection of probably hundreds of adverse mechanical, thermal and chemical stimuli. These neurons are wired to interneurons that serve as a neuronal processor with analytical power, which in turn couples to a motor response to search for or avoid certain environmental conditions. Second, mechanisms have been established in C. elegans to prevent uptake, to inactivate detrimental chemicals, or to repair the consequences of toxin exposure [1]–[4].
As part of an avoidance strategy to minimize future encounter of a toxin, it was recently reported that C. elegans surveys pathways typically disrupted by pathogens or toxins to engage in defenses. Experimental inactivation of genes in these pathways was sufficient to stimulate an aversion behavior in which the animals avoid normally attractive bacteria [3]. In this study, a large number of genes were found suggesting that this surveillance system (cSADDs) monitors the activity of core cellular components, including translation, energy metabolism, and protein degradation, and triggers food aversion, innate immunity and detoxification defenses upon detection of perturbations.
Unfolded protein responses (UPRs) are evoked when unfolded or misfolded proteins exceed the chaperone folding capacity of the cell. In eukaryotes, individual UPR pathways have evolved for distinct subcellular compartments, such as the endoplasmic reticulum (ER) or the cytosol (for review, see [5], [6]). To restore protein homeostasis, the UPRs signal from the stressed subcellular compartment to the nucleus and initiate an upregulation of a discrete set of compensatory genes, among them compartment-specific chaperones (for review, see [7], [8]). In the nematode C. elegans, reporter gene fusions of the promoters of the respective chaperones have been applied to study the UPR pathways [9].
The cytosolic UPR, also known as heat shock response, is initiated by stress interfering with the cytosolic protein folding environment (heat, e.g.) and activates genes including the cytosolic chaperone gene hsp-16.2 [10], [11]. In the endoplasmic reticulum (ER), protein folding stress can be experimentally evoked by the administration of tunicamycin, an inhibitor of protein glycosylation [12], that triggers an unfolded protein response (UPRER) to upregulate the transcription of the ER-specific chaperone gene hsp-4 [13] and results, among others, in a general blockade of translation.
Cytosolic oxidative stress elicits responses that in higher eukaryotes activate the phase II detoxification system that is triggered by the transcription factor SKN-1/Nrf2. In C. elegans, this pathway cross-talks with the DAF-2/Insulin/IGF receptor pathway, signaling to its main effector, the transcription factor DAF-16/FOXO [14]. A number of genes have been identified that are differentially regulated by SKN-1, DAF-16, or a combination of both ([15]–[20], for review see [21]).
Beside the UPR of the cytosol and the ER, more recently an unfolded protein response specific to mitochondria has been described ([22]–[25], for review see [26], [27]). The unfolded protein response of the mitochondria (UPRmt) is initiated by several modes of mitochondrial stress and activates the expression of nuclear genes, among them hsp-6 and hsp-60 encoding mitochondrial chaperones [22]. Many of the described UPRmt inducing stressors interfere directly with the mitochondrial protein folding environment: Inducing stress signals include the downregulation of the mitochondrial chaperone genes hsp-6 and hsp-60, or knockdown of spg-7 encoding a mitochondrial protease [22], or genes encoding components of the ETC which function in a cell non-autonomous way [28]. A temperature-sensitive mutation, zc32, whose corresponding gene is still enigmatic, was phenotypically characterized and shown to conditionally activate the UPRmt [23]. Several molecular components of the UPRmt pathway have been proposed and suggested a mechanistic model (for review, see [26], [27]) in which, as a first step, accumulated unfolded or misfolded proteins are cleaved by the ClpP protease in the mitochondrial matrix [24]. Partly through the HAF-1 ABC transporter, the bZip transcription factor ATFS-1 is activated, whose nuclear targeting in turn directly induces the transcription of the mitochondrial chaperone genes hsp-6 and hsp-60 [25], [29]. The homeobox transcription factor DVE-1 and the ubiquitin-like protein UBL-5 are also part of this UPRmt model and induce, independently of ATFS-1, mitochondrial chaperone expression upon peptide efflux from HAF-1 [23]–[25], [30], [31]. Recently, a much simpler mechanism was suggested by the same researchers. Under non-stress conditions, atfs-1 mRNA in the cytosol generates a transcription factor which, by default, is transported via the TIM-TOM import complexes into the mitochondria and there is proteolytically inactivated. Stress that alters the mitochondrial membrane potential blocks protein import of ATFS-1, resulting in its cytosolic accumulation and subsequent nuclear transport, where it can activate hsp-6 and hsp-60 genes [29].
Mutations in proteins of the mitochondrial electron transport chain (ETC) typically distort electron transfer to oxygen and, thus, generate reactive oxygen species (ROS). Recently, it was suggested that, in addition to recognizing protein misfolding stress, ROS in a parallel pathway may generate a signal to downregulate translation initiation via the GCN-2 dependent phosphorylation of eIF2α [32]. Thus, in analogy to the UPRER, it was proposed that activating the UPRmt has two consequences: Downregulation of translation, and selective activation of expression of chaperone genes.
Paraquat is a non-selective contact herbicide that in experimental research is frequently used to provoke the generation of reactive oxygen species in the cell, since it accepts electrons in the electron transport chain (ETC) at the inner mitochondrial membrane and transfers them to molecular oxygen, generating the superoxide anion [33]–[36]. Paraquat administration, among others, induces the mitochondrial manganese superoxide dismutase gene sod-3 [22], which is known to respond to increased ROS [37], and also the UPRmt responsive gene hsp-60 [22]. The onset of the UPRmt reporter upon paraquat-mediated accumulation of ROS may be due to consecutive protein damage, such as irreversible protein carbonylations [38]. It was shown in recent years that a moderate elevation of ROS generated in the mitochondria, such as in a loss of function mutant of the ETC component ISP-1 [34], [39], leads to an increase in lifespan [34], [40]. This effect can also be mimicked by low concentrations of paraquat [34], [40], [41]. Thus, administration of paraquat/ROS may have either detrimental or beneficial consequences for a cell or an organism.
Here, we investigate the retrograde signaling to the hsp-6 promoter initiated by an increase in mitochondrial ROS, which we trigger by low doses of paraquat. Genome-scaled RNAi screening revealed, among others, ATFS-1 as essential for the retrograde mitochondrial stress response to ROS, similar to its role in UPRmt. We also found that HAF-1 is dispensable for the paraquat induced signaling, suggesting that a peptide efflux via HAF-1 is not required after ROS induced mitochondrial stress to induce the hsp-6 promoter. We identified 54 additional genes whose downregulation prevented the activation of hsp-6. 87% of them were previously shown to encode components of cellular surveillance monitored pathways, or were found in protein complexes involved in surveillance monitored pathways (cSADDs). We postulate that cellular surveillance serves as a master regulator, activation of which inhibits the onset of the paraquat induced UPRmt. pifk-1 encodes a novel PI 4-kinase, downregulation of which blocks the UPRmt independently of the surveillance pathway and may, therefore act downstream of it. Our model suggests that C. elegans uses decisions at several levels to protect itself from external and internal stress.
Results
Paraquat induces hsp-6 reporter expression
Expression of the mitochondrial chaperone gene hsp-6 is induced upon treatment of C. elegans with paraquat (Figure 1A), which has been considered to activate oxidative stress [34], [39], [42], [43] and the mitochondrial unfolded protein response (UPRmt) [22]. We devised a new protocol for paraquat administration which allowed the detection of essential embryonic genes involved in the UPRmt, which could not have been found in previous screens to identify components of the UPRmt. In this protocol L3 stage animals were cultured with paraquat for two days. To monitor stress resistance pathways we used a previously described hsp-6::gfp reporter strain which carries the zcIs13 transgene containing about 1.7 kb of the 5′ flanking region and the first 10 codons of hsp-6 fused to GFP [22]. We performed a concentration series and observed that hsp-6::gfp induction peaked around 0.5 to 1.7 mM and faded with increasing concentrations of paraquat correlating with increased toxicity (Figure S1). To lower the possible impact of toxicity, we performed subsequent experiments using the lower concentration of 0.5 mM paraquat. This induced the hsp-6 reporter 41-fold (Figure 1B). Visual inspection by stereomicroscopy revealed that GFP expression started one day after paraquat exposure.

In addition to the expression of hsp-6, activated UPRmt can also be monitored by GFP expression from the promoter of hsp-60 [22]. Comparing the fluorescence intensity of both reporters after paraquat exposure showed that at 0.5 mM hsp-6::gfp was induced significantly while hsp-60::gfp was not (Figure S2). At a paraquat concentration of 2.0 mM, both reporters were significantly induced, as published [22], hsp-6::gfp induction, however, was twenty times stronger (Figure S2). We concluded that hsp-6::gfp is the more sensitive reporter for monitoring paraquat exposure, and, thus, performed subsequent experiments with this reporter.
Paraquat induces hsp-6 reporter expression independent of HAF-1, but requires ATFS-1
The gene haf-1 encodes a mitochondrial inner-membrane localized ABC transporter considered necessary for mitochondrial peptide release to activate the transcription factor ATFS-1, resulting in its nuclear translocation and activation of hsp-6. HAF-1 was suggested to be an essential upstream component of the UPRmt, since it was reported that in a haf-1(ok705) deletion mutant neither the hsp-6 nor the hsp-60 reporters were induced by RNAi with spg-7 or by the uncharacterized zc32 mutation, which is a standard inducer of UPRmt [25]. In the presence of 0.5 mM paraquat we observed that haf-1 was dispensable for the activation of hsp-6::gfp (Figure 2). Moreover, we observed a hyper-activation of hsp-6::gfp indicating that, at this condition, loss of haf-1 may further induce rather than block hsp-6 expression (Figure 2A). In contrast, ATFS-1, which integrates UPRmt signaling at the hsp-6 promoter [25], [32], was required for paraquat induced hsp-6 expression. Knockdown of atfs-1 by RNAi abolishes the inducibility of the reporter completely (Figure 2). These data suggest that low doses of paraquat mediated the activation of hsp-6::gfp through ATFS-1, but do not require HAF-1.

ETC impairment by ROS activates hsp-6::gfp
The UPRmt was so far primarily investigated with stressors that seem to cause unfolded protein stress by directly interfering with mitochondrial proteostasis (such as the knockdown of mitochondrial chaperones or the inactivation of mitochondrial proteases [22]–[24], [27]). Paraquat, in contrast, is a compound primarily known to cause oxidative stress impairing the ETC [42]–[46]. We wondered whether the induction of hsp-6::gfp is specific for paraquat or whether also other conditions known to increase mitochondrial ROS can activate the reporter. We exposed the hsp-6 reporter strain from early L3 stage on for two days to 0.25 µM rotenone, which targets the ubiquinone of complex I, or to 0.25 µM antimycin A, which prevents electron transfer from coenzyme Q to cytochrome C. Both substances have been shown to increase the amount of ROS [33], [36]. As with paraquat, both toxins caused an induction of the hsp-6 reporter (Figure 3A) being in line with the idea that an increase in mitochondrial ROS induces hsp-6.
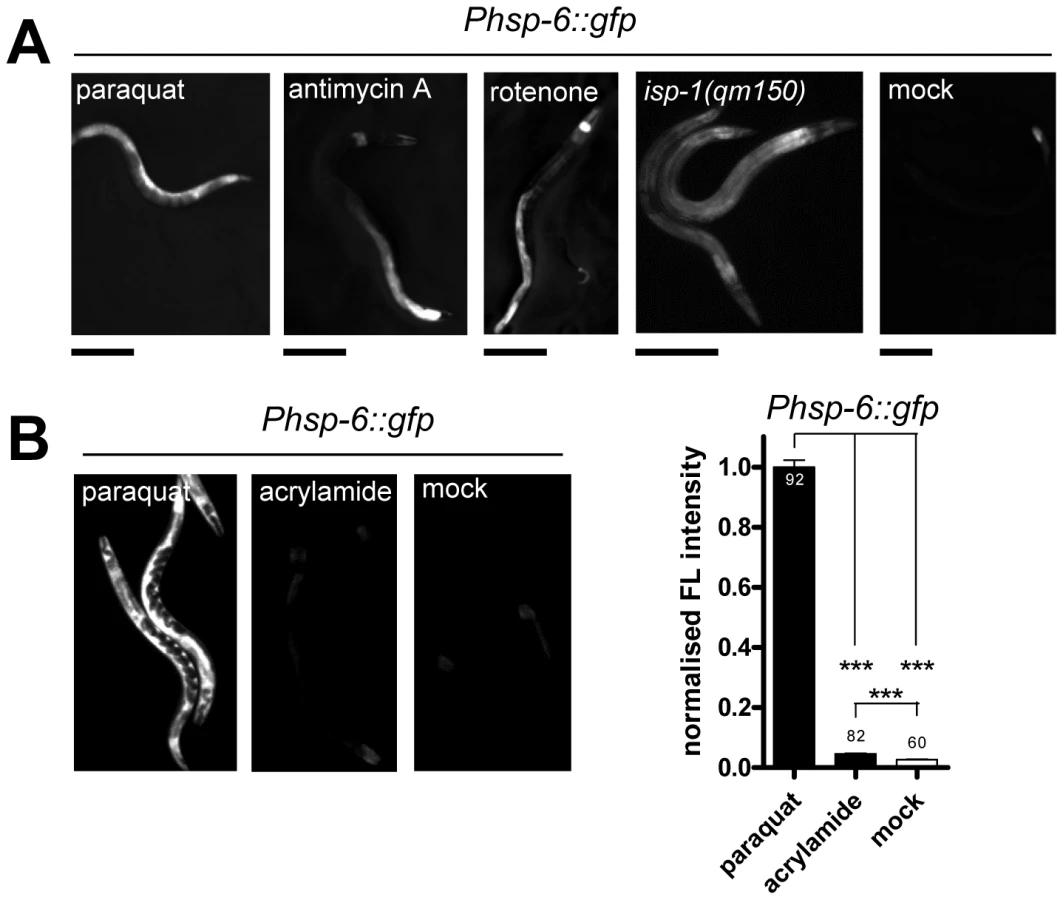
Genetic interference with the ETC by the introduction of a missense mutation in the isp-1 allele qm150 increases mitochondrial superoxide [34]. Supporting our previous findings, hsp-6::gfp was induced as well. GFP was constitutively expressed in the isp-1(qm150) mutant through all developmental stages and adulthood (Figure 3A). A similar result was observed for the mev-1(kn1) mutant, which carries a mutation in the cytochrome b of the mitochondrial respiratory chain complex II (data not shown).
We next wondered whether a compound causing oxidative stress not obviously linked to mitochondrial or ETC dysfunction can also activate hsp-6::gfp. The neurotoxin acrylamide triggers cytosolic phase II antioxidant responses in a SKN-1 dependent manner [47], [48]. Its mode of ROS production has not been associated with mitochondrial function [49]. We cultured early L3 larvae of the hsp-6 reporter strain for two days with 2.1 mM acrylamide, and observed only a slight 1.5-fold induction of the hsp-6 reporter, whereas paraquat induced the hsp-6 reporter 25 times more effectively (Figure 3B). The same acrylamide concentration, however, sufficed to activate the phase II response, monitored by the induction of gst-4::gfp. Our results suggest that hsp-6::gfp responds to both mutants and substances that are considered to increase mitochondrial ROS.
The ROS scavenger NAC reduces paraquat mediated hsp-6 induction
To analyze whether the increase in mitochondrial ROS is causative for the induction of the hsp-6 reporter, we compared the response of paraquat treated hsp-6 reporter animals with those in which paraquat treatment was paired with the addition of 10 mM of the ROS scavenger N-acetyl-L-cysteine (NAC) [50], [51]. We observed a 75% reduction in the intensity of hsp-6::gfp fluorescence in the presence of NAC (Figure 1C). This suggests that paraquat induced ROS is coupled to hsp-6::gfp induction. With this experiment we cannot distinguish whether the UPRmt is induced directly by the increased level of ROS or through a secondary protein damage caused by ROS. In the latter case, NAC treatment may also ultimately prevent protein misfolding by scavenging ROS and thus reducing hsp-6::gfp induction. Since the ROS scavenger NAC reduced hsp-6::gfp, rather than increased the induction, we consider it unlikely that paraquat induced ROS serves as a signal rather than a toxin, as has been proposed recently [32]. We conclude that ROS increase is a primary causative element in the induction of hsp-6::gfp, which, however, may also evoke the response of hsp-6 through an increase in mitochondrial unfolded proteins.
Paraquat treatment alters mitochondrial morphology
It is possible that 0.5 mM paraquat increases ROS production and signaling to hsp-6 directly or by affecting the integrity or functions of mitochondrial proteins. An effect of paraquat (0.1 mM) on protein oxidative damage has been shown, even though the abundance of mitochondria was not affected [34]. An altered mitochondrial morphology has been used as an argument for high levels of protein stress in the organelle [24]. Mitotracker is a compound that was used before in experiments to stain mitochondrial membranes [2], [16], [17]. Even at the low concentration of 0.5 mM paraquat, we found that our treatment resulted in substantial structural alterations of the hypodermal mitochondrial membrane (Figure 4A). We confirm in this experiment that 0.5 mM paraquat treatment alters mitochondrial structure, even though the general constitution and the morphology of animals is barely affected, except for a slight but obvious reduction in the body size of treated animals (see also Figure S1).

Paraquat does not induce unfolded protein responses in the ER or cytosol
Next, we investigated whether 0.5 mM paraquat induced stress responses in cellular compartments other than the mitochondria. Thus, we tested whether reporters of the unfolded protein response of the ER (hsp-4::gfp) [10] or of the cytosol (hsp-16.2::gfp) [9] also responded to paraquat treatment. We applied paraquat at early L3 stage and analyzed reporter fluorescence two days later. While both reporters were induced by their respective specific triggers tunicamycin and heat stress, no significant induction was observed with paraquat (Figure 4B, 4C). We conclude that 0.5 mM paraquat does not activate the unfolded protein response in the ER or in the cytosol.
Induction of hsp-6 by paraquat is independent of key regulators of other ROS stress responses
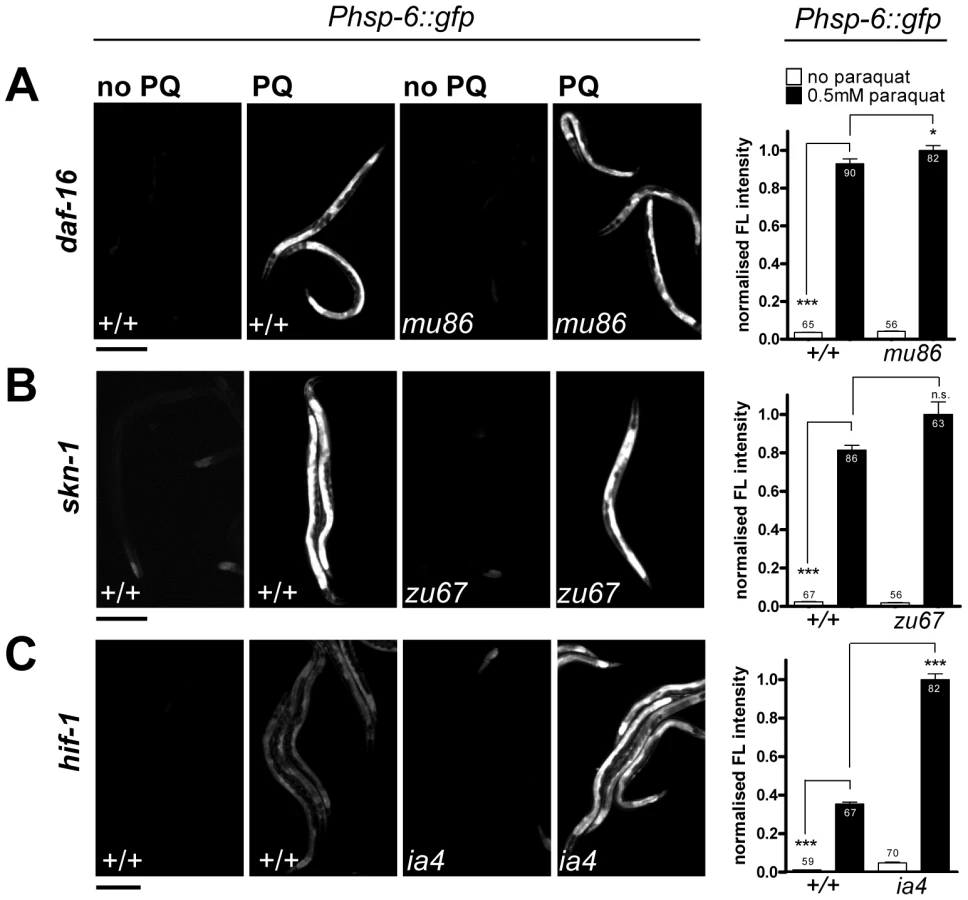
While paraquat does not activate cytosolic UPR, it is known to activate the cytosolic oxidative stress reporter gst-4::gfp [52]. In C. elegans, several key regulators of cytosolic oxidative stress responses have been described before. The transcription factors SKN-1 and DAF-16 are crucial for the induction of the phase II oxidative stress response and the defense against oxidative damage, respectively [53], [54]. Hypoxia inhibits respiration and activates HIF-1 by elevating the levels of ROS [40]. Therefore, expression of hsp-6 by paraquat could be dependent on these regulators. We therefore tested paraquat-induced expression of hsp-6::gfp in loss-of-function mutants of skn-1(zu67), daf-16(mu86) and hif-1(ia4). We found that none of the mutants prevented the induction of hsp-6 upon 0.5 mM paraquat exposure (Figure 5). This suggests that the response to paraquat triggers a pathway that does not require the transcription factors SKN-1, DAF-16 and HIF-1, and probably also not the pathways in which these factors are effectors namely the cytosolic stress response, insulin signaling, and the heat shock response.
A genome-scaled RNAi screen identifies 55 genes required for paraquat triggered hsp-6::gfp induction
To identify essential components of the paraquat mediated induction of hsp-6::gfp, we screened the ORFeome RNAi library (Open Biosystems) [55] for suppressors. Synchronized L1 larvae were allowed to develop by feeding on the respective RNAi bacteria for one day. Then, they were exposed to paraquat in order to bypass the paraquat-hypersensitive L1/early L2 stage and to benefit from the enhanced paraquat inducibility of the hsp-6 reporter in the L3 larval stage. After two days of incubation with paraquat we screened for worms that had failed to induce the hsp-6 reporter assuming that the respective RNAi clone downregulated a factor essential for hsp-6 induction (Figure S3).
We confirmed 55 genes whose knockdown led to an evident impairment of hsp-6::gfp induction (Table 1). The majority of these also showed morphological, behavioral or developmental abnormalities, among them impaired movement and developmental delay or arrest, which in each case appeared independent of paraquat administration.

Based on GO term analysis [56] the genes of all screening positives were assigned to functional groups. We found two subunits of a vacuolar H+ ATPase (Functional Group: ATP synthesis coupled proton transport), several proteasomal regulatory subunits (Functional Group: Cellular protein catabolic process (Proteolysis)), and several subunits of the cytosolic chaperonin complex, the orthologue of human TRiC/CCT (TCP-1 Ring Complex) (Functional Group: Cellular protein metabolic process (Protein folding)). We also detected several genes encoding proteins involved in intracellular protein transport, including two nuclear transport factors (Functional Group: Intracellular protein transport). Furthermore, the screen revealed three genes encoding small nuclear ribonucleoproteins (Functional Group: mRNA splicing). A large group, 26 genes, encode proteins of both ribosomal subunits (20 out of 55 screening positives) and additional genes (6 out of 55 screening positives) whose products have been associated with the translation of proteins (Functional Group: Protein translation). The mechanisms through which knockdown of these genes prevents hsp-6 reporter induction is not an attenuation of the general translation, since the inhibitory effect could not be mimicked if translation was attenuated by other means (Figure S4). Two genes (Functional Group: Regulation of transcription, DNA dependent) encode transcription factors, one being the GATA type transcription factor ELT-2, which is required for intestinal cell differentiation and maintenance [57], [58], and the other being ATFS-1, the bZip transcription factor involved in UPRmt [25]. The detection of the latter in the unbiased screen confirmed our observation that ATFS-1, in line with its previously described role in UPRmt signaling [25], [32], is also involved in the hsp-6::gfp induction by paraquat (see Figure 2). One functional group was assigned to two genes whose products are putatively involved in signaling (Functional Group: Signaling). Seven genes were not clustered in groups (Table 1). We quantified the hsp-6 reporter induction by paraquat for three screening positives, rpl-36, atfs-1, and the PI 4-kinase gene pifk-1 (F35H12.4). RNAi against each gene significantly prevented hsp-6::gfp induction (Figure 6).

Except for ATFS-1 none of the 55 screening positives had been implicated in the UPRmt before. In order to test whether we found so far not described genes in the UPRmt pathway, we performed RNAi against all in the temperature-sensitive mutant strain SJ52 [zc32; Phsp-60::gfp] used in a previous UPRmt screen. Since many of our screening positive RNAis caused larval arrest, the UPRmt screening protocol used by these authors was modified [23], [24]. We added synchronized L1 larvae to the respective RNAi plates, shifted the plates from 15°C to the restrictive temperature of 25°C when worms had developed to L4 larvae or young adults, and analyzed GFP fluorescence after two additional days. This protocol allowed the analysis of the role of our embryonic or larval lethal screening positives according to the UPRmt model. 29 candidates interfered with the activation of the hsp-60::gfp reporter in the zc32 mutant, among them atfs-1 which we considered as confirmation of the quality of our protocol (Table 1), 24 candidates did not obviously alter hsp-60::gfp expression in the background of zc32. We quantified GFP intensity in zc32 mutant animals expressing hsp-60::gfp which were treated with atfs-1, pifk-1 and rpl-36 RNAi. Confirming a previous report, knockdown of atfs-1 attenuated hsp-60 reporter induction [25] (Figure S5). Induction of the reporter was also efficiently prevented by RNAi of pifk-1 (Figure S5). In contrast, downregulation of rpl-36, which encodes a protein of the large ribosomal subunit, rather increased GFP expression of the hsp-60 reporter compared to the vector control (Figure S5). These results suggest that atfs-1 and pifk-1 are required, whereas rpl-36 may be dispensable for the zc32 triggered mitochondrial stress response (Table 1). Since the identity of the zc32 mutation is still enigmatic, it is currently not possible to interpret the difference obtained in both experimental paradigms.
ATFS-1, PIFK-1, and RPL-36 are also required for isp-1(qm150)–mediated induction of hsp-6::gfp
Beside paraquat other ROS-generating compounds and ETC mutants induce hsp-6::gfp (see Figure 3A). Here we tested whether the block of hsp-6::gfp induction is either specific for paraquat or if it would also block hsp-6::gfp induction resulting from the ETC mutant isp-1(qm150), which is a generator of mitochondrial superoxide [34]. RNAi specifically impairing paraquat uptake or metabolism would not block the isp-1(qm150) mediated signaling. We tested RNAi of atfs-1, which is known to be part of the UPRmt, pifk-1, which we found as a potential new component of the mitochondrial stress signaling, and as a third candidate rpl-36, which is not essential for the zc32, but for the paraquat-triggered hsp-6::gfp induction. GFP expressing L1 larvae were placed on the respective RNAi plates and grown until adulthood. Analyses in adulthood were possible since RNAi against these three genes did not cause larval arrest. During larval growth, GFP was continuously expressed on control plates. On the RNAi plates however, GFP fluorescence was strongly reduced. Downregulation of all three screening positives during postembryonic stages of isp-1(qm150) abrogated the induction of the hsp-6 reporter (Figure 7).

We conclude from these data that none of the three tested screening positives affects paraquat metabolism but rather mechanistic steps in the signaling cascade. However, alternatively it is also possible that these RNAis relieve the worms of ROS stress and thereby prevent the induction of hsp-6::gfp.
Knockdown of screening positive genes increases paraquat sensitivity
The expected function of a stress signaling pathway is to trigger a protective response. Screening positives may emerge for two different reasons: Either a specific stress signaling pathway could be blocked, or, alternatively, the level of stress could be reduced. In order to distinguish between these two alternatives we reasoned that a relief of stress would lead to paraquat hyposensitivity or resistance, whereas the disruption of a protective function would lead to an increased sensitivity to paraquat. We tested RNAi with three exemplary screening positives (atfs-1, rpl-36, and pifk-1) in a paraquat toxicity assay during larval development. Synchronized L1 larvae were raised on the respective RNAi plates containing 0.4 mM paraquat, and the number of animals on each plate that reached adulthood at day 5 was counted. Without paraquat, all worms developed to become adults, except for rpl-36 (RNAi) of which 1.5% did not reach adulthood within this time. Following paraquat exposure, about 50% of the controls became adults until day 5. In contrast, none of the animals subjected to RNAi against atfs-1 and rpl-36 respectively reached adulthood in this time window, but 27% of pifk-1 (RNAi) animals became adult. This data indicate that RNAi against all three genes increased, rather than decreased, paraquat sensitivity (Figure 8). Thus, for these three exemplary screening positives a relief of stress scenario can be ruled out. Furthermore, we conclude that the established UPRmt component afts-1 contributes to a protective response in line with a previous report [32].

Most genes required for ROS–dependent hsp-6 induction selectively affect the response to mitochondrial stress
We noticed that none of our screening positives encodes a mitochondrial protein. Therefore it was important to investigate, if they function either in general stress responses or have a specific role in the mitochondrial stress response. Therefore we examined their putative function in the phase II oxidative stress response, the cytosolic unfolded protein response (heat shock response) and the unfolded protein response of the ER (UPRER). All 55 screening positives were assessed qualitatively by visual inspection under the dissecting microscope to test whether the respective RNAi knockdown abrogated GFP fluorescence of the reporters tested. To get more detailed insights we quantified three candidate screening positives (afts-1, rpl-36, pifk-1) for each stress response. ATFS-1 was chosen as a known UPRmt pathway component, pifk-1 emerged as a novel gene implicated in the UPRmt and rpl-36 RNAi enhanced zc32 triggered mitochondrial stress signaling but abolished paraquat mediated induction of the hsp-6 reporter.
Acrylamide induces a SKN-1 dependent induction of gst-4::gfp [47], [52], [59]. RNAi knockdown of none of the 55 candidates blocked gst-4 expression in response to 2.1 mM acrylamide (Table 1). Quantification of three screening positives revealed that gst-4::gfp fluorescence was not suppressed by rpl-36 and afts-1 RNAi, suggesting that the inactivation of these genes does not interfere with the class II response. However, RNAi of pifk-1 reduced both the basal expression of gst-4::gfp and the acrylamide dependent induction of the gene. We suggest that either pifk-1 affects gst-4 expression in a general way, or that induction by acrylamide also involves pifk-1 function to some extent (Figure 9A).

We noticed that RNAi of cct-1, cct-5, pas-4, and pas-7 already resulted in gst-4 expression in the absence of acrylamide, confirming a previous report [48] (Table 1). Thus, for those four candidates that affect protein folding and turnover, we could not exclude that such constitutive activation of the class II detoxification system might reduce the ROS burden after paraquat administration. This would render the worms more resistant to paraquat, and could explain why hsp-6 is not induced in those four experiments. While the cct-1/-5 RNAi mediated induction of gst-4 appeared to be independent of SKN-1, knockdown of the proteasomal subunit mitigates gst-4 expression via SKN-1 [48]. We anticipated, therefore, that such an indirect effect would be SKN-1 dependent, at least in case of RNAi against a proteasomal subunit gene. Therefore, we tested paraquat mediated hsp-6 induction in skn-1(zu67) mutant animals after RNAi with pas-4, and pas-7. Loss of function of SKN-1 did not reconstitute the paraquat mediated hsp-6 induction, which argues against such an indirect effect of the SKN-1 activating RNAi experiments. However, a SKN-1 independent relief of stress cannot be excluded.
Next, we tested whether the screening positives crosstalk with the cytosolic unfolded protein response. We heat-shocked L4 staged hsp-16.2::gfp reporter worms for 4 h at 34°C and observed fluorescence one day later. Qualitative assessment of GFP fluorescence revealed no obvious impairment in any of the RNAi experiments (Table 1). Quantification showed that RNAi against pifk-1 did not affect heat-shock induction of hsp-16.2::gfp, indicating that pifk-1 is not involved in this response. Knockdown of rpl-36 and atfs-1, the two factors affecting the UPRmt response, did not prevent, but significantly decreased hsp-16.2 induction to 34% and 58%, respectively (Figure 9B). This suggests that some crosstalk between the UPRmt and the heat shock responses exists, or that these genes have dual roles in both pathways. This would make sense, since noxious heat will also result in denaturation of mitochondrial proteins, which may also increase mitochondrial ROS production. Since most of the factors involved in UPRmt are cytosolic signaling components [25], the same proteins could also help in activating the cytoplasmic heat shock response.
Next, a possible role of the screening positives in the induction of the unfolded protein response of the endoplasmic reticulum (UPRER) was tested. UPRER was triggered by incubation with 7.2 µM tunicamycin and monitored using the hsp-4::gfp reporter [9]. We found three screening positives (vha-1, snap-1, and sec-23) whose knockdown induced the hsp-4 reporter already in the absence of tunicamycin implicating that the loss of expression of those genes induces ER stress constitutively. All three candidates play a role in intracellular protein transport. With one exception, pifk-1, visual inspection revealed that none of the other RNAi treated screening positives prevented or strongly reduced hsp-4::gfp induction (Table 1). Quantification of atfs-1, rpl-36 and pifk-1, respectively, showed that RNAi with atfs-1 did not affect induction significantly, whereas rpl-36 reduced the induction to 49%, which proved to be significant (Figure 9C). Thus, it may be possible that affecting the balance of ribosomal protein expression interferes with the induction of unfolded protein responses in both ER and mitochondria. Interestingly, the observed strong impairment of the UPRER upon knockdown of pifk-1 (Table 1) was confirmed by qualitative analyses. The induction of hsp-4::gfp was reduced to 20% compared to control RNAi (Figure 9C). This is remarkable since at least to our knowledge PIFK-1 is the first protein which seems to be implied in signaling of UPRs in both organelles.
cSADDs suppresses the response of hsp-6 to paraquat
We noticed that many of the screening positives we had identified are genes also identified in a recent publication by Melo et al. (2012). There, the authors report a cellular surveillance system, which they call cSADDS (cellular surveillance activated detoxification and defenses) that monitors basic cellular functions and, if compromised, generates specific behavioral, immune, and detoxification responses, respectively [3]. Downregulation of 36 of the 55 genes (65%) identified in our screen was identified to induce the cSADDs, including food aversion behavior ([3], Table S1). In addition, of the remaining 19 genes twelve encode proteins belonging to either functional protein classes or protein complexes which activate the cellular surveillance system upon distortion [3]. Thus, in total 87% of the screening positives encode proteins belonging to processes or complexes that are monitored by the surveillance system.
Given that the cellular surveillance system is monitoring life-threatening conditions, such as toxin or pathogen exposure, we hypothesized that cSADDs may inhibit the onset of other stress responses that are evoked by milder, not life-threatening stresses, like the concentrations we have chosen for paraquat administration to induce the UPRmt. Signaling from cellular surveillance partially requires the activity of a JNK signaling cascade, in which KGB-1 is an essential component. We hypothesized that interfering with the signaling of cSADDs by a mutation in kgb-1 should at least partially release its inhibitory impact of the UPRmt. To test this idea, we crossed the hsp-6 reporter strain with the kgb-1(um3) mutant, which has been shown to partially suppress the surveillance mediated food avoidance [3]. Then, in the presence of 0.5 mM paraquat, as used in our screening protocol, kgb-1(um3); hsp-6::gfp worms were grown on elt-2(RNAi) bacteria, knockdown of which triggers cSADDs mediated aversion [3]. Whereas downregulating elt-2 in kgb-1(+) control strains eliminated the GFP induction as reported, the introduction of the kgb-1(um3) mutation released this inhibitory effect to some extent (Figure 10A). We observed the same recovery of hsp-6::gfp induction in the kgb-1(um3) mutant when worms were grown on rpl-36 RNAi (Figure S6). We conclude that elt-2 and rpl-36, and probably the other 46 genes whose RNAi activated cSADDs, contribute to cSADDs mediated inhibition of the mitochondrial stress response.

In line with this idea, loss of kgb-1 would not affect the inhibitory effect of those screening positives that do not do not evoke cSADDs. ATFS-1, the transcription factor controlling hsp-6 activation [25] and PIFK-1 (this study) have not been detected as activators of cSADDs [3]. We therefore first tested whether RNAi with these genes triggers the aversion phenotype, that served as a readout for cSADDs [3]. RNAi of neither of both genes induced food aversion, whereas RNAi of elt-2, used as a control as described [3], did (Table 2). Next, we tested whether in a kgb-1 mutant paraquat triggered hsp-6::gfp induction is relieved, as we have observed using elt-2 RNAi. In line with our hypothesis, RNAi of afts-1 or pifk-1, both in the kgb-1(+) control strain and in the kgb-1(um3) mutant, still blocked the paraquat-triggered hsp-6::gfp induction (Figure 10A, 10B).
![Knockdown of ATFS-1 and PIFK-1 does not evoke aversion behaviour <em class="ref">[3]</em>.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/1ceac92940b01a8b222938e904d97cc5.png)
From these data we conclude that we identified two functionally different groups of genes: (1) those, like atss-1 and pifk-1, that are involved in signaling from the mitochondria to the nucleus resulting in hsp-6 induction, and (2) those that are involved in processes targeted by pathogen invasion and toxin attack, whose downregulation induces the cellular surveillance system and results in cSADDs, including behavioral, immune, and detoxification responses (Figure 10C).
Discussion
Mild stress induced by paraquat evoked the UPRmt in a ROS–dependent manner
In this work we analyzed the response of C. elegans to a low, non-lethal concentration (0.5 mM) of the ROS generator paraquat by inducing the UPRmt, visualized by expression of the hsp-6::gfp reporter gene. Whereas higher concentration of paraquat resulted in a dramatic impact on the development of C. elegans, including larval arrest or rapid death, the low concentration we used in our experiments only produced a slight delay of larval development, and even extended lifespan of the animals when applied at adult stage [40]. We show here that the established ROS scavenger NAC substantially reduced hsp-6::gfp induction in our protocol, suggesting that ROS generated by paraquat constitutes a toxic activity that provokes the UPRmt. A consequence of this toxic activity is morphological alterations in the mitochondrial architecture. Downregulation of atfs-1 severely affected the viability of animals maintained at 0.4 mM paraquat starting at L1, suggesting that at these conditions the activation of UPRmt is beneficial for C. elegans. We suggest that ROS induces the UPRmt independently of haf-1, which was been proposed to be an essential component of the UPRmt after induction by ethidium bromide, zc32 and clk-1(qm30) [29].
Novel screen for genes required for paraquat induction of hsp-6 revealed preferentially cSADDs genes
We conducted a genome scaled screen employing postembryonic RNAi exposure to identify genes involved in the paraquat triggered UPRmt. This was the first systematic analysis of genes required for the paraquat/ROS induced UPRmt 22,29, and also the first protocol that allowed screening with genes with an embryonic lethal mutant phenotype [23].
Among the 55 genes we identified was atfs-1, previously suggested to encode a key regulator of the UPRmt and activator of hsp-6 and hsp-60 transcription [29]. None of the other 54 genes had previously been implicated in the ROS induced UPRmt. Most of them encode proteins involved in basic cellular functions, which include components of the protein degradation and protein folding pathways, as well as translation. Accordingly, RNAi knockdown of most of them caused a pronounced delay or arrest of larval development already in the absence of paraquat, which did not affect our screening due to the postembryonic application of RNAi, but would have prevented their identification in previously described screens.
The largest family of genes identified encodes components of the small and large ribosomal subunits, as well as several factors involved in protein translation. It has been suggested that stress of both mitochondria and the endoplasmic reticulum result in the downregulation of translation. Therefore, RNAi against the ribosomal proteins, in a simple model, may prevent general translation and, thus, result in a relief of stress. In agreement with such a model, Baker et al. [32] recently suggested that, upon protein misfolding, the activation of the kinase GCN-2 results in an inhibition of translation initiation. Thus, upon mitochondrial stress, blocking translation would reduce the load on the protein folding machinery, and thereby alleviate stress. For a number of reasons downregulation of general translation was not observed by us: First, because downregulation of ribosomal genes did not prevent the expression of other GFP reporter genes tested in this study (Figure 9, Figure S5). Second, because we found that downregulation of rpl-36, a representative member of this group of genes, showed an enhanced rather than reduced sensitivity to paraquat. Third, a general reduction of translation mediated by the ife-2(ok306) mutant in our hands was not sufficient to phenocopy the effects of RNAi against ribosomal genes. Intriguingly, we find that, in addition to genes for the ribosomal subunits, most genes identified in our screen overlap with a list of genes found in a recently published report addressing food avoidance behavior as part of cSADDs (see Table S1) [3]. There, the existence of a systemic surveillance system of basic molecular functions was proposed, which triggers defensive molecular and behavioral consequences that allows animals to detect invading pathogens or exposure to toxins. RNAi of core cellular activities, which include translation and protein turnover, induces detoxification and innate immune defense already in the absence of a pathogen or pathogenic toxin.
A total of 36 of the 55 genes (65%) identified in our screen have been linked to aversion behavior ([3], Table S1). In addition, of the remaining genes all but seven, including afts-1 and pifk-1, encode proteins belonging to functional classes or protein complexes of which genes encoding other components have been identified in aversion behavior. Since the cellular surveillance system signals through an endocrine response which involves the activity of the JNK pathway, we tested whether inactivation of kgb-1 could release the inhibitory role of cSADDs. This we could show for elt-2 and rpl-36, but not for atfs-1 and pifk-1. The knockdown of the latter two genes did not trigger cSADDs, and therefore these genes are most likely specifically involved in UPRmt signaling.
In summary, we suggest that the activation of the UPRmt by ROS is monitored by the surveillance system. Down-regulating the activity any out of a large number of cSADDs inducing genes inhibits the activation of the paraquat triggered UPRmt, but does not affect heat shock response, UPRER or the SKN-1 dependent ROS response. The repression of UPRmt by cSADDs mediated pathogen attack seems at first glance counterintuitive, but might imply that particular stress responses are handled in a prioritized way in C. elegans. It would, for example, suggest that animals experiencing damage to basic cellular functions, e.g. by exposure to strong toxins, could block the response to mild mitochondrial stress, whereas other types of stress could be preferentially handled to save energy for the execution of acutely required repair processes. Another quite attractive hypothesis is that activation of cSADDs could block UPRmt in order to actively raise ROS levels locally as part of an active defense strategy. The toxic properties of ROS are used in both plants and humans in immune responses against invaders in a process called active burst (for an overview [60]–[64]), but so far we were unable to show a local increase in ROS as a consequence of cSADDs activation. An intricate aspect of surveillance system is that it also monitors the mitochondria and their functional integrity, since we observed that established inducers of the UPRmt, most notably including paraquat, themselves can activate the cellular surveillance system and consecutively may elicit food aversion behavior. We verified that the 0.5 mM paraquat used in our screen did not itself trigger food avoidance, eliminating a direct interference between two stress responses. Concentration higher than 5–10 mM paraquat, however, inevitably induced pronounced food avoidance in the worms as a consequence of cSADDs.
A number of genes have been identified that both induce hsp-6::gfp and food avoidance behavior, when depleted by RNAi [3], [22]. These genes encode proteins essential for main mitochondrial functions, such as cytochrome oxidase, ATP synthase, and HSP-6. Because cSADDs induced by these serious mitochondrial impairment induce UPRmt instead of blocking it, we conclude, that they must activate the cellular surveillance system by a variant mechanism, which prevents blocking of UPRmt (Figure 10). In summary, during mild mitochondrial stress the cellular surveillance system suppresses the induction of UPRmt to benefit from the remaining mitochondrial activity for other stress compensatory functions, whereas in case of severe mitochondrial stress the induction of UPRmt is favored in order to maintain an essential level mitochondrial metabolism.
A recently published list suggests that genes encoding key factors of mitochondrial biogenesis, mitochondrial fission, and mitophagy are induced through the UPRmt [29]. Because these are resource consuming processes, it is conceivable that the cSADDs downregulate the UPRmt in case of residual mitochondrial function in order to allocate these resources to other defense mechanisms optimizing the benefit for the cell.
PIFK-4 is a proposed new factor in UPRmt and UPRER stress signaling
Of the seven genes found in our screen that do not obviously function in food aversion, four (C14B1.2, C18A3.3, C23G10.8, W04A4.5) have not been annotated or studied before, and, thus, could not be clustered in a functional group. The other three genes (afts-1, pifk-1 and Y47D3B.1) are the three only genes with proposed signaling functions found in our screen. Y47D3B.1 encodes a protein which resembles a G-protein coupled receptor, whereas pifk-1 encodes a protein with similarities to phosphatidylinositol 4-kinase which has not been studied before in C. elegans. The role of ATFS-1 in UPRmt signaling has been described. Since knockdown of pifk-1 did not trigger the food aversion phenotype either (see Table 2), we suggest that it also may have direct signaling roles in the UPRmt.
Animals in which pifk-1 was downregulated by RNAi are viable. Our studies revealed that pifk-1 inhibition also abrogated expression of the hsp-4::gfp reporter upon tunicamycin exposure, indicating that this is the only gene in our screen that is essential for both UPRmt and UPRER responses. This is remarkable, since so far no genes have been identified that function in the UPR of both organelles.
pifk-1 is orthologous to the membrane-bound kinase Four wheel drive (Fwd) in Drosophila and its counterpart in humans, PI4K-beta (ENSP00000271657). Fwd has been identified as a key regulator of the small G-protein Rab11. It functions in membrane trafficking during cytokinesis [65]. This resembles the proposed role of its human orthologue, PI4K-beta, which was functionally characterized as a key enzyme for Golgi disintegration and reorganization during mitosis [66]. Our observation is consistent with an essential and specific function of PIFK-1/FWD in the UPR of organelles, but not in the cytosolic UPR or phase II detoxification mediated by SKN-1. Further research will be required to reveal the mechanistic details of how PIFK-1 may exert its role in the unfolded protein responses of mitochondria and endoplasmic reticulum.
Materials and Methods
Transgenic and mutant C. elegans strains
C. elegans variety Bristol, strain N2 was used as wild type strain. All strains were maintained and raised at 20°C on NGM agar seeded with Escherichia coli OP50 [67], unless otherwise indicated. The following strains were obtained from CGC: SJ4100: zcIs13[Phsp-6::GFP], SJ4005: zcIs4[Phsp-4::GFP], CL2166: dvIs19[pAF15(gst-4::GFP::NLS)], ST66: ncIs17[Phsp-16.2::eGFP+pBluescript], RB867: haf-1(ok705)IV, KX15: ife-2(ok306)X, ZG31: hif-1(ia4)V, CF1038: daf-16(mu86)I, EU1: skn-1(zu67)IV/nT1[unc ?(n754)let ?](IV;V), DP38: unc-119(ed3)III, MQ887: isp-1(qm150)IV. The following strain was obtained by backcrossing SJ4100 seven times against laboratory N2: BR5194: zcIs13[Phsp-6::GFP]. The following strains were obtained by crossing the respective mutants (see above) with BR5194: BR6118: haf-1(ok705); zcIs13[Phsp-6::gfp], BR6019: ife-2(ok306); zcIs13[Phsp-6::gfp], BR6097: hif-1(ia4); zcIs13[Phsp-6::GFP], BR6020: daf-16(mu86); zcIs13[Phsp-6::gfp], BR6098: skn-1(zu67)/nT1; zcIs13[Phsp-6::gfp], BR 6372: isp-1(qm150); zcIs13[Phsp-6::gfp] The reporter strain SJ4058: zcIs9[Phsp-60::gfp] was obtained from C. Benedetti. The UPRmt reporter strain SJ52: [zc32 II; hsp-60::gfp V] was kindly provided by C. Haynes.
RNA interference assays
RNAi screening protocol
Genome-scaled screening was performed in duplicated 96 well liquid culture plates using the ORFeome RNAi feeding library (Open Biosystems) [55]. Day 1: RNAi bacteria from frozen glycerol stocks were inoculated in 40 µl LB supplemented with 12.5 µg/ml tetracycline and 12.5 µg/ml carbenicillin and grown at 28°C, 180 rpm overnight. Eggs were prepared from gravid C. elegans adults by alkaline sodium hypochlorite treatment and allowed to develop in M9 at 15°C overnight. Day 2 : 1.0 mM IPTG was added and the incubation was continued for another 2 h at 37°C, 180 rpm. A suspension of synchronized C. elegans L1 larvae was diluted to a concentration of 1 worm/µl in M9 supplemented with 10 µg/ml cholesterol, 50 µg/ml carbenicillin, 12 µg/ml tetracycline, 1 mM IPTG and 10 µg/ml fungizone. 20 µl of this suspension were distributed to 96 well plates after the bacterial culture had cooled down to room temperature. Day 3: paraquat (Sigma) was added to each well to a final concentration of 2.0 mM. Day 5: Plates were screened for non-GFP-expressing worms. Positive RNAi bacteria were recloned, sequenced and retested in at least one additional liquid culture test and subsequently also on RNAi NGM agar plates (see below).
RNAi on NGM plates
A 1∶50 dilution of the respective RNAi bacterial overnight culture (37°C, 150 rpm) in LB medium supplemented with 12.5 µg/ml tetracycline and 12.5 µg/ml carbenicillin was grown for another 6 h at 37°C, 150 rpm. Bacteria were then seeded on NGM plates containing 1.0 mM IPTG and 25.0 mg/ml carbenicillin.
Stress induction on NGM agar plates
Eggs were prepared from the respective gravid C. elegans adults by exposure to alkaline sodium hypochlorite and allowed to hatch in M9 [67] overnight. Synchronized L1 larvae were placed on NGM agar plates seeded with the respective bacteria.
Heat shock assay
L1 larvae (ST66) were grown on the respective RNAi bacteria for two days, subjected to 34°C for 4 h and analyzed for GFP expression one day later.
Paraquat/Acrylamide stress assays
L1 larvae (BR5194, CL2166) were grown on the respective RNAi bacteria for 24 h, subjected to 0.5 mM paraquat (Sigma), 0.25 µM rotenone (Sigma), 0.25 µM antimycin A (Sigma), or 2.1 mM acrylamide (BioRad), respectively. Chemicals were added from aqueous stock solutions (for rotenone in 0.1% dimethylsulfoxide (DMSO)) onto the plates. An influence of DMSO on the investigated stress signaling has been ruled out. GFP expression was analyzed two days later.
UPRER assay
L1 worms (SJ4005) were immediately exposed to 7.2 µM tunicamycin (Sigma) after being placed on the respective RNAi bacteria and analyzed for GFP expression three days later.
zc32 stress assay
L1 larvae (SJ52) were grown on the respective RNAi bacteria at 15°C, subjected to the restrictive temperature of 25°C as soon as animals raised on control L4440 plates had developed to L4/young adults and analyzed for GFP after two days.
NAC assay
Day 1: N-acetyl-L-cysteine (NAC) (Sigma) aqueous stock solution (200.0 mM) was distributed to NGM agar plates to a final concentration of 15 mM. Gravid adults were left to lay eggs on NAC plates for 6 h. Day 3: Paraquat was added to the plates (0.5 mM). Day 5: GFP fluorescence was quantified.
Paraquat resistance test
Eggs were prepared from gravid C. elegans adults (N2) by exposure to alkaline sodium hypochlorite and allowed to hatch in M9 [67] over night. Synchronized L1 larvae were placed on NGM agar plates seeded with the respective bacteria and containing 0.4 mM paraquat. Worms were raised at 20°C for five days. Then the number of animals that reached the adult stage and the number of animals which still remained in larval stages were determined.
Staining of mitochondria
Lyophilized Mito Tracker stain (Mitotracker Deep Red FM, Invitrogen) was suspended in anhydrous dimethylsulfoxide to a stock solution of 1 M, which was diluted further in H20 to a working solution of 10 mM. Working solution was added to the worms on NGM agar plates to a final concentration of 100 nM 8 h prior to analyses.
Microscopy and image analysis
Live worms were analyzed for GFP expression either on NGM agar plates or in 96 well microtiter plates in liquid with a stereo microscope (SZX12, Olympus). Micrographs were taken from cold-immobilized animals on NGM plates using the stereo microscope and a Zeiss MRm2 CCD camera. For quantification micrographs were taken from sodium azide-immobilized animals with an Axioimager.Z1 compound microscope with an AxioCam MRm3 CCD camera; Axiovision software version 4.8.1 (Carl Zeiss AG, Germany) was used for image analysis. Mito Tracker stained mitochondria were analyzed with a Nikon Ti A1 confocal microscope and NIS-Elements AR 4.0 64-bit software using a 60× water immersion objective with a numerical aperture of 1.2.
Food aversion assay
RNAi
Assay plates were prepared as described [3]. Synchronized L1 staged N2 were placed in the middle of the bacterial lawn. Aversion was analyzed after 48 h and scored by the quotient of the amount of animals residing outside the bacterial lawn (Noff) and the total amount of animals (Ntotal) (aversion score (AV): Noff/Ntotal). Empty vector L4440 RNAi bacteria and elt-2 RNAi expressing bacteria were used as controls [3].
Statistical analysis
Statistical analyses were performed with GraphPad Prism 4 software using unpaired t test (with Welch's correction if required), one-way analysis of variance (plus Tukey's multiple comparison test), Mann-Whitney test or Kruskal-Wallis test (plus Dunn's multiple comparison test), respectively. For the comparison of data sets with more than one parameter (RNAi and drug treatment) the background expression of the non-drug treated RNAi-fed cohort was subtracted from the respective drug-treated cohort prior to analyses if not stated otherwise.
Supporting Information
Zdroje
1. PrzybyszAJ, ChoeKP, RobertsLJ, StrangeK (2009) Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech Ageing Dev 130 : 357–369.
2. BellierA, ChenCS, KaoCY, CinarHN, AroianRV (2009) Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog 5: e1000689 doi:10.1371/journal.ppat.1000689.
3. MeloJA, RuvkunG (2012) Inactivation of Conserved C. elegans Genes Engages Pathogen - and Xenobiotic-Associated Defenses. Cell 149 : 452–466.
4. PartridgeFA, TearleAW, Gravato-NobreMJ, SchaferWR, HodgkinJ (2008) The C. elegans glycosyltransferase BUS-8 has two distinct and essential roles in epidermal morphogenesis. Dev Biol 317 : 549–559.
5. LindquistS, CraigEA (1988) The heat-shock proteins. Annu Rev Genet 22 : 631–677.
6. PatilC, WalterP (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13 : 349–355.
7. WalterP, RonD (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334 : 1081–1086.
8. SorgerPK (1991) Heat shock factor and the heat shock response. Cell 65 : 363–366.
9. LinkCD, CypserJR, JohnsonCJ, JohnsonTE (1999) Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperones 4 : 235–242.
10. LinkCD, JohnsonCJ (2002) Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. Redox Cell Biology and Genetics, Pt B 353 : 497–505.
11. SnutchTP, BaillieDL (1983) Alterations in the pattern of gene expression following heat shock in the nematode Caenorhabditis elegans. Can J Biochem Cell Biol 61 : 480–487.
12. TakatsukiA, ArimaK, TamuraG (1971) Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. J Antibiot (Tokyo) 24 : 215–223.
13. ShenX, EllisRE, LeeK, LiuCY, YangK, et al. (2001) Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107 : 893–903.
14. TulletJM, HertweckM, AnJH, BakerJ, HwangJY, et al. (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132 : 1025–1038.
15. OhSW, MukhopadhyayA, DixitBL, RahaT, GreenMR, et al. (2006) Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat Genet 38 : 251–257.
16. DongMQ, VenableJD, AuN, XuT, ParkSK, et al. (2007) Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science 317 : 660–663.
17. SchusterE, McElweeJJ, TulletJM, DoonanR, MatthijssensF, et al. (2010) DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol Syst Biol 6 : 399.
18. OliveiraRP, Porter AbateJ, DilksK, LandisJ, AshrafJ, et al. (2009) Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8 : 524–541.
19. OkuyamaT, InoueH, OokumaS, SatohT, KanoK, et al. (2010) The ERK-MAPK pathway regulates longevity through SKN-1 and insulin-like signaling in Caenorhabditis elegans. J Biol Chem 285 : 30274–30281.
20. WangJ, Robida-StubbsS, TulletJM, RualJF, VidalM, et al. (2010) RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet 6: e1001048 doi:10.1371/journal.pgen.1001048.
21. SykiotisGP, HabeosIG, SamuelsonAV, BohmannD (2011) The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr Opin Clin Nutr Metab Care 14 : 41–48.
22. YonedaT, BenedettiC, UranoF, ClarkSG, HardingHP, et al. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of Cell Science 117 : 4055–4066.
23. BenedettiC, HaynesCM, YangY, HardingHP, RonD (2006) Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174 : 229–239.
24. HaynesCM, PetrovaK, BenedettiC, YangY, RonD (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13 : 467–480.
25. HaynesCM, YangY, BlaisSP, NeubertTA, RonD (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37 : 529–540.
26. PellegrinoMW, NargundAM, HaynesCM (2012) Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 1833 (2)
410–6 doi:10.1016/j.bbamcr.2012.02.019.
27. HaynesCM, RonD (2010) The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 123 : 3849–3855.
28. DurieuxJ, WolffS, DillinA (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 : 79–91.
29. NargundAM, PellegrinoMW, FioreseCJ, BakerBM, HaynesCM (2012) Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 337 (6094)
587–90 doi:10.1126/science.1223560.
30. HartlFU, MartinJ, NeupertW (1992) Protein folding in the cell: the role of molecular chaperones Hsp70 and Hsp60. Annu Rev Biophys Biomol Struct 21 : 293–322.
31. BroadleySA, HartlFU (2008) Mitochondrial stress signaling: a pathway unfolds. Trends Cell Biol 18 : 1–4.
32. BakerBM, NargundAM, SunT, HaynesCM (2012) Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2alpha Kinase GCN-2. PLoS Genet 8: e1002760 doi:10.1371/journal.pgen.1002760.
33. NisticoR, MehdawyB, PiccirilliS, MercuriN (2011) Paraquat - and rotenone-induced models of Parkinson's disease. Int J Immunopathol Pharmacol 24 : 313–322.
34. YangW, HekimiS (2010) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 8: e1000556 doi:10.1371/journal.pbio.1000556.
35. CochemeHM, MurphyMP (2008) Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem 283 : 1786–1798.
36. Degli EspostiM (1998) Inhibitors of NADH-ubiquinone reductase: an overview. Biochim Biophys Acta 1364 : 222–235.
37. HondaY, HondaS (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13 : 1385–1393.
38. NystromT (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24 : 1311–1317.
39. FengJ, BussiereF, HekimiS (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev Cell 1 : 633–644.
40. LeeSJ, HwangAB, KenyonC (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20 : 2131–2136.
41. SchulzTJ, ZarseK, VoigtA, UrbanN, BirringerM, et al. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6 : 280–293.
42. DrechselDA, PatelM (2009) Differential contribution of the mitochondrial respiratory chain complexes to reactive oxygen species production by redox cycling agents implicated in parkinsonism. Toxicol Sci 112 : 427–434.
43. FukushimaT, TanakaK, LimH, MoriyamaM (2002) Mechanism of cytotoxicity of paraquat. Environ Health Prev Med 7 : 89–94.
44. FukushimaT, YamadaK, IsobeA, ShiwakuK, YamaneY (1993) Mechanism of cytotoxicity of paraquat. I. NADH oxidation and paraquat radical formation via complex I. Exp Toxicol Pathol 45 : 345–349.
45. ShimadaH, HiraiK, SimamuraE, HattaT, IwakiriH, et al. (2009) Paraquat toxicity induced by voltage-dependent anion channel 1 acts as an NADH-dependent oxidoreductase. J Biol Chem 284 : 28642–28649.
46. YangW, LiJ, HekimiS (2007) A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 177 : 2063–2074.
47. LinkCD, JohnsonCJ (2002) Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. Methods Enzymol 353 : 497–505.
48. KahnNW, ReaSL, MoyleS, KellA, JohnsonTE (2008) Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem J 409 : 205–213.
49. CallemanCJ, BergmarkE, CostaLG (1990) Acrylamide is metabolized to glycidamide in the rat: evidence from hemoglobin adduct formation. Chem Res Toxicol 3 : 406–412.
50. BenrahmouneM, TherondP, AbedinzadehZ (2000) The reaction of superoxide radical with N-acetylcysteine. Free Radic Biol Med 29 : 775–782.
51. AruomaOI, HalliwellB, HoeyBM, ButlerJ (1989) The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 6 : 593–597.
52. TaweWN, EschbachML, WalterRD, Henkle-DuhrsenK (1998) Identification of stress-responsive genes in Caenorhabditis elegans using RT-PCR differential display. Nucleic Acids Res 26 : 1621–1627.
53. AnJH, BlackwellTK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17 : 1882–1893.
54. LehtinenMK, YuanZ, BoagPR, YangY, VillenJ, et al. (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125 : 987–1001.
55. RualJF, CeronJ, KorethJ, HaoT, NicotAS, et al. (2004) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14 : 2162–2168.
56. Huang daW, ShermanBT, LempickiRA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57.
57. HawkinsMG, McGheeJD (1995) elt-2, a second GATA factor from the nematode Caenorhabditis elegans. J Biol Chem 270 : 14666–14671.
58. SommermannEM, StrohmaierKR, MaduroMF, RothmanJH (2010) Endoderm development in Caenorhabditis elegans: the synergistic action of ELT-2 and -7 mediates the specification–>differentiation transition. Dev Biol 347 : 154–166.
59. HasegawaK, MiwaS, IsomuraK, TsutsumiuchiK, TaniguchiH, et al. (2008) Acrylamide-responsive genes in the nematode Caenorhabditis elegans. Toxicol Sci 101 : 215–225.
60. O'BrienJA, DaudiA, ButtVS, Paul BolwellG (2012) Reactive oxygen species and their role in plant defence and cell wall metabolism. Planta 236 (3)
765–79 doi:10.1007/s00425-012-1696-9.
61. Perez-PerezME, LemaireSD, CrespoJL (2012) ROS and autophagy in plants and algae. Plant Physiol
62. SuzukiN, MillerG, MoralesJ, ShulaevV, TorresMA, et al. (2011) Respiratory burst oxidases: the engines of ROS signaling. Curr Opin Plant Biol 14 : 691–699.
63. KumarA, FarhanaA, GuidryL, SainiV, HondalusM, et al. (2011) Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med 13: e39.
64. PuertollanoMA, PuertollanoE, de CienfuegosGA, de PabloMA (2011) Dietary antioxidants: immunity and host defense. Curr Top Med Chem 11 : 1752–1766.
65. PolevoyG, WeiHC, WongR, SzentpeteryZ, KimYJ, et al. (2009) Dual roles for the Drosophila PI 4-kinase four wheel drive in localizing Rab11 during cytokinesis. J Cell Biol 187 : 847–858.
66. GrahamTR, BurdCG (2011) Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol 21 : 113–121.
67. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
68. SyntichakiP, TroulinakiK, TavernarakisN (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature 445 : 922–926.
69. TavernarakisN (2007) Protein synthesis and aging: eIF4E and the soma vs. germline distinction. Cell Cycle 6 : 1168–1171.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Fine Characterisation of a Recombination Hotspot at the Locus and Resolution of the Paradoxical Excess of Duplications over Deletions in the General Population
- Molecular Networks of Human Muscle Adaptation to Exercise and Age
- Recurrent Rearrangement during Adaptive Evolution in an Interspecific Yeast Hybrid Suggests a Model for Rapid Introgression
- Genome-Wide Association Study and Gene Expression Analysis Identifies as a Predictor of Response to Etanercept Therapy in Rheumatoid Arthritis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy