
Acute Versus Chronic Loss of Mammalian Results in Distinct Ciliary Phenotypes
Defects in cilium and centrosome function result in a spectrum of clinically-related disorders, known as ciliopathies. However, the complex molecular composition of these structures confounds functional dissection of what any individual gene product is doing under normal and disease conditions. As part of an siRNA screen for genes involved in mammalian ciliogenesis, we and others have identified the conserved centrosomal protein Azi1/Cep131 as required for cilia formation, supporting previous Danio rerio and Drosophila melanogaster mutant studies. Acute loss of Azi1 by knock-down in mouse fibroblasts leads to a robust reduction in ciliogenesis, which we rescue by expressing siRNA-resistant Azi1-GFP. Localisation studies show Azi1 localises to centriolar satellites, and traffics along microtubules becoming enriched around the basal body. Azi1 also localises to the transition zone, a structure important for regulating traffic into the ciliary compartment. To study the requirement of Azi1 during development and tissue homeostasis, Azi1 null mice were generated (Azi1Gt/Gt). Surprisingly, Azi1Gt/Gt MEFs have no discernible ciliary phenotype and moreover are resistant to Azi1 siRNA knock-down, demonstrating that a compensation mechanism exists to allow ciliogenesis to proceed despite the lack of Azi1. Cilia throughout Azi1 null mice are functionally normal, as embryonic patterning and adult homeostasis are grossly unaffected. However, in the highly specialised sperm flagella, the loss of Azi1 is not compensated, leading to striking microtubule-based trafficking defects in both the manchette and the flagella, resulting in male infertility. Our analysis of Azi1 knock-down (acute loss) versus gene deletion (chronic loss) suggests that Azi1 plays a conserved, but non-essential trafficking role in ciliogenesis. Importantly, our in vivo analysis reveals Azi1 mediates novel trafficking functions necessary for flagellogenesis. Our study highlights the importance of both acute removal of a protein, in addition to mouse knock-out studies, when functionally characterising candidates for human disease.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1003928
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003928
Summary
Defects in cilium and centrosome function result in a spectrum of clinically-related disorders, known as ciliopathies. However, the complex molecular composition of these structures confounds functional dissection of what any individual gene product is doing under normal and disease conditions. As part of an siRNA screen for genes involved in mammalian ciliogenesis, we and others have identified the conserved centrosomal protein Azi1/Cep131 as required for cilia formation, supporting previous Danio rerio and Drosophila melanogaster mutant studies. Acute loss of Azi1 by knock-down in mouse fibroblasts leads to a robust reduction in ciliogenesis, which we rescue by expressing siRNA-resistant Azi1-GFP. Localisation studies show Azi1 localises to centriolar satellites, and traffics along microtubules becoming enriched around the basal body. Azi1 also localises to the transition zone, a structure important for regulating traffic into the ciliary compartment. To study the requirement of Azi1 during development and tissue homeostasis, Azi1 null mice were generated (Azi1Gt/Gt). Surprisingly, Azi1Gt/Gt MEFs have no discernible ciliary phenotype and moreover are resistant to Azi1 siRNA knock-down, demonstrating that a compensation mechanism exists to allow ciliogenesis to proceed despite the lack of Azi1. Cilia throughout Azi1 null mice are functionally normal, as embryonic patterning and adult homeostasis are grossly unaffected. However, in the highly specialised sperm flagella, the loss of Azi1 is not compensated, leading to striking microtubule-based trafficking defects in both the manchette and the flagella, resulting in male infertility. Our analysis of Azi1 knock-down (acute loss) versus gene deletion (chronic loss) suggests that Azi1 plays a conserved, but non-essential trafficking role in ciliogenesis. Importantly, our in vivo analysis reveals Azi1 mediates novel trafficking functions necessary for flagellogenesis. Our study highlights the importance of both acute removal of a protein, in addition to mouse knock-out studies, when functionally characterising candidates for human disease.
Introduction
Centrosomes are conserved animal organelles which function as the major microtubule organising centre (MTOC), and are required for diverse processes including formation of cilia and flagella, intracellular trafficking events, cell polarity and division. Structurally, the centrosome consists of a pair of cylindrical centrioles surrounded by a proteinaceous matrix of pericentriolar material (PCM) [1]. Importantly, centrioles replicate only once per cell cycle and are essential for the formation of cilia, key signalling organelles during development and homeostasis. In post-mitotic cells, the centrosome moves to the apical surface where the mother centriole docks with the cell membrane to become a basal body and a template for the axonemal microtubules of the primary cilium. Cilia assembly and function requires diverse trafficking events, including intraflagellar transport (IFT), coordinated by the basal body ‘hub’, which regulates traffic in and out of the ciliary compartment. Enrichment of key signalling receptors and downstream effectors in cilia allows these structures to function as effective signalling organelles, with unique protein and lipid composition [2]. The transition zone, a highly specialised structure just distal to the basal body, is thought to act as an additional ciliary gate, controlling traffic into and out of the cilium [3]. Many of these aspects of ciliogenesis are highly conserved [4].
Despite these common features, cilia are also structurally and functionally diverse. Cilia play important sensory roles, acting as transducers of developmental signalling pathways, detecting fluid flow, as well as highly specialised sensory receptors [5]. Some cilia are motile, involved in generating fluid flow in the embryonic node, airways, oviduct and brain, as well as in the propulsion of sperm. How the core ciliary assembly programme is modified and elaborated on to account for these species - and cell-specific variations is not well understood [4].
Mutations in conserved cilial and centrosomal genes have been identified in a growing spectrum of clinical disorders, termed ciliopathies, with both distinct and overlapping clinical features including polydactyly, skeletal defects, situs inversus, infertility and neuropathology [6], [7]. Proteomic and genetic studies in several organisms estimate the molecular composition of cilia/centrosomes to include hundreds to thousands of putative components, many of them unknown [8], [9]. Functional dissection of the role and requirement of many of these ciliopathy candidates in cilia formation and function are often performed using cell culture [10], [11], [12] and zebrafish knock-down models [13]. Mouse mutant models are analysed less often as these are more costly in time and resources to produce. Given the phenotypic complexities of clinical features in ciliopathies [14], what is the best way to understand the underlying molecular mechanisms for candidate genes in relation to human disease?
Recently, centriolar satellites have reported to be the site of localisation of many ciliopathy proteins, and are involved in their ciliary targeting, including OFD1 (oral-facia1-digital syndrome 1), BBS4 (Bardet-Biedl Syndrome 4) and CEP290 [15]. Conserved among vertebrates, but not present in arthropods, centriolar satellites are electron dense, multi-protein complexes enriched in the area surrounding the centrosome/basal body [16], [17], [18]. These are dynamic structures trafficking along microtubules towards the centrosome utilising dynein motors [16], [18]. Centriolar satellites have been shown to regulate ciliogenesis and centriole biogenesis, in part by regulating trafficking of proteins to and/or sequestering of proteins away from the centrosome/basal body [19], [20], [21], [22]. Centriolar satellites are defined by pericentriolar material 1 (PCM1) which is a key scaffolding component of centriolar satellites and to date, all centriolar satellite-localised proteins have been shown to interact with PCM1 [17]. However, what the functional significance of vertebrate-specific centriolar satellites to mammalian development is and how they affect the function of highly conserved components in ciliogenesis and centriole biogenesis is unknown.
Here, we address the role and requirement of 5-azacytidine induced gene 1 (Azi1)/Cep131 (MGI:107440), a highly conserved centrosomal protein, in ciliogenesis. We screened a subset of cilia-enriched orthologous candidates from Drosophila melanogaster studies [23] by RNAi to identify genes involved in mammalian ciliogenesis, and identified Azi1/Cep131. This finding agrees with a previous siRNA screen, which showed a role for the human orthologue, AZI1, in ciliogenesis [10]. Both Drosophila melanogaster mutants and Danio rerio morphants of Azi1 (dila/CG1625 and cep131, respectively) phenocopy mutations of known ciliary genes [24], [25], suggesting Azi1 plays a conserved function in ciliogenesis. AZI1 was described as a centrosomal protein (Centrosomal protein 131: Cep131) in a large scale proteomics screen and this localisation was recently refined to the centriolar satellites [26], [27], [28]. The mouse protein is highly expressed in the testes in germ cells during the period of flagellar formation [29]. More recently, additional roles for AZI1 include involvement in genome stability and centriole duplication. Knock-down of AZI1 leads to an increase in double-stranded DNA breaks, indicated by γH2AX staining, as well as a slight increase in cells with extra centrioles [28], [30]. However, little is known of the in vivo role of mouse Azi1 and its requirement for development.
Here we utilise knock-down, localisation and live-imaging techniques, to further investigate the role of Azi1 in mammalian ciliogenesis at the cellular level. To determine the requirement for Azi1 in mouse development, we generated Azi1 null mutant mice and focused on the in vivo role of Azi1 in ciliogenesis and genome stability. Our analysis of Azi1 knock-down (acute loss) versus gene deletion (chronic loss) suggests that Azi1 plays a conserved, but non-critical trafficking role in ciliogenesis. Importantly, our in vivo analysis reveals Azi1 mediates novel trafficking events necessary for spermiogenesis and male fertility.
Results
Functional cell-based screening of putative ciliary candidates from Drosophila identifies Dila orthologue Azi1 as required for mammalian ciliogenesis
Using a set of forty orthologous putative ciliary genes identified as highly expressed in ciliated cell types in Drosophila melanogaster [23], we carried out an siRNA screen in a mouse fibroblast cell line to identify genes involved in mammalian ciliogenesis. Cilia formation was assayed as the percentage of cells with a cilium, marked by anti-Arl13b, a ciliary membrane marker [31], and anti-acetylated α-Tubulin, a ciliary axoneme marker, using an automated imaging and image analysis system.
We identified Azi1/Cep131 as the top hit for genes involved in cilia formation, with at least two of four siRNAs giving a significant reduction in ciliogenesis across three independent assays (data not shown). This observation is supported by the study of Graser et al. (2007), who found a reduction in ciliogenesis on AZI1 knock-down in human hTERT-RPE1 cells [10]. To exclude off-target effects of the siRNAs, we co-transfected a different pool of four siRNAs, specifically targeting the 3′ untranslated region (UTR) of only Azi1, along with a plasmid encoding either GFP or Azi1-GFP. The Azi1-GFP plasmid lacks the 3′ UTR of Azi1 and so is resistant to these siRNA. Transfection of Azi1 3′ UTR siRNA leads to a reduction in Azi1 protein to 10% of wild type levels, which can be partially rescued by co-transfection with Azi1-GFP (Figure 1A). Azi1 knock-down leads to a 50% reduction of transfected cells with cilia (Figure 1B, D and F), similar to that seen with a positive control siRNA targeting Ift88, a gene essential for ciliogenesis [32], [33]. Importantly, co-expression of Azi1-GFP rescues the phenotype back to control levels demonstrating that the ciliary phenotype observed upon addition of Azi1 siRNA is not due to off-target effects of the siRNA (Figure 1C, E and F). We conclude that Azi1 is involved in mammalian cilia formation.

Azi1 traffics along microtubules towards the centrosome/ciliary base, where it localises to the transition zone
AZI1 was originally identified as a centrosomal protein (Cep131 [26]). We have spatially refined the localisation of mouse endogenous and GFP-tagged Azi1 to centriolar satellites, marked by anti-PCM1, which confirms recent human AZI1 immunofluorescence reports (Figure S1A and G) [28], [34]. We identified a further pool of human and mouse Azi1 at the transition zone (Figures 2A, B and S1B–C), indicated by co-staining with anti-polyglutamylated tubulin, which stains the ciliary axoneme and basal body but, importantly, is absent from the transition zone [35]. The transition zone is an area at the base of the cilia involved in regulating traffic into the cilium [3]. Co-staining with anti-NPHP1, a marker of the transition zone [36], [37], confirms this localisation (Figures 2C, D and S1D). This is consistent with the observation that in D. melanogaster ciliated sensory neurons Azi1 homologue dila localises distal to the basal body at the putative transition zone [24].

Recently, CEP290 has also been reported to localise to both centriolar satellites and the transition zone [38], [39], [40], raising the possibility this could be a general trend for centriolar satellite proteins. To test this, we investigated the localisation of PCM1, the core component of centriolar satellites at the transition zone. Indeed, PCM1 localises to the transition zone of most, but not all cilia, as indicated by the gap between the basal body and axoneme on anti-polyglutamylated tubulin staining (Figure 2E and F). Previous reports had shown OFD1 and PCM1 to similarly localise to the distal portion of basal bodies [15]. Interestingly, the putative functional orthologue of OFD1, UNC, is also found at the putative transition zone of Drosophila mechanosensory neurons [24], [41]. Together, this suggests that docking at the transition zone may be a conserved feature of components of mammalian centriolar satellites.
We used live imaging of Azi1-GFP to address the dynamics of Azi1 localisation. In interphase cells, centriolar satellites have been proposed to function in dynein motor-dependent, microtubule-based trafficking of proteins to the centrosome [19], [40], and it has been shown recently that the pericentriolar localisation of AZI1 is microtubule dependent [28]. Similarly, trafficking of cargo associated with IFT motors along microtubules into the ciliary compartment is selectively regulated in part by the transition zone [42]. To examine Azi1 trafficking more directly we imaged Azi1-GFP movement in live mouse NIH-3T3 cells. Azi1-GFP was observed to traffic along microtubules, co-labelled with Map4-RFP (Figure 2G). Azi1-GFP traffics with a dynamic saltatory motion, with periods of fast movement, for an average distance of 3.4 µm (range 1.3–10.2 µm) interspersed with sometimes long stationary periods. Azi1-GFP was observed to move both towards and away from the centrosome, similar to observations of PCM1-GFP [18] (Movie S1). Although average speeds varied according to direction, 1.8±0.2 µm/s (mean ± SEM) towards the minus end of microtubules at the centrosome, and 1.0±0.3 µm/s away from the centrosome, they were consistent with speeds observed previously for microtubule-based motors in vivo [43], [44]. This suggests Azi1 is involved in microtubule-based trafficking to and from the centrosome/basal body.
Higher levels of endogenous AZI1 staining are observed around basal bodies and surrounding centriolar satellites of ciliated cells compared to non-ciliated cells (Figure 2H and I). However, total levels of AZI1 do not change upon serum starvation (Figure 2J) indicating that under these conditions to induce ciliogenesis, there is a redistribution of AZI1 towards the basal body area.
It has been proposed that centriolar satellites act as proteinaceous scaffolds to physically restrict access of proteins to the centrosome/cilium complex [22]. Disruption of core components results in dissolution or dispersal of centriolar satellites, and relocalisation of associated centriolar satellite proteins to the centrosome/basal body [15], [20], [22], [40]. Despite its localisation, Azi1 siRNA knock-down in mouse cells does not alter centriolar satellite integrity as shown by Pcm1 localisation, consistent with recent reports for human AZI1 [28]. This suggests Azi1 is not required for mammalian centriolar satellite integrity nor retention of Pcm1 to these structures (Figure S1E–H).
Azi1 is dispensable for mouse embryonic development
Given the high conservation of Azi1 among ciliates (Table S1), including arthropods which lack centriolar satellites, together with phenotypic mutant data from diverse organisms [24], [25], [45], [46], we predicted Azi1 would have a central role in mammalian cilia biology in vivo and thus generated mouse mutants null for Azi1. Azi1Gt(CCOG35)Wtsi embryonic stem (ES) cells, which have a gene trap inserted into intron 2 of Azi1 (Figure 3A), were used to generate Azi1+/Gt(CCOG35)Wtsi mice (referred to as Azi1Gt/+ throughout this manuscript). Azi1Gt/Gt mice are born at sub-Mendelian ratios, with approximately two thirds of the expected numbers of Azi1Gt/Gt mice remaining after weaning (see Table 1, P = 0.0025). A significant reduction in Azi1Gt/Gt numbers was observed at embryonic day 11.5–13.5 (E11.5–13.5), suggesting roughly a third of mutants are lost before mid-gestation, although further work is needed to determine exactly when this loss occurs (Table 1). Azi1Gt/Gt mice that are born appear morphologically normal, and are the same weight as wild type littermates (Figure S5G and H). Viable Azi1 mutant mice showed none of the gross abnormalities associated with cilia dysfunction in mice, including failure to thrive, hydrocephalus, situs inversus, and chronic airway infections.
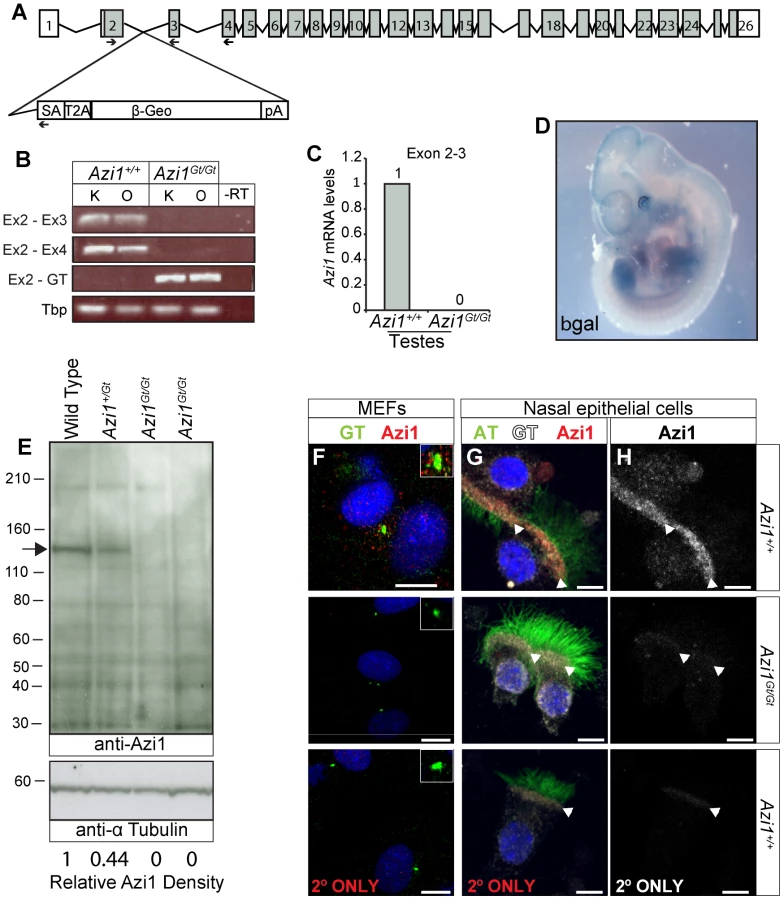

Azi1 has several coiled-coil domains as well as a predicted t-SNARE domain (IPR010989) implicated in membrane fusion events during vesicular transport (Figure S2B). The gene trap is predicted to truncate Azi1 after the initial 69 amino acids such that any remaining Azi1 trapped protein in Azi1Gt/Gt mice will lack all the predicted domains in the more highly conserved C terminus (Figure S2, Table S1), and hence unlikely to be functional. To confirm that the gene trap eliminated gene expression, we examined Azi1 mRNA expression levels across the gene trap insertion site (Figure 3B and C). No expression across the insertion site was detected in kidneys, ovaries or testes from Azi1Gt/Gt mice by RT-PCR or qRT-PCR (Figure 3B and C), whereas robust expression of the gene trap was detected (Figure 3B). X-Gal staining of E11.5 Azi1Gt/+ embryos further confirmed expression of the gene trap β-geo gene, and showed Azi1 expression is ubiquitous during development, with higher expression in tissues with high levels of cilia-dependent developmental signalling such as the limbs, eyes, somite derivatives and brain (Figure 3D).
We detected some low level expression of the 3′ end of Azi1 in Azi1Gt/Gt mice; when quantified by qRT-PCR this was less than 2% of wild type levels (Figure S2C–E). Importantly, no Azi1 protein was detected in Azi1Gt/Gt mice when probing with an antibody raised against the C terminal of Azi1 (Figure 3E and S2B), despite a single strong band of the expected size in Azi1+/+ and a band of roughly 50% intensity in Azi1Gt/+ samples. Furthermore, anti-Azi1 immunofluorescence analysis of Azi1Gt/Gt mutant mouse embryonic fibroblasts (MEFs) or multiciliated airway epithelial cells detected no signal, despite clear centrosomal/basal body localisation of Azi1 in littermate controls (Figure 3F–H). This suggests that any low level transcription detected at the 3′ end of Azi1 in Azi1Gt/Gt mice is untranslated; indeed there are several predicted untranslated transcripts at the 3′ end of Azi1 (ENSMUST00000156075, ENSMUST00000150463, ENSMUST00000144128 and ENSMUST00000145641). We conclude that Azi1Gt/Gt is a null allele of Azi1.
Cilia, centrioles and centriolar satellites of Azi1 null MEFs are grossly normal
Primary cilia are required for mammalian development, so the fact most Azi1 null mice survive without any patterning defects suggests Azi1 is not required for mammalian ciliogenesis in vivo. In contrast, transient siRNA knock-down of Azi1 leads to a two-fold reduction in ciliogenesis (Figure 1). On careful examination of cilia formation in Azi1 null primary MEFs we found normal numbers of cilia, marked by anti-Arl13b (Figure 4A, F and K). Moreover, cilia compartmentalisation appeared normal, with correct distributions of the ciliary membrane protein Arl13b, the IFT-B protein Ift88, and components of the transition zone Nphp1 and Mks1 (Figure 4A–D, F–I). Post-translational modifications of tubulin also appeared normal: acetylated α-Tubulin is present at the basal body and axoneme, polyglutamylated tubulin is present at the axoneme and basal body but absent from the transition zone, and γ-Tubulin is present at the basal body (Figure 4A–J). Localisation of Rab8, a small GTPase involved in trafficking to the cilium [21], whose localisation has been shown to be centriolar satellite-dependent [40], was also localised normally in Azi1Gt/Gt MEFs (Figure 4E and J). Together, this analysis suggests that Azi1 is dispensable for primary cilia formation and compartmentalisation both in vivo and in primary cells.

Given its enrichment at centriolar satellites in mouse and human cells, where Azi1 interacts with Pcm1 (Figure S1) [28], [34], we examined formation of centriolar satellites in the absence of Azi1. Similar to Azi1 knock-down, Pcm1 is correctly localised in Azi1Gt/Gt MEFs (Figure S3A–C), as are centriolar satellite components Cep72 and Cep290, involved in modulating localisation and composition of centriolar satellites [22], [40] (data not shown). This confirms Azi1 is not required for centriolar satellite formation, or localisation of key structural components like Pcm1, nor regulatory components like Cep290 or Cep72.
As centriolar satellites are proposed to have a role in regulating centrosome and centriole biogenesis, and AZI1 knock-down in human cells leads to increased Centrin2-positive foci [17], [19], [28], [47], we examined the numbers of centrioles (marked by Centrin2-GFP and anti-Centrin3) and centrosomes (marked by anti-γ Tubulin) in cells lacking Azi1 (Figure 4A, F, L–R). We found both centrosome and centriole numbers were normal in Azi1Gt/Gt MEFs (Figure 4A, F and N). Multiciliated cells have the ability to assemble hundreds of centrioles through two parallel pathways [48], [49], in both of which, protein-rich fibrous granules, akin to centriolar satellites, marked by PCM1, are found surrounding the elongating centrioles [18]. After assembly in the cytoplasm, these centrioles move apically to dock at the plasma membrane and each extend a ciliary axoneme. Ultrastructural analysis of motile multiciliated epithelia of adult trachea revealed normal numbers and docking of basal bodies and appendage formation in Azi1 null mice (Figure S3D and G). Confirming the immunofluorescent analysis in Azi1 mutant cells, transition zone ultrastructure appeared morphologically normal in Azi1 null mice (Figure S3E and H).
In summary, and in contrast with the acute Azi1 knock-down in both human and mouse cells (Figure 1) [10], [28], cilia and centriole structure appears grossly normal in Azi1 null cells.
Azi1 null MEFs have compensated for the loss of Azi1
The difference in ciliary phenotypes observed with acute Azi1 knock-down versus its chronic absence in Azi1Gt/Gt MEFs is intriguing. To rule out differences between the cell line used for the screen and primary cells, we co-transfected wild-type MEFs with siRNA against Azi1 along with plasmids encoding either GFP or Azi1-GFP and examined cilia formation. Similar to the results obtained in the embryonic fibroblast cell line (Figure 1), Azi1 knock-down in primary MEFs led to a robust reduction in ciliogenesis which was rescued by the siRNA-resistant Azi1-GFP (Figure 5A–D and I). To rule out any residual Azi1 function in our mutant Azi1Gt/Gt cells, and to further eliminate off-target effects, we transfected Azi1Gt/Gt MEFs with Azi1 siRNA. While our positive control Ift88 siRNA gave a robust reduction in ciliogenesis in Azi1Gt/Gt MEFs, ciliogenesis was unaffected upon Azi1 knock-down (Figure 5E–I), demonstrating that Azi1 null MEFs have compensated for the loss of Azi1. Discrepancies between the phenotypic severity observed with siRNA knock-down versus genetic deletion has previously been attributed to the acute nature of knock-down, allowing less time for compensation to occur [50], [51]. We conclude that although Azi1 is involved in cilia formation in mouse, compensation during embryogenesis in the absence of Azi1 allows ciliogenesis to proceed normally in most tissues of Azi1 null mice.

A global DNA damage response is not observed in Azi1 null mice
A role for cilia/centrosomal proteins in genome stability and DNA damage response pathways has been proposed [52], and was recently reported for AZI1 [28], [30]. Unlike the reported AZI1 knock-down [28], [30], we saw no increase in the number or intensity of γH2AX foci in Azi1 null MEFs (Figure S4A, F and K and data not shown). To test whether Azi1 mutants were more susceptible to DNA damaging agents, we challenged MEFs with either hydroxyurea (HU) or ionising radiation and examined γH2AX foci. No significant difference was observed between genotypes at lower concentrations of HU or upon challenge with ionising radiation, although Azi1 null MEFs were more susceptible to high doses of HU (5 mM) (Figure S4B–E, and G–K). As Staples et al. (2012) reported an increase in micronuclei in AZI1 depleted U2OS cells [28], we examined Azi1Gt/Gt mice using a peripheral blood micronuclei assay - a highly sensitive method for detecting in vivo DNA damage [53]. Whilst positive control Mcph1Gt/Gt mice show a marked increase in micronucleated erythrocytes as previously documented (http://www.sanger.ac.uk/mouseportal/phenotyping/MBGX/micronuclei/) [54], Azi1 null mice show no such increase, suggesting there is no elevation in DNA damage in the peripheral blood of Azi1 null mice (Figure S4L and M). Taken together, there is no gross evidence for chromosomal instability in Azi1 null animals, neither in vivo nor in primary cell culture. Once again it is possible that the differences observed between siRNA knock-down and genetic null of Azi1 are due to compensation for Azi1 loss in the genetic null, as seen for the ciliogenic phenotypes observed.
Azi1 function is not compensated in the sperm flagella resulting in male infertility
We examined Azi1 null mice carefully for adult-onset ciliopathic phenotypes to determine whether cilia formation and function was normal in all tissues. Azi1 mutant males display complete infertility with no evidence of pregnancy or pups born from more than 25 plugged dams. In contrast, Azi1Gt/Gt female mice have normal litter numbers and sizes (Figure 6A). Azi1Gt/Gt males have reduced testes weight (Azi1Gt/Gt: 185 mg+/−23.9 mg vs. Azi1Gt/+: 127 mg+/−9.7 mg, P<0.05, Student's t-test) corresponding to a drastic reduction in sperm density (less than 2% of wild type) (Figure 6B). Male infertility is a common symptom of ciliopathies, often in conjunction with airway dysfunction and late-onset phenotypes including retinal degeneration, kidney cysts and obesity in mouse mutants of cilia genes [55], [56], [57]. Immunofluorescent and ultrastructural analysis of postnatal multiciliated airway epithelium revealed mutant cilia to be morphologically normal, consistent with the lack of chronic airway infections in these mice (Figure 3G–H and S3D–I). In aged cohorts of littermates, no signs of retinal degeneration were observed in Azi1 null eyes either by ophthalmoscopic examination (data not shown) or histologically at six months (n = 7, Figure S5A–C). No cysts were observed in Azi1 null kidneys aged 6 months or older (n = 6, Figure S5D–F), nor was any obesity observed in aged Azi1Gt/Gt mice (n = 8 at 3 months, n = 5 at 6 months, Figure S5G and H). In support of our cellular analysis, these in vivo studies of Azi1 null mice demonstrate that Azi1 is not required for cilia formation or function in general but is required for formation and function of the specialised cilia derivative, the sperm flagella.

Spermiogenesis arrests at Step 9 in Azi1 null tubules, with severe flagellar defects and teratozoospermia
To determine when spermatogenesis is disrupted in Azi1 mutant mice, we examined periodic acid-Schiff (PAS) stained sections of testes. Azi1Gt/Gt mutant testes show a significant reduction in tubule lumen size (Lumen diameter: 15.1+/−1.2 µm in Azi1Gt/Gt, compared to 36.7+/−1.4 µm in Azi1+/+, mean +/ − SEM, P<0.001, Student's t-test, n = 3), with a drastic reduction in the number of sperm flagella visible in the lumens of Azi1Gt/Gt testes (Figure 6C–L). Anti-acetylated α-Tubulin, which marks the flagellar axonemes, reveals that whilst control tubule lumens are filled with sperm flagella, almost none were detected in mutant tubules, and any flagella seen appeared shorter and morphologically abnormal, suggesting Azi1 is necessary for flagella formation (Figure 6M and N). Light microscopy revealed the pre-spermiogenic stages of spermatogenesis, up to Step 7–8 spermatids, appeared to be normal (Figure 6E–L, O–P). However, from Step 9, spermatid morphology is highly abnormal, with mutant elongating spermatids mislocalised and misorientated within the tubule. In addition to axonemal defects, mutant elongating spermatids also exhibit teratozoospermia, where sperm heads show an abnormal club-shaped nuclear morphology, as opposed to the normal hook-shaped head in wild type elongate spermatids (Figure 6E–L, P). Very few spermatids reach maturity and are successfully passed into the epididymis (Figure 6B, S6A and B). Importantly these phenotypes were observable from the first wave of spermatogenesis (Figure S6C–J). Whereas Azi1Gt/Gt tubules resemble Azi1+/+ tubules at postnatal day 20 (P20) and P25 (Figure S6C–F), by P30, defects such as lack of flagella and misorientated spermatids displaying teratozoospermia become apparent. These results confirm observations in the adult mutant tubules that defects in spermiogenesis arise as the spermatids begin to elongate (P25–30) (Figure S6G–J).
To investigate whether the reduction in mature sperm was due to an increase in spermatid death in Azi1Gt/Gt tubules, we analysed levels of activated Caspase 3a and TUNEL staining, both indicators of apoptosis, in adult mutant tubules. Azi1 mutant tubules showed increases in both activated Caspase 3a staining and TUNEL positive cells (Figure S6K–P). The restricted spatial distribution of activated Caspase 3a positive cells, combined with the cell counts showing a reduction in elongating spermatids (Figure 6O) indicate Azi1Gt/Gt spermatids undergo apoptosis from Step 9 onwards.
The germline is particularly sensitive to DNA damage and given the suggested role for AZI1 in genome stability we considered whether Azi1 null male infertility is due to an increase in DNA damage. Infertility due to defects in DNA damage response pathways generally presents as an early arrest in spermiogenesis, with spermatocytes not progressing through meiosis [58], [59], [60]. This is in contrast to the relatively late arrest in post-meiotic spermatogenesis seen in Azi1 null mice, reminiscent of other ciliopathic mouse models [55], [56], [57], [61], [62]. To affirm that the arrest in spermatogenesis in Azi1 mutant mice is not due to genome instability, we stained Azi1 null testes with anti-γH2AX. In Azi1 mutant tubules, we observed anti-γH2AX staining comparable to controls, emphasising that there is no increase in DNA double-stranded breaks in Azi1 null testes (Figure S4N and O). Together with the previous in vitro and in vivo data (Figure S4), this shows there is no increase in DNA damage in the absence of Azi1 under physiological conditions.
Loss of Azi1 results in multiple microtubule-dependent trafficking defects in mutant spermatids
Cauda or caput epididymides from Azi1 mutant mice only contained debris and degenerating sperm, none of which were motile (Figure 6B, 7A and B, Movie S2 and S3, and data not shown). Even in the testes, flagella were rarely observed by ultrastructure analysis of mutant tubules, which exhibited drastically reduced lumen diameters filled with vacuolar cells and proteinaceous cell debris, in contrast to the open, flagella filled wild type tubule lumens (Figure 7C and D). The rare flagella that remained showed evidence of abnormal trafficking, with swollen flagellar lumens and ectopic mistrafficked outer dense fibres, occasionally associated with microtubules (Figure 7E and F, S7). Remaining sperm were morphologically abnormal with flagella of severely reduced length (Figure 7G–I). The processes involved in extension of the mammalian sperm flagella are not well understood, but are thought to involve IFT-mediated trafficking [63], [64], [65]. Truncated Azi1 mutant axonemes exhibit abnormal post-translational modifications of microtubules and irregular distributions of IFT trains, as marked by anti-Ift88 (Figure 7G and H). Extended axonemal structures are rarely detected by longitudinal TEM sections of mutant spermatids, instead occasional swollen shortened flagellar remnants with build-up of IFT cargo can be seen (Figure S7). Together these data suggest disruptions in regulated IFT account for the failure of flagellar axoneme extension.

However, some of the abnormalities in Azi1 mutant spermatid development, such as teratozoospermia cannot easily be explained by defects in IFT. These defects are first observed at step 9, when spermatids undergo a series of complex morphological changes, including nuclear remodelling and formation of the transient microtubule structure of the manchette [66], [67]. This microtubular “sleeve” structure surrounds the head and is assembled concurrently with the elongation and condensation of the spermatid nucleus, as well as growth of the centrosome-derived axoneme [68]. As in IFT, motor-driven trafficking along this track of microtubules delivers cargo from the Golgi-derived acrosome toward the centrosome and nascent sperm tail, in a process of intramanchette transport (IMT) [67]. Abnormal club shaped nuclei were previously observed in mutants with defects in manchette formation and function [69], [70]. Ultrastructural analysis revealed the manchette to be present in Azi1 mutant spermatids but it often appears kinked, and is occasionally misnucleated away from the spermatid head (Figure 8A and B, S8A and B). Late stage spermatids exhibited abnormal nuclear morphologies, consistent with the histological analyses (Figure 6 and S6), often with detachment of the acrosome from the nucleus (Figure S8C–E). Formation of the sperm tail involves the migration and modification of a peripheral pair of centrioles to the caudal pole of the nucleus, opposite the acrosome, where they become lodged to form the neck or head-tail coupling apparatus (HTCA). In early spermatid differentiation, modifications to the proximal centriole, lodging of the centrioles into the implantation fossa of the nuclear membrane and formation of the centriolar adjunct appear grossly normal in Azi1 mutant spermatids [71] (Figure 8C and D). A range of HTCA phenotypes are observed in later stage mutant spermatids, including misalignment of HTCA with the nucleus and/or displaced implantation fossa [72] (Figure 8E and F, S8F). Together, these results suggest Azi1 may be required for maturation and functional integrity of the HTCA.

To understand these spermiogenic phenotypes observed in Azi1 mutants, we examined Azi1 localisation during sperm development. Azi1 is found at the Golgi-derived acrosome (Step 10–12 spermatids: Figure 8G), then in the centrosome-containing HTCA at the flagellar base in later stage spermatids (Figure 8I). Importantly, no Azi1 staining is detected in any stage of mutant spermatids, confirming specificity of this Azi1 localisation (Figure 8H and J). This dynamic stage-specific redistribution of Azi1 is consistent with Azi1 undergoing IMT, although we failed to detect Azi1 specifically in the manchette, possibly due to dispersal of Azi1 below the detection threshold during IMT. This redistribution of Azi1 to the HTCA is consistent with a role in the maturation and functional integrity of this structure, and is reminiscent of enrichment of Azi1 around basal bodies upon ciliation in somatic cells.
We next examined localisation of an IMT cargo, Hook1, a coiled-coil protein implicated in vesicular transport which is mobilised progressively from the acrosome to HTCA during spermiogenesis [73], [74]. Post-translational modification of microtubules in the manchette are proposed to determine trafficking events by motors [74]. Manchette microtubules in wild type spermatids, although stabilised, are not labelled by the usual microtubule stabilising modifications, such as acetylation [75] (Figure 8K–M). However, strong anti-acetylated α-Tubulin staining is observed in Hook1-positive Azi1 mutant manchettes at step 9–12 (Figure 8N and O). Subsequently, Hook1 is prematurely lost from Step 14–15 mutant manchettes (Figure 8M and P). These results suggest Azi1 mutant spermatids exhibit altered microtubule dynamics and IMT cargo localisation. While intramanchette transport (IMT) is essential for both normal sperm head morphology and flagella formation [66], [73], [76], mouse mutations of components trafficked by IMT, like Hook1 [68], [69], [73] and RIM-BP3 [70], do not block extension of the flagellar axoneme completely.
These results suggest that loss of Azi1 disrupts the microtubule-based trafficking of both flagellar-directed IMT, and intraflagellar transport, resulting in both abnormal sperm head morphology together with the lack of flagella in Azi1 mutant spermatids.
Discussion
Genomic and proteomic studies on the centrosome/cilium complex (the “ciliome”) have estimated that thousands of gene products are involved in the formation and function of this important structure [8], [9]. Although many of the components are conserved across ciliates, it has generally been assumed that extrapolation from diverse genetic model organisms such as photosensitive, motile flagella of Chlamydomonas and ciliated mechanosensory chordotonal organs of Drosophila can be confidently extended to vertebrate cilia. In particular, there has been a growing trend of morpholino-based knock-down in the D. rerio model to functionally analyse putative ciliopathy candidate genes [13], [25]. Several recent mammalian cell culture-based knock-down screens have identified novel components involved in regulating cilia biology [10], [11], [12]. Facing a flood of next-generation sequencing detecting human disease variants, the daunting challenge is how best to investigate the requirement and function of novel and poorly characterised genes during development and disease.
Although Azi1/Cep131 was predicted to be essential for cilia from studies in fly and zebrafish [24], [25], we present here detailed analysis of both acute loss (by siRNA) and chronic absence (by genetic mutation), and show that Azi1 has a conserved but non-essential role in mammalian ciliogenesis, and is essential for the formation of sperm flagella (Figure 9).

Acute versus chronic lack of Azi1: Lessons for functional characterisation of putative human disease genes
We and others have shown transient knock-down of mammalian Azi1 leads to a reduction in ciliogenesis [10]. Importantly, we show this Azi1 knock-down ciliogenesis defect is rescued by overexpressing Azi1-GFP, emphasising the phenotype is “on-target” (Figure 1). Surprisingly, following genetic deletion of all gene function, cilia develop and function normally in vivo and in primary cells (Figure 3, 4, S3 and S5). Ruling out any sub-detectable “leakiness” of our Azi1 gene-trap, transfection of Azi1 siRNA into Azi1Gt/Gt MEFs does not affect ciliogenesis (Figure 5), suggesting that compensation for the lack of Azi1 has occurred. It has been previously suggested that acute knock-down of proteins can give a more severe phenotype than long-term deletions due to compensation in vivo [50], [51]. We present the first demonstration of a lack of phenotype in null cells treated with siRNA against the gene of interest, proving this functional compensation exists.
In the absence of Azi1, mammalian cilia develop and function properly in all mouse tissues except for the developing sperm (Figure 3, 4 S3 and S5). Azi1 is not essential for ciliogenesis and any involvement it may have in cilia biology can be compensated for in most tissues, aside from the modified cilia of the sperm flagella. Azi1 null mice are born at sub-Mendelian ratios and a third of mutants appear to be lost before mid-gestation (Table 1), suggesting that a proportion of mutant embryos may fail to compensate for the loss of Azi1 and die earlier in development. Several ciliopathy mouse models are also born at sub-Mendelian ratios, suggesting stochastic events may affect the requirement for ciliary proteins during embryogenesis [55], [56], [57], [77].
Our study highlights the importance of functional follow-up studies of siRNA data, and cautions against direct extrapolation of siRNA phenotypes to the genetic in vivo phenotype. On the other hand, acute knock-down by siRNA can expose roles for genes that would otherwise be overlooked due to compensation or redundancy in long-term deletion studies in vivo.
Azi1 null male mice are infertile, suggesting the loss of Azi1 cannot be compensated for during spermiogenesis. Azi1 null mice exhibit post-meiotic defects in spermatogenesis with misorientation and abnormal morphology of elongating and elongated spermatids, including teratozoospermia, from Step 9 of spermatid development onwards (Figure 6 and S6). Sperm flagella are mostly absent and any remaining axonemes are truncated and immotile, with swollen flagella lumens and evidence of mistrafficking of IFT components and cargo (Figure 7 and S7), suggesting Azi1 is required for IFT during the formation of the sperm flagella.
Additional roles for ciliary proteins in spermatid development outside the axoneme have been suggested, with parallels drawn between intraflagellar transport (IFT) and intramanchette transport (IMT) [67]. The abnormal club-shaped sperm head morphology is similar to mutants with defective manchettes [68], [69], [70]. Azi1 mutant manchettes are structurally abnormal, and exhibit altered microtubule post-translational modifications, which are implicated in motor-selection for cargo delivery [74] (Figure 8 and S8). Altered expression of Hook1, an effector of IMT implicated in vesicular transport [74], is also observed in these mutant structures, suggesting the process of IMT is disrupted in Azi1 mutants. Many motors and associated proteins, some linked to IFT, have been localised to the manchette and the testicular phenotype of Ift88orpk/orpk hypomorphic mice phenocopies the Azi1-dependent trafficking defects [67], [74].
Although the role of centriolar satellite or transition zone proteins in spermiogenesis is not well understood, it is tempting to draw parallels between the microtubule-based trafficking of centriolar satellites towards the centrosome/basal body of primary cilia, and the trafficking of proteins along the transient microtubular structure of the manchette to the highly specialised motile sperm flagellum. The dynamic relocalisation of Azi1 from the acrosome to the HTCA during spermatid development suggests Azi1 undergoes IMT, as this is the main transport route between these structures (Figure 8). Interestingly, components of the dynactin complex, which localise to centriolar satellites and is required for their peri-centrosomal localisation, have also been localised to the manchette [19], [47], [74], [78].
It is a conundrum for the cilia field as to why broadly expressed “core” cilia genes have clinically restricted phenotypes when mutated. This “tissue-specific” requirement is demonstrated by the limited phenotypes of late-onset ciliopathies, and many centriolar satellite and transition zone-specific mouse mutants display tissue specific ciliary phenotypes [56], [61], [62], [77], [79], [80]. In the case of the transition zone complex components, Nphp4 and Nphp1 null mutant mice display male infertility without kidney phenotypes, in contrast to the human disease in which patients have severe nephronophthisis [61], [62], [81]. This discrepancy could reflect differences in species and/or functional nature of the mutation, but the severity in humans could also reflect the mutational load in other components of the complex [82], [83]. Importantly primary cilia formation and function appears grossly normal in these mutants, like in our Azi1 null mice, emphasising that compensation for function of key cilia genes is likely to be a recurrent theme given the central importance of primary cilia in mammalian development and patterning.
Evolutionary function and divergence of the requirement for Azi1
Azi1/Cep131 is a conserved protein found in all ciliates except for nematodes, leading to the suggestion that Azi1 may be involved in cilia motility (Table S1) [46]. Alternatively, Azi1/Cep131 could be required to build a canonical nine-triplet centriole, as nematodes build specialized, non-canonical centrioles. However, an Azi1/Cep131 orthologue is present in the Toxoplasma gondii genome which also build non-canonical centrioles [84]. Furthermore, knock-down of Azi1/Cep131 in planaria does not affect centriole formation, but affects cilia motility [45]. While deletion mutations of dila in D. melanogaster [24] lead to defects in specialised “motile” mechanosenory Type I neurons and sperm, cilia do form with defects in mechanosensory neuronal cilia morphology characteristic of IFT mutants, suggesting defective trafficking. Similarly, Azi1 D. rerio morphants phenocopy IFT morphants with cilia forming but displaying tissue-specific reductions in length [25]. Interestingly, in trypanosomes TzAZI1 localises to the flagellar pocket, a dynamic endo-exocytic organelle implicated in membrane trafficking surrounding the flagellar base, and TzAZI1 RNAi affects flagellar function, as opposed to its formation or maintenance [46]. These studies suggest a conserved trafficking function for Azi1 in regulating ciliary-bound cargo.
We confirmed mammalian Azi1 localises to centriolar satellites and provide the first direct observation of Azi1 trafficking along microtubules, both towards and away from the centrosome (Figure 2G, Supplementary Movie 1), similar to the movement observed for PCM1-GFP [18]. The transport away from the centrosome may involve kinesin motors, and it has been shown that CEP290, which binds AZI1, interacts with both Dynactin components and the kinesin motor KIF3a [28], [79], supporting the theory that these proteins could undergo bidirectional trafficking along microtubules.
Centriolar satellites are thought to spatially restrict centrosomal access of proteins involved in basal body maturation and ciliogenesis [22]. While centriolar satellites and the key centriolar satellite protein, Pcm1, are not found in Drosophila, tight transcriptional control of dila mRNA limits expression to just before the onset of ciliogenesis where the protein can localise to the PCM [24]. Although the regulatory mechanisms are different, in both mammals and flies, it appears Azi1/dila recruitment is involved in centrosome to basal body maturation. In D. melanogaster, mutations in two other coiled-coil proteins YURI, conserved only among the Drosophila genus [85], [86], and UNC, also insect-specific but for which centriolar satellite protein OFD1 is proposed to be a functional orthologue [87] partially phenocopy dila mutants and genetically interact with dila [24]. These proteins are involved in the proper maturation and anchoring of the sperm basal bodies to the nuclear membrane [24], [86], [87]. Formation of Drosophila sperm flagella axoneme is unusual in that it is IFT-independent, forming instead in cytoplasmic cysts. Sperm axonemes do form in dila mutant flies, although the sperm display defective HTCA formation. This is reminiscent of the defects we observe in compromised integrity of HTCA in Azi1 null sperm (Figure 8 and S8), although the requirement for IFT in mammalian sperm axonemal formation may explain the more severe IFT-based flagellar phenotypes observed in the mouse mutant spermatids.
Interestingly, similar to Ma et al. (2011) who showed transition zone localisation for DILA and UNC in D. melanogaster [24], we show Azi1 and PCM1 also localise to transition zone of primary cilium. While this manuscript was in preparation, OFD1 was also shown to localise to the transition zone [41]. Transition zone and centriolar satellite localisation has previously been described for CEP290, which was recently shown to interact with AZI1 [28], [38], [39]. Our data supports that redistribution of centriolar satellite proteins to the transition zone during ciliogenesis is of functional significance. It is possible that centriolar satellite-associated cargo is transported to the centrosome as it matures into a basal body with a functional transition zone ready to be trafficked into the cilium. Alternatively, Azi1/CEP290 could also have a role in the gating function at the transition zone, regulating protein content in the cilium, accounting for the IFT-like phenotypes reported in dila Drosophila mutants [24] and Cep290 Chlamydomonas mutants [42]. Given that centriolar satellites are scaffolds for controlling activity of ciliopathy-associated proteins [15], [22], it will be important to define the composition of these specific sub-complexes that move to the transition zone, and ask whether they are part of the compensation mechanism observed in Azi1 null animals. If true, we propose that these interactions among the centriolar satellite proteins could extend to multi-allelic mutational load, including AZI1, in a subset of human ciliopathies with diverse clinical presentations beyond male infertility.
Materials and Methods
Cell culture and transfection
ShhLIGHT-II (ATCC, genetically modified NIH-3T3) and NIH-3T3 cells were maintained in DMEM (Life Technologies), hTERT-RPE cells were maintained in DMEM-F12 (Life Technologies), all supplemented with 1.5 g/L sodium bicarbonate (Sigma), 10% foetal calf serum, 5×108 U/L penicillin and 11 mM streptomycin at 37°C, 5% CO2. Early passage MEFs were maintained in Optimem (Life Technologies) plus 0.5 mM beta-mercaptoethanol (Sigma), 10% foetal calf serum, 5×108 U/L penicillin and 11 mM streptomycin at 37°C, 5% CO2, 3% O2. To induce ciliogenesis, serum was removed for 48 hours. ShhLIGHT-II cells were co-transfected with 25 nM Dharmacon OnTarget Plus siRNA and 1 µg/mL plasmid DNA with Dharmafect Duo (Dharmacon), serum was removed after 24 h and samples were analysed 72 hours after transfection. MEFs were co-transfected with 50 nM Dharmacon OnTarget Plus siRNA and 1.6 µg/mL plasmid DNA using the Invitrogen Neon, according to the manufacturer's protocol.
siRNA sequences used: Ctrl: 5′UGGUUUACAUGUCGACUAA3′; Ift88 #3 : 5′-CGGAGAAUGUUGAAUGUUU-3′; Ift88 #4 : 5′-GCUUGGAGCUUAUUACAUU-3′; Azi1 3′UTR Pool: Equimolar pool of: 5′-AGACACAGGGCUAAGGGUA-3′, 5′-CAGCUGUUCUAUAGUAAAA-3′, 5′-CCCUUGGGAUGACGAGCCA-3′ and 5′-GUGUCCAGGUCACGCUCCA-3′.
For live imaging of Azi1-GFP on Map4-RFP microtubules, NIH-3T3 cells were transduced with 30 particles per cell (PPC) of CellLIGHT MAP4-RFP BacMan 2.0 (Life Technologies), left for 24 hours then transfected with Azi1-GFP using Lipofectamine 2000 (Life Technologies) and imaged 24 hours later.
For DNA damage assays, MEFs were challenged with hydroxyurea (Sigma) at given concentrations for 18 hours. Alternatively, MEFs were irradiated in culture medium at 2Gy/minute using a Faxitron CellRad cabinet X-ray system (Faxitron Bioptics), cultured for 3 hours and then fixed and analysed.
Generation of plasmids
Azi1 was PCR amplified from mouse cDNA and TA cloned into pcDNA6.2-C-Em-GFP-GW-TOPO cDNA plasmid (Life Technologies). Centrin2 was PCR amplified from mouse cDNA, adding EcoR1 and Sal1 sites. This was restriction enzyme cloned into pEGFP-N1 (Clontech).
Western blot and densitometry
Cells or testes were homogenised in cell lysis buffer (Cell Signaling Technology) plus 1 mM phenylmethylsulfonyl fluoride (PMSF) (Thermo Scientific) and Complete Protease Inhibitor Cocktail (Roche), sonicated for 2×30 seconds. Testes extracts were concentrated with Amicon Ultra-0.5 mL 30 kDa centrifugal filters (Millipore). Samples were separated on Novex 3–8% Tris Acetate gels (Life Technologies) then transferred to Hybond nitrocellulose membrane (GE Healthcare), which were blocked in 5% milk. Membranes were incubated in primary antibodies (Table S4) washed, incubated with horse radish peroxidase (HRP) -conjugated secondaries (Table S5) and developed with Amersham ECL-plus western blotting detection system. Densitometry was performed using ImageJ.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde/phosphate buffered solution (PFA/PBS) for 10 minutes at room temperature, or 4% PFA/PHEM (120 mM PIPES, 140 mM HEPES, 20 mM EGTA, 16 mM MgSO4, pH7) for 10 minutes at 37°C. Alternatively, pre-extraction was performed for 30 seconds on ice in 0.1 M PIPES pH 6.8, 2 mM EGTA and 1 mM MgSO4, then cells were fixed in ice cold methanol on ice for 10 minutes. Cells were blocked in 10% donkey serum/0.1% Triton-X in Tris buffered solution (TBS). Primary antibodies were added (Table S4), cells were washed then incubated in Alexa Fluor-conjugated secondaries (Table S5) and slides were mounted using Prolong Gold (Life Technologies).
Generation of Azi1 mutant mice
Azi1+/Gt(CCOG35)Wtsi 129Ola embryonic stem cells, which have a gene trap inserted into intron 2 of Azi1, were ordered from the Mutant Mouse Regional Resource Centre (MMRRC). ES cells were injected into C57BL/6J blastocysts and implanted into a recipient C57BL/6J female. These were backcrossed onto C57BL/6J for at least 5 generations for most analyses. Animals were maintained in SPF environment and studies carried out in accordance with the guidance issued by the Medical Research Council in “Responsibility in the Use of Animals in Medical Research” (July 1993) and licensed by the Home Office under the Animals (Scientific Procedures) Act 1986. Genotyping was performed using gene trap specific primers (GT Forward: 5′-GGTCCCGAAAACCAAAGAAG-3′ and GT Reverse: 5′-AGTATCGGCCTCAGGAAGATCG-3′) and primers to Azi1 intron 2, spanning the insertion site which fail to amplify in the mutant (In Forward: 5′-GAGGAACCTGGGTGAGACCT-3′ and In Reverse: 5′-GCAGCAGATCTTTGGTCCAC-3′). Details of primers used for characterisation of the Azi1GT allele by RT-PCR and RT-qPCR are provided in Tables S2 and S3.
Histology and immunohistochemistry
Tissue samples were collected, kidneys were fixed in 4% PFA/PBS, testes were fixed in Bouin's fixative, and eyes were fixed in Davidson's fixative according to standard protocols. Tissues were serially dehydrated and embedded in paraffin. Microtome sections of 8 µm thickness were examined histologically via haematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) staining.
For immunofluorescent analysis, paraffin sections were dewaxed and re-hydrated via ethanol series. Antigen retrieval was performed by boiling the sections for 15 minutes in the microwave in citrate buffer. Sections were blocked in 10% donkey serum/0.1% Triton-X in PBS and primary antibodies were diluted in 1% donkey serum/PBS (Table S4). Slides were washed and incubated in Alexafluor conjugated secondary antibodies (Table S5), washed and mounted in ProLong Gold (Life technologies).
For immunohistochemistry, the same procedure was used, with the addition of one step after the re-hydration. Slides were immersed in 3% H2O2 in PBS for 20 minutes to block endogenous peroxidases. Slides were incubated in primary antibody, washed, then incubated in biotin-conjugated secondary antibody (Vector laboratories). This was detected using the Vector ABC kit and Vector NovaRed peroxidase substrate kit.
For TUNEL, after dewaxing, sections were incubated in 0.25% Triton-X and labelling was performed using Click-IT TUNEL staining kit following manufacturer's instructions (Life Technologies).
LacZ staining using X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside)
E11.5 Azi1+/Gt embryos were fixed in 4% PFA/PBS for 20 minutes, rinsed in PBS and washed 3 times in detergent buffer (0.1 M phosphate buffer, 2 mM MgCl2, 0.1% sodium deoxycholate and 0.02% NP-40 (IGEPAL CA-630)). Embryos were then stained overnight in detergent buffer containing 50 mg/ml X-gal, 5 mM K3 and 5 mM K4 at 37°C, protected from light, washed twice in detergent buffer and post fixed overnight in 4% PFA.
Sperm preparation
Testes, cauda and caput epididymides were dissected into M2 media (Invitrogen). For live imaging, sperm were imaged in M2 media or 1% methyl cellulose (Sigma), in capillary tubes (Vitrotubes Mountain Leaks) sealed with Cristaseal (Hawskley). Sperm counts were performed on sperm from the cauda epididymides, diluted in H2O using a haemocytometer, only counting intact sperm (with both head and tail). For fixed samples, either sperm spreads or testes squashes were prepared. For sperm spreads, testes were placed through a 100 µm nylon mesh (BD Biosciences). Sperm from the caput epididymus and testes were then placed on a 20–40% Percoll gradient (GE Healthcare) and spun at 3,000 g for 10 minutes. The sperm were spread on Poly-D-lysine slides (BD Biosciences) and fixed 4% PFA/PBS. Cauda sperm was spread directly on Poly-D-Lysine slides and fixed with 4% PFA/PBS. In all cases, sperm were permeabilised with 0.4% Triton-X in PBS and immunofluorescence was performed as described. For testes squashes, tubules were dissected as described [88], placed on a slide and squashed with a coverslip. Slides were snap frozen in liquid nitrogen, coverslips removed and samples fixed and permeabilised by 10 minutes in −20°C methanol, 30 seconds in acetone and then 15 minutes in 4% PFA/PBS. Immunofluorescence was then performed as described.
Nasal brushing
Nasal brushing was performed as described [89]. Cells were fixed for 30 minutes on ice in 4% PFA, cytospun onto slides, fixed with −20°C methanol for 10 minutes then immunofluorescence was performed as described.
Transmission electron microscopy
Samples were dissected into PBS. Samples were fixed in 2% PFA/2.5% gluteraldehyde/0.1 M Sodium Cacodylate Buffer pH7.4 + 0.04% CaCl2. Testes capsules were removed prior to immersion in fix. After 30 minutes at room temperatures, samples were cut into 1 mm cubes and fixed overnight or longer at 4°C. Tissue was rinsed in 0.1 M sodium cacodylate buffer, post-fixed in 1% OsO4 (Agar Scientific) for one hour and dehydrated in sequential steps of acetone prior to impregnation in increasing concentrations of resin (TAAB Lab Equipment) in acetone followed by 100%, placed in moulds and polymerised at 60°C for 24 hours.
Ultrathin sections of 70 nm were subsequently cut using a diamond knife on a Leica EM UC7 ultramicrotome. Sections were stretched with chloroform to eliminate compression and mounted on Pioloform filmed copper grids prior to staining with 1% aqueous uranyl acetate and lead citrate (Leica). They were viewed on a Philips CM100 Compustage Transmission Electron Microscope with images collected using an AMT CCD camera (Deben).
Micronuclei assay
Mice were tail tipped and blood was collected using a microhematocrit capillary tube with heparin coating (Globe Scientific) into heparin (Sigma). This was fixed in super-chilled methanol. Saline was added and then blood was pelleted at 600 g for 5 minutes. Pellets were treated with RNAseA and anti-CD71primary antibody (Lifespan Biosciences). Cells were incubated on ice for 30 minutes then at room temperature for 30 minutes. Propidium iodide was added and flow cytometry was performed on a FACScalibur (BD Biosciences). Data was analysed using FlowJo software (v7.6.1, Tree Star) as described by [53].
Imaging and image analysis
The initial siRNA screen imaging was carried out on the Olympus ScanR microscope, imaging 16 frames per well. Image analysis, including identification and counting of cells and cilia was performed using the Olympus ScanR Analysis Software.
Confocal images were captured with a Nikon A1R confocal microscope, comprising a Nikon Eclipse TiE inverted microscope and four laser modules: 405 (laser diode), 457, 488, 514 (multiline Argon) 561 (diode-pumped solid-state) and 638 nm (laser diode). For live imaging of Azi1-GFP and MAP4-RFP, a Zeiss Axiovert 200 fluorescence microscope was used equipped with 100×/1.4 plan apochromat objective (Carl Zeiss, Welwyn, UK), Lambda LS 300W Xenon source with liquid light guide and 10-position excitation, neutral density and emission filterwheels (Sutter Instrument, Novato, CA), ASI PZ2000 3-axis XYZ stage with integrated piezo Z-drive (Applied Scientific Instrumentation, Eugene, OR) and a Photometrics Coolsnap HQ2 CCD camera (Roper Scientific, Tucson, AZ). Brightfield images were captured with a Coolsnap HQ CCD camera (Photometrics Ltd, Tucson, AZ) on a Zeiss Axioplan II fluorescence microscope with Plan-neofluar objectives (Carl Zeiss, Welwyn Garden City, UK). Colour additive filters (Andover Corporation, Salem, NH) installed in a motorised emission filter wheel (Prior Scientific Instruments, Cambridge, UK) were used sequentially to collect red, green and blue images that were then superimposed to form a colour image.
For live imaging of sperm, a Qimaging Retiga camera running at 30 frames per second (bin 2×2 half frame) captured image sequences with a 5× objective at zoom 5 on a Nikon AZ100 macroscope. Still figures show all time points superimposed into one image to depict the movement, or lack thereof, during the movie.
Apart from the initial screen, image analysis including intensity profiling was performed in ImageJ.
Statistics
Throughout P<0.05 is considered significant. Statistics were carried out in Microsoft Excel or GraphPad Prism (La Jolla, CA) and the test used is specified in the text/figure legend.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NiggEA, RaffJW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139 : 663–678.
2. SinglaV, ReiterJF (2006) The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313 : 629–633.
3. CzarneckiPG, ShahJV (2012) The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol 22(4): 201–10.
4. SilvermanMA, LerouxMR (2009) Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol 19 : 306–316.
5. IshikawaH, MarshallWF (2011) Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol 12 : 222–234.
6. GerdesJM, DavisEE, KatsanisN (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137 : 32–45.
7. BakerK, BealesPL (2009) Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 151C: 281–295.
8. GhermanA, DavisEE, KatsanisN (2006) The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat Genet 38 : 961–962.
9. InglisPN, BoroevichKA, LerouxMR (2006) Piecing together a ciliome. Trends Genet 22 : 491–500.
10. GraserS, StierhofYD, LavoieSB, GassnerOS, LamlaS, et al. (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 179 : 321–330.
11. KimJ, LeeJE, Heynen-GenelS, SuyamaE, OnoK, et al. (2010) Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 464 : 1048–1051.
12. LaiCK, GuptaN, WenX, RangellL, ChihB, et al. (2011) Functional characterization of putative cilia genes by high-content analysis. Mol Biol Cell 22 : 1104–1119.
13. ZaghloulNA, KatsanisN (2011) Zebrafish assays of ciliopathies. Methods Cell Biol 105 : 257–272.
14. HildebrandtF, BenzingT, KatsanisN (2011) Ciliopathies. N Engl J Med 364 : 1533–1543.
15. LopesCA, ProsserSL, RomioL, HirstRA, O'CallaghanC, et al. (2011) Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J Cell Sci 124 : 600–612.
16. BalczonR, VardenCE, SchroerTA (1999) Role for microtubules in centrosome doubling in Chinese hamster ovary cells. Cell Motil Cytoskeleton 42 : 60–72.
17. BarenzF, MayiloD, GrussOJ (2011) Centriolar satellites: busy orbits around the centrosome. Eur J Cell Biol 90 : 983–989.
18. KuboA, SasakiH, Yuba-KuboA, TsukitaS, ShiinaN (1999) Centriolar satellites: molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J Cell Biol 147 : 969–980.
19. DammermannA, MerdesA (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol 159 : 255–266.
20. KimJC, BadanoJL, SiboldS, EsmailMA, HillJ, et al. (2004) The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet 36 : 462–470.
21. NachuryMV, LoktevAV, ZhangQ, WestlakeCJ, PeranenJ, et al. (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129 : 1201–1213.
22. StoweTR, WilkinsonCJ, IqbalA, StearnsT (2012) The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol Biol Cell 23 : 3322–3335.
23. CacheroS, SimpsonTI, Zur LagePI, MaL, NewtonFG, et al. (2011) The gene regulatory cascade linking proneural specification with differentiation in Drosophila sensory neurons. PLoS Biol 9: e1000568.
24. MaL, JarmanAP (2011) Dilatory is a Drosophila protein related to AZI1 (CEP131) that is located at the ciliary base and required for cilium formation. J Cell Sci 124 : 2622–2630.
25. WilkinsonCJ, CarlM, HarrisWA (2009) Cep70 and Cep131 contribute to ciliogenesis in zebrafish embryos. BMC Cell Biol 10 : 17.
26. AndersenJS, WilkinsonCJ, MayorT, MortensenP, NiggEA, et al. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426 : 570–574.
27. JakobsenL, VanselowK, SkogsM, ToyodaY, LundbergE, et al. (2011) Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 30 : 1520–1535.
28. StaplesCJ, MyersKN, BeveridgeRD, PatilAA, LeeAJ, et al. (2012) The centriolar satellite protein Cep131 is important for genome stability. J Cell Sci 125 : 4770–4779.
29. AotoH, TsuchidaJ, NishinaY, NishimuneY, AsanoA, et al. (1995) Isolation of a novel cDNA that encodes a protein localized to the pre-acrosome region of spermatids. Eur J Biochem 234 : 8–15.
30. PaulsenRD, SoniDV, WollmanR, HahnAT, YeeMC, et al. (2009) A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35 : 228–239.
31. CasparyT, LarkinsCE, AndersonKV (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 12 : 767–778.
32. MurciaNS, RichardsWG, YoderBK, MucenskiML, DunlapJR, et al. (2000) The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127 : 2347–2355.
33. PazourGJ, DickertBL, VucicaY, SeeleyES, RosenbaumJL, et al. (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol 151 : 709–718.
34. AkimovV, RigboltKT, NielsenMM, BlagoevB (2011) Characterization of ubiquitination dependent dynamics in growth factor receptor signaling by quantitative proteomics. Mol Biosyst 7 : 3223–3233.
35. LechtreckKF, GeimerS (2000) Distribution of polyglutamylated tubulin in the flagellar apparatus of green flagellates. Cell Motil Cytoskeleton 47 : 219–235.
36. WinkelbauerME, SchaferJC, HaycraftCJ, SwobodaP, YoderBK (2005) The C. elegans homologs of nephrocystin-1 and nephrocystin-4 are cilia transition zone proteins involved in chemosensory perception. J Cell Sci 118 : 5575–5587.
37. SangL, MillerJJ, CorbitKC, GilesRH, BrauerMJ, et al. (2011) Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145 : 513–528.
38. ChihB, LiuP, ChinnY, ChalouniC, KomuvesLG, et al. (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 14 : 61–72.
39. Garcia-GonzaloFR, CorbitKC, Sirerol-PiquerMS, RamaswamiG, OttoEA, et al. (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43 : 776–784.
40. KimJ, KrishnaswamiSR, GleesonJG (2008) CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet 17 : 3796–3805.
41. WangWJ, TayHG, SoniR, PerumalGS, GollMG, et al. (2013) CEP162 is an axoneme-recognition protein promoting ciliary transition zone assembly at the cilia base. Nat Cell Biol 15 : 591–601.
42. CraigeB, TsaoCC, DienerDR, HouY, LechtreckKF, et al. (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol 190 : 927–940.
43. CaiD, McEwenDP, MartensJR, MeyhoferE, VerheyKJ (2009) Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol 7: e1000216.
44. Flores-RodriguezN, RogersSS, KenwrightDA, WaighTA, WoodmanPG, et al. (2011) Roles of dynein and dynactin in early endosome dynamics revealed using automated tracking and global analysis. PLoS One 6: e24479.
45. AzimzadehJ, WongML, DownhourDM, Sanchez AlvaradoA, MarshallWF (2012) Centrosome loss in the evolution of planarians. Science 335 : 461–463.
46. Gabernet-CastelloC, DuboisKN, NimmoC, FieldMC (2011) Rab11 function in Trypanosoma brucei: identification of conserved and novel interaction partners. Eukaryot Cell 10 : 1082–1094.
47. KodaniA, TonthatV, WuB, SutterlinC (2010) Par6 alpha interacts with the dynactin subunit p150 Glued and is a critical regulator of centrosomal protein recruitment. Mol Biol Cell 21 : 3376–3385.
48. HagiwaraH, OhwadaN, TakataK (2004) Cell biology of normal and abnormal ciliogenesis in the ciliated epithelium. Int Rev Cytol 234 : 101–141.
49. DirksenER (1991) Centriole and basal body formation during ciliogenesis revisited. Biol Cell 72 : 31–38.
50. FreudenbergJM, GhoshS, LackfordBL, YellaboinaS, ZhengX, et al. (2012) Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res 40 : 3364–3377.
51. PulversJN, BrykJ, FishJL, Wilsch-BrauningerM, AraiY, et al. (2010) Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc Natl Acad Sci U S A 107 : 16595–16600.
52. ChakiM, AirikR, GhoshAK, GilesRH, ChenR, et al. (2012) Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150 : 533–548.
53. ReinholdtL, AshleyT, SchimentiJ, ShimaN (2004) Forward genetic screens for meiotic and mitotic recombination-defective mutants in mice. Methods Mol Biol 262 : 87–107.
54. LiangY, GaoH, LinSY, PengG, HuangX, et al. (2010) BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet 6: e1000826.
55. FathMA, MullinsRF, SearbyC, NishimuraDY, WeiJ, et al. (2005) Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet 14 : 1109–1118.
56. MykytynK, MullinsRF, AndrewsM, ChiangAP, SwiderskiRE, et al. (2004) Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci U S A 101 : 8664–8669.
57. NishimuraDY, FathM, MullinsRF, SearbyC, AndrewsM, et al. (2004) Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A 101 : 16588–16593.
58. CelesteA, PetersenS, RomanienkoPJ, Fernandez-CapetilloO, ChenHT, et al. (2002) Genomic instability in mice lacking histone H2AX. Science 296 : 922–927.
59. RuzankinaY, Pinzon-GuzmanC, AsareA, OngT, PontanoL, et al. (2007) Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1 : 113–126.
60. XuX, AprelikovaO, MoensP, DengCX, FurthPA (2003) Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development 130 : 2001–2012.
61. JiangST, ChiouYY, WangE, LinHK, LeeSP, et al. (2008) Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Hum Mol Genet 17 : 3368–3379.
62. WonJ, Marin de EvsikovaC, SmithRS, HicksWL, EdwardsMM, et al. (2011) NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum Mol Genet 20 : 482–496.
63. PazourGJ, BakerSA, DeaneJA, ColeDG, DickertBL, et al. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157 : 103–113.
64. BakerSA, FreemanK, Luby-PhelpsK, PazourGJ, BesharseJC (2003) IFT20 links kinesin II with a mammalian intraflagellar transport complex that is conserved in motile flagella and sensory cilia. J Biol Chem 278 : 34211–34218.
65. LoJC, JamsaiD, O'ConnorAE, BorgC, ClarkBJ, et al. (2012) RAB-like 2 has an essential role in male fertility, sperm intra-flagellar transport, and tail assembly. PLoS Genet 8: e1002969.
66. ToshimoriK, ItoC (2003) Formation and organization of the mammalian sperm head. Arch Histol Cytol 66 : 383–396.
67. KierszenbaumAL (2002) Intramanchette transport (IMT): managing the making of the spermatid head, centrosome, and tail. Mol Reprod Dev 63 : 1–4.
68. MeistrichML, Trostle-WeigePK, RussellLD (1990) Abnormal manchette development in spermatids of azh/azh mutant mice. Am J Anat 188 : 74–86.
69. ColeA, MeistrichML, CherryLM, Trostle-WeigePK (1988) Nuclear and manchette development in spermatids of normal and azh/azh mutant mice. Biol Reprod 38 : 385–401.
70. ZhouJ, DuYR, QinWH, HuYG, HuangYN, et al. (2009) RIM-BP3 is a manchette-associated protein essential for spermiogenesis. Development 136 : 373–382.
71. FawcettDW, PhillipsDM (1969) The fine structure and development of the neck region of the mammalian spermatozoon. Anat Rec 165 : 153–164.
72. ChemesHE, PuigdomenechET, CarizzaC, OlmedoSB, ZanchettiF, et al. (1999) Acephalic spermatozoa and abnormal development of the head-neck attachment: a human syndrome of genetic origin. Hum Reprod 14 : 1811–1818.
73. Mendoza-LujambioI, BurfeindP, DixkensC, MeinhardtA, Hoyer-FenderS, et al. (2002) The Hook1 gene is non-functional in the abnormal spermatozoon head shape (azh) mutant mouse. Hum Mol Genet 11 : 1647–1658.
74. KierszenbaumAL, RivkinE, TresLL (2011) Cytoskeletal track selection during cargo transport in spermatids is relevant to male fertility. Spermatogenesis 1 : 221–230.
75. HermoL, OkoR, HechtNB (1991) Differential post-translational modifications of microtubules in cells of the seminiferous epithelium of the rat: a light and electron microscope immunocytochemical study. Anat Rec 229 : 31–50.
76. KierszenbaumAL, RivkinE, TresLL, YoderBK, HaycraftCJ, et al. (2011) GMAP210 and IFT88 are present in the spermatid golgi apparatus and participate in the development of the acrosome-acroplaxome complex, head-tail coupling apparatus and tail. Dev Dyn 240 : 723–736.
77. KulagaHM, LeitchCC, EichersER, BadanoJL, LesemannA, et al. (2004) Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet 36 : 994–998.
78. FouquetJ, KannM, SouesS, MelkiR (2000) ARP1 in Golgi organisation and attachment of manchette microtubules to the nucleus during mammalian spermatogenesis. J Cell Sci 113 (Pt 5) 877–886.
79. ChangB, KhannaH, HawesN, JimenoD, HeS, et al. (2006) In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet 15 : 1847–1857.
80. LancasterMA, GopalDJ, KimJ, SaleemSN, SilhavyJL, et al. (2011) Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med 17 : 726–731.
81. HildebrandtF, OttoE, RensingC, NothwangHG, VollmerM, et al. (1997) A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet 17 : 149–153.
82. KatsanisN (2004) The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet 13 Spec No 1: R65–71.
83. KatsanisN, AnsleySJ, BadanoJL, EichersER, LewisRA, et al. (2001) Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293 : 2256–2259.
84. ChengYZ, EleyL, HynesAM, OvermanLM, SimmsRJ, et al. (2012) Investigating embryonic expression patterns and evolution of AHI1 and CEP290 genes, implicated in Joubert syndrome. PLoS One 7: e44975.
85. ArmstrongJD, TexadaMJ, MunjaalR, BakerDA, BeckinghamKM (2006) Gravitaxis in Drosophila melanogaster: a forward genetic screen. Genes Brain Behav 5 : 222–239.
86. TexadaMJ, SimonetteRA, JohnsonCB, DeeryWJ, BeckinghamKM (2008) Yuri gagarin is required for actin, tubulin and basal body functions in Drosophila spermatogenesis. J Cell Sci 121 : 1926–1936.
87. BakerJD, AdhikarakunnathuS, KernanMJ (2004) Mechanosensory-defective, male-sterile unc mutants identify a novel basal body protein required for ciliogenesis in Drosophila. Development 131 : 3411–3422.
88. KotajaN, KimminsS, BrancorsiniS, HentschD, VoneschJL, et al. (2004) Preparation, isolation and characterization of stage-specific spermatogenic cells for cellular and molecular analysis. Nat Methods 1 : 249–254.
89. HolderE, StevensonB, FarleyR, HilliardT, WodehouseT, et al. (2010) Detection of CFTR transgene mRNA expression in respiratory epithelium isolated from the murine nasal cavity. J Gene Med 12 : 55–63.
90. HamerG, Roepers-GajadienHL, van Duyn-GoedhartA, GademanIS, KalHB, et al. (2003) DNA double-strand breaks and gamma-H2AX signaling in the testis. Biol Reprod 68 : 628–634.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy