
Dual Regulation of Gene Expression Mediated by Extended MAPK Activation and Salicylic Acid Contributes to Robust Innate Immunity in
Network robustness is a crucial property of the plant immune signaling network because pathogens are under a strong selection pressure to perturb plant network components to dampen plant immune responses. Nevertheless, modulation of network robustness is an area of network biology that has rarely been explored. While two modes of plant immunity, Effector-Triggered Immunity (ETI) and Pattern-Triggered Immunity (PTI), extensively share signaling machinery, the network output is much more robust against perturbations during ETI than PTI, suggesting modulation of network robustness. Here, we report a molecular mechanism underlying the modulation of the network robustness in Arabidopsis thaliana. The salicylic acid (SA) signaling sector regulates a major portion of the plant immune response and is important in immunity against biotrophic and hemibiotrophic pathogens. In Arabidopsis, SA signaling was required for the proper regulation of the vast majority of SA-responsive genes during PTI. However, during ETI, regulation of most SA-responsive genes, including the canonical SA marker gene PR1, could be controlled by SA-independent mechanisms as well as by SA. The activation of the two immune-related MAPKs, MPK3 and MPK6, persisted for several hours during ETI but less than one hour during PTI. Sustained MAPK activation was sufficient to confer SA-independent regulation of most SA-responsive genes. Furthermore, the MPK3 and SA signaling sectors were compensatory to each other for inhibition of bacterial growth as well as for PR1 expression during ETI. These results indicate that the duration of the MAPK activation is a critical determinant for modulation of robustness of the immune signaling network. Our findings with the plant immune signaling network imply that the robustness level of a biological network can be modulated by the activities of network components.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1004015
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004015
Summary
Network robustness is a crucial property of the plant immune signaling network because pathogens are under a strong selection pressure to perturb plant network components to dampen plant immune responses. Nevertheless, modulation of network robustness is an area of network biology that has rarely been explored. While two modes of plant immunity, Effector-Triggered Immunity (ETI) and Pattern-Triggered Immunity (PTI), extensively share signaling machinery, the network output is much more robust against perturbations during ETI than PTI, suggesting modulation of network robustness. Here, we report a molecular mechanism underlying the modulation of the network robustness in Arabidopsis thaliana. The salicylic acid (SA) signaling sector regulates a major portion of the plant immune response and is important in immunity against biotrophic and hemibiotrophic pathogens. In Arabidopsis, SA signaling was required for the proper regulation of the vast majority of SA-responsive genes during PTI. However, during ETI, regulation of most SA-responsive genes, including the canonical SA marker gene PR1, could be controlled by SA-independent mechanisms as well as by SA. The activation of the two immune-related MAPKs, MPK3 and MPK6, persisted for several hours during ETI but less than one hour during PTI. Sustained MAPK activation was sufficient to confer SA-independent regulation of most SA-responsive genes. Furthermore, the MPK3 and SA signaling sectors were compensatory to each other for inhibition of bacterial growth as well as for PR1 expression during ETI. These results indicate that the duration of the MAPK activation is a critical determinant for modulation of robustness of the immune signaling network. Our findings with the plant immune signaling network imply that the robustness level of a biological network can be modulated by the activities of network components.
Introduction
How network properties, such as robustness against network perturbations, emerge from biological networks has been a central question in systems biology [1], [2]. Possible modulation of network robustness in a biologically relevant context and mechanisms underlying the modulation are areas of study that have rarely been explored.
Innate immunity, in which defense responses are induced through signaling events initiated by recognition of pathogen attack, composes a major part of plant immunity [3]. PAMP/Pattern-Triggered Immunity (PTI) and Effector-Triggered Immunity (ETI) are modes of plant innate immunity defined by the way pathogens are detected [4], [5]. PTI is triggered by recognition of microbe/pathogen-associated molecular patterns (MAMPs/PAMPs) by the cognate pattern-recognition receptors (PRRs), which are typically receptor-like kinases or receptor-like proteins [6]. For example, Arabidopsis thaliana FLS2 is the PRR for flg22, an elicitor-active epitope of flagellin from Gram-negative bacteria [7]. While most non-adapted pathogens cannot overcome PTI, adapted pathogens deliver effectors into the plant cell that manipulate plant cell functions to facilitate their infection by, for instance, interfering with PTI signaling [8], [9]. ETI is triggered by specific recognition of effectors by resistance (R) proteins, which are often nucleotide-binding leucine-rich repeat (NB-LRR) proteins [10]. For example, the Arabidopsis intracellular NB-LRR R proteins RPS2 and RPM1 indirectly recognize perturbations of the PTI signaling component RIN4 by the effectors AvrRpt2 and AvrRpm1/AvrB, respectively, of a Gram-negative bacterial pathogen, Pseudomonas syringae [3]. In addition to proteinaceous effectors, some P. syringae strains deliver coronatine, which is a jasmonic isoleucine mimic, in order to suppress plant immunity [11]. Recently, it was shown that coronatine suppresses immune responses dependent on salicylic acid (SA) as well as independent of SA [12], [13]. Thus, there are evolutionary arms races between hosts and pathogens. Pathogens evolve much faster than hosts, rapidly changing effector repertoires, thereby changing points of attack in host immune networks. As hosts cannot match the speed of pathogen evolution, it is important that hosts develop robust immune networks that remain functional in the face of effector attack. Mechanisms underlying network robustness are thus a critical aspect of immunity.
SA is a signal molecule controlling a major portion of immunity against biotrophic and hemibiotrophic pathogens, including P. syringae [14]. SID2 encodes a key enzyme for SA biosynthesis in response to pathogen infection [15]. In Arabidopsis sid2 mutants, pathogen-induced SA accumulation is almost undetectable [14]. Hundreds of genes are transcriptionally regulated by SA signaling, mediated mainly by a positive regulator of SA signaling, NPR1 [14]. PR1 is one SA-inducible gene used as a canonical SA marker [14].
Arabidopsis has 20 mitogen-activated protein kinases (MAPKs) [16], and four of them, MPK3, MPK4, MPK6 and MPK11, have been described as immune signaling components [17]. MPK3 and MPK6 are associated with immune responses, such as reactive oxygen species (ROS) production, ET production/signaling, phytoalexin production and cell death [17]. For instance, ethylene production is positively controlled by dual regulation of enzymes (ACS) synthesizing the ethylene precursor 1-amino-cyclopropane-1-carboxylic acid. MPK6 stabilizes ACS2 and ACS6 by their phosphorylation, and MPK3 and MPK6 control gene expression through a transcription factor, WRKY33, which is activated by the MAPKs [18], [19]. The same cascade is required for production of a phytoalexin, camalexin, by controlling expression of a biosynthetic gene, PAD3 [20]. A double mutant deficient in MPK3 and MPK6 is embryonic lethal but the single mutants are viable, suggesting functional redundancy between them in development [21]. MPK3 phosphorylates the bZIP type transcription factor VIP1 whose phosphorylation is required for its nuclear translocation [22]. Transient over-expression of VIP1 led to weak induction of PR1 in Arabidopsis protoplasts although involvement of SA in this PR1 induction is not known [23].
The overall spectra of induced defense responses are overlapping between PTI and ETI whereas the kinetics and intensity of the responses seem different [4], [24]. In Arabidopsis, knocking out the hub genes of four major signaling sectors abolished 80% of flg22-triggered PTI (flg22-PTI) and AvrRpt2-triggered ETI (AvrRpt2-ETI), indicating extensively shared signaling network machinery between PTI and ETI [25]. Relationships among these signaling sectors are part compensatory and part synergistic in flg22-PTI but are predominantly compensatory in AvrRpt2-ETI, which explains a high level of robustness in the ETI level against network perturbations [25]. Single mutations (dde2, ein2, pad4 and sid2) weakly but significantly compromised flg22-PTI but not AvrRpt2-ETI while the quadruple mutation largely abolished both. These observations demonstrated differences in the robustness of the highly overlapping signaling networks during the two modes of plant immunity. However, the molecular mechanism controlling modulation of the network robustness is not known.
Here we report a molecular mechanism that affects the robustness of the plant immune signaling network. Although Arabidopsis MPK3 and MPK6 are activated during both PTI and ETI, the duration of the activation was much longer during ETI than PTI. Only sustained activation of the MAPKs supported expression of a majority of SA-responsive genes in the absence of SA. The roles of MPK3 and SA signaling during AvrRpt2-ETI were compensatory, contributing to network robustness against perturbations during ETI. Our findings demonstrate that a biologically important differential network property, robustness, can emerge from duration of the activity of a network component.
Results
Most SA-responsive genes were properly regulated in the absence of SA during ETI
We previously reported that ETI is more robust against network perturbations than PTI due to a higher level of network compensation [25]. We hypothesized that this compensation occurred at the level of gene regulation. To test this hypothesis, we examined expression of a canonical SA marker gene, PR1, during ETI. Transcriptional induction of PR1 was completely dependent on SID2, which is a key SA biosynthetic enzyme, and hence completely dependent on SA signaling during PTI [26]. We found that PR1 induction was only partially dependent on SID2 and NPR1 at a late time point of 24 hours post inoculation (hpi) with ETI-triggering P. syringae pv. tomato DC3000 (Pto) strains expressing the effectors AvrRpt2 (Pto AvrRpt2) or AvrRpm1 (Pto AvrRpm1) (Figure 1A and Figure S1). While AvrRpt2 and AvrRpm1 are recognized by the CC-type NB-LRR proteins RPS2 and RPM1, AvrRps4 is recognized by the TIR-type NB-LRR protein RPS4 [3]. We also observed SID2 - and NPR1-independent PR1 induction during AvrRps4-triggered ETI although induction levels were lower compared to AvrRpt2 - and AvrRpm1-ETI (Figure S1). In contrast, PR1 induction was completely dependent on SID2 in the case of the non-ETI triggering Pto strain carrying an empty vector (Pto EV). Inoculation of the ETI-triggering strains at a high dose can trigger a form of programmed cell death called a hypersensitive response (HR) [3]. The inoculation dose used in this experiment was relatively low (OD600 = 0.001), and we did not observe a macroscopic HR within 24 hpi. To test the possibility that the SA level increased independently of SID2 during ETI, we measured the SA level in these tissues. The increased SA accumulation was completely dependent on SID2 in all conditions (Figure 1B). These results indicate that some SA-independent mechanism(s) can activate PR1 during ETI. At an earlier time point of 6 hpi, only SA-dependent PR1 induction was observed with all three strains (Figure 1A), suggesting that this SA-independent mechanism(s) during ETI requires more than 6 hours to be effective.

SA-independent mechanism(s) for PR1 induction during ETI prompted us to investigate the possibility that other SA-responsive genes can also be transcriptionally regulated in an SA-independent manner during ETI. For this purpose, mRNA profiles were analyzed using a whole genome DNA microarray. Leaves of wild type (Col) or sid2 plants were inoculated with water (mock), Pto hrcC, Pto EV, or Pto AvrRpt2, and were collected at 24 hpi for mRNA profiling. The Pto hrcC strain is deficient in the type III secretion system used to transport effectors into plant cells. It elicits the PTI response by presenting various MAMPs [11]. Among 2828 genes that were significantly up - or down-regulated (with q values<0.01 and more than 2-fold changes) in both Pto EV and Pto AvrRpt2 infection in Col, regulation of 187 genes showed strong SID2-dependence in Pto EV infection (Figure 2A and Table S1). These genes are designated SA-responsive genes hereafter. Remarkably, regulation of most SA-responsive genes, including PR1, at 24 hpi with Pto AvrRpt2 is largely SID2-independent although SA contributes to their full expression, indicating that SA-independent signaling mechanism(s) can regulate most SA-responsive genes during AvrRpt2-ETI. The SID2-dependency of gene regulation after Pto hrcC inoculation was similar to that after Pto EV inoculation, although the overall extent of up - or down-regulation was lower, and distinct from that after Pto AvrRpt2 inoculation (Figure S2 and Table S2). Thus, initiation of ETI appears to be the key for activation of this SA-independent mechanism(s).

Activation of MPK3 and MPK6 was sustained in ETI but transient in non-ETI
We hypothesized that a kinetic difference in activation of network components is responsible for activation of SA-independent mechanism(s). A prior study suggested that the duration of MPK3 and MPK6 activation is longer during ETI than non-ETI [27]. We compared the duration of MAPK activation in ETI and PTI. When wild-type seedlings in a liquid medium were treated with the PTI inducer flg22, activation of the MAPKs was observed after 10 min and returned to the basal level within one hour (Figure 3A), confirming previous observations [28]. The possibility that flg22 was rapidly degraded in the liquid culture was excluded since the MAPKs were activated similarly when fresh seedlings were placed in the liquid medium containing flg22 that had been incubated with other seedlings for 3 hours (Figure 3A). Thus, MAPK activation is truly transient after flg22 treatment. We employed transgenic seedlings carrying an estradiol-inducible AvrRpt2 transgene (XVE-AvrRpt2) to measure MAPK activation during ETI in the absence of PTI. The MAPKs were activated by three hours and remained active for at least 7 hours after estradiol treatment (Figure 3B). This sustained MAPK activation was ETI-specific as no such activation was observed in the rps2 mutant background, which lacks the corresponding receptor (Figure 3C). PR1 induction during AvrRpt2-ETI was independent of SA in XVE-AvrRpt2 transgenic seedlings (Figure S3), which is consistent with the results obtained using adult leaves inoculated with a Pto strain expressing AvrRpt2 (Figure 1). Similar trends in MAPK activation duration were observed when adult leaves were inoculated with Pto strains: sustained activation of the MAPKs was observed with Pto AvrRpt2 in a manner dependent on the R gene RPS2, but not with the strains that do not trigger ETI (Figure 4). While the amounts of activated MPK3 and MPK6 were similar during AvrRpt2-ETI triggered in XVE-AvrRpt2 transgenic plants (Figure 3), there was more activated MPK3 than activated MPK6 during AvrRpt2-ETI triggered by Pto AvrRpt2 (Figures 4, S4 and S5), suggesting that MPK3 plays a major role during AvrRpt2-ETI in bacterial infection. We also observed sustained MAPK activation during AvrRps4-ETI although levels of activation were weaker compared to AvrRpt2-ETI (Figure S4). Since there are 20 MAPKs in Arabidopsis [16], we determined the identities of the activated MAPKs. Indeed, the activated MAPKs during AvrRpt2 - and AvrRps4-ETI were MPK3 and MPK6 (Figure S4). Previously, Beckers et al (2009) reported that an SA analog, benzo(1,2,3,)thiadiazole-7-carbothioic acid S-methyl ester (BTH), induced priming of MPK3 activation by inducing expression of MPK3 [29]. In contrast, sustained activation of MPK3 during AvrRpt2-ETI was independent of SA (Figure S5). The sustained activation was not due to an increased amount of MPK3 as we did not observe obvious changes in the MPK3 protein level during AvrRpt2-ETI (Figure 3). Taken together, our data show that sustained activation of the MAPKs is SA-independent and occurs during ETI but not during non-ETI responses.


Sustained activation of MPK3 and MPK6 is sufficient for PR1 induction in the absence of SA
To test if sustained activation of MPK3 and MPK6 can induce PR1 in an SA-independent manner, transgenic plants expressing constitutively active forms of MKK4 (MKK4DD) or MKK5 (MKK5DD) under the control of a dexamethasone (DEX)-inducible promoter were employed (DEX-MKK4DD and DEX-MKK5DD). MKK4 and MKK5 are MAP kinase kinases, whose activated forms phosphorylate and activate MPK3 and MPK6 [17]. DEX-induced expression of MKK4DD or MKK5DD leads to sustained activation of MPK3 and MPK6 (Figure S6) [30]. Induction of PR1 was observed 9 hours after DEX treatment (Figure 5A), suggesting that sustained activation of MPK3 and MPK6 is sufficient for induction of PR1. Induction of FRK1 is thought to be a good marker for activation of MPK3 and MPK6 [31] and was observed 3 hours after DEX treatment while PR1 was not (Figure 5B). FRK1 was strongly induced 30 minutes after flg22 treatment [32], and the induction did not require SA accumulation (Figure S7). Thus, although transient MAPK activation of MPK3 and MPK6 is sufficient for FRK1 induction, sustained MAPK activation is necessary and sufficient for SA-independent PR1 induction. The sustained activation of MPK3 and MPK6 by DEX-induced MKK4DD or MKK5DD did not increase the level of SA (Figure 6A). Furthermore, a wild-type-like PR1 induction 24 hours after DEX treatment was observed in plants deficient in SID2 or NPR1 (Figure 6B). Since PR1 induction was not observed in a DEX-inducible ß-glucuronidase (GUS, an arbitrary reporter gene) line after DEX treatment, PR1 induction was not caused by the DEX-inducible system or DEX but by induced expression of MKK4DD or MKK5DD. Although MPK4 was activated as well as MPK3 and MPK6 during PTI and ETI (Figure 4; [17]), expression of MKK4DD or MKK5DD does not lead to strong activation of MPK4 [30]. Therefore, it is unlikely that MPK4 plays a role. We conclude that sustained activation of MPK3 and/or MPK6 causes PR1 induction in an SA-independent manner.


We tested whether mpk3 and mpk6 single mutations had effects on PR1 induction by MKK4DD or MKK5DD expression. PR1 induction was unaffected in mpk6 but strongly reduced in mpk3 plants (Figure S8A). MKK4DD induction was also strongly reduced in mpk3 plants (Figure S8B), so the reduction of PR1 induction in DEX-MKK4DD/mpk3 may be due to reduction of MKK4DD expression. MKK5DD induction in DEX-MKK5DD/mpk3 was reduced compared to DEX-MKK5DD/Col yet 10 times higher than MKK4DD induction in DEX-MKK4DD/mpk3 while PR1 induction was similarly compromised in both plant lines. Thus, these results suggest that MPK3 is required for SA-independent PR1 induction conferred by forced MKK5 activation while MPK6 is dispensable.
Sustained activation of MPK3 and MPK6 supported transcriptional regulation of most SA-responsive genes
We tested whether sustained activation of MPK3 and/or MPK6 also regulates other SA-responsive genes. Leaves of the DEX-MKK4DD transgenic lines in wild type (Col) or sid2 backgrounds were treated with DEX or mock control and were collected for mRNA profiling at 24 hours after treatment. The transcriptomic changes caused by DEX treatment were very similar between Col and sid2 (Figure S9 and Table S3), indicating that gene regulation by sustained activation of the MAPKs is mostly independent of SA. Therefore, only the mRNA profile from the DEX-MKK4DD sid2 line was included in the following analysis. The heatmap in Figure 2A shows that a majority of the SA-responsive genes responded in the DEX-treated DEX-MKK4DD sid2 line similarly to sid2 plants during AvrRpt2-ETI: most up-regulated or down-regulated SA-responsive genes in sid2 during AvrRpt2-ETI were up-regulated or down-regulated, respectively, in the DEX-treated DEX-MKK4DD sid2 line. This suggests that sustained activation of the MAPKs regulates a majority of SA-responsive genes in an SA-independent manner during AvrRpt2-ETI.
Three gene clusters were selected for further analysis (Clusters I–III in Figure 2A). The expression level changes of genes in each cluster were averaged and shown in Figure 2B–D. Clusters I and III include genes up - or down-regulated, respectively, in a SID2-independent manner during AvrRpt2-ETI and by sustained activation of the MAPKs. Thus, these genes appear to be regulated by sustained activation of the MAPKs during ETI. Cluster II includes genes that were up-regulated in a largely SID2-independent manner during ETI but not up-regulated by sustained activation of the MAPKs. Thus, up-regulation of the Cluster II genes during ETI is supported by a mechanism(s) other than the mechanism mediated by the MAPKs. When the GO terms associated with the clusters were examined, Cluster I, but none of the other clusters, was enriched with genes related to biological stresses (response to biotic stimulus, P = 2.8×10−5; response to other organism, P = 1.1×10−4; multi-organism process, P = 5.9×10−4). The results imply that genes induced by both SA and the MAPKs are important for biological stress responses. The regulatory trends for the clusters were confirmed by qRT-PCR analysis of one gene from each cluster (Figure S10).
Compensatory relationships between the MAPKs and SA signaling confer robustness to AvrRpt2-ETI
We investigated if compensation between MPK3/MPK6 and SA signaling could be detected in the PR1 expression level during ETI. Leaves of wild type (Col), mpk3, mpk6, sid2, mpk3 sid2 and mpk6 sid2 plants were inoculated with Pto AvrRpt2 or Pto AvrRpm1, and PR1 expression levels were determined 24 hpi (Figure 7A). While PR1 expression was compromised in sid2 but not in mpk3 or mpk6 during AvrRpt2-ETI, it was compromised in mpk3 sid2 more than in sid2 (blue bar), suggesting compensation between MPK3 and SID2 on PR1 expression during AvrRpt2-ETI. To quantify the level of compensation between MPK3 and SID2 on PR1 expression, a signaling allocation analysis was applied [25]. In this analysis, the effects of the genes and their interactions were estimated for contribution to the PR1 expression level after inoculation. We estimated the individual contribution of MPK3 on the PR1 expression level as the difference in expression levels between sid2 and mpk3 sid2, that of SID2 as the difference in PR1 expression levels between mpk3 and mpk3 sid2 and their combined contribution as the difference in PR1 expression levels between the wild type and mpk3 sid2. The value of the genetic interaction between MPK3 and SID2 was calculated by subtracting the sum of the individual contributions of MPK3 and SID2 from their combined contribution. Their combined contribution in the wild type was less than the sum of the individual contributions of SA and MPK3, which is signified by the negative interaction between them. We previously defined this less-than-additive combined contribution as compensation [25]. Such compensation was observed for AvrRpt2-ETI (Figure 7B, top). Thus, signaling mediated by MPK3 and SA is compensatory on PR1 expression during AvrRpt2-ETI. No significant effects of MPK6 or the interaction (MPK6:SID2) on PR1 expression were detected during AvrRpt2-ETI (Figure 7A and B). No significant effects of MPK3, MPK6 or their interactions (MPK3:SID2 and MPK6:SID2) on PR1 expression (Figure 7A and B, red bar) or resistance (Figure S11) were detected during AvrRpm1-ETI, suggesting a divergence in the mechanisms that modulate network robustness between different cases of ETI.

A similar trend was observed with the effects of MPK3 and MPK6 on bacterial resistance in AvrRpt2-ETI (Figure 7C). AvrRpt2-ETI is defined as the difference in in planta growth of Pto EV and Pto AvrRpt2 on a log10-scale [25]. The compensation between MPK3 and SID2 was clear from the signaling allocation analysis, as both had positive effects and their interaction was negative (Figure 7D, left). We did not detect significant effects of MPK6 or the interaction (MPK6:SID2), although we observed a similar pattern to the case of MPK3 (Figure 7D, right). Thus, compensation of SA signaling by a signaling mechanism involving MPK3 exists in inhibition of bacterial growth, as well as in PR1 expression, during AvrRpt2-ETI.
Lethality of the double mutants mpk3 mpk6 [21] does not allow us to determine combined contributions of MPK3 and MPK6 to compensation of SA signaling during ETI. It is possible that MPK6 is not a major factor in SA signaling compensation during ETI and that a signaling mechanism(s) other than that involving MPK3 or MPK6 is important during AvrRpm1-ETI. Nonetheless, these results clearly demonstrate that at least during AvrRpt2-ETI, SA signaling can be compensated by MPK3-mediated signaling in regulation of SA-responsive gene expression and that this compensation increases the robustness of the network output. This allows immunity to be maintained even if the major network sector, SA signaling, is compromised.
Discussion
In this study, we identified a mechanism that can increase the robustness of the plant immune signaling network during AvrRpt2-ETI. Our results demonstrate that (1) MPK3 and MPK6 are activated in a sustained manner during ETI and in a transient manner during non-ETI; (2) Transient MAPK activation during non-ETI such as PTI does not contribute to SA-independent regulation of the SA-responsive genes; (3) Sustained MAPK activation activates an SA-independent alternative mechanism that regulates the SA-responsive genes; (4) SA-independent alternative mechanisms which regulate the SA-responsive genes were activated during AvrRpt2-ETI; (5) SA signaling compensation by the signaling sector involving MPK3 contributes to increased robustness against network perturbations during AvrRpt2-ETI (Figure 8).
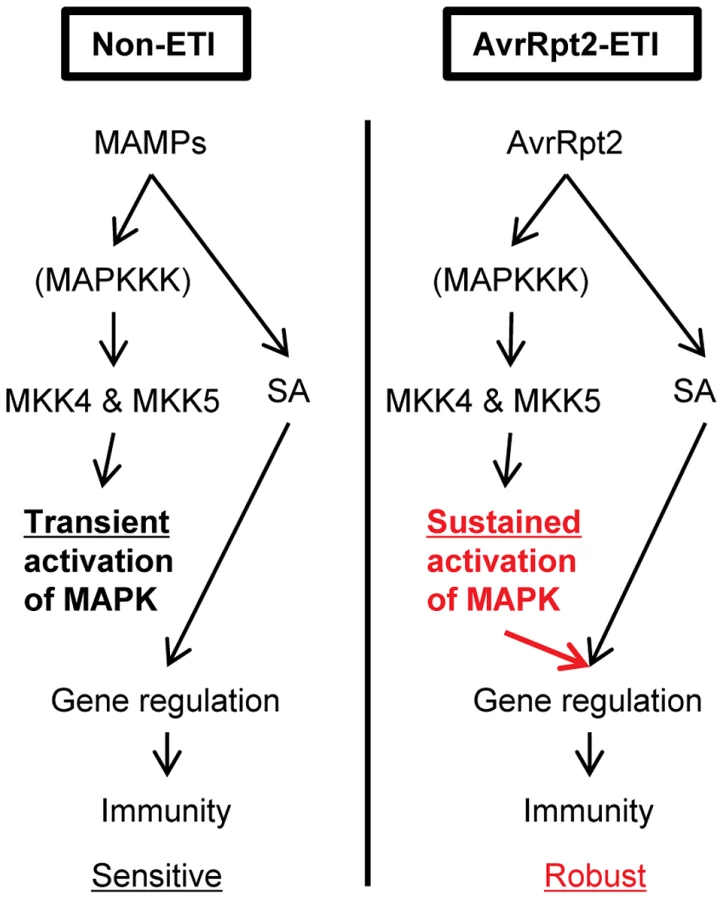
Factor(s) controlling differential activation duration of the MAPKs
A prior study implied that the duration of MPK3 and MPK6 activation is longer during ETI compared to during non-ETI upon P. syringae infection [27]. However, it did not rule out the possibility that the effector AvrRpt2 caused sustained MAPK activation through a mechanism independent of recognition of AvrRpt2 via RPS2. We clearly demonstrated that sustained MAPK activation occurs when ETI is triggered (Figures 3 and 4). The duration of MPK3 and/or MPK6 activation is the determinant for activation of the SA-independent alternative mechanism to regulate the SA-responsive genes: only sustained MAPK activation results in activation of the alternative mechanism. One potential cause of the differential activation duration is rapid turnover of PTI receptors, PRRs. FLS2 is rapidly degraded and disappears within one hour upon exposure to flg22 [33], [34]. Although turnover rates of other PRRs are not known, if many PRRs turn over rapidly upon activation, this could explain transient activation of the MAPKs by Pto hrcC (Figure 4), which presents multiple MAMPs [11]. The turnover rates of R proteins, the ETI receptors, upon their activation are largely unknown. Whether turnover rate is involved or not, this hypothesis that the duration of MAPK activation and, consequently, the robustness of the network can be tuned to each receptor is attractive because it would enable network robustness to be evolutionarily adapted according to what pathogen-derived signals are recognized by the receptors. Another potential but not mutually exclusive cause of the differential activation duration is involvement of protein phosphatases that dephosphorylate and inactivate the MAPKs: activation of the MAPKs may be negatively regulated by a phosphatase(s) during non-ETI responses while the phosphatase may be inactivated during ETI, resulting in the sustained activation of the MAPKs. Multiple types of such phosphatases including MAPK phosphatases are known in Arabidopsis [35]. Differential regulation of these phosphatases during ETI and non-ETI responses may explain the differential duration of MAPK activation.
Decoding of the activation duration information
Switching of downstream signaling by differential duration of MAPK activation is known in animals and yeast [36]–[38]. In one case, it is explained by nuclear translocation of a MAPK that occurs only after its sustained activation [36]. In this way, sets of substrates available to the MAPK are distinct between its transient and sustained activation, which could lead to distinct downstream signaling. In plants, it has also been reported that MAPKs are translocated to the nucleus upon stimulation [39], [40]. Investigation of potential subcellular localization changes of Arabidopsis MPK3 and MPK6 during PTI and ETI will provide insight into this possibility. Another appealing explanation is involvement of a feed-forward network motif [41]. For example, activation of a transcription factor TF-X may mediate the alternative mechanism regulated by sustained MAPK activation. The activation of TF-X may require signal Y in addition to active MPK3 and/or MPK6. Signal Y may be slowly generated as a consequence of the activation of the MAPKs (e.g., 5 hours). The MAPKs would need to be activated for a long time to simultaneously have both signal Y and the active MAPKs to activate TF-X and regulate the SA-responsive genes. In either scenario, discovery of the signaling components downstream of the sustained MAPK activation will be the key to elucidate the mechanism that decodes duration of MAPK activation. Multiple transcription factors, such as TGAs, WRKYs, TBF1 and VIP1 [14], [22], [23], [42]–[44], are involved in regulation of PR1. These transcription factors may provide a good starting point for a search for the decoding mechanism.
ETI may provide robustness under perturbation by coronatine
Pto produces the small molecule coronatine, which is a molecular mimic of the JA-Ile conjugate and promotes virulence by suppressing SA signaling [13]. Pto is highly virulent on Arabidopsis plants while ETI-triggering strains of Pto, such as Pto AvrRpt2, are much less virulent. Nevertheless, coronatine could suppress SA signaling. Therefore, SA-independent alternative mechanism(s) to regulate expression of the SA-responsive genes, such as that mediated by the MAPKs, may have a substantial role against perturbation of the immune signaling network by coronatine. This hypothesis is consistent with our observation that loss of both MPK3 and SA led to increased susceptibility to Pto AvrRpt2 (Figure 7).
Can AvrRpt2-ETI overcome suppression effects by other effectors?
Pto DC3000 possesses type III effectors which directly or indirectly suppress MAPK activation [45]–[48]. However, we observed sustained activation of MPK3 and MPK6 during AvrRpt2-ETI when AvrRpt2 was delivered from Pto DC3000 (Figure 4). We speculate that the amounts of such MAPK-inhibiting type III effectors delivered and/or the kinetics of their delivery are not optimal to effectively suppress MAPK activation when the type III effectors are delivered from Pto DC3000, which represents a relatively natural context.
The effector HopAI1 from Pto DC3000 can physically interact with and inactivate MPK3 and MPK6 by removing the phosphate group from phosphothreonine via a phosphothreonine lyase activity [45]. HopAI1 also targets MPK4 and decreases MPK4 activity [48]. Decreased MPK4 activity appears to be monitored by the NB-LRR protein SUMM2, resulting in triggering ETI. Overexpression of HopAI1 in wild-type Col-0 plants but not summ2 mutant plants leads to dwarfism and constitutive activation of immune responses [48]. However, Pto DC3000 does not trigger SUMM2-mediated ETI. Consistently, HopAI1 of Pto DC3000 is disrupted by an insertion in its promoter region [49]. Thus, the amount of HopAI1 delivered from Pto DC3000 appears insufficient for effective inhibition of MPK3 and MPK6 activation during AvrRpt2-ETI.
Another effector, HopF2, from Pto DC3000 can also suppress activity of MPK3, MPK4 and MPK6 by targeting the upstream MKK5 and likely other MKKs as well [46], [47]. When overexpressed in plants, HopF2 interferes with AvrRpt2-ETI by inhibiting AvrRpt2-mediated RIN4 degradation [50]. Again, the reason that HopF2 cannot suppress sustained activation of MPK3 and MPK6 triggered by AvrRpt2 when it is delivered from Pto DC3000 (Figure 4) is likely insufficient HopF2 or inappropriate timing of its delivery. Delivery of AvrRpt2 may precede that of HopF2 [50].
Is the low level of robustness required during PTI?
One enigma is why plants need to make the robustness of the immune signaling network lower during PTI when the network itself has the capacity to be highly robust. If the network output during PTI were as robust as during ETI, the chance that “true” pathogens will overcome PTI would be much lower. We speculate that the lower robustness during PTI is selected through evolution as trade-offs with other requirements. Many MAMPs are shared among pathogens and benign microbes and provide low quality information about pathogen attack. It is probably not adaptive for plants to respond to a MAMP with strong and sustained immune responses similar to those during ETI since in many cases, plants encounter benign microbes and ETI-type responses cost fitness. A strategy apparently selected is to respond weakly first and wait to intensify the response until further information increases the probability that a true pathogen is present [24]. In contrast, since effectors are a hallmark of true pathogens and provide high quality information, during ETI plants can induce rapid and strong immune responses with a very low chance of needless fitness costs.
Concluding remarks
The signaling sector activated by sustained activation of the MAPKs during ETI and the SA signaling sector can regulate the common set of genes. This is one of the mechanisms underlying robustness of the immunity level against network perturbations during ETI. This modulation of the network robustness is controlled by signaling kinetics of a network component. Our findings imply that properties of biological networks can be modulated through network component activities.
Materials and Methods
Free SA measurement, MAP kinase assays, bacterial growth assays and the signaling allocation analysis were performed as described previously [25], [26].
Plant materials and growth conditions
Arabidopsis plants were grown in a controlled environment at 22°C with a 12 h photoperiod and 75% relative humidity. Arabidopsis thaliana accession Col-0 was the background of all mutants used in this study. Arabidopsis mpk3-1 (SALK_151594) [21], mpk6-2 (SALK_073907) [18], npr1-1 [51], rps2 101C [52] and sid2-2 [15] were previously described. We generated the double mutants mpk3 sid2 and mpk6 sid2 by standard genetic crosses. Estradiol-inducible AvrRpt2 transgenic lines [53] and the DEX-MKK4DD and -MKK5DD transgenic lines [30] were previously described. We crossed DEX-MKK4DD and -MKK5DD into the mutant backgrounds mpk3, mpk6, npr1, sid2 and vip1. Primers and restriction enzymes used for screening of the mutants are listed in Table S4.
Chemicals
Flg22 peptide was purchased from EZBiolab Inc (Westfield, IN, USA). Estradiol (E8875) and DEX (D1756) were purchased from Sigma (Saint Louis, MO, USA).
Quantitative RT-PCR analysis
Pto DC3000 strains (or water for mock) or 2 µM DEX (or 0.1% ethanol for mock) were infiltrated into leaves of 4-week-old plants. Leaves were collected at the indicated time points. Total RNA isolation and qRT-PCR analysis were carried out as described previously [54], [55]. The following models were fit to the relative Ct value data compared to Actin2 using the lme function in the nlme package in the R environment: Ctgytr = GYTgyt+Rr+εgytr, where GYT, genotype:treatment:time interaction, and random factors; R, biological replicate; ε, residual; Ctgyr = GYgy+Rr+εgytr, where GY, genotype:treatment interaction; Ctgtr = GTgt+Rr+εgtr, where GT, genotype:time interaction. The mean estimates of the fixed effects were used as the modeled relative Ct values and visualized as the relative log2 expression values and compared by two-tailed t-tests. For the t-tests, the standard errors were calculated using the variance and covariance values obtained from the model fitting. Primers used in the study are listed in Table S4.
DNA microarrays
Four-week-old Arabidopsis Col-0 and sid2 leaves were infiltrated with Pto hrcC, Pto pLAFR (EV), Pto AvrRpt2 or water (mock). Independently, leaves of four-week-old DEX-MKK4DD plants in Col-0 or a sid2 background were infiltrated with 2 µM DEX or 0.1% ethanol (mock). Samples were collected at 24 hpi. Total RNA was extracted as described previously [26] and profiled using the NimbleGen DNA microarray (A. thaliana Gene Expression 12×135K array TAIR9.0) following the manufacturer's protocol (Roche Applied Science, Indianapolis, IN, USA). Three independent experiments (biological replicates) were performed. The microarray data were submitted to Gene Expression Omnibus (Accession, GSE40555). Probe signal values were subjected to the robust multi-array average (RMA) summarization algorithm [56] using the standard NimbleGen software to obtain the expression level values of the transcripts. Among transcripts of a single gene, those with higher expression values were selected as the representative transcripts of the genes. The following models were fit to log2 expression values using the lmFit function in the limma package in the R environment: Sgyr = GYgyt+Rr+εgyr, where S, log2 expression value, GY, genotype:treatment interaction, and random factors; R, biological replicate; ε, residual. The eBayes function in the limma package was used for variance shrinkage in calculation of the p-values and the Storey's q-values were calculated from the p-values using the qvalue function in the qvalue package. First, genes whose expression was up-regulated or down-regulated (q values<0.01 and more than 2 fold change) in both Pto EV and Pto AvrRpt2-infected Col compared to mock were selected (2828 genes). Second, SID2-dependent genes in Pto EV infection (inductions/suppression in sid2 are less than 20% compared to Col) were selected (187 “SA-responsive” genes) for the clustering analysis. Heatmaps were generated by CLUSTER [57] using uncentered Pearson correlation and complete linkage, and visualized by TREEVIEW [57].
Accession numbers
The accession numbers for the Arabidopsis genes discussed in this article are as follows: Actin2 (At2g18780), Chitinase (At1g02360), CHS (At5g13930), FRK1 (At2g19190), MKK4 (At1g51660), MKK5 (At3g21220), MPK3 (At3g45640), MPK4 (At4g01370), MPK6 (At2g43790), NPR1 (At1g64280), RPM1 (At3g07040), RPS2 (At4g26090) and SID2 (At1g74710).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MaselJ, SiegalML (2009) Robustness: mechanisms and consequences. Trends Genet 25 : 395–403.
2. ShinarG, FeinbergM (2011) Design principles for robust biochemical reaction networks: what works, what cannot work, and what might almost work. Math Biosci 231 : 39–48.
3. JonesJD, DanglJL (2006) The plant immune system. Nature 444 : 323–329.
4. TsudaK, KatagiriF (2010) Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr Opin Plant Biol 13 : 459–465.
5. SpoelSH, DongX (2012) How do plants achieve immunity? Defence without specialized immune cells. Nat Rev Immunol 12 : 89–100.
6. SchwessingerB, RonaldPC (2012) Plant innate immunity: perception of conserved microbial signatures. Annu Rev Plant Biol 63 : 451–482.
7. Gomez-GomezL, BollerT (2000) FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell 5 : 1003–1011.
8. HannDR, Gimenez-IbanezS, RathjenJP (2010) Bacterial virulence effectors and their activities. Curr Opin Plant Biol 13 : 388–393.
9. de JongeR, BoltonMD, ThommaBP (2011) How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr Opin Plant Biol 14 : 400–406.
10. TakkenFL, GoverseA (2012) How to build a pathogen detector: structural basis of NB-LRR function. Curr Opin Plant Biol 15 : 375–384.
11. Penaloza-VazquezA, PrestonGM, CollmerA, BenderCL (2000) Regulatory interactions between the Hrp type III protein secretion system and coronatine biosynthesis in Pseudomonas syringae pv. tomato DC3000. Microbiology 146 (Pt 10) 2447–2456.
12. GengX, ChengJ, GangadharanA, MackeyD (2012) The coronatine toxin of Pseudomonas syringae is a multifunctional suppressor of Arabidopsis defense. Plant Cell 24 : 4763–4774.
13. ZhengXY, SpiveyNW, ZengW, LiuPP, FuZQ, et al. (2012) Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe 11 : 587–596.
14. VlotAC, DempseyDA, KlessigDF (2009) Salicylic Acid, a multifaceted hormone to combat disease. Annu Rev Phytopathol 47 : 177–206.
15. WildermuthMC, DewdneyJ, WuG, AusubelFM (2001) Isochorismate synthase is required to synthesize salicylic acid for plant defence. Nature 414 : 562–565.
16. DocziR, OkreszL, RomeroAE, PaccanaroA, BogreL (2012) Exploring the evolutionary path of plant MAPK networks. Trends Plant Sci 17 : 518–525.
17. RasmussenMW, RouxM, PetersenM, MundyJ (2012) MAP Kinase Cascades in Arabidopsis Innate Immunity. Front Plant Sci 3 : 169.
18. LiuY, ZhangS (2004) Phosphorylation of 1-aminocyclopropane-1-carboxylic acid synthase by MPK6, a stress-responsive mitogen-activated protein kinase, induces ethylene biosynthesis in Arabidopsis. Plant Cell 16 : 3386–3399.
19. LiG, MengX, WangR, MaoG, HanL, et al. (2012) Dual-Level Regulation of ACC Synthase Activity by MPK3/MPK6 Cascade and Its Downstream WRKY Transcription Factor during Ethylene Induction in Arabidopsis. PLoS Genet 8: e1002767.
20. RenD, LiuY, YangKY, HanL, MaoG, et al. (2008) A fungal-responsive MAPK cascade regulates phytoalexin biosynthesis in Arabidopsis. Proc Natl Acad Sci U S A 105 : 5638–5643.
21. WangH, NgwenyamaN, LiuY, WalkerJC, ZhangS (2007) Stomatal development and patterning are regulated by environmentally responsive mitogen-activated protein kinases in Arabidopsis. Plant Cell 19 : 63–73.
22. DjameiA, PitzschkeA, NakagamiH, RajhI, HirtH (2007) Trojan horse strategy in Agrobacterium transformation: abusing MAPK defense signaling. Science 318 : 453–456.
23. PitzschkeA, DjameiA, TeigeM, HirtH (2009) VIP1 response elements mediate mitogen-activated protein kinase 3-induced stress gene expression. Proc Natl Acad Sci U S A 106 : 18414–18419.
24. KatagiriF, TsudaK (2010) Understanding the plant immune system. Mol Plant Microbe Interact 23 : 1531–1536.
25. TsudaK, SatoM, StoddardT, GlazebrookJ, KatagiriF (2009) Network properties of robust immunity in plants. PLoS Genet 5: e1000772.
26. TsudaK, SatoM, GlazebrookJ, CohenJD, KatagiriF (2008) Interplay between MAMP-triggered and SA-mediated defense responses. Plant J 53 : 763–775.
27. UnderwoodW, ZhangS, HeSY (2007) The Pseudomonas syringae type III effector tyrosine phosphatase HopAO1 suppresses innate immunity in Arabidopsis thaliana. Plant J 52 : 658–672.
28. ChinchillaD, ZipfelC, RobatzekS, KemmerlingB, NurnbergerT, et al. (2007) A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448 : 497–500.
29. BeckersGJ, JaskiewiczM, LiuY, UnderwoodWR, HeSY, et al. (2009) Mitogen-activated protein kinases 3 and 6 are required for full priming of stress responses in Arabidopsis thaliana. Plant Cell 21 : 944–953.
30. RenD, YangH, ZhangS (2002) Cell death mediated by MAPK is associated with hydrogen peroxide production in Arabidopsis. J Biol Chem 277 : 559–565.
31. TenaG, BoudsocqM, SheenJ (2011) Protein kinase signaling networks in plant innate immunity. Curr Opin Plant Biol 14 : 519–529.
32. ZipfelC, RobatzekS, NavarroL, OakeleyEJ, JonesJD, et al. (2004) Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428 : 764–767.
33. RobatzekS, ChinchillaD, BollerT (2006) Ligand-induced endocytosis of the pattern recognition receptor FLS2 in Arabidopsis. Genes Dev 20 : 537–542.
34. LuD, LinW, GaoX, WuS, ChengC, et al. (2011) Direct ubiquitination of pattern recognition receptor FLS2 attenuates plant innate immunity. Science 332 : 1439–1442.
35. BartelsS, Gonzalez BesteiroMA, LangD, UlmR (2010) Emerging functions for plant MAP kinase phosphatases. Trends Plant Sci 15 : 322–329.
36. TraverseS, GomezN, PatersonH, MarshallC, CohenP (1992) Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J 288 (Pt 2) 351–355.
37. SabbaghWJr, FlatauerLJ, BardwellAJ, BardwellL (2001) Specificity of MAP kinase signaling in yeast differentiation involves transient versus sustained MAPK activation. Mol Cell 8 : 683–691.
38. GlotinAL, CalipelA, BrossasJY, FaussatAM, TretonJ, et al. (2006) Sustained versus transient ERK1/2 signaling underlies the anti - and proapoptotic effects of oxidative stress in human RPE cells. Invest Ophthalmol Vis Sci 47 : 4614–4623.
39. LeeJ, RuddJJ, MacioszekVK, ScheelD (2004) Dynamic changes in the localization of MAPK cascade components controlling pathogenesis-related (PR) gene expression during innate immunity in parsley. J Biol Chem 279 : 22440–22448.
40. AhlforsR, MacioszekV, RuddJ, BroscheM, SchlichtingR, et al. (2004) Stress hormone-independent activation and nuclear translocation of mitogen-activated protein kinases in Arabidopsis thaliana during ozone exposure. Plant J 40 : 512–522.
41. ManganS, AlonU (2003) Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100 : 11980–11985.
42. Pajerowska-MukhtarKM, WangW, TadaY, OkaN, TuckerCL, et al. (2012) The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Curr Biol 22 : 103–112.
43. ZhangY, YangY, FangB, GannonP, DingP, et al. (2010) Arabidopsis snc2-1D activates receptor-like protein-mediated immunity transduced through WRKY70. Plant Cell 22 : 3153–3163.
44. WangD, AmornsiripanitchN, DongX (2006) A genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. PLoS Pathog 2: e123.
45. ZhangJ, ShaoF, LiY, CuiH, ChenL, et al. (2007) A Pseudomonas syringae effector inactivates MAPKs to suppress PAMP-induced immunity in plants. Cell Host Microbe 1 : 175–185.
46. WangY, LiJ, HouS, WangX, LiY, et al. (2010) A Pseudomonas syringae ADP-ribosyltransferase inhibits Arabidopsis mitogen-activated protein kinase kinases. Plant Cell 22 : 2033–2044.
47. WuS, LuD, KabbageM, WeiHL, SwingleB, et al. (2011) Bacterial effector HopF2 suppresses arabidopsis innate immunity at the plasma membrane. Mol Plant Microbe Interact 24 : 585–593.
48. ZhangZ, WuY, GaoM, ZhangJ, KongQ, et al. (2012) Disruption of PAMP-induced MAP kinase cascade by a Pseudomonas syringae effector activates plant immunity mediated by the NB-LRR protein SUMM2. Cell Host Microbe 11 : 253–263.
49. LindebergM, CartinhourS, MyersCR, SchechterLM, SchneiderDJ, et al. (2006) Closing the circle on the discovery of genes encoding Hrp regulon members and type III secretion system effectors in the genomes of three model Pseudomonas syringae strains. Mol Plant Microbe Interact 19 : 1151–1158.
50. WiltonM, SubramaniamR, ElmoreJ, FelsensteinerC, CoakerG, et al. (2010) The type III effector HopF2Pto targets Arabidopsis RIN4 protein to promote Pseudomonas syringae virulence. Proc Natl Acad Sci U S A 107 : 2349–2354.
51. CaoH, GlazebrookJ, ClarkeJD, VolkoS, DongX (1997) The Arabidopsis NPR1 gene that controls systemic acquired resistance encodes a novel protein containing ankyrin repeats. Cell 88 : 57–63.
52. MindrinosM, KatagiriF, YuGL, AusubelFM (1994) The A. thaliana disease resistance gene RPS2 encodes a protein containing a nucleotide-binding site and leucine-rich repeats. Cell 78 : 1089–1099.
53. TsudaK, QiY, Nguyen leV, BethkeG, TsudaY, et al. (2012) An efficient Agrobacterium-mediated transient transformation of Arabidopsis. Plant J 69 : 713–719.
54. IgarashiD, TsudaK, KatagiriF (2012) The peptide growth factor, phytosulfokine, attenuates pattern-triggered immunity. Plant J 71 : 194–204.
55. HeidrichK, WirthmuellerL, TassetC, PouzetC, DeslandesL, et al. (2011) Arabidopsis EDS1 connects pathogen effector recognition to cell compartment-specific immune responses. Science 334 : 1401–1404.
56. IrizarryRA, HobbsB, CollinF, Beazer-BarclayYD, AntonellisKJ, et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4 : 249–264.
57. EisenMB, SpellmanPT, BrownPO, BotsteinD (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95 : 14863–14868.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy