
Autozygome Sequencing Expands the Horizon of Human Knockout Research and Provides Novel Insights into Human Phenotypic Variation
The use of autozygosity as a mapping tool in the search for autosomal recessive disease genes is well established. We hypothesized that autozygosity not only unmasks the recessiveness of disease causing variants, but can also reveal natural knockouts of genes with less obvious phenotypic consequences. To test this hypothesis, we exome sequenced 77 well phenotyped individuals born to first cousin parents in search of genes that are biallelically inactivated. Using a very conservative estimate, we show that each of these individuals carries biallelic inactivation of 22.8 genes on average. For many of the 169 genes that appear to be biallelically inactivated, available data support involvement in modulating metabolism, immunity, perception, external appearance and other phenotypic aspects, and appear therefore to contribute to human phenotypic variation. Other genes with biallelic inactivation may contribute in yet unknown mechanisms or may be on their way to conversion into pseudogenes due to true recent dispensability. We conclude that sequencing the autozygome is an efficient way to map the contribution of genes to human phenotypic variation that goes beyond the classical definition of disease.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1004030
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004030
Summary
The use of autozygosity as a mapping tool in the search for autosomal recessive disease genes is well established. We hypothesized that autozygosity not only unmasks the recessiveness of disease causing variants, but can also reveal natural knockouts of genes with less obvious phenotypic consequences. To test this hypothesis, we exome sequenced 77 well phenotyped individuals born to first cousin parents in search of genes that are biallelically inactivated. Using a very conservative estimate, we show that each of these individuals carries biallelic inactivation of 22.8 genes on average. For many of the 169 genes that appear to be biallelically inactivated, available data support involvement in modulating metabolism, immunity, perception, external appearance and other phenotypic aspects, and appear therefore to contribute to human phenotypic variation. Other genes with biallelic inactivation may contribute in yet unknown mechanisms or may be on their way to conversion into pseudogenes due to true recent dispensability. We conclude that sequencing the autozygome is an efficient way to map the contribution of genes to human phenotypic variation that goes beyond the classical definition of disease.
Introduction
Autozygosity, or the biparental inheritance of the identical founder haplotypes, is a genomic signature of a limited reproductive pool as in the case of populations with a high rate of consanguinity or a significant founder effect; although it is also observed but at a much smaller scale in outbred populations [1]. Historically, autozygosity has been used as a mapping tool in the search for autosomal recessive disease genes variants because it unmasks the recessiveness of mutations in these genes. It facilitates variant identification because the entire set of autozygous intervals (autozygome) can be mapped as homozygous blocks upon genome wide genotyping using polymorphic markers [2], [3]. More recently, autozygome analysis became a useful clinical diagnostic tool. Others have even shown that the occurrence of heterozygous markers within the autozygome can point to de novo events thus facilitating the calculation of the frequency of such events in the human genome [4], [5].
Although the role of autozygosity in unmasking disease-causing recessive alleles is well established, little attention has been paid to its potential in unmasking recessive alleles that do not result in human disease. In a proof of concept paper, we have shown that studying copy number alterations within the autozygome can uncover entire segments of DNA that are nullizygous i.e. completely absent in apparently healthy individuals [6]. Many of these segments span genes, which raises interesting questions about the extent to which humans tolerate the complete loss of function of some protein-coding genes [6]. While systematic mapping of “non-essential” genes has been possible in lower organisms [7], a different approach is needed in humans by searching for naturally occurring inactivating mutations. This has only been possible recently, thanks to the advent of next generation sequencing that allows the unbiased examination of all genes in an individual. Indeed, a recent study by the 1000 Genomes Consortium has provided valuable insight into the occurrence of loss of function (LoF) variants in “healthy” humans [8]. However, most of the LoF variants reported in that study were in the heterozygous state so those individuals who harbor them do not represent a true “knockout” of the genes involved. On the other hand, LoF variants within the autozygome may fully inactivate the involved genes due to their biallelic presence in a homozygous state. Thus, next generation sequencing of individuals born to first cousin parents (in order to maximize the size of sequenced autozygome) is expected to enrich for the occurrence of homozygous LoF variants, which can then be studied in the context of phenotypic consequences.
The systematic identification of genes with biallelic LoF variants will provide an important resource that is likely to inform various lines of investigation into the human genome. First, the fundamental question of “what constitutes the bare minimum genetic material to sustain life” which is now being asked at the level of single cells in the context of synthetic biology is only going to grow more relevant to more complex forms of life with time [9]. Cataloging genes with a presumed complete knockout and yet are compatible with live birth and at least early survival of human subjects will help address this question. Second, with the race to crack the code of Mendelian genes at its highest pace, it is extremely helpful for researchers to have a catalog of genes that can accumulate biallelic loss of function without causing a discernible Mendelian phenotype. Many labs are doing this individually by putting less emphasis on genes they frequently observe to accumulate inactivating mutations. However, the ability to identify such low priority genes relies heavily on the frequency of such inactivating mutations and genes that only rarely accumulate inactivating mutations will require a prohibitively large number of individuals to be screened. Even then, finding the rare individual carrier of one such allele is not very helpful because his/her presumed benign haploinsufficiency does not rule out an adverse outcome caused by a biallelic loss of function of that same gene. On the other hand, even very rare LoF variants can effectively become homozygous through autozygosity, thus making autozygome sequencing an appealing shortcut to identify such rare homozygous occurrences without resorting to sequencing a very large number of individuals. Third, with so much debate about the role of rare variants in shaping the risk for common diseases, we believe that the identification of biallelic loss of function mutations in some genes can accelerate their identification as risk loci, as we have shown recently for systemic lupus erythematosus and inflammatory bowel disease [10], [11].
In an attempt to test the effectiveness of autozygome sequencing in addressing the above questions, we conducted exome sequencing of 77 well phenotyped individuals all born to first cousin parents. This approach provided the highest yield of LoF variants per individuals tested, and revealed a list of biallelically inactivated genes comparable to the list published by the 1000 Genomes Consortium with less than half the number of individuals studied by the consortium (77 vs. 185) and with a very strict definition for loss of function. Further, we show that many such genes may participate in modulating important phenotypic aspects and are therefore important candidates in human phenotypic variation.
Results
Identification of homozygous LoF variants
In total, 77 individuals were enrolled in this study. As expected from the consanguineous nature of the parents, there was preponderance of autozygosity in these individuals (on average 7.7% per genome, ranging from <1% to 21.6%, based on ROHs 2MB or longer). Since this is the largest collection of exome data on Arabs, and given the intense interest in learning about Arab-specific SNPs, particularly in the homozygous state, to inform future disease mapping projects involving Arab patients, we list all autosomal SNPs and indels detected in our cohort in Supp. Table S1.
The focus of this study, however, is on genes that appear to be fully inactivated as a result of harboring biallelic LoF variants. The NGS reported over 6.2 million SNPs and indels across the 77 exomes. Of these we identified 214,774 alleles that were homozygous variants in autosomal chromosomes. However, only 678 candidate alleles fit our criteria for a complete LoF allele: either a frameshifting insertion or deletion or a stop-gain SNP. We specifically chose not to consider other highly disruptive genetic mutations so we could focus solely on what can be considered a full knockout of the gene. Upon closer manual inspection of the candidate alleles (see methods), many were found to potentially be artifacts and were excluded: some arose from the NGS pipeline but many also appeared to be sequence errors or rare variants in the reference genome itself (listed in Supp. Table S7). We also excluded alleles where the candidate LoF variant interacted with other mutations such that the overall effect was more local than expected (for example: two complementary frameshifting indels in nearby positions, or a stop-gain SNP together with another SNP in the same codon). We then performed over a thousand Sanger sequencing reactions and manually examined sequencing trace files to confirm the variants reported by NGS (see methods). Combining the output of several computational and experimental highly conservative filters, we report 175 highly reliable LoF variants found in 169 distinct genes in 77 individuals, at an average of 22.8 knocked out genes per genome. Most genes had a single LoF variant but some had two (Supp. Table S2).
In order to rank the potential for the reported allele to actually fully ablate the expected gene product, we manually assigned each LoF variant a simple integer score from 1 (least likely) to 3 (most likely). The score is essentially based on the location of the allele within the gene's open reading frame (ORF) and whether or not the allele was in an alternatively spliced exon (see methods). More than 53% of our reported variants have a score of 3 and only about 7% fall in the group least likely to fully knockout the gene function (due to the presence of unaffected alternative transcripts and that the mutation only affects the last 10% of the ORF for the affected transcript). Clearly, the biological impact of an LoF variant is also strongly affected by the cellular state and environment. However, those are elements which are well beyond the scope of this work.
Distribution of homozygous LoF variants
As expected, most of the homozygous LoF variants were low in frequency (median: 3.9%) and were significantly lower in frequency than nonsynonymous (median: 6.5%, p<0.0001; Mann-Whitney non-parametric test) and synonymous (median: 9.1%, p<0.0001; MW test) variants observed in the same gene set (Figure 1). Because homozygous LoF variants are expected to be found within a block of homozygous sequence (a run of homozygosity; ROH) we examined the relationship between the enrichment for homozygous LoF alleles and ROH length; especially longer ROH since these are more likely to be autozygous (identical-by-descent; IBD) compared to the shorter ROH (possibly arising from homozygosity for long, but common haplotypes, less likely to contain LoF mutations, or through identity-by-state; IBS). Starting from ROHs 0.7 megabases (Mb) or longer we found homozygous LoF allele enrichment to be 100% higher than random expectation (that is, if LoF alleles were distributed evenly randomly across the individuals' genomes without regard to ROHs), reaching a peak of >110% for ROHs 1 Mb or longer and leveling at about 60% above random expectation for ROH lengths of 7 Mb or longer (Figure 2). Specifically, when restricting ROHs to a minimum length cutoff of 2 Mb we find that 14.4% of all our confirmed homozygous LoF alleles reside within an ROH block that is likely autozygous. Considering that these ROHs span only about 7.4% of the entire genome size (averaged over all individuals), the enrichment above random expectation is thus 95% (Figure 2; see supp. methods for details).


Because autozygosity can render homozygous variants that arose as recent as two generations ago, we hypothesized that LoF variants within the autozygome are more likely to be private or at least of lower frequency compared to those outside the autozygome and that was indeed confirmed (median freq.: 11.7% vs. 60.4% respectively, p<0.0005; MW test. Figure 3).

Given the detailed phenotyping of the individuals in our cohort, we expected to see no additional homozygous LoF variants in genes known to cause Mendelian disorders, other than those that we had identified as causative for each individual. However, seven genes were found to harbor an LoF variant and were also listed in OMIM as causative of autosomal recessive diseases. Yet, the diseases were absent from the individuals carrying the defective genes (Supp. Table S3). Upon closer examination we found that for the two most frequent alleles (in VPS13B and SETBP1), the LoF was in fact on an alternatively spliced exon and near the end of the protein, only affecting its C-terminus and so the effect might not be severe or equal in all cells. Interestingly, these alleles were also reported on the Exome Variant Server (EVS) at relatively high frequencies and the LoF allele actually recovers the ancestral allele. For the two rarest alleles (occurring only in one patient each; one in NLRP12 and one in TRPM1) we found the LoF to either occur on an alternatively spliced exon or near the end of the amino acid chain. Both alleles were also found on the EVS at very low frequencies. Interestingly, the CLDN16 LoF variant, affecting 6 patients did not seem phenotypic even though it was a single base deletion in the ORF that appeared to affect all transcripts of the gene (assigned a score of 3). We found this LoF to be also reported in the 1000 Genomes project (rs56086318; no frequency information) and on the EVS (relatively low frequencies). It is somewhat close to the start of the protein (codon 55 of 305) at a position that is not highly conserved. In the rat and the wild boar, the orthologous open reading frame starts at a nearby alternative Methionine downstream of the variant, and so this mutation would be in the 5′UTR in these organisms. Additionally, all pathogenic mutations reported in OMIM for this gene were much further downstream in the sequence. We therefore suspect that in this case the protein remains functional even with the presence of what strongly appears to be an inactivating biallelic LoF variant. There are, however, other possibilities, such as a compensating pathway or environmental interaction, which require further focused experiments to resolve. Lastly, in two more genes (ACY1 and UPB1) an LoF highly expected to be effective (assigned a score 3) was detected and confirmed in only one individual each and was never reported in a homozygous state in the EVS. It turns out however that even though the genes were reported in OMIM as causative for certain phenotypes, their pathogenicity was in fact contested in the literature, with reports of many cases where the genes where fully knocked out in patients that lack the described neurological phenotypes [12], [13]. These two cases provide a good illustration of the complexity of the genotype-phenotype link and the imperfect nature of some causality reports.
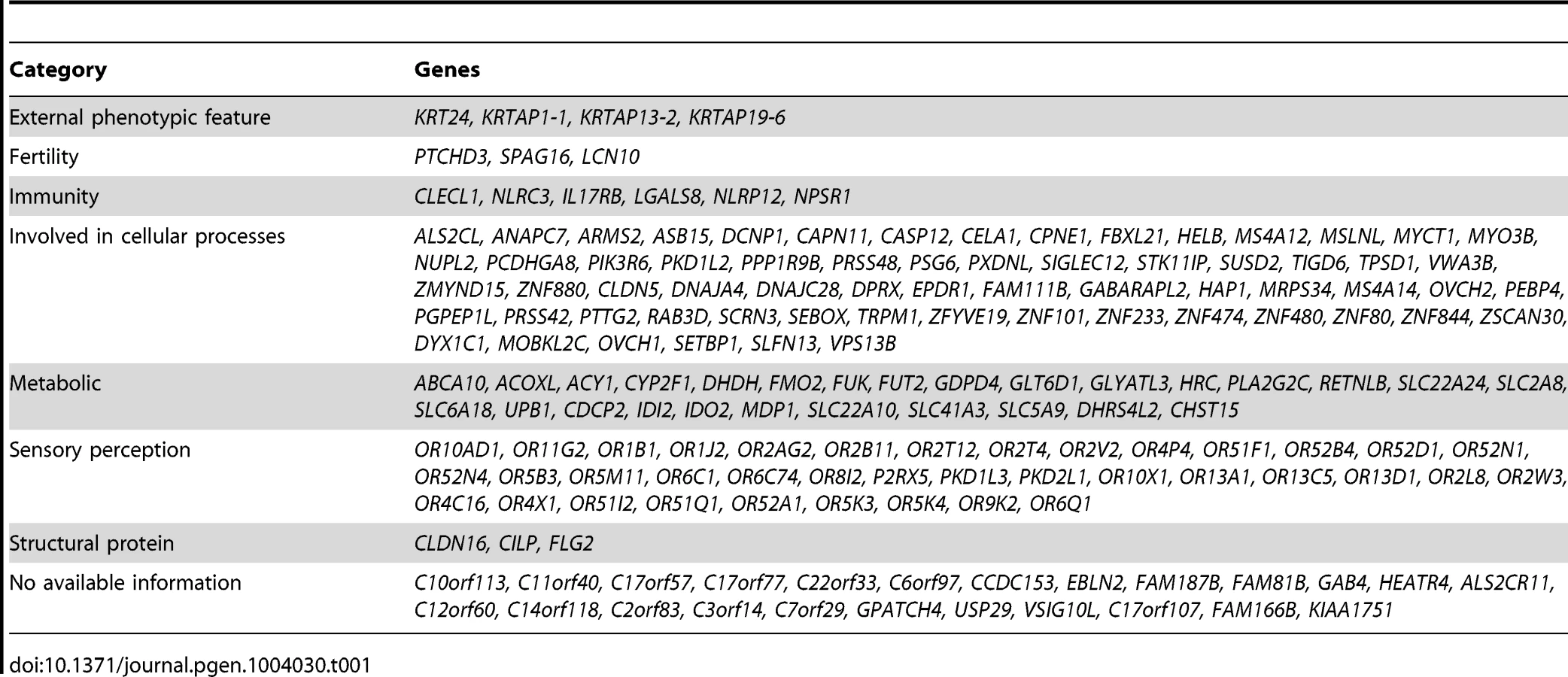
We examined each of the 169 genes in our list for potential phenotypic consequences as a result of complete inactivation. Interestingly, many of these genes (>85%) are indeed predicted to have consequence when inactivated and the resulting phenotypes comprised hematological, immunological, metabolic and external phenotypic variation, among other categories (Table 1).
Estimation of carrier capacity for disease associated genes
In addition to the set of fully inactivating biallelic LoF variants, we also examined heterozygous LoF alleles in autosomal recessive disease-associated genes (ARDGs). By combining our NGS data to OMIM annotation, we identified 327 ARDGs with at least one heterozygous LoF variant in at least one of our patients. However, after attempting to validate >70% of the NGS reported alleles by Sanger sequencing, we could confirm LoF variants in only 43 genes (Supp. Table S6). Most of the unconfirmed alleles were of low frequency and likely to be NGS or genome artifacts. Based on the confirmed variants only, we find that on average, each genome is a carrier for 1.9±0.2(s.e.) ARDGs (median is 1.0). In comparison, extracting comparable data from the published 1000 Genomes Consortium [8] data yields an average of 18.0 ARDGs per genome, but only 160 ARDGs were identified in total. Notably, if the 1000 Genomes dataset is restricted to the high confidence LoF variants only, the figures drop sharply to 34 genes averaging 1.3 ARDGs per genome, with only 3 genes in common with our list.
Discussion
Historically, the study of the relationship between genes and phenotypes has been largely driven by the quest to identify genes that cause diseases (which can be considered as extreme phenotypes) while the role genes play in modulating more subtle phenotypes was unknown, mostly due to technical limitations. The advent of high density SNP arrays in the first decade following the completion of the Human Genome Project made it possible for the first time to interrogate human phenotypes like height, ear wax type, etc. in a hypothesis-free genome wide fashion (Genome Wide Association Studies or GWAS). However, GWAS essentially miss the contribution of rare variants even though some of these are likely to have a large effect size [14]. Indeed, we show in this study that LoF variants tend to be low in frequency and many can be considered private. Therefore, sequencing-based strategies are now widely viewed as the way forward to more comprehensively study the relationship between genes and phenotypes [14].
A recent study by the 1000 Genomes Consortium showed that LoF variants are more common than previously thought, although in the majority of cases their frequency is still too low for inclusion in the commonly used SNP platforms [8]. They reported that individuals of Northern and Western European ancestry had (counting only stop-gain and frameshift variants) 14.4 homozygous LoF variants per individual, Chinese and Japanese individuals had 15.9, and Yoruban individuals from Nigeria had 14.3. Their list of 221 genes that harbor 233 biallelic LoF variants (all LoF mutations included) was based on sequencing 185 individuals. On the other hand, we show that by sequencing just 77 individuals enriched for autozygosity we are able to generate a comparable list of 169 genes, without including splice-site mutations which we opted to exclude since their validation would require RT-PCR assays in a variety of cell/tissue types. Another important advantage of our study is that all variants reported were verified by direct Sanger sequencing in at least three individuals (or all reported individuals if below 4). Therefore, we believe our list represents a conservative lower estimate as we have clearly missed many additional LoF variants by design and by conservative filtering.
Consistent with the low frequency of many of these LoF, we find that less than one third of our list overlaps with that published by the 1000 Genomes Consortium. This suggests that many more individuals will have to be sequenced to generate a more comprehensive list of genes that are “non-essential” and yet likely to contribute to other phenotypic aspects. We plan to expand the number of sequenced individuals in the future. Further, we suggest that sequencing of the autozygome is an efficient way to achieve this goal, as it allows a major reduction in the size of the sequencing target without a significant reduction in the expected yield of homozygous LoF. More importantly, the rarer LoF variants are more likely to be found within the autozygome than outside of it.
Closely examining the list of genes reported, we find that several interesting phenotypes can be linked to the genes that we showed to harbor homozygous LoF variants and some examples are highlighted below.
FUT2 encodes a fucosyltransferase that participates in the H antigen synthesis but with different tissue specificity. LoF alleles in FUT2 have been observed (at the same locus as in our report and at another), with a non-secretor phenotype that was found to be linked to lower plasma vitamin B(12) level [15]. Interestingly, the subtlety of the phenotype associated with inactivation of this gene (confirmed hematologically in these cases) is a likely explanation for how these LoF alleles have reached a relatively high frequency (23.4% for FUT2).
Twenty six of the genes in our list with an established function code for proteins involved in metabolic pathways (the above examples on H antigen synthesis and modification can also be viewed as such). FUK codes for the enzyme fucokinase (FUK), which mediates the salvage of L-fucose molecules produced from the degradation of cellular glycoproteins and glycolipids as well as dietary L-fucose, and this pathway is considered an alternative pathway for the production of GDP-Fucose [16]. Lack of an apparent phenotype in individuals with LoF variants in this gene, therefore, is expected due to a likely redundancy between the two pathways and may be limited to an intermittent presence of reducing substance in urine.
Similarly, ACY1 codes for the enzyme aminoacylase 1, which is involved in the salvage pathway of acylated L-amino acids, producing L-amino acids and an acyl group. Individuals with biallelic LoF mutations in ACY1 have been reported with high levels of acetylated amino acids in urine, and while some of those patients demonstrated variable neurological symptoms, others had normal development [13]. This enzyme appears to be less important in states of good supply of amino acids and may serve as an example of context-specific dispensability, in this case nutritional state. A closely related examples is the enzyme dihydrodiol dehydrogenase DHDH, encoded by DHDH, which is involved in the intoxication of naphthalene and benzene and oxidation of certain pentose and hexose monosaccharides [17]. In humans, DHDH is expressed in the intestine suggesting a role in dietary metabolism of dihydrodiol of aromatic hydrocarbons and free radical formation from such molecules [18]. We identified four individuals with a homozygous LoF variant in DHDH with apparently benign consequences, and several other individuals were also reported by the 1000 Genomes Consortium. It is possible that this deficiency is only consequential upon the ingestion of dietary substrates of DHDH.
An interesting category of genes with homozygous LoF are those involved in perception. The best example is genes involved in olfaction since we show that 35 olfactory receptor genes harbor homozygous LoF variants. Another example is LoF alleles in genes regulating taste perception, namely PKD1L3 and PKD2L1, also reported by the 1000 Genomes Consortium, that code for a transient receptor potential ion channel shown to function as sour taste receptor [19]. Interestingly, very recent data suggest that these genes may be under selection pressure in certain areas with special dietary preferences [20]. Additional homozygous LoF alleles were found in P2RX5 (reported by the 1000 Genomes paper) that codes for the purinergic receptor P2x5 that was observed to have a potentiating effect on membrane current produced by the acid-activated sodium channel Asic3, which in turn mediates sensation of ischemic pain in exercising muscle [21]. It will be interesting to test individuals with biallelic inactivating mutation in P2RX5 for variation in pain threshold in response to intense exercise.
Another interesting phenotypic aspect is that of external appearance and this category involves homozygous LoF variants in genes coding for keratins and keratin associated proteins KRT24, KRTAP1-1, KRTAP13-2 (this variant was reported by the 1000 Genomes paper) and KRTAP19-6. These proteins are critical structural proteins of hair and nail, and variations in these genes are of great economic importance in the sheep wool industry [22]–[24]. More recently, variations in these genes in humans have also been studied in the context of hair texture [25], [26]. Thus, it is possible that the inactivation of these genes will have an effect on hair texture but the number of individuals we have with such mutations is too small to allow for statistical analysis.
Our study also provides an opportunity to study the effect of complete inactivation in genes that have been proposed to modulate the risk of multifactorial disorders. For instance, we have 28 individuals with homozygous LoF in CLECL1, which encodes a novel C-type lectin–like molecule expressed by antigen presenting cells and has been hypothesized to modulate risk of autoimmune diseases. Interestingly, follow up assessment revealed that three of these 28 individuals have elevated antinuclear antibody (ANA) titers including two who meet the clinical definition of systemic lupus erythematosus. Similarly, NLRC3 codes for a member of a family of proteins that modulate the inflammatory response by negatively regulating vital inflammatory proteins, it has been shown that Nlrc3 attenuates the activation of macrophages brought upon by toll-like receptor stimulation. Surprisingly, Nlrc3-null mice did not show any marked difference from wild type mice across a wide range of parameters, but when challenged with lipopolysaccharide (LPS) they exhibited an exaggerated response characterized by higher body temperature and delayed recovery of lost weight compared to wild type controls [27]. By extending this observation to the individual reported in this study with bi-allelic LoF in NLRC3, it is possible that the phenotype is context-dependent e.g. exposure to LPS. Furthermore, the gene CILP codes for a protein involved in cartilage structure, that is expressed abundantly in intervertebral discs and a SNP in this gene was shown to be a modulator of susceptibility to lumbar disc disease [28]. The individuals with homozygous LoF variants in this gene may be considered at high risk for this very common multifactorial phenotype and longitudinal follow up data on them will be informative. We also identified homozygous LoF variants in Resistin like protein beta (RETNLB) which is known to promote liver insulin resistance and increase glucose production in spite of the presence of physiologic insulin levels [29]. Although in our study the young individuals with presumed deficiency in this protein do not have evidence of impaired glucose homeostasis, longitudinal follow up data will be needed to better understand the role such genes may have, if any, in diabetes risk.
In addition to the above examples, we provide a comprehensive list of functions and proposed phenotypic consequence of the remaining genes in Supp. Table S4. Those genes for which we could not provide a hypotheses about the effect of inactivation may indeed be involved in modulating important yet unrecognized aspects of the phenotype and we hope that future discovery of additional LoF variants in these genes will make such proposed effects more tractable.
This work is strongly connected to several important yet unanswered questions. Knowledge about the “minimally required” genes to support early human development is only likely to be more important as the question of the minimum requirement for life in a single cell that has been pursued recently in a synthetic biology approach becomes more relevant to more complex organisms. There is also need to identify genes whose biallelic LoF does not appear to cause a discernible phenotype in order to inform projects that focus on the mapping of Mendelian genes. Obviously, these genes will not be a priority when investigators filter through sequencing data to identify their most likely candidate disease-causing mutation. Finally, we believe the mapping of genes that accumulate biallelic LoF variants addresses an important aspect about human evolution by highlighting genes that are likely to be on their way to true dispensability and converting into pseudogenes. It is unclear if the allele frequency is the best indicator of the latter possibility or if it is the “burden” of LoF for a given gene; because if a gene is truly dispensable it is predicted to freely accumulate various LoF alleles. Since most of the genes in our list are homozygous for just one LoF allele, it will be very interesting to follow up on our list in different populations to investigate whether some may indeed be accumulating various LoF alleles that would suggest true dispensability, particularly if the frequency of such alleles is high.
In summary, we show that focusing sequencing efforts on the autozygome is an efficient way to catalog human LoF variants in the homozygous states in contexts that go beyond the traditional definition of health and disease. The extent to which complete inactivation of genes contributes to human phenotypic variability has just begun to be appreciated and we hope this study will stimulate continued interest in this very important research field.
Materials and Methods
Human subjects
This study was conducted on a subset of our patients with autosomal recessive disorders whose underlying causal mutation we resolved via exome sequencing. These patients typically have had a thorough medical and family history, physical examination and a number of tests that include basic hematological and metabolic parameters. All patients were recruited in Riyadh, Saudi Arabia (but came from nearly all regions of the country), were of Arabic heritage and were born to first cousin parents. A written informed consent was obtained from all participants in a protocol approved by the IRB at King Faisal Specialist Hospital and Research Center.
Exome sequencing
Exome capture was performed using TruSeq Exome Enrichment kit (Illumina) following the manufacturer's protocol. Blood samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using the Illumina HiSeq 2000 Sequencer. Sequence quality, depth and target alignment results are summarized and listed in Supp. Table S5.
Variant validation
In order to confirm that none of the candidate homozygous LoF variants that are used for subsequent analysis was an NGS artifact, PCR amplification followed by Sanger sequencing was attempted for each homozygous LoF allele in a sample of the individuals where the allele was detected as follows. For alleles detected in up to three individuals, all instances were Sanger sequenced. For alleles detected at a higher frequency, a random sample of three or more individuals was selected. In some cases, we sequenced the allele amplicon in all 77 individuals. Amplicons targeting a region of about 400 bases around each candidate allele were manually selected followed by primer design using Primer3. For some targets it was not possible to design suitable primers and so the alleles were disqualified and excluded from any further analysis. Genomic DNA was extracted from blood samples and PCR amplified and the resulting amplicons were then Sanger sequenced; all following standard protocols. Each sequence trace file was examined manually to confirm concordance with the NGS exome results. In about 45% of the tested alleles, the trace files in at least two individuals (or one if the variant was reported only once by NGS) showed either a heterozygous locus or signal that could not reliably support the NGS report. These alleles were thus deemed unreliable and also excluded from any further analysis. For the remaining 55%, trace files strongly supported the NGS reports in >98% of all tested individuals. In the few cases where the allele could not be reliably confirmed in only one individual but confirmed in two or more other individuals the NGS report was considered reliable and reported.
Bioinformatic analysis
The overall flow of the analysis is depicted in Figure S1 and briefly described here. Full details are provided in Text S1. For efficient management, all data were loaded onto an MS Access (http://www.microsoft.com) relational database and manipulated via SQL queries. Some plots and supplementary tables were produced using MS Excel (http://www.microsoft.com) spreadsheet software and statistical analysis was performed using Prism5 (http://www.graphpad.com) statistics software.
After exome sequencing the 77 samples with NGS, the sequences were aligned to the NCBI reference human genome (GRCh37/hg19) using BWA (http://bio-bwa.sourceforge.net/). The variants (small insertions/deletions and SNPs) were then called using SAMTOOLS (http://samtools.sourceforge.net/) by comparison against the reference genome. Functional prediction of the impact of each variant was produced using ANNOVAR (http://www.openbioinformatics.org/annovar/) by comparison against RefSeq mRNA sequences. The tool also linked variants to dbSNP reports if known. We then filtered the resulting variants for autosomal, homozygous LoF alleles, where an LoF was specifically defined as a stop-gain SNP or a frameshifting (non-mod 3) indel in a protein coding gene. The full set of resulting variants was aggregated by position and mutation type. This set of candidate LoF alleles was then manually examined on the UCSC genome browser (http://genome.ucsc.edu) to verify the accuracy of each allele in a broader genomic and transcriptomic context. The examination mainly utilized the UCSC genes, conservation and dbSNP tracks, as well as a various other tracks as needed. The examination aimed to verify the SAMTOOLS annotation, detect artifacts, put the allele within the alternative transcription perspective and identify potential rescue mechanisms that might prevent the candidate variant from becoming a LoF allele. A separate contextual variations report was generated to allow examination of each variant against all other variants on the same gene in the same individual. This was used to detect variant interactions that might also prevent the candidate allele from being an effective LoF variant.
After filtering out artifacts, counteracting interactions and suspected highly polymorphic loci, the resulting set of alleles were examined on the genome browser to select a suitable target region. PCR primers were designed using Primer3 (http://primer3.wi.mit.edu/) web tool. The results of variant validation by Sanger sequencing were entered into the database to produce a subset of now validated and verified variants; our confirmed variants list (CVL).
The CVL was cross linked to EVS (http://evs.gs.washington.edu/EVS/; ESP6500SI data), OMIM (http://www.omim.org/) and HGNC (http://www.genenames.org/) to provide additional information on known genotype frequencies, disease association and gene families, respectively. HGNC was also utilized to resolve naming ambiguities resulting from the use of multiple resources for annotation.
Prior to NGS, each sample was also genotyped on the Axiom Genome-Wide Population-Optimized Human SNP chips (CEU version, about 600K total SNPs). That data was collected and formatted for input into PLINK (v1.07; http://pngu.mgh.harvard.edu/purcell/plink/) to detect ROH blocks. After filtering the probes for quality and stability, we called as ROH all blocks of 100,000 bases or longer and entered the results into the database for crosslinking against the CVL.
Heterozygous variants were filtered and validated following essentially the same methods but were not subjected to a thorough manual contextual examination. The initial candidates list was additionally filtered based on a minimum NGS call quality score to reduce false positives. The final validated heterozygous variants list is restricted to genes annotated in OMIM with an autosomal recessive disease connection only, and does not represent the full list of all heterozygous LoF variants detected.
Further details for these methods and other subsections of this work are available in the Text S1.
Supporting Information
Zdroje
1. McQuillanR, LeuteneggerAL, Abdel-RahmanR, FranklinCS, PericicM, et al. (2008) Runs of homozygosity in European populations. Am J Hum Genet 83 : 359–372.
2. LanderES, BotsteinD (1987) Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236 : 1567–1570.
3. AlkurayaFS (2010) Autozygome decoded. Genet Med 12 : 765–771.
4. CampbellCD, ChongJX, MaligM, KoA, DumontBL, et al. (2012) Estimating the human mutation rate using autozygosity in a founder population. Nat Genet 44 : 1277–1281.
5. AlkurayaFS (2010) Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genet Med 12 : 236–239.
6. KhalakHG, WakilSM, ImtiazF, RamzanK, BazB, et al. (2012) Autozygome maps dispensable DNA and reveals potential selective bias against nullizygosity. Genet Med 14 : 515–519.
7. GlassJI, Assad-GarciaN, AlperovichN, YoosephS, LewisMR, et al. (2006) Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A 103 : 425–430.
8. MacArthurDG, BalasubramanianS, FrankishA, HuangN, MorrisJ, et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science 335 : 823–828.
9. GibsonDG, GlassJI, LartigueC, NoskovVN, ChuangRY, et al. (2010) Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329 : 52–56.
10. Al-MayoufSM, SunkerA, AbdwaniR, AbrawiSA, AlmurshediF, et al. (2011) Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 43 : 1186–1188.
11. AlangariA, AlsultanA, AdlyN, MassaadMJ, KianiIS, et al. (2012) LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol 130 : 481–488 e482.
12. Yaplito-LeeJ, PittJ, MeijerJ, ZoetekouwL, MeinsmaR, et al. (2008) Beta-ureidopropionase deficiency presenting with congenital anomalies of the urogenital and colorectal systems. Mol Genet Metab 93 : 190–194.
13. SassJO, OlbrichH, MohrV, HartC, WoldsethB, et al. (2007) Neurological findings in aminoacylase 1 deficiency. Neurology 68 : 2151–2153.
14. EichlerEE, FlintJ, GibsonG, KongA, LealSM, et al. (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11 : 446–450.
15. HazraA, KraftP, SelhubJ, GiovannucciEL, ThomasG, et al. (2008) Common variants of FUT2 are associated with plasma vitamin B12 levels. Nat Genet 40 : 1160–1162.
16. ParkSH, PastuszakI, DrakeR, ElbeinAD (1998) Purification to apparent homogeneity and properties of pig kidney L-fucose kinase. J Biol Chem 273 : 5685–5691.
17. CarboneV, HaraA, El-KabbaniO (2008) Structural and functional features of dimeric dihydrodiol dehydrogenase. Cell Mol Life Sci 65 : 1464–1474.
18. ZepedaS, MonasterioO, UretaT (1990) NADP(+)-dependent D-xylose dehydrogenase from pig liver. Purification and properties. Biochem J 266 : 637–644.
19. IshimaruY, InadaH, KubotaM, ZhuangH, TominagaM, et al. (2006) Transient receptor potential family members PKD1L3 and PKD2L1 form a candidate sour taste receptor. Proc Natl Acad Sci U S A 103 : 12569–12574.
20. PatzkeS, RedickS, WarsameA, Murga-ZamalloaCA, KhannaH, et al. (2010) CSPP is a ciliary protein interacting with Nephrocystin 8 and required for cilia formation. Molecular biology of the cell 21 : 2555–2567.
21. BirdsongWT, FierroL, WilliamsFG, SpeltaV, NavesLA, et al. (2010) Sensing muscle ischemia: coincident detection of acid and ATP via interplay of two ion channels. Neuron 68 : 739–749.
22. RogersGR, HickfordJG, BickerstaffeR (1994) Polymorphism in two genes for B2 high sulfur proteins of wool. Anim Genet 25 : 407–415.
23. GongH, ZhouH, HickfordJG (2010) Polymorphism of the ovine keratin-associated protein 1-4 gene (KRTAP1-4). Mol Biol Rep 37 : 3377–3380.
24. GongH, ZhouH, HickfordJG (2011) Diversity of the glycine/tyrosine-rich keratin-associated protein 6 gene (KAP6) family in sheep. Mol Biol Rep 38 : 31–35.
25. ShimomuraY, AokiN, SchweizerJ, LangbeinL, RogersMA, et al. (2002) Polymorphisms in the human high sulfur hair keratin-associated protein 1, KAP1, gene family. J Biol Chem 277 : 45493–45501.
26. KariyaN, ShimomuraY, ItoM (2005) Size polymorphisms in the human ultrahigh sulfur hair keratin-associated protein 4, KAP4, gene family. J Invest Dermatol 124 : 1111–1118.
27. SchneiderM, ZimmermannAG, RobertsRA, ZhangL, SwansonKV, et al. (2012) The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol 13 : 823–831.
28. SekiS, KawaguchiY, ChibaK, MikamiY, KizawaH, et al. (2005) A functional SNP in CILP, encoding cartilage intermediate layer protein, is associated with susceptibility to lumbar disc disease. Nat Genet 37 : 607–612.
29. RajalaMW, ObiciS, SchererPE, RossettiL (2003) Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest 111 : 225–230.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy