
Detection of Slipped-DNAs at the Trinucleotide Repeats of the Myotonic Dystrophy Type I Disease Locus in Patient Tissues
Slipped-strand DNAs, formed by out-of-register mispairing of repeat units on complementary strands, were proposed over 55 years ago as transient intermediates in repeat length mutations, hypothesized to cause at least 40 neurodegenerative diseases. While slipped-DNAs have been characterized in vitro, evidence of slipped-DNAs at an endogenous locus in biologically relevant tissues, where instability varies widely, is lacking. Here, using an anti-DNA junction antibody and immunoprecipitation, we identify slipped-DNAs at the unstable trinucleotide repeats (CTG)n•(CAG)n of the myotonic dystrophy disease locus in patient brain, heart, muscle and other tissues, where the largest expansions arise in non-mitotic tissues such as cortex and heart, and are smallest in the cerebellum. Slipped-DNAs are shown to be present on the expanded allele and in chromatinized DNA. Slipped-DNAs are present as clusters of slip-outs along a DNA, with each slip-out having 1–100 extrahelical repeats. The allelic levels of slipped-DNA containing molecules were significantly greater in the heart over the cerebellum (relative to genomic equivalents of pre-IP input DNA) of a DM1 individual; an enrichment consistent with increased allelic levels of slipped-DNA structures in tissues having greater levels of CTG instability. Surprisingly, this supports the formation of slipped-DNAs as persistent mutation products of repeat instability, and not merely as transient mutagenic intermediates. These findings further our understanding of the processes of mutation and genetic variation.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1003866
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003866
Summary
Slipped-strand DNAs, formed by out-of-register mispairing of repeat units on complementary strands, were proposed over 55 years ago as transient intermediates in repeat length mutations, hypothesized to cause at least 40 neurodegenerative diseases. While slipped-DNAs have been characterized in vitro, evidence of slipped-DNAs at an endogenous locus in biologically relevant tissues, where instability varies widely, is lacking. Here, using an anti-DNA junction antibody and immunoprecipitation, we identify slipped-DNAs at the unstable trinucleotide repeats (CTG)n•(CAG)n of the myotonic dystrophy disease locus in patient brain, heart, muscle and other tissues, where the largest expansions arise in non-mitotic tissues such as cortex and heart, and are smallest in the cerebellum. Slipped-DNAs are shown to be present on the expanded allele and in chromatinized DNA. Slipped-DNAs are present as clusters of slip-outs along a DNA, with each slip-out having 1–100 extrahelical repeats. The allelic levels of slipped-DNA containing molecules were significantly greater in the heart over the cerebellum (relative to genomic equivalents of pre-IP input DNA) of a DM1 individual; an enrichment consistent with increased allelic levels of slipped-DNA structures in tissues having greater levels of CTG instability. Surprisingly, this supports the formation of slipped-DNAs as persistent mutation products of repeat instability, and not merely as transient mutagenic intermediates. These findings further our understanding of the processes of mutation and genetic variation.
Introduction
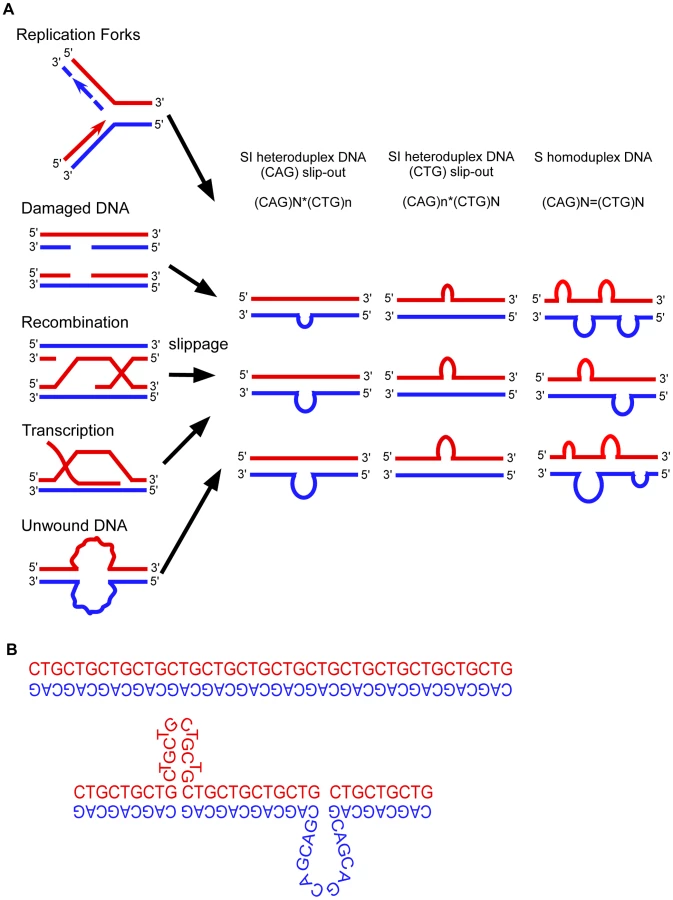
All models proposed to explain the instability of trinucleotide repeats involve DNA slippage at the repeats (Fig. 1) [1]–[12]. Slipped-DNAs were first hypothesized to exist in 1958 [13]. Slipped-DNAs are thought to contribute to more than 30 neuromuscular/neurodegenerative diseases caused by unstable microsatellite repeats, including myotonic dystrophy type 1 (DM1) and numerous cancers that show microsatellite instability [1]–[3], hence understanding slipped-DNAs in patient tissues is of great importance [14], [15]. Expansion mutations continue in DM1 patients as they age, coinciding with worsening symptoms. Patients exhibit inter-tissue repeat length differences as great as 5,770 repeats, with large expansions occurring in affected tissues such as brain, muscle and heart, indicating high levels of continuing expansions [4], [5]. The formation and aberrant repair of slipped-DNAs is a likely source of repeat instability and progressive disease severity in patients (Fig. 1) [6], [7]. An understanding of these DNA mutagenic intermediates in patients should provide insight as to how they may be processed and lead to mutations. The important questions demanding answers are 1) Do slipped-DNAs form at disease loci? 2) Do their levels vary in patient tissues that undergo variable levels of repeat expansion within a given individual? And, 3) What is the biophysical structure of these slipped-DNAs? These questions cannot be answered in a heterologous model system that shows repeat instability that does not reflect the instability ongoing in a patient, nor one lacking tissues. While slipped-DNAs have been characterized in vitro [8], [9], data supporting the presence of slipped-DNAs at an endogenous locus in biologically relevant tissues has been lacking. Previously it was reported that hairpin DNAs can form in a model cell line which contain CTG/CAG repeats integrated at an ectopic locus, where instability was contraction biased as opposed to the expansion bias present in affected patients (10). Although the authors report hairpin formation in living cells, that report could not comment upon either structure formation at an endogenous disease locus, their variation between tissues showing variable instability, upon their persistence in patients, or on the structural features of the DNAs. Furthermore, in that system, instability and hairpin detection depended upon DNA replication, contrasting with the high levels of instability arising in post-mitotic tissues of patients. It is imperative to study instability in patient tissues since repeat mutations are expansion-biased, arising by processes distinct from contractions, coupled with the known locus-specific effects, the tissue-specific variations of instability, and the fact that most expansions arise in non-replicating cells. To identify slipped-DNAs at a disease locus in patient tissues we devised a DNA-immunoprecipitation (IP) protocol that uses a highly specific monoclonal anti-DNA junction antibody (2D3) that recognizes 3-way DNA junctions [9], [11], [12], a structural feature of slipped-DNAs [8], [9]. The 2D3 antibody has been characterized extensively (summarized in Text S1 and citations therein). Briefly, 2D3 binds specifically to junction-containing DNAs with no sequence preference. Footprinting has mapped the 2D3 binding site at DNA junctions [9], [11], [12] where binding can occur from either of the angles sub-tended by the junction arms. For each junction 1–2 antibodies can bind per DNA junction. 2D3 does not bind single-stranded DNA, hairpins, Z-DNA, triplex or quadruplex, with no off-target binding reported in in vitro systems used (see Text S1 and citations therein). 2D3 binds best to slipped-DNAs [9], strengthening its use to isolate these structures.
Results
To verify that the anti-junction DNA antibody recognizes slipped-DNA structures with varying slip-out sizes and slip-out numbers, we assessed 2D3 binding to various slipped-DNAs using an electrophoretic mobility shift assay. Various slipped-DNA structures can form at trinucleotide repeats, and our lab has structurally characterized them in detail by electrophoresis, chemical and enzymatic probing, and electron microscopy [8], [9], [16]–[18]. While a slip-out does not translationally move along the repeat tract (slip or slide), multiple junction conformations occur and these are in dynamic equilibrium [8], [9], [16]–[18]. Previously, we demonstrated that 2D3 binds homoduplex slipped-DNAs of 50 CTG/CAG repeats on complementary strands as well as slipped intermediate heteroduplex DNAs (with an excess of 20 CAGs or 20 CTGs) [9]. These putative mutagenic intermediates may involve isolated slip-outs of various sizes, determined by the length difference between the two repeat-containing strands. Slipped-DNAs can also arise in tracts where the number of repeats between complementary strands does not differ, where each molecule contains multiple clustered short slip-outs [8], [9], [16]–[18]. For example each slipped molecule formed by (CTG)50•(CAG)50 contained 2–62 short slip-outs (1–31 on each strand), each composed of one to three repeat units [8], [9], [16]–[18]. To determine if 2D3 will recognize and bind to smaller slip-outs or clustered slip-outs, DNAs were made with isolated slip-outs of 1 - or 3 - or 20-excess repeat units of either the CTG or CAG strand, as well as a substrate with multiple clustered slip-outs on both strands along a tract of 50 repeats (Fig. 2A, see schematics). The antibody was incubated with each of these radioactively labeled DNAs, and resolved on polyacrylamide gels to visualize antibody-DNA complexes, evident as slower migrating than the protein-free DNA. Each of the substrates gave rise to slow-migrating species, where only the highest concentration led to partial shifting of the fully-duplexed control (leftmost lanes, see hollow arrowheads). These non-specific complexes were completely lost upon the addition of increasing amounts of competitor DNA (25-fold of non-specific competitor; linearized plasmid). This low-level non-specific binding, typical of many DNA-binding proteins/antibodies, has previously been observed to be readily competed-out [9], [11], [12]. In contrast, the lowest concentrations of 2D3 bound and shifted all of the slip-outs with an excess of 20 repeats (50×30 and 30×50) evident as several species (hollow and gray arrowheads). With the addition of competitor, these substrates remained completely shifted, but migrated faster (black arrowheads), indicating slip-out specific interaction. The 3 - and 1-excess slip-outs with variable 2D3 concentrations also yielded shifted species, which were partially resistant to non-specific competition. The multiple shifted species may reflect different antibody-junction conformations [8], [9], [16]–[18], since each antibody can bind at either of the three junction angles, each of these could migrate differently and yield a broad and smeared appearance. The multiple species may also reflect different numbers of antibodies, as it was previously demonstrated that one to two 2D3 antibodies could bind a DNA-junction [9], [11], [12]. The clustered slip-outs were readily shifted by 2D3 and progressively shifted with increasing amounts of 2D3; indicative of increasing numbers of bound antibodies to the many slip-outs/molecule. We have previously shown this interaction to be resistant to competition [9], [11], [12]. Thus, 2D3 bound all sizes of slip-outs tested, with only very minor off-target binding to linear DNA that is eliminated after competition (Fig. 2A). 2D3 binds most effectively to larger slip-outs and clustered slip-outs. Importantly, the anti-DNA junction antibody did not induce the formation of slipped-DNA in fully-duplexed (CTG)50•(CAG)50 DNA (Fig. 2A, leftmost lanes), consistent with its inability to induce cruciform or hairpin extrusion [11], [12], [19]. Further control experiments with longer repeats are described below. 2D3 binding to slipped-DNAs was sensitive to proteinase K. Non-specific antibody-DNA binding was not seen with an isotype-matched anti-actin antibody control, which should not bind DNA (detailed in Supplementary Fig. S1). Thus, the anti-junction DNA antibody specifically recognizes a variety of slip-out sizes as well as DNA containing isolated and multiple clustered slip-outs; supporting the antibody as a useful tool to detect slipped-DNA structures.

To determine whether slipped-DNAs are present in a disease locus of patient tissues we developed a DNA-IP strategy (Fig. 2C). To be sure that we were not inducing slipped-DNA formation through manipulations (genomic isolation, IP etc.) we used several precautionary measures and performed a series of controls. Precautionary measures included preparing genomic DNAs under conditions that avoided DNA denaturation or shearing. Genomic DNAs were prepared from patient tissues under non-denaturing conditions to avoid inducing the formation of unusual DNA structures (see Fig. S1 and Text S1). Slipped-DNAs in CAG/CTG repeats cannot be induced by DNA supercoiling, and their biophysical stability does not depend upon supercoiling [8], [20]. Slipped DNAs can stably exist in linear unrestrained DNA [8]. To reduce DNA shear, enhance immunoprecipitation and eliminate the binding of 2D3 to supercoil-dependent DNA structures (cruciforms), genomic DNAs were restriction digested with BbsI and BamHI (which cut 180 bp upstream and 244 bp downstream of the CTG repeat, respectively, Fig. 2C), and then IP'd using 2D3 antibody and protein G agarose beads. While the 2D3 antibody does not induce cruciform or hairpin extrusion (see above) or slipped-DNAs in short fragments (less than 50 repeats) (Fig. 2A) it was important to assess its effect upon disease-relevant CTG expansions. Control experiments using in vitro prepared DNAs with (CTG)500•(CAG)500 showed that the 2D3 antibody did not induce slipped-DNAs into the expanded tracts (Fig. S2A, detailed in Text S1). Furthermore, we tested the possibility that structures might arise during genomic isolation in two ways; first, by adding the synthetically formed, fully-paired (CTG)500•(CAG)500 or (CTG)50•(CAG)50 DNAs to tissues to follow them through the genomic isolation protocol (Fig. S1), which did not induce slipped-DNA formation, and second, by assembling and disassembling nucleosomes on a supercoiled (CTG)250•(CAG)250 repeat plasmid, which again did not induce slipped-DNAs (Fig. S1). Thus, removal of chromatin from supercoiled repeats and the IP conditions used does not structurally alter the repeats from a fully-paired Watson-Crick duplex, consistent with previous reports [20]. Using these conditions and controls, we proceeded to assess the presence of slipped-DNA in patient tissues.
Towards determining whether the anti-junction antibody could recognize slip-outs that may have arisen at the expanded locus in human patient DNAs we first characterized the CTG tract length in a set of tissues from DM1 patients and a control non-DM1 individual by Southern blot (summarized in Fig. 2B, this LNA-Southern blot analyses have been previously published in [4]). A tissue-specific length variation of the expanded allele was evident in both patients used throughout this study, with many tissues showing a heterogeneous range of repeat sizes (Fig. 2B). Increased length heterogeneity within a tissue and larger expansions between tissues are indicative of high levels of active CTG instability, while limited length heterogeneity and shorter expansions reflect less instability [4]. The largest and most heterogeneous expansions were present in the most affected tissues (cortex, muscle and heart; indicated by broad repeat size ranges), while the cerebellum showed the shortest expansion with little or no length heterogeneity (as indicated by a relatively distinct sized fragment). Control non-DM1 samples showed only non-expanded alleles. These tissues were used throughout the study.
To determine whether 2D3 would recognize and bind to slipped-DNAs formed at the endogenous DM1 locus, we immunoprecipitated (IP'd) DNAs from various tissues, using the IP protocol (Fig. 2C) which would not be expected to either introduce or remove any pre-existing slipped-DNA structures. IP'd DNAs were subsequently characterized by various means, as described below.
Immunoprecipitation of slipped-DNA structures would be expected to enrich for the expanded disease-DM1 allele, as the non-expanded allele would not be expected to contain slipped-DNAs. Slipped-DNAs were in fact present on the expanded but not the non-expanded allele, as outlined: Because the expanded CTG tract in DM1 patient tissues is beyond the PCR amplifiable size range (1310–6550 repeats), we devised a multiplex PCR to distinguish the presence of the disease-allele from the non-disease allele in the immunoprecipitated DNAs. As outlined in Figure 3A, two sets of PCR primers amplifying adjacent overlapping regions of the DM1 locus were used in the same PCR reaction. The regions included the CTG repeat, and the downstream CTCF binding site. In the presence of both the expanded disease allele and the non-expanded allele, we expect four PCR products of distinct sizes. The largest two products would be unique to the non-expanded template derived from amplification across both the short CTG tract and the downstream CTCF site (259 bp), and the short CTG tract alone (211 bp). Amplification of the CTCF site only (204 bp) or the product in the overlapping region (156 bp) could arise from either allele. We did not expect either a PCR product from across the very large CTG expansion or from across the expansion and the adjacent CTCF site. Thus, when only the expanded CTG allele is present, we would not expect PCR products unique to the non-expanded allele (259 bp and 211 bp), whereas when both expanded and non-expanded alleles are present we would expect all four PCR products. This multiplex PCR assay was applied to 2D3-IP'd samples derived from various tissues of a DM1 patient (ADM5) with an expanded CTG tract of 1310–6550 repeats, and a non-expanded allele of (CTG)4 (Fig. 3B). Each genomic DNA prior to IP revealed all four multiplex PCR products, expected from both DM1 alleles (Fig. 3B, all lanes indicated by “−”). The IP'd DNAs revealed only the CTCF and overlapping PCR products (Fig. 3B, all lanes indicated by “+”). The absence of the upper two products, unique to the non-expanded allele provides strong support for the IP'd DNA to be enriched for the expanded DM1 allele, and deficient in the non-expanded allele. Multiplex PCR analysis of 2D3-IP'd DNAs from a non-DM1 control individual with DM1 alleles of (CTG)5 and (CTG)25 failed to detect any PCR products (Fig. S3A) – indicating that the 2D3 antibody could not detect any slipped-DNAs at either of the non-expanded alleles. The specificity of the 2D3-IP protocol to enrich for slipped-DNAs was supported by the inability to detect DNAs derived from a control region (lamin B2) that is devoid of unusual structure forming DNA sequence motifs (Fig. S3B). Thus, the ability to detect only those multiplex PCR products common to both the expanded and non-expanded alleles, but not those unique to the non-expanded allele, supports a specific enrichment of the expanded DM1 allele through anti-DNA junction IP. This strongly supports the presence of slipped-DNA in only the mutant expanded DM1 allele.

We used an independent direct method to determine the enrichment of the expanded DM1 allele in the IP'd material. Triplet-primed PCR (TP-PCR), which has been used as a DM1 diagnostic tool, allows for amplification of short stretches of a large expanded CTG tract [21]. TP-PCR uses a primer that hybridizes to the repeat and a primer that hybridizes downstream of the repeat (Fig. 3C). TP-PCR of the expanded CTG allele typically yields a heterogeneous range of CTG sizes in the PCR products, electrophoretically visible as a smear. This range of PCR products arises by PCR amplification of the repeat-specific primer hybridized randomly along the CTG expansion but relatively close to the flanking primer. TP-PCR across the non-expanded allele yields a distinct but shorter size range of PCR products, due to the limited locations to which the repeat-primer can hybridize (Fig. 3C). TP-PCR analysis of the DM1 patient genomic DNAs prior to IP revealed both the expected smear of products for the expanded allele and the distinct band for the non-expanded allele (Fig. 3D, “Gen.” lanes). However, TP-PCR of the IP'd sample revealed predominantly the longer range smeared products (“IP” lanes) derived from the expanded allele, while the DNA in the supernatant following IP from the same experiment revealed predominantly the shorter products derived from the non-expanded allele (“SN” lanes). These results directly support the enrichment of the expanded DM1 disease allele by the anti-DNA junction antibody, consistent with the interpretation that slipped-DNAs are present along the expanded allele.
The levels of DM1 mutant alleles containing slipped-DNA might be expected to correlate with the levels of CTG instability, which varies between tissues of the same DM1 individual [4]. We quantified the amount of DM1 DNA being IP'd from two tissues from the same patient harboring high and low levels of CTG instability, indicated by the larger and heterogeneous CTG lengths in the heart (ranging from 4100–6000 repeats), and the more discrete and shorter length in the cerebellum (1100–1500 repeats, predominantly 1200 repeats), respectively (Fig. 2B). Quantification was accomplished using the highly accurate competitive quantitative PCR [22], [23] which involves the coamplification of a target DNA sample with known amounts of a cloned competitor DNA that shares most of the nucleotide sequence and primer sites with the target (detailed in Fig. S4A). Competitive PCR permits quantification of the absolute number of target molecules (a few representative examples are shown in Fig. S4B and S4C). Anti-DNA junction antibody IP'd DNA levels were significantly greater in the heart over the cerebellum (relative to input) of a DM1 individual (Fig. 4A), an enrichment that is consistent with a greater proportion of DM1 alleles with slipped-DNA present in tissues having greater levels of CTG instability. These findings indicate more DM1 alleles with at least one slipped-out, but cannot reveal the number of slip-outs per allele. DNA from a non-DM1 individual was not enriched for slipped-DNAs in either cerebellum or heart (Fig. 4A). These findings reveal a trend of increased levels of slipped-DNAs in tissues with higher levels of instability.

As a further test for the presence of slipped-DNAs in DM1 patient tissues, DNA was treated with enzymes that specifically digest features of slipped-DNA structures; a treatment that should eliminate slipped-DNAs. Mung bean nuclease (MBN) and T7 endonuclease I (T7endoI) have been shown to specifically cleave the single-stranded regions in slip-outs and across DNA-junctions of slipped-DNAs [8], [16] (Fig. S5D). Following enzyme digestions, DNAs were subjected to TP-PCR to assess the potential loss of the expanded alleles (Fig. 4B,C,D; additional examples in Fig. S5). If the expanded allele contains slipped-DNA, cleavage by these enzymes should decrease the amount of the smear representing the expanded allele. The amount of expanded repeat products in several DM1 patient tissues was significantly reduced following treatment with MBN or T7endoI (Fig. 4D, Fig. S5C). Non-DM1 control DNAs showed no difference after enzyme treatment (Fig. S5B,C). The significant reduction of the expanded allele by either structure-specific enzyme is consistent with a portion of the DM1 disease alleles being in the slipped-DNA conformation.
It was of interest to know if the slipped DNAs were present within tissues in the native chromatin context. Furthermore, the detection of slipped-DNAs directly on chromatinized DNA in tissues, where freeze and thawing of tissues does not affect chromatin packaging relative to fresh tissues [24], provides further evidence for the existence of slipped-DNAs prior to chromatin removal during DNA isolation in the above experiments. To assess the presence of slipped-DNAs in tissues, we used a slight modification of an established protocol of DNA nuclease accessibility assay on tissues, which assesses DNA in its native chromatin context [24] (see references in Text S1). Unusual DNA conformations have been detected in native chromatin through nuclease digestion [25]–[27]. For example, cruciform and stem-loops structures, similar to slipped-DNAs, were susceptible to nuclease digestion and found to stably reside in the inter-nucleosomal region [26], [27]. DM1 patient and control tissues were treated directly with either buffer only as a negative control, or both T7endonucleaseI and mung bean nuclease to test for digestion, or AluI restriction enzyme to test for off-target digestion (Fig. 5A, B; Fig. S6). Following treatment, DNA was isolated and subjected to TP-PCR to assess the potential loss by digestion of the expanded alleles. There was a significant reduction in the expanded allele from patient ADM9 muscle tissue when treated with T7 and MBN nucleases (p = 0.0038), but not when treated with AluI (p = 0.255). There was no significant reduction in either control cerebellum or heart, or patient cerebellum, after treatment with either T7 and MBN, or AluI (Fig. 5A,B; Fig. S6). These results are consistent with the above results in that the tissue with greater CTG instability contains more slipped-DNAs, and control tissues do not. Thus, slipped-DNAs were present at the mutant DM1 locus, in the native chromatin in DM1 tissues and at levels that correlated with levels of instability.

Towards biophysically characterizing the slipped-DNAs, we visualized these by electron microscopy (EM). EM analysis of the IP'd slipped-DNAs revealed multiple bends, kinks, bulges, branched arms, and regions with increased thickness relative to control DNA (Fig. 5C, D, Fig. S7). A significantly increased number of molecules with visibly detectable slip-outs were identified in tissues showing high levels of CTG instability compared to the cerebellum, which showed the lowest level of instability (skeletal muscle vs. cerebellum, two-sided t-test; p = 0.005; pancreas vs. cerebellum, two-sided t-test; p = 0.0202; all unstable tissues (heart, liver, pancreas, cortex, skeletal muscle) vs. cerebellum; p = 0.0386; Fig. S6C). The variation of visibly detectable slip-outs between IP'd material of different tissues might indicate a slip-out size variation between tissues. The size of the slip-outs presented a bimodal distribution ranging from 1–100 repeats with peaks at ∼30 and <10 repeats (Fig. 5D). Multiple slip-outs were clustered along a given DNA, with distances of <100 bp between slip-outs (Fig. 5D). These features were also present in in vitro induced slipped-DNAs in synthetic (CTG)800•(CAG)800 (Fig. S7A) and are consistent with previous electron micrographs showing multiple short slip-outs in (CTG)•(CAG) tracts with 50–250 repeats [16], [17]. The IP'd DNAs were shorter than expected, possibly due to IP-induced DNA shearing of the very long repeat expansions that we studied, a phenomenon previously observed for DNAs enriched in single-strand nicks, gaps or DNAse I hypersensitive sites. An enrichment of such strand breaks may be expected for arrested repair of clustered slip-out lesions [28], a sensitivity to site fragility [29], and susceptibility of slipped-DNAs to double-strand breaks [30].
Discussion
We report the isolation of slipped-DNAs present at an endogenous disease locus from various DM1 patient tissues. Our observations are supported by multiple independent, complementary experimental approaches. To better understand processes leading to disease-causing mutations, it is crucial to study these structures at specific genomic loci and in relevant patient tissues. This is particularly important for trinucleotide repeat mutations as these events are expansion-biased, arising by processes distinct from contractions, with instability patterns varying between disease loci, and between mitotic and non-mitotic tissues. An understanding of these DNA mutagenic intermediates occurring in patients should provide insight as to how they may be processed and lead to mutations, as model systems do not accurately reflect what is ongoing in affected individuals, especially on a tissue-specific level. Here, we reveal slipped-DNAs at the expanded CTG/CAG DM1 disease locus in tissues including the brain and heart. The brain in humans is essentially post-mitotic after birth [31], with both the cerebellum and cortex developing from very early on in embryogenesis and continuing up until the first years after birth [32], [33]. Similarly, the majority of cells in the heart are post-mitotic in nature, with cell division ceasing in cardiac myocytes soon after birth and the dividing cell population in adults being less than 1% of the total number of cells [34]–[36]. Since CTG expansions continue to expand at a greater rate following birth compared to prenatal expansion [4], our detection of slipped-DNAs in non-mitotic tissues supports that they arose in the absence of DNA replication. However, it is also possible that slipped-DNAs could form during early development and persist throughout the lifetime of an individual. Despite the similarity in timing of the post-mitotic nature of the frontal cortex, cerebellum and the heart, the cortex and heart showed significantly more slipped-DNA in patients compared to cerebellum, further supporting the hypothesis of the presence of tissue-specific instability factors. In DM1 and other CAG/CTG repeat diseases the tissue selectivity of repeat expansions correlates with the tissue selectivity of pathogenesis in the brain regions [37]–[41], the heart [42] and muscle [4], [43]. Our evidence at a patient locus from various tissues serves as the only evidence supporting the existence of slipped-DNAs in patients. Our observations of slipped-DNAs at a disease locus in post-mortem patient tissues, coupled with the report of hairpin DNAs at exogenously integrated repeats in cultured cell models [10] supports the formation of slipped-DNAs in patients. Importantly, we reveal a strong difference in the allelic levels of slipped-DNA between tissues of the same individual, and these levels correlate with the levels of instability in the tissues assessed.
Surprisingly, slipped-DNAs are present as tandem clusters of multiple small slip-outs, with increased levels in DM1 patient tissues showing high repeat instability. This contrasts with the long-presumed concept that slipped-DNAs arise as isolated slip-outs on either strand of the duplex. That slipped-DNAs are clusters of short slip-outs gives new insight into the role they may play in the mutation process. The majority of analyses addressing the potential roles of slip-outs have centered upon the presumption that single isolated slip-outs may form transiently and be processed to fully-duplexed repeat-length mutations. Complementing our isolation of clustered slipped-DNA structures, it has been shown previously that clustered structures are poorly repaired compared to isolated single slip-outs [28]. The most striking implication of our findings is that slipped-DNAs do not appear to be merely transient in nature, as previously believed [44], but may be persistent in vivo products of instability, consistent with their poor ability to be repaired [28]. We propose that intrastrand slippage may occur after attempted repair events on slipped-DNAs become arrested due to adjacent slip-outs (Fig. 6). This shifts the repeats further out of register leaving a gap that, when filled, results in an increased number of repeats in that strand and producing slipped-DNAs with an excess of repeats on one or both strands. Reiterations of such events in the absence of proper repair would lead to instability, where products are slipped-DNAs.

Our IP protocol has identified slipped-DNAs at a CTG/CAG disease locus in patient tissues, structures that were not detected in appropriate controls (i.e., the antibody did not induce structure formation). While our findings are not definitive in vivo evidence, the fact that the allelic levels of slipped-DNAs varied between DM1 patient tissues, which showed varying levels of instability supports that these were formed in the patient. This concept is further supported by the detection of hairpins at integrated CTG/CAG repeats in cell models [10]. Different mutational paths involving slipped-DNAs may involve specific DNA repair pathways. For example, the mismatch repair complex MutSβ is required for CAG/CTG expansions [1]–[3]. Similarly, the levels and biophysical structure of slipped-DNAs forming at mono - and dinucleotide repeats may be distinct between tumors showing microsatellite instability compared to those that are microsatellite stable. Slipped-DNA identification and characterization could provide mechanistic and prognostic insights into medically important mutations in the many repeat-disease loci, hypermutable viruses, mitochondria, hypervariable genomic regions, and fragile sites [45]–[49] as it has done herein for DM1 repeat instability.
Materials and Methods
Human tissues analyzed in this study are listed in Fig. 2B. Autopsy tissues from a non-affected individual (ADN1) were obtained snap-frozen from the National Disease Research Interchange. DNA extractions from human tissues were carried out under conditions to minimize DNA denaturation, to avoid inducing unusual DNA structure formation. Procedures were performed in low binding tubes, which avoid DNA denaturation. Complete protocol can be found in Text S1. Human-CTG repeat lengths were assessed by Southern blot using an LNA probe [DIG-labeled (CAG)7-5′-gcAgCagcAgCagCagcAgca-3′], as described previously [4]. Non-expanded CTG alleles were sized by sequencing of the products obtained after PCR amplification (forward primer 409, reverse primer 407). Slipped-DNA structures for bandshift were made as previously described [9] with minor changes (see Text S1). Individual structures and varying concentrations of antibody were run on a 4% (w/v) polyacrylamide gel in 1× TBE buffer at a constant 150 V for 1.5 hours. Following electrophoresis gels were dried and autoradiographed. Fully-duplexed (CTG)50•(CAG)50 were used to test for specificity of binding, with linearized p-Bluescript plasmid additionally being used as a competitor during antibody binding. DNA-IP of slipped-structures was carried out using the anti-DNA junction monoclonal antibody 2D3. 1 ug of genomic DNAs were restriction digested with BamHI and BbsI overnight at 37°C. 50 ul of hybridoma 2D3 culture supernatant containing ∼5 ug/ml immunoglobulin was diluted 1∶1 with PBS and added to the restriction digested patient DNA the following day and incubated on ice for one hour. 80 ul Protein G beads were prepared and resuspended in 250 ul 1× TE, which were then added to the antibody-DNA mixture and incubated on ice for 1 hour, mixing occasionally. The antibody/DNA/bead complex was washed free of unbound DNA and the affinity purified DNA was eluted using TE+2% SDS. DNA was further purified by phenol-chloroform-isoamyl alcohol extraction, followed by 100% ethanol precipitation. IP'd DNA samples were analyzed by electron microscopy essentially as described [16]. Multiplex PCR and TP-PCR protocol conditions are described in Text S1. For structure specificity experiments, patient DNA was digested with MBN or T7EndoI overnight and used for TP-PCR with either a fluorescently labeled primer after which the products were analyzed by GeneScan, or with a non-fluorescently labeled primer after which the products were run on a 1% agarose gel for visual analysis. DNAs digested within their native chromatin context were subjected to a modified nuclease accessibility protocol before digestion and DNA isolation (Text S1). EM and nucleosome assembly and disassembly were carried out as previously reported (Text S1, references 9 and 42, respectively).
Supporting Information
Zdroje
1. Lopez CastelA, ClearyJD, PearsonCE (2010) Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11 : 165–170.
2. PearsonCE, Nichol EdamuraK, ClearyJD (2005) Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6 : 729–742.
3. WangYH, AmirhaeriS, KangS, WellsRD, GriffithJD (1994) Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science 265 : 669–671.
4. Lopez CastelA, NakamoriM, TomeS, ChitayatD, GourdonG, et al. (2011) Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues. Hum Mol Genet 20 : 1–15.
5. ClearyJD, TomeS, Lopez CastelA, PanigrahiGB, FoiryL, et al. (2010) Tissue - and age-specific DNA replication patterns at the CTG/CAG-expanded human myotonic dystrophy type 1 locus. Nat Struct Mol Biol 17 : 1079–1087.
6. PanigrahiGB, LauR, MontgomerySE, LeonardMR, PearsonCE (2005) Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat Struct Mol Biol 12 : 654–662.
7. HouC, ChanNL, GuL, LiGM (2009) Incision-dependent and error-free repair of (CAG)(n)/(CTG)(n) hairpins in human cell extracts. Nat Struct Mol Biol 16 : 869–875.
8. PearsonCE, SindenRR (1996) Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry 35 : 5041–5053.
9. TamM, Erin MontgomeryS, KekisM, StollarBD, PriceGB, et al. (2003) Slipped (CTG).(CAG) repeats of the myotonic dystrophy locus: surface probing with anti-DNA antibodies. J Mol Biol 332 : 585–600.
10. LiuG, ChenX, BisslerJJ, SindenRR, LeffakM (2010) Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat Chem Biol 6 : 652–659.
11. FrappierL, PriceGB, MartinRG, Zannis-HadjopoulosM (1989) Characterization of the binding specificity of two anticruciform DNA monoclonal antibodies. J Biol Chem 264 : 334–341.
12. SteinmetzerK, Zannis-HadjopoulosM, PriceGB (1995) Anti-cruciform monoclonal antibody and cruciform DNA interaction. J Mol Biol 254 : 29–37.
13. FelsenfeldG (1958) Theoretical Studies of the Interaction of synthetic polyribonucleotides. Biochim Biophys Acta 29 : 133–144.
14. DeJesus-HernandezM, MackenzieIR, BoeveBF, BoxerAL, BakerM, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72 : 245–256.
15. RentonAE, MajounieE, WaiteA, Simon-SanchezJ, RollinsonS, et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72 : 257–268.
16. PearsonCE, WangYH, GriffithJD, SindenRR (1998) Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n. (CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res 26 : 816–823.
17. PearsonCE, TamM, WangYH, MontgomerySE, DarAC, et al. (2002) Slipped-strand DNAs formed by long (CAG)*(CTG) repeats: slipped-out repeats and slip-out junctions. Nucleic Acids Res 30 : 4534–4547.
18. SleanMM, ReddyK, WuB, Nichol EdamuraK, KekisM, et al. (2013) Interconverting conformations of slipped-DNA junctions formed by trinucleotide repeats affect repair outcome. Biochemistry 52 : 773–785.
19. FrappierL, PriceGB, MartinRG, Zannis-HadjopoulosM (1987) Monoclonal antibodies to cruciform DNA structures. J Mol Biol 193 : 751–758.
20. BacollaA, GellibolianR, ShimizuM, AmirhaeriS, KangS, et al. (1997) Flexible DNA: genetically unstable CTG.CAG and CGG.CCG from human hereditary neuromuscular disease genes. J Biol Chem 272 : 16783–16792.
21. WarnerJP, BarronLH, GoudieD, KellyK, DowD, et al. (1996) A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet 33 : 1022–1026.
22. ZentilinL, GiaccaM (2007) Competitive PCR for precise nucleic acid quantification. Nat Protoc 2 : 2092–2104.
23. ZentilinL, GiaccaM (2010) The renaissance of competitive PCR as an accurate tool for precise nucleic acid quantification. Methods Mol Biol 630 : 233–248.
24. GaubatzJ, EllisM, ChalkleyR (1979) Nuclease digestion studies of mouse chromatin as a function of age. J Gerontol 34 : 672–679.
25. LarsenA, WeintraubH (1982) An altered DNA conformation detected by S1 nuclease occurs at specific regions in active chick globin chromatin. Cell 29 : 609–622.
26. NickolJ, MartinRG (1983) DNA stem-loop structures bind poorly to histone octamer cores. Proc Natl Acad Sci U S A 80 : 4669–4673.
27. NobileC, NickolJ, MartinRG (1986) Nucleosome phasing on a DNA fragment from the replication origin of simian virus 40 and rephasing upon cruciform formation of the DNA. Mol Cell Biol 6 : 2916–2922.
28. PanigrahiGB, SleanMM, SimardJP, GileadiO, PearsonCE (2010) Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci U S A 107 : 12593–12598.
29. FreudenreichCH, KantrowSM, ZakianVA (1998) Expansion and length-dependent fragility of CTG repeats in yeast. Science 279 : 853–856.
30. MarcadierJL, PearsonCE (2003) Fidelity of primate cell repair of a double-strand break within a (CTG).(CAG) tract. Effect of slipped DNA structures. J Biol Chem 278 : 33848–33856.
31. DobbingJ, SandsJ (1979) Comparative aspects of the brain growth spurt. Early Hum Dev 3 : 79–83.
32. ten DonkelaarHJ, LammensM, WesselingP, ThijssenHO, RenierWO (2003) Development and developmental disorders of the human cerebellum. J Neurol 250 : 1025–1036.
33. ShankleWR, LandingBH, RafiiMS, SchianoA, ChenJM, et al. (1998) Evidence for a postnatal doubling of neuron number in the developing human cerebral cortex between 15 months and 6 years. J Theor Biol 191 : 115–140.
34. Cruz-MunozW, KalairW, CosentinoL, HeddleJA (2000) ENU induces mutations in the heart of lacZ transgenic mice. Mutat Res 469 : 23–34.
35. EngermanRL, PfaffenbachD, DavisMD (1967) Cell turnover of capillaries. Lab Invest 17 : 738–743.
36. MarinoTA, HaldarS, WilliamsonEC, BeaversonK, WalterRA, et al. (1991) Proliferating cell nuclear antigen in developing and adult rat cardiac muscle cells. Circ Res 69 : 1353–1360.
37. TeleniusH, KremerB, GoldbergYP, TheilmannJ, AndrewSE, et al. (1994) Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet 6 : 409–414.
38. De RooijKE, De Koning GansPA, RoosRA, Van OmmenGJ, Den DunnenJT (1995) Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum Genet 95 : 270–274.
39. OnoderaO, IdezukaJ, IgarashiS, TakiyamaY, EndoK, et al. (1998) Progressive atrophy of cerebellum and brainstem as a function of age and the size of the expanded CAG repeats in the MJD1 gene in Machado-Joseph disease. Ann Neurol 43 : 288–296.
40. RomeoV, PegoraroE, FerratiC, SquarzantiF, SoraruG, et al. (2010) Brain involvement in myotonic dystrophies: neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol 257 : 1246–1255.
41. LeroyO, WangJ, MaurageCA, ParentM, CooperT, et al. (2006) Brain-specific change in alternative splicing of Tau exon 6 in myotonic dystrophy type 1. Biochim Biophys Acta 1762 : 460–467.
42. GrohWJ, LoweMR, ZipesDP (2002) Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol 13 : 444–448.
43. ThorntonCA, JohnsonK, MoxleyRT3rd (1994) Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol 35 : 104–107.
44. StreisingerG, OkadaY, EmrichJ, NewtonJ, TsugitaA, et al. (1966) Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday. Cold Spring Harb Symp Quant Biol 31 : 77–84.
45. BergI, NeumannR, CederbergH, RannugU, JeffreysAJ (2003) Two modes of germline instability at human minisatellite MS1 (locus D1S7): complex rearrangements and paradoxical hyperdeletion. Am J Hum Genet 72 : 1436–1447.
46. BoyerJC, BebenekK, KunkelTA (1992) Unequal human immunodeficiency virus type 1 reverse transcriptase error rates with RNA and DNA templates. Proc Natl Acad Sci U S A 89 : 6919–6923.
47. BuardJ, CollickA, BrownJ, JeffreysAJ (2000) Somatic versus germline mutation processes at minisatellite CEB1 (D2S90) in humans and transgenic mice. Genomics 65 : 95–103.
48. GugliettaS, PantaleoG, GraziosiC (2010) Long sequence duplications, repeats, and palindromes in HIV-1 gp120: length variation in V4 as the product of misalignment mechanism. Virology 399 : 167–175.
49. WangL, ParadeeW, MullinsC, ShridharR, RosatiR, et al. (1997) Aphidicolin-induced FRA3B breakpoints cluster in two distinct regions. Genomics 41 : 485–488.
50. DiviaccoS, NorioP, ZentilinL, MenzoS, ClementiM, et al. (1992) A novel procedure for quantitative polymerase chain reaction by coamplification of competitive templates. Gene 122 : 313–320.
51. MenzoS, BagnarelliP, GiaccaM, ManzinA, VaraldoPE, et al. (1992) Absolute quantitation of viremia in human immunodeficiency virus infection by competitive reverse transcription and polymerase chain reaction. J Clin Microbiol 30 : 1752–1757.
52. SestiniR, OrlandoC, ZentilinL, GelminiS, PinzaniP, et al. (1994) Measuring c-erbB-2 oncogene amplification in fresh and paraffin-embedded tumors by competitive polymerase chain reaction. Clin Chem 40 : 630–636.
53. GiaccaM, ZentilinL, NorioP, DiviaccoS, DimitrovaD, et al. (1994) Fine mapping of a replication origin of human DNA. Proc Natl Acad Sci U S A 91 : 7119–7123.
54. PelizonC, DiviaccoS, FalaschiA, GiaccaM (1996) High-resolution mapping of the origin of DNA replication in the hamster dihydrofolate reductase gene domain by competitive PCR. Mol Cell Biol 16 : 5358–5364.
55. AbdurashidovaG, DanailovMB, OchemA, TrioloG, DjeliovaV, et al. (2003) Localization of proteins bound to a replication origin of human DNA along the cell cycle. EMBO J 22 : 4294–4303.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy