
An Regulatory Circuit Modulates /Wnt Signaling and Determines the Size of the Midbrain Dopaminergic Progenitor Pool
MicroRNAs regulate gene expression in diverse physiological scenarios. Their role in the control of morphogen related signaling pathways has been less studied, particularly in the context of embryonic Central Nervous System (CNS) development. Here, we uncover a role for microRNAs in limiting the spatiotemporal range of morphogen expression and function. Wnt1 is a key morphogen in the embryonic midbrain, and directs proliferation, survival, patterning and neurogenesis. We reveal an autoregulatory negative feedback loop between the transcription factor Lmx1b and a newly characterized microRNA, miR135a2, which modulates the extent of Wnt1/Wnt signaling and the size of the dopamine progenitor domain. Conditional gain of function studies reveal that Lmx1b promotes Wnt1/Wnt signaling, and thereby increases midbrain size and dopamine progenitor allocation. Conditional removal of Lmx1b has the opposite effect, in that expansion of the dopamine progenitor domain is severely compromised. Next, we provide evidence that microRNAs are involved in restricting dopamine progenitor allocation. Conditional loss of Dicer1 in embryonic stem cells (ESCs) results in expanded Lmx1a/b+ progenitors. In contrast, forced elevation of miR135a2 during an early window in vivo phenocopies the Lmx1b conditional knockout. When En1::Cre, but not Shh::Cre or Nes::Cre, is used for recombination, the expansion of Lmx1a/b+ progenitors is selectively reduced. Bioinformatics and luciferase assay data suggests that miR135a2 targets Lmx1b and many genes in the Wnt signaling pathway, including Ccnd1, Gsk3b, and Tcf7l2. Consistent with this, we demonstrate that this mutant displays reductions in the size of the Lmx1b/Wnt1 domain and range of canonical Wnt signaling. We posit that microRNA modulation of the Lmx1b/Wnt axis in the early midbrain/isthmus could determine midbrain size and allocation of dopamine progenitors. Since canonical Wnt activity has recently been recognized as a key ingredient for programming ESCs towards a dopaminergic fate in vitro, these studies could impact the rational design of such protocols.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1003973
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003973
Summary
MicroRNAs regulate gene expression in diverse physiological scenarios. Their role in the control of morphogen related signaling pathways has been less studied, particularly in the context of embryonic Central Nervous System (CNS) development. Here, we uncover a role for microRNAs in limiting the spatiotemporal range of morphogen expression and function. Wnt1 is a key morphogen in the embryonic midbrain, and directs proliferation, survival, patterning and neurogenesis. We reveal an autoregulatory negative feedback loop between the transcription factor Lmx1b and a newly characterized microRNA, miR135a2, which modulates the extent of Wnt1/Wnt signaling and the size of the dopamine progenitor domain. Conditional gain of function studies reveal that Lmx1b promotes Wnt1/Wnt signaling, and thereby increases midbrain size and dopamine progenitor allocation. Conditional removal of Lmx1b has the opposite effect, in that expansion of the dopamine progenitor domain is severely compromised. Next, we provide evidence that microRNAs are involved in restricting dopamine progenitor allocation. Conditional loss of Dicer1 in embryonic stem cells (ESCs) results in expanded Lmx1a/b+ progenitors. In contrast, forced elevation of miR135a2 during an early window in vivo phenocopies the Lmx1b conditional knockout. When En1::Cre, but not Shh::Cre or Nes::Cre, is used for recombination, the expansion of Lmx1a/b+ progenitors is selectively reduced. Bioinformatics and luciferase assay data suggests that miR135a2 targets Lmx1b and many genes in the Wnt signaling pathway, including Ccnd1, Gsk3b, and Tcf7l2. Consistent with this, we demonstrate that this mutant displays reductions in the size of the Lmx1b/Wnt1 domain and range of canonical Wnt signaling. We posit that microRNA modulation of the Lmx1b/Wnt axis in the early midbrain/isthmus could determine midbrain size and allocation of dopamine progenitors. Since canonical Wnt activity has recently been recognized as a key ingredient for programming ESCs towards a dopaminergic fate in vitro, these studies could impact the rational design of such protocols.
Introduction
MicroRNAs regulate gene expression in various aspects of central nervous system (CNS) and peripheral nervous system (PNS) development and function, including neurogenesis, glial differentiation, fate specification, synaptogenesis, spine formation and plasticity [1]–[7]. Less studied is their role in modulating the most critical developmental signaling molecules in the embryonic CNS – morphogens. Recent studies have suggested that morphogen function is not simply based on a concentration gradient, but rather an integral of concentration as well as the time of exposure [8]–[10]. Thus, mechanisms must exist to control the dose and time of morphogen expression and function. MicroRNAs have been shown to target key elements of morphogen pathways in the early embryo [11]. We considered it plausible that microRNAs may be involved in modulating morphogen function in the developing CNS.
Wnts are key morphogens in the developing and adult CNS that are involved in proliferation, survival, patterning, and neurogenesis [12]–[14]. Wnt1 is the prototypical canonical Wnt and its function has been documented particularly in the midbrain region [14]. Wnt1 is dynamically expressed in the midbrain, being expressed in a broad swath at 8.5 days post coitum (dpc) and ultimately restricting to the Roof Plate (RP), Isthmic Organizer (IsO) and Floor Plate (FP) regions. Loss of Wnt1 leads to a drastic decrease in midbrain size, as well as reduction and misspecification of midbrain dopamine neurons (mDAs), and this is exacerbated by loss of Wnt5a [15], [16]. Studies that have interrupted Wnt/beta-catenin signaling have revealed that this pathway is critical for specification and neurogenesis of mDAs [17]–[19]. Wnt signaling is required for the expression of key mDA determinants Lmx1a, Otx2, and Ngn2 and for the downregulation of Shh [17]. Counterintuitively, excessive Wnt signaling is also detrimental for mDA production [20], adding to the general notion that morphogen dosage must be tightly regulated [21].
In the ventral midbrain, the Foxa2+, Shh+ FP is roughly allocated into two main progenitor domains: a medial Lmx1a/b+/Msx+ progenitor domain that gives rise to many mDAs (this domain may be further subdivided [22], [23]), and a lateral Nkx6.1+/Sim1+ domain that gives rise to many Brn3a+ neurons, populating among others, the red nucleus [22], [24]–[28]. During early development, however, Nkx6.1 is expressed at the midline and Lmx1b is expressed more broadly than Lmx1a/Msx. Between 9.0–9.5 dpc, Lmx1a/Msx expression is initiated, and expands laterally [27]. At the midline, Lmx/Msx ultimately subsumes Nkx6.1, resulting in a medial Lmx/Msx and lateral Nkx6.1/Sim1 domain demarcated by a sharp boundary. Lineage-based progenitor labeling studies have suggested that the expansion of the Lmx1a domain, in part occurs by inductive mechanisms [22], [28]. This induction is likely, at least in part, mediated by Wnt1/beta-catenin signaling, which is important in the ventral midbrain [17], [29]–[31], and both necessary and sufficient for Lmx1a expression in the FP [17], [18], [32]. Indeed, this capacity to elicit drastic gene expression changes makes the ventral midbrain a particularly good model to interrogate Wnt1/Wnt signaling and its modulators.
In this study we have identified an autoregulatory loop involving Lmx1b and miR135a2 that is critical for determining mDA allocation. We show that Lmx1b promotes mDA progenitor fate, whereas miR135a2 delimits the mDA domain. Forced maintenance of Lmx1b results in expanded mDA progenitors whereas loss of Lmx1b results in diminished mDA progenitors. MicroRNA studies show the opposite effects. Conditional removal of Dicer1 from ESCs results in expanded mDA progenitors at the expense of Nkx6.1 progenitors. In contrast, increased miR135a2 levels, only during an early window, result in a reduction in the proportion of mDA progenitors. In addition to progenitor allocation defects, we observed changes in midbrain size in these mutants. Both progenitor allocation and midbrain size phenotypes may be caused, at least in part, by alterations in Wnt1/Wnt signaling. While Lmx1b promotes Wnt1/Wnt signaling, miR135a2 appears to negatively regulate Lmx1b/Wnt1/Wnt signaling in the context of the embryonic midbrain.
Results
miR135a2 is enriched in the ventral midbrain and embedded within a novel intron of Rmst
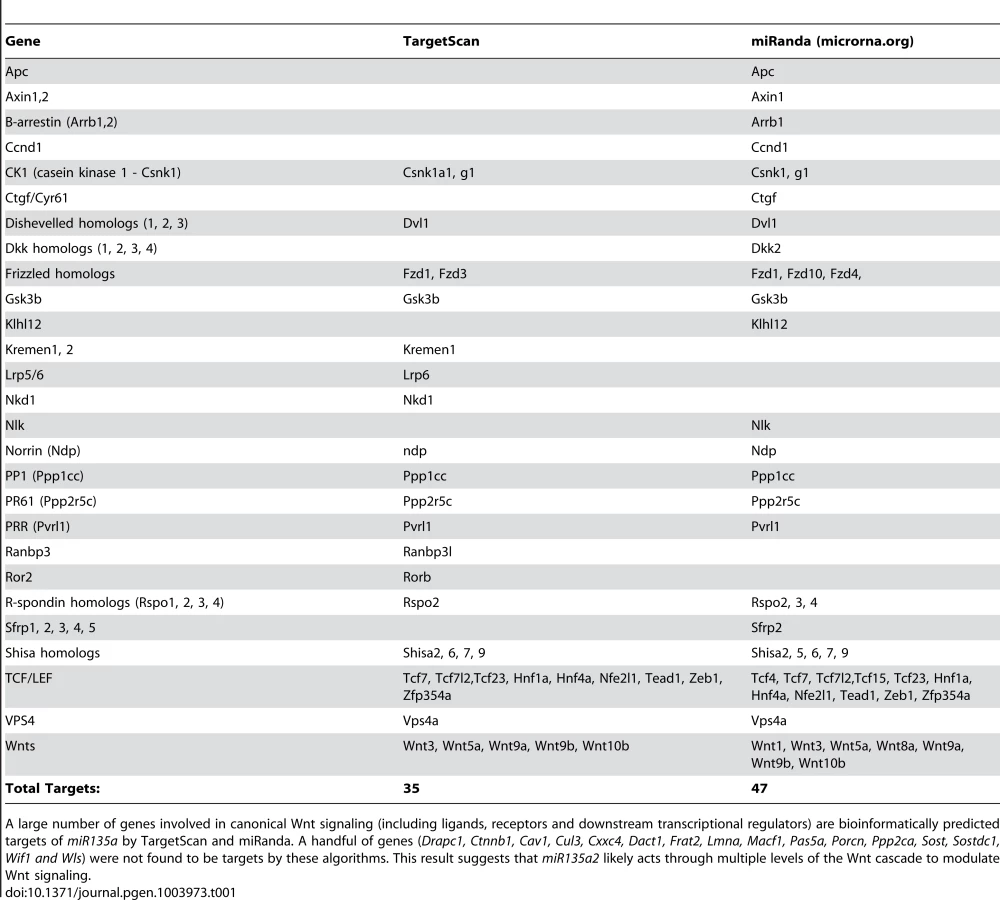
In order to identify microRNAs (miRs) in the Wnt1–rich mDA progenitor domain, ventral midline and dorsal lateral tissues were microdissected from 11.5 dpc mouse embryos and used to perform a microRNA array, followed by qRT-PCR validation with select individual TaqMan assays (Applied Biosystems). We reasoned that functionally relevant microRNAs would be 1) robustly expressed 2) differentially expressed and 3) bioinformatically predicted to target genes in the Lmx/Wnt axis. From five candidates that fit these criteria, we focused on mmu-miR-135a (miR135a), which was robustly expressed and showed a greater than 3-fold increase in the ventral midline compared to dorsal lateral tissue. The closely related mmu-miR-135b (miR135b) was also increased in the ventral midbrain, but this increase was not statistically significant (Figure 1A). Ventral midbrain enrichment of miR135a was further confirmed by Locked Nucleic Acid (LNA, Exiqon) in situ hybridization, which is designed to specifically detect mature microRNAs (Figure 1B). In addition, miR135a was predicted to target Lmx1b and several genes of the Wnt pathway through evolutionarily conserved binding sites in the 3′UTR [33](Table 1), thereby warranting further study.

To extend the expression data, we designed a reporter transgene (“sensor”) to verify the functional activity of miR135a in the ventral midbrain [34]. This transgene, comprised of eGFP with several sequences complementary to miR135a in the 3′UTR, is designed to broadly express eGFP. In regions of high miR135a activity, however, the eGFP levels should be suppressed. Since we did not design bulges in the microRNA binding sites, this transgene will not serve as a microRNA “sponge”, but only as a “sensor”. A control transgene, comprised of tdTomato with no complementary miR135a sites in the 3′UTR, was designed to broadly express tdTomato regardless of microRNA activity (Figure 1C). The transgenes were co-injected and transient transgenic embryos were harvested at 11.5 or 12.5 dpc. In ventral midline progenitors, eGFP was markedly reduced in the region predicted to have high miR135a activity, equivalent to that detected by the 135a LNA probe. In contrast, tdTomato showed little to no reduction at the ventral midline compared to neighboring regions (Figure 1D–G).
In mice, there are two miR135a family members, mmu-miR135a-1 (miR135a1) and mmu-miR135a-2 (miR135a2). Below we provide evidence suggesting that miR135a2, is expressed in the midbrain. Although miR135a2 was predicted to be intergenic on the miRBase Sequence Database and UCSC genome browser, a separate screen [35] coupled with further bioinformatic analysis revealed that miR135a2 was likely located between two exons of a previously uncharacterized gene. Based on its proximity to the 3′ end of nearby non-coding RNA, Rhabdomyosarcoma 2 associated transcript (Rmst)(Ensembl browser), which is known to be expressed in the midbrain [36], we hypothesized that miR135a2 was embedded in this gene. Thus, we performed RT-PCR on 11.5 dpc ventral midbrain RNA using a forward primer in Rmst and a reverse primer in the downstream flanking exon of miR135a2. This experiment yielded two predominant bands of approximately 700 bp and 900 bp (Figure S1A), indicating possible splice variants that will be further characterized in a future study. The most prominent fragment was sequenced and a BLAST search revealed that a) a variant of the Rmst transcript exists, which excludes exon 13, and has at least three additional exons and b) miR135a2 is located in the final detected intron of this transcript (Figure 2 – top panel). Moreover, in situ hybridizations with two separate probes (probe A and probe B) designed against this region showed similar expression in the midbrain and hindbrain FP, RP from the hindbrain to the telencephalon, and the IsO (Figure 2H; Figure S1B), equivalent to that detected by the 135a LNA probe (Figure 1B). Together, these results suggest that miR135a2 is coexpressed with Rmst in the midbrain. Although a separate internal promoter for the microRNA remains an alternative possibility, it is likely processed from an intron, a finding common for more than 50% of microRNAs [37].

miR135a2/Rmst expression is dynamic and resembles that of Lmx1b and Wnt1
The presence of miR135a2/Rmst in the FP, RP, and IsO resembles previously described expression patterns of two important genes, Lmx1b and Wnt1 [38], [39]. Thus, we compared the spatio-temporal relationship of these genes throughout early midbrain development. We observed similar and widespread expression with all three probes, as well as the miR135a LNA probe at 8.0 dpc, likely in the prospective midbrain, based on pan-midbrain reporter expression in Wnt1::Cre fate maps (Figure S1C). By 9.5 dpc all three genes were no longer detected throughout the midbrain, but rather were restricted to the FP, RP, and IsO (Figure 2A–L). The expression of these three genes correlates until ∼11.5 dpc, but at this age miR135a2/Rmst is also observed, at lower levels, in cells appearing to exit the ventricular zone throughout the midbrain (Figure S1B). This expression likely contributes to the low levels of miR135a observed in the dorsolateral samples in Figure 1A and is likely independent of Lmx1b. Between 12.5 dpc and 14.5 dpc, the microRNA transcriptional unit is maintained in dopamine progenitors, whereas Lmx1b and Wnt1 are severely downregulated, consistent with the phenomenon of “temporal exclusion” that has been described for many microRNAs and their cognate targets (Figure 3) [40]. On the other hand, Lmx1a, which is closely related to, and has partially overlapping function with Lmx1b, is not an in silico predicted target of miR135a2 and remains easily detectable in dopamine progenitors (Figure 3D–F).

These dynamic and tightly correlated expression patterns suggested miR135a2/Rmst as a potential component of the Lmx1b/Wnt1 regulatory network. At this point, we designed experiments to elucidate the details of this network by testing a) the hierarchical relationship between Lmx1b, miR135a2/Rmst, and Wnt1 b) the role of Lmx1b in midbrain development, and c) whether microRNAs, and specifically miR135a2 levels, were important for early midbrain development.
Lmx1b is necessary and sufficient for normal Wnt1/Wnt signaling and miR135a2/Rmst expression in the midbrain
Given the overlapping expression patterns of Lmx1b, miR135a2/Rmst and Wnt1 in the FP, RP and IsO, we first tested the hierarchical relationship between these genes. To do this, we utilized a mouse strain designed to conditionally express Lmx1b coding sequence, but lacking greater than 95% of the 3′UTR, under control of robust CAG regulatory elements. Using this strain, we generated embryos in which Lmx1b was conditionally activated throughout the midbrain and rhombomere 1 from ∼8.0 dpc onward, with Engrailed 1 (En1) Cre recombination [41], [42]. Forced maintenance of Lmx1b in En1Cre/+; Rosa26Lmx1b/+ (En1::Cre;Lmx1bOE) led to ectopic mir135a2/Rmst and Wnt1 expression in the midbrain, although both of these mRNAs were expressed more robustly in dorsal compared to ventral regions (Figure 4A–H). To determine whether changes in Wnt1 expression correlated with changes in Wnt signaling, we employed an Axin2::d2eGFP reporter allele that provides a transcriptional readout of this pathway [43], [44]. In En1::Cre;Lmx1bOE,Axin2::d2eGFP embryos d2eGFP fluorescence is detectable throughout the midbrain, whereas in controls it is predominantly confined to the ventral midline and the region surrounding the roof plate (Figure 4I–J and data not shown).

Conversely, in embryos wherein Lmx1b was conditionally deleted (En1::Cre;Lmx1bcKO) [45], miR135a2/Rmst and Wnt1 were significantly reduced in mDA progenitors (Figure 4K–N), undetectable in the isthmus (Figure 4Q–T), and mildly reduced in the dorsal midbrain (Figure S2A–D). Axin2::d2eGFP transgene expression was drastically reduced in the ventral midbrain (Figure 4O–P) and barely detectable in the isthmus (Figure 4U–V), but appeared only slightly diminished in the dorsal midbrain (Figure S2E–F). These data suggest that Lmx1b is upstream of both genes and promotes Wnt signaling, but that in regions where Lmx1a is expressed, it can at least partially compensate for Lmx1b. This finding is consistent with in vitro studies wherein both Lmx1a and Lmx1b were shown to drive Wnt1 expression [46].
Finally, to determine whether the observed changes in miR135a2/Rmst corresponded to the mature microRNA, we used qRT-PCR to quantify mature miR135a levels. In En1::Cre,Lmx1bOE midbrain, mature microRNA was increased 2-fold, whereas in En1::Cre,Lmx1bcKO midbrain it was modestly reduced (Figure S2G). These results suggest that miR135a2 is coexpressed with the Rmst transcript and responsive to Lmx1b manipulations.
Lmx1b determines the size of the midbrain, the FP, and the mDA progenitor domain
To determine the role of Lmx1b in midbrain development, we first investigated the functional consequences of maintaining Lmx1b in the early embryonic midbrain. Coronal sections through En1::Cre;Lmx1bOE embryos revealed an overall increase in third ventricle size and morphogenetic abnormalities (Figure S3A–B, G–H). In the ventral midbrain of 9.5–11.5 dpc En1::Cre;Lmx1bOE embryos we observed the dorsal-ventral (DV) extent of the FP marker Foxa2 to be significantly expanded, although depressed in level. The DV extent of the transcription factor Lmx1a was also significantly expanded throughout most of the Foxa2 progenitor domain. Lmx1a levels at the midline were consistently reduced, and in lateral regions were at even lower and graded levels (Figure 5A–F, Figure S3C–D, and data not shown). This result is consistent with ectopic Wnt signaling in the midbrain, in which Lmx1a is initially induced but ultimately attenuated [18]. Alternatively, progenitors might compensate for the overexpression of Lmx1b by downregulating the closely related Lmx1a.

To determine whether these domain changes were merely a consequence of overall increase in midbrain size, we measured the DV extent of the Lmx1a progenitor domain, the Foxa2 progenitor domain, and the length of the third ventricle (3V). By normalizing the DV extent of Lmx1a to the DV extent of Foxa2, and the DV extent of Foxa2 to the length of the third ventricle we determined that these domains are specifically expanded, rather than a consequence of the general increase in midbrain size (Figure 5A–I; Foxa2/3V shows a 20% increase, n = 3, control mean = 0.29±0.006, mutant mean = 0.35±0.02; p-value = .009). Moreover, the entirety of the Nkx6.1 progenitor domain is expanded, but intermingled with Lmx1a progenitors in these mutants (Figure 5J–K). As reported in previous studies [27], [47], [48] Lmx1a/b expansion led to repression of Nkx6.1, but likely because Lmx1a levels in lateral regions were not as robust as at the midline, this repression was only partial (i.e. several progenitors coexpressed Lmx1a and Nkx6.1). As a result, ectopic Lmx1a+ neurons, which appear to have increased Lmx1a levels after exiting the ventricular zone, were seen emanating from lateral aspects of the Foxa2 domain in addition to Nkx6.1+ neurons (Figure 5J–K). Further, we observed an increase in the DV extent of Shh, but a slight reduction in levels (Figure 5L–M). This result is consistent with an increase in Wnt signaling (see Figure 4) [17].
The early expansion of the Lmx1a/Foxa2 domain ultimately led to severe disruptions in ventral midbrain neuron types at later stages. Quantification of 13.5 dpc sections showed a 194% increase in Lmx1a+ cells and an 80% increase in TH+ mature mDAs. Brn3a+ red nucleus neurons were increased by 60%, likely because the overall Nkx6.1 progenitor domain was also expanded despite partial repression by Lmx1a. In contrast, a dramatic loss (90%) of Islet+ oculomotor neurons was observed (Figure 5N–U).
At 14.5 dpc, in addition to ectopic TH+ neurons in lateral regions, we observed a decrease in TH+ neurons at the midline, particularly in rostral sections (Figure S3I–R). Many cells at the midline appear to be stalled at the Nurr1+TH − state. This is likely because excess Lmx1b leads to increased Wnt1/Wnt signaling, too much of which is detrimental for normal dopamine neuron differentiation [20]. Alternatively, since a small Nurr1+/TH − population does exist in the wildtype postnatal midbrain, the increase in this population could indicate a change in fate. These data collectively demonstrate that failure to restrict Lmx1b during early embryogenesis drastically increases the third ventricle size and alters patterning of the midbrain. Further, excessive Lmx1b within the dopamine progenitor domain (see Figure S3E–F) is detrimental for normal dopamine differentiation, suggesting a need for careful modulation of its expression level.
Next, to determine whether Lmx1b was required for normal midbrain development, we examined En1::Cre;Lmx1bcKO embryos. Such embryos generated a complementary phenotype to the En1::Cre;Lmx1bOE in that the length of the third ventricle and midbrain size were reduced (Figure S4K–L). This reduction is at least in part due to apoptosis, as activated Caspase-3+ cells were increased, particularly in lateral midbrain regions (Figure S4A–B). Additionally, in En1::Cre;Lmx1bcKO mutants some Otx2+ cells were detected across the isthmic boundary in the hindbrain (Figure S4M–N). Fgf8 was drastically reduced in the isthmic region (Figure S4O–P).
Upon examination of the ventral midbrain of En1::Cre;Lmx1bcKO embryos, we observed that the DV extent of the Shh+ FP domain was reduced (Figure 6A–B). However, Shh appeared to be maintained rather than downregulated at the midline in mutant embryos, in accordance with the fact that Wnt1/Wnt signaling is reduced (see Figure 4N and P) [17]. Further, the DV extent of the Foxa2+ FP was reduced (Figure 6C–E, Figure S4C–D). After normalizing the DV extent of Foxa2 to the 3V, this reduction was found to be proportionate to the reduction in midbrain size (Foxa2/3V shows a 12% decrease, n = 3, control mean = 0.31±0.01, mutant mean = 0.28±0.01; p-value = .066). Within this domain, the Lmx1a+ dopamine progenitor domain was selectively reduced (Figure 6F–K, Figure S4E–H), similar to mice with defects in Wnt1/Wnt signaling [15], [17]. Likely, however, due to some residual Wnt1/Wnt signaling in En1::Cre;Lmx1bcKO, the morphological changes seen in the Wnt1 knockout and Shh::Cre;Ctnnb1cKO are not observed, and the distribution of remnant Lmx1a+ cells is different. Moreover, outside the main mDA progenitor domain, some Lmx1a+ cells were stranded within Nkx6.1+ territory, likely reflecting a failed attempt to establish a broader dopamine progenitor domain in this mutant. The Nkx6.1 territory was also reduced albeit not as drastically (22% reduction, n = 3, p-value = 0.06)(Figure 6L–M).

The early reduction of the Lmx1a/Foxa2 domain ultimately led to the diminution of many ventral midbrain neuron types at later stages. TH, a mature mDA marker, was drastically reduced, and quantification of 13.5 dpc sections revealed a 68% reduction of Lmx1a+ nascent mDAs. Brn3a+ red nucleus neurons showed a milder 39% reduction, likely reflective of a slight decrease in Nkx6.1+ progenitors. Only a few Islet+ oculomotor neurons were detected at 9.5 dpc, and these were virtually undetectable by 13.5 dpc (Figure 6N–T, Figure S4I–J). Altogether these data suggest that in the early embryo, Lmx1b is a key determinant of midbrain size, isthmic integrity, FP size, mDA progenitor domain size, and ventral neuron numbers.
microRNAs are necessary for proper allocation of FP progenitors
Since the proper dosage of transcription factors in the FP is imperative for determining progenitor allocation between the Lmx and Nkx6.1 domains, we next tested whether microRNAs played a role in regulating this process. To do this, we used an ESC line that harbored a CAG::CreERT2 construct and Dicer1floxed/floxed alleles, such that Dicer1, the key microRNA processing enzyme, could be deleted upon 4-hydroxy tamoxifen (4OHT) administration [5]. We next developed an optimized protocol to derive dopamine progenitors from embryoid body aggregates (Figure 7A) [49]. Remarkably, in controls, we were able to achieve conditions that mimic the in vivo midbrain FP, wherein most progenitors were Foxa2+, and of these roughly equal numbers were Lmx1a/b+ and Nkx6.1+. We quantified the proportion of Foxa2+ cells that were Lmx1a+, Lmx1b+ and Nkx6.1+ in controls and mutants. In both controls and mutants, large numbers of Foxa2+ cells were observed. In controls, the proportion of cells that were Lmx1a+, Lmx1b+, or Nkx6.1+ was roughly equivalent. In 4OHT treated cultures, however, the proportion of Lmx1a/b+ cells was drastically increased, while the proportion of Nkx6.1+ cells was decreased (Figure 7B–C). These data suggest that microRNAs are involved in progenitor cell allocation between the Lmx1a/b+ and Nkx6.1+ domain in ESC derived cultures, and under these conditions, de-repression of target genes through the loss of microRNAs expands mDA progenitors.

Early miR135a2 overexpression reduces Lmx1b/Wnt1/Wnt signaling and phenocopies loss of Lmx1b
We sought to determine if miR135a2 is sufficient to repress Lmx1b, and can thereby modulate midbrain development. To test this hypothesis, we first performed a luciferase assay in HEK293 cells, and found that miR135a2 was able to repress a construct harboring a fragment of the Lmx1b 3′UTR, but not constructs harboring mutations within the evolutionarily conserved miR135a2 binding site of the Lmx1b 3′UTR (Figure 8A–B and data not shown).

Next, we generated transgenic mice that conditionally express a mmu-miR-135a-2 precursor under control of CAG elements (CAG-loxP-STOPr-loxP-miR135a2-IRESeGFP; see Materials and Methods for description); in dissected 8.5 dpc midbrain, miR135a was detected in controls, and was approximately 3 fold increased in En1::Cre;135a2OE mutants (Figure S5D), although eGFP activity was not detectable (see Materials and Methods). In situ hybridizations showed a reduction in the domain size and levels of Lmx1b (Figure 8C–F), which was confirmed by qRT-PCR (Figure 8G) and indicated that miR135a2 is sufficient to repress Lmx1b in vivo. Otx2, which harbors a binding site for the closely related miR135b, is also decreased by both in situ hybridization and q-RT-PCR (Figure S6I–K); however, it remains to be determined whether Otx2 levels are decreased due to direct microRNA mediated repression, or in response to reduced Wnt1/beta-catenin signaling, or both (Figure S7).
We reasoned that overexpressing miR135a2 should, at least in part, phenocopy Lmx1b-deficient embryos. Indeed, in En1::Cre;135a2OE embryos obtained from three separate transgenic lines (Figure S5A–C, and data not shown), we observed an overall reduction in midbrain size (Figure S6A–B and J–K), albeit not as drastic as En1::Cre;Lmx1bcKOs. In 9.5 dpc En1::Cre;135a2OE embryos, we detected an increase in the apoptotic marker activated Caspase-3 (Figure S6A–B). Large numbers of apoptotic cells were observed in lateral regions of the midbrain, whereas few were detected in the ventral midbrain. In 10.5 dpc and 11.5 dpc embryos, apoptosis is markedly reduced relative to 9.5 dpc embryos (data not shown). Increased apoptosis, particularly at 9.5 dpc, could at least in part underlie the size reduction of the midbrain.
We next examined the progenitor domains in the ventral midbrain of En1::Cre;135a2OE. The DV extent of the Shh domain was decreased, and in many mutants analyzed (n = 6/10), Shh was not as robustly downregulated at the midline as in controls (Figure 8H–I). In one particularly severe mutant, Shh was maintained at the midline in a manner identical to En1::Cre;Lmx1bcKO embryos (Figure S6P–Q). The DV extent of the Foxa2 domain was also decreased (Figure 8J–L and S6C–D). After normalizing the DV extent of Foxa2 to the 3V, this reduction was found to be proportionate to the reduction in midbrain size (Foxa2/3V revealed an 8% decrease, n = 4, control mean = 0.28±0.02, mutant mean = 0.24±0.01; p-value = .28). Further analysis of the progenitor populations within the Foxa2 domain showed disproportionate changes. We observed a dramatic reduction in the dimensions of the Lmx1a/b+ domain, both in width (i.e. dorsal – ventral dimension) and thickness (i.e. ventricular – pial dimension). This reduction in the Lmx1a domain was not solely due to a reduction in FP size. When measured relative to Foxa2, the Lmx1a domain still appeared selectively reduced, as the width of the neighboring Nkx6.1+ domain was largely unaffected (Figure 8M–T and S6E–H).
Since Lmx1b is upstream of Wnt1/Wnt signaling, and Wnt1/Wnt signaling is important in mDA progenitor specification, we examined alterations in Wnt1/Wnt signaling in En1::Cre;135a2OE embryos. From 9.5–11.5 dpc, the levels of Wnt1 and the size of the Wnt1 expression domain, which is included within the Lmx1b domain, appeared to be reduced (Figure 9A–D). Although the reduction was not as drastic as in the En1::Cre;Lmx1bcKO, and is unlikely to singularly explain this phenotype, these results suggested that the amount of available Wnt1 ligand in the ventral midbrain was restricted.

We next examined ongoing Wnt signaling in the ventral midbrain using the Axin2::d2eGFP allele described above, which provides a transcriptional readout of robust canonical Wnt signaling [43], [44]. In control 10.5 and 11.5 dpc embryos harboring an Axin2::d2eGFP transgene, d2eGFP is observed in the ventral midbrain in two stripes adjacent to the midline. At 10.5 dpc, GFP labeling is predominantly observed outside of the Lmx1a domain, although some is detected within the domain. At 11.5 dpc, GFP labeling still exceeds the Lmx1a domain by several cell diameters along the DV axis. In comparison to controls, En1::Cre;135a2OE,Axin2::d2eGFP mutants showed diminished GFP labeling intensity within the ventral midbrain, which was nearly extinguished in particularly severe embryos (Figure 9E–H). Further, consistent with the reduction in Wnt1 ligand, the total range of GFP labeled cells in En1::Cre;135a2OE,Axin2::d2eGFP embryos was reduced along the DV axis (Figure 9I). Little to no reduction of GFP was observed in the dorsal midbrain (data not shown). Although not as severe as the En1::Cre;Lmx1bcKO, reduced canonical Wnt signaling, which is critical for early activation and maintenance of Lmx1a [17], [46], could in part, explain the diminished mDA progenitor domain. At 11.5 dpc, reduced Wnt signaling is likely to be important for aspects of neurogenesis rather than the establishment of the mDA progenitor domain.
As Lmx1b and Wnt1 are also present in the isthmus, we examined this region in En1::Cre;135a2OE embryos. Analysis of the midbrain/hindbrain junction revealed differences between the En1::Cre;Lmx1bcKO and En1::Cre;135a2OE mutants. While Lmx1b, Wnt1, and Fgf8 were modestly, but consistently, narrower in En1::Cre;135a2OE mutants, the isthmic boundary, as determined by Otx2, was unchanged (Figure S8A–H). In contrast, in the isthmus of En1::Cre;Lmx1bcKO embryos, both Wnt1 (Figure 4T) and Fgf8 (Figure S4P) were drastically reduced, and Otx2+ cells were found in the rostral hindbrain (Figure S4N).
We next examined later stage embryos to determine the consequences of early reduction of the mDA progenitor population. In 13.5 dpc En1::Cre;135a2OE embryos we observed a drastic decrease of TH+ mDAs. Quantification revealed a 66% reduction in Lmx1a+ nascent mDAs, as well as a 48% reduction in Islet+ oculomotor neurons. In contrast, Brn3a+ neuron numbers in the red nucleus [24], were not altered (Figure 10A–I), likely reflective of the largely unaffected Nkx6.1+ progenitor domain. Moreover, several Brn3a+ cells were observed at the midline in the dopaminergic field (Figure 10F–G and Figure S9A–D), which is consistent with a reduction of the Lmx1b+ mDA progenitor domain [50], [51]. Further analysis at 16.5 dpc confirmed a drastic reduction of TH+ dopamine neurons throughout the anterior-posterior axis (Figure 10J–O). At 19.5 dpc we observed that the remnant neurons express mature mDA markers including Nurr1 and DAT (Figure 10P–S); this finding is different from reported En1::Cre;Lmx1bcKO studies [52] likely because in En1::Cre;135a2OE mutants, Lmx1b is reduced but not abolished. Consistent with the reduction of dopamine neuron numbers, a concomitant decrease of mDA projections to the striatum was observed (Figure 10T–U).

Next, we reasoned that because deletion of Lmx1b with a slightly later and more specific Shh::Cre driver reveals no changes in the size of the mDA domain [51], if miR135a2 is indeed acting through the Lmx1b/Wnt axis, then using Shh::Cre to overexpress miR135a2 should have no effect on mDA domain size. To determine whether miR135a2, could elicit mDA progenitor domain changes when activated at later stages (i.e. after the early restriction to the RP, FP, and IsO) we used both Shh::Cre and Nestin (Nes)::Cre recombination. When miR135a2 overexpression was initiated at ∼8.5–9.5 dpc with Shh::Cre or ∼10.5 dpc with Nes::Cre, the DV extent of the Foxa2 and Lmx1a domains were unchanged (Figure 11).

To interpret these results, we carefully considered the spatiotemporal expression of the different Cre drivers. En1 expression is initiated at the 2 somite stage and encompasses the prospective midbrain and rhombomere 1 regions, inclusive of the IsO, by the 4–6 somite stage (8.0 dpc) [53], [54]. In contrast, Shh is initiated slightly later at the 8-somite stage (8.5 dpc) [55], specifically at the ventral midline. At this early stage, however, Shh expression is confined to a small group of medially located progenitors, which does not encompass the entire prospective mDA progenitor domain [22], [28]. Subsequently, between 8.75 and 9.5 dpc, Shh expression flares out laterally, and encompasses the entire mDA domain [22]. If one considers the recombination of the entire prospective mDA progenitor domain, there is a significant time difference between En1::Cre and Shh::Cre recombination. Nestin::Cre is active throughout this region, including the IsO, between ∼10.5–11.5 dpc [56]. Considering these recombination kinetics, our results indicate that increased miR135a2 levels are sufficient to restrict the mDA progenitor domain size only during an early critical window. Since Lmx1b is broadly expressed at the ventral midline during this time window [27], we posit that miR135a2 expression aids in the DV restriction of Lmx1b and the refinement of the mDA progenitor domain; within the mDA domain, the outer edges appear more sensitive to increased miR135a2 levels. An alternative interpretation of these results is that the spatial differences in recombination between En1::Cre and Shh::Cre, rather than solely the timing differences, lead to the differing effects on the mDA progenitor domain. Particularly, since En1::Cre recombines the entire midbrain-rhombomere 1 region, while Shh::Cre does not, it is possible in En1::Cre;135a2OE repression of the Lmx1b/Wnt1 axis results in a partial breakdown of the Wnt-Fgf loop [18], [57]. Thus, early defects in IsO activity cannot be ruled out, and could also contribute to the phenotype observed in En1::Cre;135a2OE.
By eight criteria: total midbrain size, apoptosis, Wnt1/Wnt signaling, DV extent of the FP (Foxa2/Shh domain size), mDA progenitor domain size, mDA numbers, oculomotor neuron numbers, and temporal sensitivity, the miR135a2OEs, at least partially resemble the Lmx1bcKOs (Figure S10). In several criteria tested, the En1::Cre;Lmx1bcKO is more affected than the En1::Cre;135a2OE, likely because Lmx1b levels are not completely abolished in the latter. Together with the in vitro luciferase assays, our data suggests that the En1::Cre;135a2OE phenotype is at least in part dependent on Lmx1b repression in the early embryo. Further, the En1::Cre;Lmx1bcKO and the En1::Cre;135a2OE phenotypes are similar because they may both, in part, be due to net deficits in Wnt signaling. In the En1::Cre;Lmx1bcKO, reductions in Wnt signaling are likely due to a massive deficit of Wnt1 ligand (Figure 4N). In the En1::Cre;135a2OE, reductions in Wnt signaling are likely due to modest reductions in several targets including multiple levels of the Wnt pathway (Figure 9, Figure S7, and Table 1), altogether resulting in a significant net Wnt signaling deficit.
Discussion
In this study we have identified a novel transcription factor/microRNA negative feedback loop that critically impacts Wnt1/Wnt signaling and midbrain development. Feedback circuitry inherently possesses a powerful buffering capacity, such that fluctuations in gene expression can be stabilized and protein expression in dynamically changing environments can be fine-tuned [58]. In this case, widespread Lmx1b/Wnt1/Wnt signaling must be sharply restricted over a short period (between 8.0 and 9.5 dpc), as maintenance of Lmx1b/Wnt1/Wnt signaling leads to several unwanted consequences. Here, we provide evidence that Lmx1b is upstream of miR135a2/Rmst, and that miR135a2 represses Lmx1b/Wnt1/Wnt signaling (Figure 12A). Thus, in the early midbrain, these factors may determine mDA progenitor allocation and midbrain size.

Our data indicate that microRNAs play a critical role in determining the size of the mDA progenitor pool. We found that loss of the key microRNA processing enzyme, Dicer1, in embryoid body aggregates skews the proportion of Foxa2+ progenitors in favor of an Lmx1a/b+ mDA fate over an Nkx6.1+ fate. Conversely, we found that early miR135a2 overexpression in vivo led to a disproportionate reduction in the size of the Lmx1a/b+ mDA progenitor domain. We propose that miR135a2 might influence net Lmx stoichiometry in two ways: first, by directly repressing Lmx1b, and second, by lowering Wnt1/Wnt signaling and therefore Lmx1a levels. In the early embryo, these alterations most prominently affect the outer edges of the Lmx domain, which mainly express Lmx1b, until Lmx1a is induced in this region [27]. Additionally, it is possible that lowering Wnt signaling alters proliferation, and that can in part account for the reduced mDA progenitor domain size.
We postulate that because of the reduction in Lmx1b and Wnt1/Wnt signaling in En1::Cre;135a2OE mutants, the Lmx1a+ domain fails to expand, ultimately resulting in a region where both Lmx1a and Lmx1b are extinguished and instead occupied by Nkx6.1. Supporting the notion that the boundaries are most vulnerable to changes in Lmx levels, even complete removal of Lmx1b in the En1::Cre;Lmx1bcKO results in the selective failure to establish the outer edges of the mDA progenitor pool. Taken together, our data indicate that the precise balance of Lmx1b and miR135a2 at early time points in the embryonic midbrain determines the size of the dopamine progenitor domain (Figure 12B), and ultimately affects the organization and number of neurons found within different ventral midbrain populations.
Our data show that both increased or decreased Lmx1b expression in the early embryo results in reduced oculomotor neuron numbers. In the early midbrain, Lmx1b is transiently expressed in the oculomotor primordium [50]. Based on this, one possible result was that prolonged maintenance of Lmx1b would lead to increased oculomotor neurons in En1::Cre;Lmx1bOE mutants, but we found the opposite (Figure 5N–P). Thus, we conclude that Lmx1b downregulation is critical for production of normal oculomotor numbers. miR135a2 may play a role in the timely regulation of Lmx1b in this context.
Precise and dynamic expression of the Lmx1b-miR135a2 duo is also important for determining overall midbrain size. Failure to restrict Lmx1b leads to an enlargement of the third ventricle and morphogenetic abnormalities, while excess miR135a2-mediated repression or loss of Lmx1b, leads to a reduction in third ventricle and midbrain size. Since Lmx1b drives Wnt1 expression, the phenotypes in En1::Cre;Lmx1bOE, En1::Cre;Lmx1bcKO and En1::Cre;135a2OE mutants can at least in part be attributed to alterations in early Wnt signaling. In accordance with this notion, Wnt1/Wnt signaling has been shown to determine the overall size of the midbrain by influencing cell survival and proliferation, [38], [54], [59]–[62]. The potency of Wnt1/Wnt signaling in midbrain development suggests that a method to control its stoichiometry is critical; we propose that the dose and spatiotemporal expression of Wnt1/Wnt signaling is, at least in part, determined by Lmx1b and miR135a2.
miR135a2 is predicted to regulate a large set of target genes in addition to Lmx1b. Lmx1a, a related transcription factor, is not a predicted target by any algorithm we encountered. However, some of the genes in the predicted miR135a2 target set, including Wnt1, Wnt5a, several molecules in the canonical Wnt signaling pathway (Figure 9, Figure S7, and Table 1), and Msx1/2 (Anderegg, unpublished observations), may play a critical role in midbrain development and contribute to the observed phenotypes. Moreover, Otx2, a transcription factor also upstream and downstream of Wnt1/beta-catenin signaling [17], [29], [63], whose dosage determines the size of the mDA progenitor pool [63], is a predicted target of the closely related miR135b (Anderegg, unpublished observations). Since the seed is identical in these two microRNAs, it is possible that Otx2 dosage is also controlled, in part, by miR135a2, although our luciferase assays could not detect a significant interaction (see Figure S7B).
It also remains possible that miR135a2 acts on other signaling pathways important for midbrain development including TGF-beta/Bmp [64], [65], Shh [66], and Fgf [54], [67], either directly or indirectly through points of crosstalk. In fact, the mDA phenotype observed in En1::Cre;135a2OE mutants bears some resemblance to that of En1::Cre;Fgfr triple knockout mice [67]. However, parallel data obtained from miR135a2 overexpression in the forebrain resulting in phenotypes that overlap Wnt mutants, supports the notion that defects in Wnt signaling contribute to the observed midbrain phenotype (Caronia-Brown, unpublished observations). Additionally, in colorectal cancers, miR135a targets the Wnt pathway, although in that context, with net positive effect [68]. By potentially targeting multiple levels of the Wnt pathway (see Table 1), including upstream transcription factors (Lmx1b and Otx2), ligands, positive and negative modifiers, and downstream targets it is likely that miR135a2 confers precision to the stability of the overall early Wnt regulatory network responsible for midbrain size and dopamine progenitor specification. In this context, it is conceivable that the modest increase in miR135a2 levels in En1::Cre;135a2OE modulates multiple Wnt pathway targets resulting in a net negative Wnt signal; in contrast, in En1::Cre;Lmx1bcKO, a net negative Wnt signal is primarily obtained by severe downregulation of Wnt1.
Our model is based on several lines of evidence derived from the mutants presented here. Our data suggest that miR135a2 is sufficient to modulate canonical Wnt signaling and the mDA progenitor phenotype, but it does not address whether miR135a2 is necessary for regulation of these phenotypes. Future loss of function studies are important to address this question and are currently underway. Since single microRNA knockouts often have subtle or no phenotypes, a double knockout of miR135a2 and closely related miR135b will likely be required to unmask the functions of this microRNA family [69]. Given the importance of the Wnt pathway in disease and cancer, however, the gain of function experiments described here are an important advance, as they provide a novel modality for targeting this pathway.
The Lmx1b-miR135a2 pair appears to be an integral component in the molecular circuitry governing establishment of mDA progenitors. Here we have demonstrated that this pair determines early aspects of mDA progenitor domain allocation, likely via direct effects on the ventral midbrain but possibly through IsO activity as well. However, after the size and spatial boundaries of the dopamine progenitor domain are firmly established, it is likely that miR135a2 has a distinct, later role of tuning, and ultimately downregulating Lmx1b/Wnt1 expression within the mDA progenitor pool (see Figure 3). This later role could be to tune optimal levels of Wnt signaling in the mDA domain, as well as aspects of neurogenesis. Thus, understanding this circuit may shed light on dopamine neuron numbers, and may therefore be relevant for understanding dopamine related disorders and susceptibilities to these conditions.
Generating large numbers of bona fide mDAs is a key goal of stem cell based approaches towards Parkinson's disease treatment. Previously, this process was limited by the inability to produce authentic mDAs that survived grafting. Recently this problem was overcome, using Wnt1/Wnt agonists to program hESCs towards a bona fide mDA fate [70], [71]. In this context, understanding agents that modulate Wnt1/Wnt signaling is of critical importance. It is conceivable, that just as iPS cells can now be programmed using microRNAs [72], mDAs will be derived from stem cells with the aid of a rationally designed cocktail of microRNAs and anti-microRNAs.
Materials and Methods
Ethics statement
All mouse work was done in accordance with Northwestern University's ACUC guidelines.
MicroRNA array
Tissue was microdissected from the ventral midline and dorsal lateral midbrain of unfixed, coronally sectioned 11.5 dpc Swiss Webster embryos. Total RNA, including small RNAs, was extracted using the mirVana kit (Ambion). The TaqMan Rodent MicroRNA A+B Cards Set v2.0 (4400239) was used to perform the array and validation of individual miRs was done with TaqMan PCR Assays for miR135a (ID 000460), miR135b (ID 002261), and normalized to snoRNA202 (ID 001232)(Applied Biosystems)(see below).
Quantitative Real Time RT-PCR (qRT-PCR)
For miR135a analysis in various mutant lines, tissue was dissected from littermates and snap frozen on liquid nitrogen. The meso-diencephalic region was dissected from older embryos (9.75 dpc En1::Cre;Lmx1bOE, 9.75 dpc En1::Cre;Lmx1bcKO, and 11.5 dpc Nes::Cre;135a2OE), while the whole head was used for younger embryos (8.75 dpc En1::Cre;135a2OE). Total RNA, including small RNAs, was extracted using the mirVana kit (Ambion). The TaqMan PCR Assay for miR135a (ID 000460) was used and normalized to snoRNA202 (ID 001232)(Applied Biosystems).
For transcription factor analysis, ∼8.5 dpc En1::Cre;135a2OE littermates were used. A cut was made just caudal to the future rhombomere 1, and the tissue from the head was snap frozen on liquid nitrogen. Total RNA was extracted using the mirVana kit (Ambion) then the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to generate cDNA. The TaqMan gene assay for Lmx1b (Mm00440209_m1) was used to analyze RNA from 8–14 somite embryos, and the TaqMan gene assay for Otx2 (Mm00446859_m1) was used to analyze RNA from 13–23 somite embryos. Gene expression was normalized to Gapdh (Mm99999915_g1)(Applied Biosystems).
For all qRT-PCR experiments, the amount of total RNA per reaction was the same for each sample. The delta Ct method was used to calculate the relative fold changes in gene expression. All statistical values were calculated using a two-sample equal variance, two tailed, Student's t-Test.
Rmst RT-PCR
Ventral midbrain tissue was dissected from 11.5 dpc Swiss Webster embryos and flash frozen on liquid nitrogen. Total RNA, including small RNAs, was extracted using the mirVana kit (Ambion). A cDNA library was synthesized using SuperScript II Reverse Transcriptase (Invitrogen) and random hexamers. PCR was performed with the following primers:
exon 12 (F) GCTAGCTCGAGAGCCACGCTCTTTCCCAACAC
exon 15 (F) GCTAGCTCGAGATGAATGTTGTCGACACTGTCCCATC
exon 16 (R) TCAGGAAGCTTTCTTTCTCAAAGGTCCAGCTTAGATCC
In Situs (LNA, whole mount, and section)
For locked-nucleic acid (LNA) in situ, the GEISHA project microRNA detection protocol version 1.1 (http://geisha.arizona.edu) was used. For whole mount in situ, embryos were harvested and fixed in 4% DEPC PFA in PBS overnight, rinsed in PBS, put through a methanol series, and then stored in 100% methanol at −20°C until used. For section in situ, embryos were harvested and fixed in 4% DEPC PFA in PBS overnight and prepared for cryosectioning at 20 µm. In situ hybridization was performed with single-stranded digoxigenin labeled riboprobes directed against the following mRNAs: FGF8 (C. Tabin), Lmx1a (K. Millen), Lmx1b (C. Tabin), LNA probe 135a (Exiqon 39037-01), Otx2 (C. Tabin), miR135a2/Rmst probe A (Genbank AI853140; B. Harfe), miR135a2/Rmst probe B (Genbank BI734849; B. Harfe), Shh (A. McMahon), and Wnt1 (A. McMahon).
Embryonic stem cell differentiation and quantification
An optimized protocol for dopamine neuron differentiation from embryoid body aggregates was modified from motor neuron differentiation protocol [49] and as described below. An ESC line that harbored a CAG::CreERT2 construct and Dicer1floxed/floxed alleles [5] was transiently treated with the FGF inhibitors PD173074 (Sigma, 50 nM as final concentration) from days 2–4 of culture. Then, on day 4 of culture, Smoothened agonist (EMD 566660, 15 nM as final concentration) and mouse recombinant FGF8b protein (Cell Signaling, 100 ng/ML as final concentration) were added for 4 days. Concomitantly, either control solvent (Ethanol) or 4-hydroxy tamoxifen (4OHT) (Sigma, 250∼500 nM as final concentration) was added for 2 days. On day 9, embryoid bodies were fixed in 4% PFA, cryosectioned, and immunolabeled. Images were collected on a Zeiss LSM510 confocal microscope. MetaMorph Software (Molecular Devices) was used to quantify Lmx1a, Lmx1b, and Nkx6.1 levels in Foxa2+ cells of 15 embryoid bodies (5 each from 3 independent experiments, n = 3).
Luciferase assays
The stem-loop of mmu-miR-135a-2 (accession MI0000715) plus ∼100 bp 5′ and 3′ flanking sequence was PCR purified from genomic DNA and then cloned into the pCAG-RFP-int vector (Addgene plasmid 19822). A fragment of each target gene 3′UTR, which corresponds to the region predicted to contain a mmu-miR-135a-2 seed match, was PCR purified from genomic DNA and then cloned into the pmirGLO vector (Promega E1330). The predicted miR135a2 binding site in the Lmx1b 3′UTR was directly mutated by using the Phusion site-directed mutagenesis kit (Thermo Scientific) to create constructs with scrambled or deleted binding sites. The success of mutagenesis was confirmed by sequencing. The following primers, including restriction sites, were used for cloning:
135a2 (F) GCTAGGCGATCGCGTGTGCTTTGTGTCCCTTACATGTAGC
135a2 (R) TTAGGACGCGTGACACTCAAGGAACACCAAAGAGG
Lmx1b (F) GGATCTAGATCCATGCAGAGCTCCTACTTTG
Lmx1b (R) CAGTCTAGAAAGTGACTGTCCAAGAGCTCTGGGTC
Lmx1b-M1 (F): TCAGACTCTTCAGACCAATCAGCGGTGCCCTCCCCT
Lmx1b-M1 (R): GCGGGTGGTGGGCTGGGGG
Lmx1b-M2 (F): CGCTCAGACTCTTCAGACTAGGCTACGGTGCCCT
Lmx1b-M2 (R): GGTGGTGGGCTGGGGGGCC
Lmx1b-del (F): ACGGTGCCCTCCCCTCGGCCAGCC
Lmx1b-del (R): GTCTGAAGAGTCTGAGCGGGTGGTGGGCT
Ccnd1 (F): GCTAGGCTAGCGGTCTGCTTGACTTTCCCAACC
Ccnd1 (R): TCAGGCTCGAGTGACAGGACGATCGCCATCAG
Gsk3b (F): GCTAGGCTAGCAAGGATCATGGCAGGATCCCAG
Gsk3b (R): TCAGGCTCGAGTGTGGAGTGGGCAAAGGTGC
Otx2 (F): GCTAGGCTAGCAGGTTTTGTGAAGACCTGTAGAAGC
Otx2 (R): TCAGGCTCGAGTAGGACAATCAGTCGCACAATC
Tcf7l2 (F): GCTAGGCTAGCCTTGCTGTACCTGTATGTCCGTCC
Tcf7l2 (R): TCAGGCTCGAGCACAGGGCAGTTGACTAGGAGGT
Wnt1 (F): GCTAGCTCGAGTTCTGCACGAGTGTCTATGAGGTG
Wnt1 (R): TTAGGGTCGACAAGGGCGCCTATGAGAAGCTG
The plasmids were co-transfected into 24-well plates of HEK293 cells using Lipofectamine 2000 (Invitrogen #11668). After 24 hours, the cells were harvested for a luciferase assay (Promega E1910) and measured with the Clarity Luminescence Microplate Reader (Bio-Tek). Each Firefly reading was normalized to Renilla and replicated 8 times each in 2 separate experiments.
Generating and testing the miR135a2OE transgene
To generate the 135a2OE transgene, we isolated a 324 bp fragment of genomic DNA that contains the precursor sequence of miR135a2, using the 135a2 forward and reverse primers described above, and cloned it into different mammalian expression vectors. Ultimately, after testing four different vectors in vitro, CAG-loxP-STOPr-loxP-miR135a2-IRESeGFP was determined to have the least leakiness even though the STOP cassette was in the reverse orientation. The transgene was microinjected into B6SJL at Northwestern University's Transgenic and Targeted Mutagenesis Laboratory. Six founder lines were obtained, and three separate lines (En1::Cre;135a2OE #2, #3, and #5) were used for experiments. All three show a very similar phenotype, in terms of overall midbrain size as well as reduced mDA progenitors, thus ruling out the possibility of site-of-integration dependent phenotypes.
To assess the miR135a expression levels in different mutant scenarios we performed qRT-PCR (see above). When En1::Cre is used to recombine the transgene, we detect a ∼3 fold increase of miR135a in 8.75 dpc mutants compared to littermate controls. When Nes::Cre is used to recombine the transgene a few days later, we detect modest overexpression of miR135a in 11.5 dpc mutants (See Figure S5D), but the quantification in mDAs is complicated by high endogenous levels of miR135a in controls at this stage, particularly in cells exiting the ventricular zone throughout the midbrain (See Figure 4K and Figure S1B). Separate analysis indicates that endogenous miR135a levels increase ∼8 fold between 8.5 dpc control samples and 11.5 dpc control samples (data not shown). eGFP activity was undetectable from this transgene in all scenarios tested, as it is likely degraded during microRNA processing.
Generation of sensor transgenics
For the miR135a2 “sensor” experiment, we designed a transgene, CAG-eGFP-WPRE-5XmiR135a2rc, which contained five exact miR135a2 reverse complement repeats, each separated by a GGCCGGCC spacer. For a control, we designed a similar transgene, CAG-tdTomato-WPRE with no complementary miR135a2 sites in the 3′UTR. The transgenes were co-injected into BL6SJL embryos and transient transgenics were harvested at 11.5 dpc or 12.5 dpc.
Mouse husbandry and genotyping
For Lmx1b overexpression experiments, male mice harboring an allele in which Cre recombinase was knocked in to the En1 locus [41] were bred to mice harboring the Rosa26Lmx1b/+ allele [42]. For Lmx1b deletion experiments, mice harboring an allele of Lmx1b with loxP sites flanking the homeodomain in exons 4–6 were used [45]. En1::Cre;Lmx1bF/+ males were bred to Lmx1bF/F females to obtain mutant embryos. Both En1::Cre- and En1::Cre+;Lmx1bF/+ littermates were used as controls, as no phenotype was observed in heterozygotes. For miR135a2 overexpression experiments, we bred females harboring the 135a2OE allele (described above) with males harboring 1) the En1::Cre knock-in allele, 2) an allele in which a GFP-Cre fusion cassette was knocked in to the Shh locus [8] or 3) a Nes::Cre transgene, expressed under control of the rat Nestin promoter and enhancer [73]. To assess Wnt signaling, we used mice harboring an allele in which destabilized eGFP was placed under control of the Axin2 promoter, exon 1 and intron 1 (Axin2::d2eGFP) [43]. For all matings, the morning when a vaginal plug was detected was designated as 0.5 days post coitum (dpc). Mice were maintained and sacrificed according to the protocols approved by the Northwestern University Animal Care and Use Committee.
Antibody labeling
Embryos were harvested and fixed in 0.2%–4% PFA in PBS for various amounts of time depending upon embryonic ages, and sectioned at 20 µm. Tissue sections were post-fixed in 1%–4% PFA in PBS, rinsed in PBS, antigen retrieved depending on the antibody (Vector Labs H-3301), blocked in 5% donkey serum, 0.1% Triton X-100 in PBS, and incubated overnight at 4°C with primary antibodies diluted in blocking solution: rabbit Active Caspase-3 (Cell Technology; 1∶1000), rat DAT (Santa Cruz; 1∶50), goat Foxa2 (Santa Cruz; 1∶50), rabbit GFP (Molecular Probes; 1∶1500), mouse Islet-1 (Developmental Studies Hybridoma Bank; 1∶100), guinea pig Lmx1a (Y-C. Ma; 1∶20,000), rabbit Lmx1b (custom; 1∶5,000), mouse Nkx6.1 (Developmental Studies Hybridoma Bank; 1∶100), rabbit Nurr1 (Santa Cruz; 1∶500), rabbit Pitx3 (Zymed; 1∶500), and sheep TH (Pel Freeze; 1∶250). Sections were rinsed in PBS and incubated with appropriate Alexa 488, 555, 647 (Molecular Probes) or Cy3 and Cy5 (Jackson Immuno Research) secondary antibodies diluted 1∶250 in blocking solution, rinsed in PBS, covered with DAPI (1 mg/mL; Sigma) in PBS, rinsed in PBS, and coverslipped followed by epifluorescent (Leica) or confocal microscopy (Zeiss LSM 510 META laser scanning or Leica DM6000 CFS). Images were processed in Adobe Photoshop CS2.
Measurements, neuron counts, and statistical analysis
To measure the intensity of eGFP and tdTomato fluorescence in 11.5 dpc transient transgenic embryos, images of unstained coronal sections were taken with a Leica DM6000 CFS microscope. After alignment in Adobe Photoshop, such that the sections were equally matched along the anterior-posterior axis, a representative rostral, midlevel and caudal section was selected from each embryo. Using the line scan measurement tool in the Leica Application Suite Advanced Fluorescence Software, two lines were drawn in the medial and lateral domain of each section and the mean fluorescence intensity was recorded. For measurements of the third ventricle and overall midbrain area, coronal sections from En1::Cre;Lmx1bOE, En1::Cre;Lmx1bcKO, or En1::Cre;135a2OE littermates were aligned in Adobe Photoshop, such that they were equally matched along the anterior-posterior axis. For four representative sections, the edges of the ventricular and pial surfaces were traced, and then both the perimeter and area were calculated using the “Analyze” function in NIH ImageJ. To calculate tissue area, the ventricular area was subtracted from the pial area. To determine FP and mDA progenitor domain size, coronal sections from En1::Cre;Lmx1bOE, En1::Cre;Lmx1bcKO, En1::Cre;135a2OE, Shh::Cre;135a2OE, or Nes::Cre;135a2OE littermates were immunostained for Foxa2/Lmx1a/Nkx6.1 and then aligned in Adobe Photoshop, such that they were equally matched along the anterior-posterior axis. For every third section, the distances along the ventricular surface from 1) the ventral midline to the dorsal edge of the Lmx1a domain, and 2) the ventral midline to the dorsal edge of the Foxa2 domain were calculated using the “segmented line measure” function in ImageJ. To account for changes in overall midbrain size, the dorsal-ventral extent of the Lmx1a domain was normalized to the extent of the Foxa2 domain by dividing the first measurement by the second. To quantify the amount of different ventral neuron types in En1::Cre;Lmx1bOE, En1::Cre;Lmx1bcKO, and En1::Cre;135a2OE littermates, coronal sections were immunostained with combinations of Brn3a/Islet/Lmx1a/TH and then aligned in Adobe Photoshop, such that they were equally matched along the anterior-posterior axis. One-half of a representative rostral, midlevel and caudal section were chosen for manual counting from each embryo, except where noted below. Islet was counted from every third section in En1::Cre;Lmx1bOE and En1::Cre;135a2OE, and Brn3a was counted from every sixth section in En1::Cre;Lmx1bOE. All statistical values were calculated using a two-sample equal variance, two tailed, Student's t-Test.
Bioinformatic analysis
To determine miR135a targets, we used TargetScan, MicroCosm, miRanda, and miRDB algorithms. Wnt pathway genes were obtained from the following references [13], [74], [75].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KhudayberdievS, FioreR, SchrattG (2009) MicroRNA as modulators of neuronal responses. Commun Integr Biol 2 : 411–413.
2. FinebergSK, KosikKS, DavidsonBL (2009) MicroRNAs potentiate neural development. Neuron 64 : 303–309.
3. HeX, YuY, AwatramaniR, LuQR (2012) Unwrapping myelination by microRNAs. Neuroscientist 18 : 45–55.
4. YunB, AndereggA, MenichellaD, WrabetzL, FeltriML, et al. (2010) MicroRNA-deficient Schwann cells display congenital hypomyelination. J Neurosci 30 : 7722–7728.
5. ChenJA, HuangYP, MazzoniEO, TanGC, ZavadilJ, et al. (2011) Mir-17-3p controls spinal neural progenitor patterning by regulating Olig2/Irx3 cross-repressive loop. Neuron 69 : 721–735.
6. Shi Y, Zhao X, Hsieh J, Wichterle H, Impey S, et al. MicroRNA regulation of neural stem cells and neurogenesis. J Neurosci 30 : 14931–14936.
7. PengC, LiN, NgYK, ZhangJ, MeierF, et al. (2012) A unilateral negative feedback loop between miR-200 microRNAs and Sox2/E2F3 controls neural progenitor cell-cycle exit and differentiation. J Neurosci 32 : 13292–13308.
8. HarfeBD, ScherzPJ, NissimS, TianH, McMahonAP, et al. (2004) Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118 : 517–528.
9. DessaudE, YangLL, HillK, CoxB, UlloaF, et al. (2007) Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450 : 717–720.
10. AhnS, JoynerAL (2004) Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 118 : 505–516.
11. MartelloG, ZacchignaL, InuiM, MontagnerM, AdornoM, et al. (2007) MicroRNA control of Nodal signalling. Nature 449 : 183–188.
12. InestrosaNC, ArenasE (2010) Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci 11 : 77–86.
13. Alves dos SantosMT, SmidtMP (2011) En1 and Wnt signaling in midbrain dopaminergic neuronal development. Neural Dev 6 : 23.
14. UlloaF, BriscoeJ (2007) Morphogens and the control of cell proliferation and patterning in the spinal cord. Cell Cycle 6 : 2640–2649.
15. AnderssonER, SaltoC, VillaescusaJC, CajanekL, YangS, et al. (2013) Wnt5a cooperates with canonical Wnts to generate midbrain dopaminergic neurons in vivo and in stem cells. Proc Natl Acad Sci U S A 110: E602–610.
16. YangJ, BrownA, EllisorD, PaulE, HaganN, et al. (2013) Dynamic temporal requirement of Wnt1 in midbrain dopamine neuron development. Development 140 : 1342–1352.
17. JoksimovicM, YunBA, KittappaR, AndereggAM, ChangWW, et al. (2009) Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat Neurosci 12 : 125–131.
18. ChilovD, SinjushinaN, Saarimaki-VireJ, TaketoMM, PartanenJ (2010) beta-Catenin regulates intercellular signalling networks and cell-type specific transcription in the developing mouse midbrain-rhombomere 1 region. PLoS ONE 5: e10881.
19. TangM, MiyamotoY, HuangEJ (2009) Multiple roles of beta-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development 136 : 2027–2038.
20. TangM, VillaescusaJC, LuoSX, GuitarteC, LeiS, et al. (2010) Interactions of Wnt/beta-catenin signaling and sonic hedgehog regulate the neurogenesis of ventral midbrain dopamine neurons. J Neurosci 30 : 9280–9291.
21. CajanekL, RibeiroD, ListeI, ParishCL, BryjaV, et al. (2009) Wnt/beta-catenin signaling blockade promotes neuronal induction and dopaminergic differentiation in embryonic stem cells. Stem Cells 27 : 2917–2927.
22. JoksimovicM, AndereggA, RoyA, CampochiaroL, YunB, et al. (2009) Spatiotemporally separable Shh domains in the midbrain define distinct dopaminergic progenitor pools. Proc Natl Acad Sci U S A 106 : 19185–19190.
23. Blaess S, Bodea GO, Kabanova A, Chanet S, Mugniery E, et al. Temporal-spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev 6 : 29.
24. AgarwalaS, RagsdaleCW (2002) A role for midbrain arcs in nucleogenesis. Development 129 : 5779–5788.
25. NakataniT, MinakiY, KumaiM, OnoY (2007) Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development 134 : 2783–2793.
26. PrakashN, PuellesE, FreudeK, TrumbachD, OmodeiD, et al. (2009) Nkx6-1 controls the identity and fate of red nucleus and oculomotor neurons in the mouse midbrain. Development 136 : 2545–2555.
27. AnderssonE, TryggvasonU, DengQ, FrilingS, AlekseenkoZ, et al. (2006) Identification of intrinsic determinants of midbrain dopamine neurons. Cell 124 : 393–405.
28. BlaessS, BodeaGO, KabanovaA, ChanetS, MugnieryE, et al. (2011) Temporal-spatial changes in Sonic Hedgehog expression and signaling reveal different potentials of ventral mesencephalic progenitors to populate distinct ventral midbrain nuclei. Neural Dev 6 : 29.
29. PrakashN, BrodskiC, NaserkeT, PuellesE, GogoiR, et al. (2006) A Wnt1-regulated genetic network controls the identity and fate of midbrain-dopaminergic progenitors in vivo. Development 133 : 89–98.
30. TangM, MiyamotoY, HuangEJ (2009) Multiple roles of {beta}-catenin in controlling the neurogenic niche for midbrain dopamine neurons. Development 136 : 2027–2038.
31. Castelo-BrancoG, WagnerJ, RodriguezFJ, KeleJ, SousaK, et al. (2003) Differential regulation of midbrain dopaminergic neuron development by Wnt-1, Wnt-3a, and Wnt-5a. Proc Natl Acad Sci U S A 100 : 12747–12752.
32. JoksimovicM, PatelM, TaketoMM, JohnsonR, AwatramaniR (2012) Ectopic wnt/beta-catenin signaling induces neurogenesis in the spinal cord and hindbrain floor plate. PLoS ONE 7: e30266.
33. LewisBP, BurgeCB, BartelDP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120 : 15–20.
34. MansfieldJH, HarfeBD, NissenR, ObenauerJ, SrineelJ, et al. (2004) MicroRNA-responsive ‘sensor’ transgenes uncover Hox-like and other developmentally regulated patterns of vertebrate microRNA expression. Nat Genet 36 : 1079–1083.
35. RockJR, LopezMC, BakerHV, HarfeBD (2007) Identification of genes expressed in the mouse limb using a novel ZPA microarray approach. Gene Expr Patterns 8 : 19–26.
36. UhdeCW, VivesJ, JaegerI, LiM (2010) Rmst is a novel marker for the mouse ventral mesencephalic floor plate and the anterior dorsal midline cells. PLoS ONE 5: e8641.
37. GromakN (2012) Intronic microRNAs: a crossroad in gene regulation. Biochem Soc Trans 40 : 759–761.
38. GuoC, QiuHY, HuangY, ChenH, YangRQ, et al. (2007) Lmx1b is essential for Fgf8 and Wnt1 expression in the isthmic organizer during tectum and cerebellum development in mice. Development 134 : 317–325.
39. AdamsKA, MaidaJM, GoldenJA, RiddleRD (2000) The transcription factor Lmx1b maintains Wnt1 expression within the isthmic organizer. Development 127 : 1857–1867.
40. StarkA, BrenneckeJ, BushatiN, RussellRB, CohenSM (2005) Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123 : 1133–1146.
41. KimmelRA, TurnbullDH, BlanquetV, WurstW, LoomisCA, et al. (2000) Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev 14 : 1377–1389.
42. LiY, QiuQ, WatsonSS, SchweitzerR, JohnsonRL (2010) Uncoupling skeletal and connective tissue patterning: conditional deletion in cartilage progenitors reveals cell-autonomous requirements for Lmx1b in dorsal-ventral limb patterning. Development 137 : 1181–1188.
43. JhoEH, ZhangT, DomonC, JooCK, FreundJN, et al. (2002) Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22 : 1172–1183.
44. ZengYA, NusseR (2010) Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 6 : 568–577.
45. ZhaoZQ, ScottM, ChiechioS, WangJS, RennerKJ, et al. (2006) Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci 26 : 12781–12788.
46. ChungS, LeungA, HanBS, ChangMY, MoonJI, et al. (2009) Wnt1-lmx1a forms a novel autoregulatory loop and controls midbrain dopaminergic differentiation synergistically with the SHH-FoxA2 pathway. Cell Stem Cell 5 : 646–658.
47. LinW, MetzakopianE, MavromatakisYE, GaoN, BalaskasN, et al. (2009) Foxa1 and Foxa2 function both upstream of and cooperatively with Lmx1a and Lmx1b in a feedforward loop promoting mesodiencephalic dopaminergic neuron development. Dev Biol 333 : 386–396.
48. NakataniT, KumaiM, MizuharaE, MinakiY, OnoY (2010) Lmx1a and Lmx1b cooperate with Foxa2 to coordinate the specification of dopaminergic neurons and control of floor plate cell differentiation in the developing mesencephalon. Dev Biol 339 : 101–113.
49. WichterleH, LieberamI, PorterJA, JessellTM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110 : 385–397.
50. DengQ, AnderssonE, HedlundE, AlekseenkoZ, CoppolaE, et al. (2011) Specific and integrated roles of Lmx1a, Lmx1b and Phox2a in ventral midbrain development. Development 138 : 3399–3408.
51. YanCH, LevesqueM, ClaxtonS, JohnsonRL, AngSL (2011) Lmx1a and lmx1b function cooperatively to regulate proliferation, specification, and differentiation of midbrain dopaminergic progenitors. J Neurosci 31 : 12413–12425.
52. SmidtMP, AsbreukCH, CoxJJ, ChenH, JohnsonRL, et al. (2000) A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat Neurosci 3 : 337–341.
53. DavisCA, JoynerAL (1988) Expression patterns of the homeo box-containing genes En-1 and En-2 and the proto-oncogene int-1 diverge during mouse development. Genes Dev 2 : 1736–1744.
54. ChiCL, MartinezS, WurstW, MartinGR (2003) The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 130 : 2633–2644.
55. EchelardY, EpsteinDJ, St-JacquesB, ShenL, MohlerJ, et al. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75 : 1417–1430.
56. VernayB, KochM, VaccarinoF, BriscoeJ, SimeoneA, et al. (2005) Otx2 regulates subtype specification and neurogenesis in the midbrain. J Neurosci 25 : 4856–4867.
57. LiuA, LososK, JoynerAL (1999) FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development 126 : 4827–4838.
58. HornsteinE, ShomronN (2006) Canalization of development by microRNAs. Nat Genet 38 Suppl: S20–24.
59. InestrosaNC, ArenasE (2010) Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci 11 : 77–86.
60. PanhuysenM, Vogt WeisenhornDM, BlanquetV, BrodskiC, HeinzmannU, et al. (2004) Effects of Wnt1 signaling on proliferation in the developing mid-/hindbrain region. Mol Cell Neurosci 26 : 101–111.
61. MatsunagaE, KatahiraT, NakamuraH (2002) Role of Lmx1b and Wnt1 in mesencephalon and metencephalon development. Development 129 : 5269–5277.
62. McMahonAP, BradleyA (1990) The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 62 : 1073–1085.
63. OmodeiD, AcamporaD, MancusoP, PrakashN, Di GiovannantonioLG, et al. (2008) Anterior-posterior graded response to Otx2 controls proliferation and differentiation of dopaminergic progenitors in the ventral mesencephalon. Development 135 : 3459–3470.
64. CaiJ, SchleidtS, Pelta-HellerJ, HutchingsD, CannarsaG, et al. (2013) BMP and TGF-beta pathway mediators are critical upstream regulators of Wnt signaling during midbrain dopamine differentiation in human pluripotent stem cells. Dev Biol 376 : 62–73.
65. FalkS, WurdakH, IttnerLM, IlleF, SumaraG, et al. (2008) Brain area-specific effect of TGF-beta signaling on Wnt-dependent neural stem cell expansion. Cell Stem Cell 2 : 472–483.
66. Perez-BalaguerA, PuellesE, WurstW, MartinezS (2009) Shh dependent and independent maintenance of basal midbrain. Mech Dev 126 : 301–313.
67. Saarimaki-VireJ, PeltopuroP, LahtiL, NaserkeT, BlakAA, et al. (2007) Fibroblast growth factor receptors cooperate to regulate neural progenitor properties in the developing midbrain and hindbrain. J Neurosci 27 : 8581–8592.
68. NagelR, le SageC, DiosdadoB, van der WaalM, Oude VrielinkJA, et al. (2008) Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res 68 : 5795–5802.
69. MendellJT, OlsonEN (2012) MicroRNAs in stress signaling and human disease. Cell 148 : 1172–1187.
70. KriksS, ShimJW, PiaoJ, GanatYM, WakemanDR, et al. (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480 : 547–51.
71. KirkebyA, GrealishS, WolfDA, NelanderJ, WoodJ, et al. (2012) Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep 1 : 703–714.
72. Anokye-DansoF, TrivediCM, JuhrD, GuptaM, CuiZ, et al. (2011) Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8 : 376–388.
73. TroncheF, KellendonkC, KretzO, GassP, AnlagK, et al. (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23 : 99–103.
74. MacdonaldBT, SemenovMV, HeX (2007) SnapShot: Wnt/beta-catenin signaling. Cell 131 : 1204.
75. TanakaSS, KojimaY, YamaguchiYL, NishinakamuraR, TamPP (2011) Impact of WNT signaling on tissue lineage differentiation in the early mouse embryo. Dev Growth Differ 53 : 843–856.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy