
A Hereditary Spastic Paraplegia Mouse Model Supports a Role of ZFYVE26/SPASTIZIN for the Endolysosomal System
Hereditary spastic paraplegias (HSPs) are characterized by progressive weakness and spasticity of the legs because of the degeneration of cortical motoneuron axons. SPG15 is a recessively inherited HSP variant caused by mutations in the ZFYVE26 gene and is additionally characterized by cerebellar ataxia, mental decline, and progressive thinning of the corpus callosum. ZFYVE26 encodes the FYVE domain-containing protein ZFYVE26/SPASTIZIN, which has been suggested to be associated with the newly discovered adaptor protein 5 (AP5) complex. We show that Zfyve26 is broadly expressed in neurons, associates with intracellular vesicles immunopositive for the early endosomal marker EEA1, and co-fractionates with a component of the AP5 complex. As the function of ZFYVE26 in neurons was largely unknown, we disrupted Zfyve26 in mice. Zfyve26 knockout mice do not show developmental defects but develop late-onset spastic paraplegia with cerebellar ataxia confirming that SPG15 is caused by ZFYVE26 deficiency. The morphological analysis reveals axon degeneration and progressive loss of both cortical motoneurons and Purkinje cells in the cerebellum. Importantly, neuron loss is preceded by accumulation of large intraneuronal deposits of membrane-surrounded material, which co-stains with the lysosomal marker Lamp1. A density gradient analysis of brain lysates shows an increase of Lamp1-positive membrane compartments with higher densities in Zfyve26 knockout mice. Increased levels of lysosomal enzymes in brains of aged knockout mice further support an alteration of the lysosomal compartment upon disruption of Zfyve26. We propose that SPG15 is caused by an endolysosomal membrane trafficking defect, which results in endolysosomal dysfunction. This appears to be particularly relevant in neurons with highly specialized neurites such as cortical motoneurons and Purkinje cells.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1003988
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003988
Summary
Hereditary spastic paraplegias (HSPs) are characterized by progressive weakness and spasticity of the legs because of the degeneration of cortical motoneuron axons. SPG15 is a recessively inherited HSP variant caused by mutations in the ZFYVE26 gene and is additionally characterized by cerebellar ataxia, mental decline, and progressive thinning of the corpus callosum. ZFYVE26 encodes the FYVE domain-containing protein ZFYVE26/SPASTIZIN, which has been suggested to be associated with the newly discovered adaptor protein 5 (AP5) complex. We show that Zfyve26 is broadly expressed in neurons, associates with intracellular vesicles immunopositive for the early endosomal marker EEA1, and co-fractionates with a component of the AP5 complex. As the function of ZFYVE26 in neurons was largely unknown, we disrupted Zfyve26 in mice. Zfyve26 knockout mice do not show developmental defects but develop late-onset spastic paraplegia with cerebellar ataxia confirming that SPG15 is caused by ZFYVE26 deficiency. The morphological analysis reveals axon degeneration and progressive loss of both cortical motoneurons and Purkinje cells in the cerebellum. Importantly, neuron loss is preceded by accumulation of large intraneuronal deposits of membrane-surrounded material, which co-stains with the lysosomal marker Lamp1. A density gradient analysis of brain lysates shows an increase of Lamp1-positive membrane compartments with higher densities in Zfyve26 knockout mice. Increased levels of lysosomal enzymes in brains of aged knockout mice further support an alteration of the lysosomal compartment upon disruption of Zfyve26. We propose that SPG15 is caused by an endolysosomal membrane trafficking defect, which results in endolysosomal dysfunction. This appears to be particularly relevant in neurons with highly specialized neurites such as cortical motoneurons and Purkinje cells.
Introduction
Hereditary spastic paraplegias (HSPs/SPGs) are a heterogeneous group of neurodegenerative movement disorders, which present with progressive weakness and spasticity of the legs. The unifying neuropathology is a time - and length-dependent degeneration of cortical motoneuron axons. Whereas in pure HSP other cells of the central nervous system are usually spared, neurodegeneration in complex HSP is more widespread and results in additional clinical symptoms. The age at onset and the course of disease are highly variable [1]–[3]. The clinical variability is mirrored by more than 50 spastic paraplegia gene loci, characterizing HSP as a genetically highly heterogeneous disease. HSP is therefore considered a model disease for unraveling the various requirements for long-term axon survival [4].
SPG15 is a complex autosomal recessive form of HSP associated with mutations in ZFYVE26 [5]. In addition to the spastic gait disorder, patients also suffer from progressive thinning of the corpus callosum, cognitive impairment, cerebellar ataxia, Parkinsonism, bladder dysfunction, and macular degeneration [5], [6]. However, there is considerable variability of the clinical presentation between affected individuals from different families and even within families [5]–[7]. The ZFYVE26 gene encodes the 285 kD protein ZFYVE26 (also called SPASTIZIN or FYVE-CENT) that is predicted to contain a coiled-coil and a FYVE domain [5]. Coiled-coil domains often mediate protein-protein interactions [8], whereas FYVE domains target proteins to intracellular membranes enriched for phosphatidylinositol 3-phosphate [9], such as endosomal membranes. In agreement with this prediction, a GST fusion protein of the FYVE domain of ZFYVE26 bound to phosphatidylinositol 3-phosphate in vitro [10]. In HeLa cells, however, no co-localization with endosomes was found for ZFYVE26. Instead, it was described to localize to centrosomes and to play a crucial role in mitosis, as knockdown of ZFYVE26 resulted in a defect of cytokinesis [10].
From the clinical presentation SPG15 cannot be distinguished from SPG11, which is caused by mutations in SPATACSIN, also a protein of largely unknown function [11]. In zebrafish, the knockdown of either Zfyve26/Spastizin or Spatacsin caused motor impairment and abnormal branching of spinal cord motoneurons at the neuromuscular junction [12]. As the partial knockdown of either protein did not result in an overt phenotype, but the partial depletion of both proteins at the same time caused motor impairment, it was further concluded that both proteins might be involved in the same cellular pathway [12]. Indeed, ZFYVE26/SPASTIZIN co-immunoprecipitated with SPATACSIN as well as the three other largely uncharacterized proteins C20orf29, DKFZp761E198, and KIAA0415 [13]. By phylogenetic and structure prediction analyses these proteins were recently identified as subunits of a new adaptor protein 5 (AP5) complex, which is proposed to be involved in cargo trafficking in the endolysosomal compartment [14]. In accordance AP5, ZFYVE26, and SPATACSIN were shown to co-localize with lysosomal markers in HeLa cells [15]. In this cell type the siRNA mediated knockdown of either members of the AP5 complex [14], SPATACSIN, or ZFYVE26 [15] resulted in the formation of early endosome clusters positive for the cation-independent mannose 6-phosphate receptor.
To gain deeper insight into the physiological function of the AP5 complex-interacting protein ZFYVE26 and to elucidate the pathophysiology of SPG15 we disrupted the Zfyve26 gene in mice. Knockout mice developed a progressive spastic gait disorder closely resembling SPG15 disease. Preceding the loss of cortical motoneurons and Purkinje cells, we detected pathological accumulations of autofluorescent, electron-dense material in lysosomal structures of Zfyve26-deficient neurons. The data suggest that Zfyve26 plays a crucial role in the trafficking within the endolysosomal pathway of specialized neurons.
Results
Zfyve26 is broadly expressed in the brain including the motor cortex and the cerebellum
To characterize the protein encoded by the Zfyve26 gene we immunized rabbits either against an N-terminal or C-terminal epitope of Zfyve26 and affinity-purified the resulting antisera. In agreement with the predicted molecular mass of Zfyve26 the anti-C-terminus antibody was reactive with a 285 kD polypeptide in a variety of tissues such as brain, liver, lung, and kidney (Fig. 1A). The anti-N-terminus antibody recognized the 285 kD Zfyve26 protein as well (Fig. 2C).


Since both the anti-N - and anti-C-terminus antibodies did not detect endogenous Zfyve26 in immunofluorescence studies, we next performed non-radioactive in situ hybridizations to study the expression of Zfyve26 transcript abundance in the brain in more detail. No signal was detected with the sense control (Fig. 1B). With the antisense probe Zfyve26 transcripts were broadly expressed in the brain (Fig. 1C) including the olfactory bulb (Fig. 1C′), cortical neurons (Fig. 1C″), the hippocampus (Fig. 1C′″), the cerebellum including Purkinje cells (Fig. 1C″″), and spinal cord neurons (Fig. 1D,D′).
Absence of Zfyve26 transcripts and Zfyve26 protein upon exon 15 deletion
To study the physiological role of Zfyve26 in more detail we generated Zfyve26 deficient mice by deleting exon 15 of the Zfyve26 gene (Fig. 2A). Deletion of exon 15 is predicted to cause a frame-shift (p.Val843GlyfsX10). Northern blot analysis of brain tissue suggested that the aberrant transcript is subjected to nonsense-mediated mRNA decay, as no Zfyve26 transcripts were detected in homozygous knockout (KO) mice (Fig. 2B). The absence of the 285 kD band in brain lysates prepared from knockout mice with either the antibody against the N-terminal (Fig. 2C) or C-terminal epitope (Fig. 2D) confirmed the absence of the Zfyve26 protein in the knockout. Since no additional Zfyve26-specific bands of lower size were detected with our anti-Zfyve26 antibodies raised against the anti-N-terminus of Zfyve26 in knockout compared to wild-type (WT) brain lysates, it can be excluded that exon 15 deletion led to the expression of a variant truncated Zfyve26 protein.
Because of the close interaction between ZFYVE26 and SPATACSIN, we assessed whether disruption of Zfyve26 might affect the expression of Spatacsin. Indeed, the intensity of the band of the expected molecular mass of Spatacsin of 250 kD was reduced by roughly 40% compared to wild-type in brain lysates of Zfyve26 knockout mice (Fig. 2E), whereas Spatacsin mRNA levels were comparable (Fig. 2F).
Zfyve26 knockout mice develop a progressive spastic and ataxic gait disorder
Zfyve26 knockout mice were viable and their survival did not differ from control littermates (data not shown). Young knockout mice did not show any obvious abnormalities or altered body weight compared to wild-type littermates up to 8 months of age. At 16 months of age, however, the body weight of knockout mice was reduced (17% for male knockout mice; Fig. 3A). Moreover, in the Morris water maze paradigm performed at 5 months of age time and distance traveled to detect the hidden platform did not differ between genotypes (Fig. 3B,C).

At 12 months of age Zfyve26 knockout animals displayed a progressive gait disorder and motor deficits, as quantified by measuring the foot-base-angle (FBA) at toe-off positions of the hind-paws (Fig. 3D–F and movie S1 and movie S2). In 16-month-old knockout mice the foot-base-angle was decreased to values around 50° in contrast to values around 75° in wild-type mice. The motor deficits were accompanied by a progressive decline in the beam walking performance (Fig. 3G). Knockout mice already had a tendency to fall more often than their wild-type littermates at 8 months of age. The number of falls as a correlate of impaired motor coordination was significantly increased at 16 months, when knockout mice fell off the beam in about 1 of 3 trials (Fig. 3G). At that age knockout mice displayed an abnormal posture with kyphosis of the spine. Finally, Zfyve26 knockout mice also suffered from dysfunction of the bladder, as evident from an enlarged bladder at post mortem analysis after 16 months of age (data not shown).
Disruption of Zfyve26 causes severe cortical and cerebellar neuron loss
Importantly, analysis of different brain structures at 2 months of age did not reveal any differences between genotypes, thus brain development appears not to be grossly impaired by disruption of Zfyve26. In line with a neurodegenerative disease, the brain size was significantly reduced in the knockout cohort at 16 months of age (Fig. 4A–C), when knockout mice displayed severe motor deficits. To quantify cortical neurons we labeled neurons by NeuN-immunostaining at different ages (Fig. 4D–F). The number of neurons in the motor cortex was not changed in Zfyve26 knockout mice at 2 or 8 months of age (data not shown), in 16-month-old knockout mice, however, the number of neurons in cortical layers V and VI was significantly reduced (Fig. 4F). Neuron loss was accompanied by activation of astrocytes as shown by the increase of GFAP-positive cells (Fig. 4D,E and S1A,B). Consistent with neuron loss being restricted to deep cortical layers V and VI, astrogliosis predominantly occurred in layers V and VI (Fig. 4D,E) and was not yet observed at 2 months of age (data not shown).

As the cerebellum plays a key role for motor coordination, we assessed whether the neurodegenerative process also affects the cerebellum. In accordance with the ataxic gait disorder, Purkinje cells were strongly reduced to 26% of controls in 16-month-old Zfyve26 knockout mice (Fig. 4G–I). Similarly to the cortex, the Purkinje cell loss was accompanied by a marked astrocytosis of the cerebellum (Fig. 4G–H and S1E,F).
Neither neurodegeneration nor obvious activation of astrocytes were observed in the hippocampus (Fig. S1I,J) or the olfactory bulb (Fig. S1M,N). Moreover, we did not observe a thinning of the corpus callosum in 16-month-old knockout mice (Fig. S2A,B).
At 8 months of age we noted ongoing axonal degeneration in the corticospinal tract of knockout mice, which was not yet observed at 2 months of age (Fig. S2C–F). At 16 months of age, when cortical motoneurons were already reduced, large diameter axons were almost absent from horizontal lumbar spinal cord sections of knockout mice (Fig. 4J–K). The ultrastructure of a degenerating axon is shown in Fig. 4L. At that age we also noted a clear activation of astrocytes in the spinal cord, mainly in the white matter (Fig. S1Q,R). The number of neurons in the gray matter of the spinal cord appeared to be normal (data not shown).
To assess whether disruption of Zfyve26 affects axon outgrowth, we cultured embryonic motoneurons of both genotypes. Under these conditions the overall survival of motoneurons was not affected (data not shown). After 4 days in culture the length of the axons identified by immunoreactivity for the axonal marker protein Tau (Fig. 4M) was measured. The length of the outgrowing axons (Fig. 4N) as well as the bidirectional axonal transport rate of mitochondria (Fig. S3A,B) were significantly reduced in Zfyve26 knockout motoneurons. Axonal branching, however, was not altered upon disruption of Zfyve26 (Fig. 4O).
Loss of Zfyve26 leads to accumulation of large irregular, autofluorescent, membrane-surrounded deposits
At 2 months of age we first noted the accumulation of autofluorescent intracellular material (emission wavelength between 460 and 630 nm) in Purkinje cells of knockout mice compared to wild-type (WT) mice (Fig. 5A,E). At 16 months of age autofluorescent material was present in Purkinje cells of both genotypes, however, in knockout tissues the particles were more frequent and larger, both in deep cortical layers (Fig. S4A,C) as well as in Purkinje cells (Fig. S4E,G and Fig. 5B,F), i.e. the sites of ongoing neuronal loss. In addition to differences in size and number, the localization of autofluorescent deposits appeared to be altered in Zfyve26 knockout neurons and extended to atypical subcellular regions including the dendrites of Purkinje cells. A moderate increase of autofluorescent material was also observed in brain regions not affected by neuron loss like the hippocampus (Fig. S1K,L) or the olfactory bulb (Fig. S1O,P). Autofluorescent particles observed in Zfyve26 knockout mice corresponded with deposits stained positive with Sudan Black (Fig. S4), a dye that predominantly stains lipopigments.

Ultrastructural analyses revealed that the large deposits either consisted of smaller particles of variable electron density surrounded by membranes (arrowheads, Fig. 5H), which looked different from regular lipofuscin particles (Fig. 5C,G, black arrows in Fig. 5D,H) or represented larger structures of variable electron density enclosed by a membrane (Fig. 5I). In addition to lysosomal vesicles of normal appearance (Fig. 5J,K), in knockout brains some lysosomal structures were reminiscent of fingerprint bodies (Fig. 5L) as e.g. observed in juvenile neuronal ceroid lipofuscinosis [16].
Autofluorescent deposits are positive for the lysosomal marker Lamp1
To investigate the identity of the autofluorescent lipopigment-like deposits, cerebellar sections from 10-month-old mice of both genotypes were stained for different subcellular marker proteins. A spectral analysis was performed to distinguish the spectrum of the respective labeled compartment from the autofluorescence of the material by a linear unmixing algorithm [17], [18]. Importantly, the autofluorescent deposits rarely co-localized with EEA1 (Fig. 6A–B′″), a marker for early endosomes, the cis-Golgi compartment marker Giantin (Fig. S5A–B′″), or late endosomal 300 kD mannose 6-phosphate-receptor (M6PR) (Fig. S5C–D′″). Instead, the majority of autofluorescent lipopigment material was found to be co-localized in enlarged Lamp1-positive organelles representing endolysosomal structures, shown independently with two different Lamp1 antibodies (Fig. 6C–D′″ and Fig. S5E–F′″).
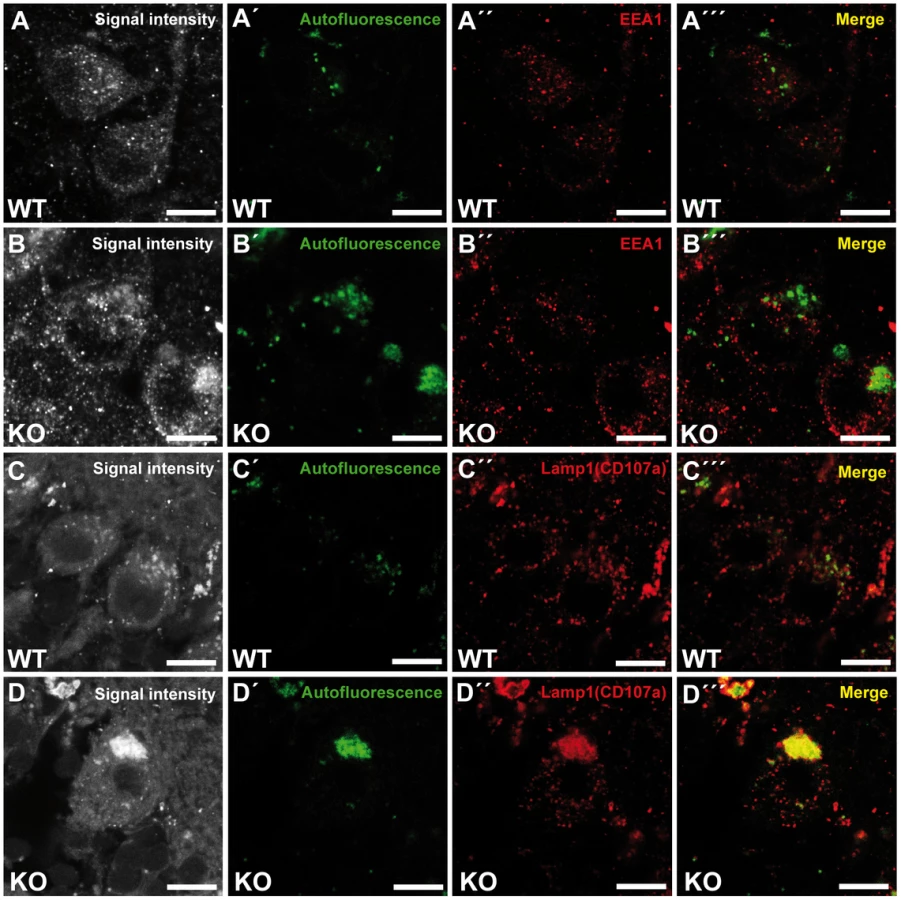
Zfyve26 localizes to the endolysosomal system
To examine the subcellular localization of Zfyve26, we followed two different approaches. First, a ZFYVE26-GFP fusion construct was expressed in mouse 3T3 cells. The fusion protein localized to a limited number of punctate structures centered around the nucleus (Fig. 7A,D). These Zfyve26-GFP positive structures showed a considerable spatial co-staining with early endosomal EEA1-positive membranes (50±10%; Fig. 7A–C) that have been reported to contain phosphatidylinositol 3-phosphate [9]. There was also some co-staining with Lamp1 (8±5%; Fig. 7D–F). To test whether the FYVE domain is crucial for the proper subcellular targeting of the full-length ZFYVE26 protein, we introduced a point mutation into the FYVE domain (p.His1834Ala). This mutation resulted in a diffuse cytosolic distribution of the protein indicating that this mutation is sufficient to disrupt the typical localization of ZFYVE26 (Fig. 7G–I). Moreover, incubation of cells with wortmannin, a known inhibitor of phosphatidylinositol-3-kinases, resulted in a more diffuse distribution of the ZFYVE26-GFP signal accompanied with the loss of EEA1-positive vesicular structures (Fig. 7J–L). There was no overlap of ZFYVE26-GFP with γ-adaptin, a marker for clathrin-coated structures at the TGN and vesicle there of (Fig. S6A–C). Some overlap was observed with the M6PR (Fig. S6D–F). Although reported previously [10], we could not detect co-localization with γ-tubulin, which labels centrosomes (Fig. S6G–I).

In our second approach, the localization of endogenous Zfyve26 in the mouse brain was analyzed by subcellular fractionation. Immunoblot analysis of these fractions detected a 285 kD polypeptide band representing Zfyve26 predominantly in the light membrane fraction, which was also positive for EEA1 and the AP5M1/μ5 subunit of the newly discovered AP5 complex (Fig. 7M). No Zfyve26 immunoreactive material was found in Lamp1-enriched heavy membrane fractions (Fig. 7M). The light membrane fraction isolated from Zfyve26 knockout brains lacked the 285 kD band.
Loss of Zfyve26 impacts on the endolysosomal compartment
Because of the endolysosomal localization of ZFYVE26, we studied basic endolysosomal function in mouse embryonic fibroblasts (MEFs) isolated from wild-type and knockout embryos (embryonic day E13.5). Stainings of MEFs from either genotype did not reveal gross structural abnormalities in the distribution of Lamp1 (Fig. S7A,D), Cathepsin D (CtsD) (Fig. S7B,E), EEA1 (Fig. S7G,J), the M6PR (Fig. S7H,K), or the AP5Z1/ζ subunit (Fig. S7M,N). Western blot analyses of the lysosomal protease CtsD and Lamp1 showed similar amounts and processed CtsD forms in wild-type and Zfyve26 knockout MEFs at steady state (Fig. S8A). This was in agreement with normal activities of the lysosomal hydrolases in MEFs (Fig. S8B).
We also labeled MEFs of both genotype with [35S]-methionine and either harvested (Fig. S8C, lanes 1 and 3) or chased for 5 h (Fig. S8C, lanes 2 and 4) in non-radioactive medium followed by immunoprecipitation of the lysosomal protease Cathepsin Z (CtsZ). The fluorography showed similar synthesis rates of the CtsZ precursor (p) (Fig. S8C, lanes 1 and 3). In addition the proteolytic processing to the mature (m) lysosomal form (Fig. S8C, lanes 2 and 4) and the M6PR-mediated sorting efficiency in the trans Golgi network shown by the amounts of CtsZ precursor in the medium after 5 h chase (Fig. S8C, lanes 5 and 6) were not affected. Moreover, both the internalization and the proteolytic processing of the arylsulfatase B (ASB) precursor form by lysosomal proteases were comparable in wild-type and knockout MEFs (Fig. S8D). Taken together these data indicate that in MEFs the transport of lysosomal enzymes along the biosynthetic and the endocytic pathway are not affected by the loss of Zfyve26.
Of course, the cellular pathology which finally results in the accumulation of autofluorescent material and neuron loss can be neuron-specific and may be time-dependent. To study whether alterations of the endolysosomal compartment are present in the brain before neurodegeneration starts, we performed subcellular fractionations of brain lysates from 20-day-old wild-type and knockout mice. Confirming that the endolysosomal system is compromised upon disruption of Zfyve26, the Lamp1-positive compartment was shifted towards fractions of higher densities compared to wild-type before (Fig. 8A,B) and after the appearance of large autofluorescent deposits (Fig. 8C). No shift was detected for the AP5M1/μ5 subunit (Fig. 8D). Lamp1 and EEA1 levels were unchanged in brain lysates of 16 month old knockout mice. Only Cathepsin D was significantly increased (Fig. 8E). Furthermore, increases in the overall activities of β-hexosaminidase (Fig. 8F) and β-galactosidase (Fig. 8G) in extracts of 16-month-old Zfyve26 knockout mouse brains suggested an altered composition/function of lysosomes upon disruption of Zfyve26.

Discussion
In addition to progressive spastic paraplegia, which usually starts within the first decades of life, SPG15 patients also suffer from ataxia, retinopathy, bladder incontinence, progressive cognitive decline, and atrophy of the corpus callosum and the cerebellum [5], [7], [19]. While it is clear that HSP/SPG15 is associated with mutations in the ZFYVE26 gene, the cellular pathomechanisms of the disease were largely unknown.
Here we demonstrate that Zfyve26-deficient mice develop a progressive gait disorder starting at 12 months of age, which is further complicated by bladder dysfunction and ataxia. Corresponding with the progressive gait disorder, we observed a degeneration of axons within the corticospinal tract at 8 months of age, a finding in line with mouse models of other types of HSP [20]–[22]. This degeneration mainly affected large diameter axons of the lumbar corticospinal tract. At 16 but not at 8 months of age we also observed a loss of neuronal somata in cortical layers V/VI, where the large projection motoneurons reside. This suggests that the cellular pathology is not restricted to motoneuron axons, but also affects their somata in the course of the disease. This is in sharp contrast to mouse models of pure HSP like SPG4, SPG7 or SPG31, where no cortical motoneuron loss had been reported [20]–[22] and corresponds with reports from SPG15 patients, which also display variable degrees of brain atrophy [5], [6], [19]. Moreover, neurodegeneration in Zfyve26 knockout mice was not restricted to cortical motoneurons, but, like in SPG15 patients, also included the cerebellum, where Purkinje cells were almost lost in aged knockout mice. No degeneration was found in the hippocampus or the olfactory bulb. Thus, it appears that large elaborate neurons like cortical motoneurons and Purkinje cells are particularly sensitive to the disruption of Zfyve26. As Purkinje cells play a critical role for motor coordination, the loss of Purkinje cells at least contributes to the cerebellar ataxia phenotype observed in both Zfyve26 knockout mice and SPG15 patients.
Although axon outgrowth of cultured embryonic Zfyve26 knockout motoneurons was delayed, young Zfyve26 knockout mice did not show any obvious phenotypes or motor impairment and brain and spinal cord of knockout mice did not reveal gross structural abnormalities at 2 months of age. Moreover, hippocampal spatial learning as assessed by the Morris water maze was not impaired before onset of the motor phenotype. This suggests that Zfyve26 is largely dispensable for normal brain and spinal cord development in mice, though knockdown studies in HeLa cells suggested alterations in cytokinesis [10] and knockdown studies in zebrafish resulted in abnormal branching of spinal cord motor neurons [12]. In Zfyve26 knockout mice, however, axonal branching of cultured motoneurons was not altered.
Taken together our mouse data are fully consistent with the view that HSP generally manifests as a neurodegenerative rather than a neurodevelopmental disease [23]. The very similar phenotypes of aged Zfyve26 knockout mice and SPG15 patients further confirm that SPG15 is caused by Zfyve26 loss-of-function and that Zfyve26-deficient mice are a valid model for the complex form of HSP.
What is the cellular pathology behind this neurodegeneration? Starting around 2 months of age we observed the progressive accumulation of autofluorescent intracellular material in neurons of Zfyve26 knockout mice. Notably, autofluorescent material was also detected in the retina of SPG15 patients suffering from macular defects [5]. The lipofuscin-like particles in Zfyve26-deficient mice were stained by Sudan Black, which particularly labels lipopigments [24] and which thus appear to be a major constituent of the deposits. At the ultrastructural level, the deposits were revealed as abnormal conglomerates of vesicular material of variable electron density and shape. Lipofuscin is a common finding in aged post mitotic neurons and is considered as non-degradable intralysosomal material [25]. Indeed, the large autofluorescent deposits in knockout neurons, which we never observed in wild-type tissues, were intensely labeled by the lysosomal marker Lamp1 and differed drastically in terms of time of onset, localization, and size compared to normal lipofuscin particles. Our biochemical analysis showed that Lamp1-positive membrane-compartments of unusually high density were already formed in pre-symptomatic young mice. In accordance with a progressive alteration of the endolysosomal compartment we observed an increase in the activity of β-hexosaminidase or β-galactosidase in brain lysates of aged knockout mice. In brain lysates from 16 month-old knockout mice levels of Lamp1 and the early endosome marker EEA1 were unchanged, whereas Cathepsin D levels were increased. Similar increases in the activity of specific enzymes have been reported for other neurodegenerative lysosomal disorders like e.g. neuronal ceroid lipofuscinosis [26].
Although lipofuscin is generally considered as harmless inert intracellular debris, it is increasingly suspected to interfere with various cellular functions and to thereby promote age-related pathologies [25]. Due to binding of transition metals, such as iron and copper, lipofuscin is for example considered to sensitize cells to oxidative stress [27].
What causes the pathological accumulation of non-degradable material in Zfyve26-deficient neurons? To get an idea we readdressed the subcellular localization of Zfyve26, which was still controversial. Whereas ZFYVE26 has been reported to localize to centrosomes with recruitment to the midbody during cytokinesis [10], others reported localization to early endosomes and the endoplasmic reticulum [5], or even the nucleus [28]. Part of the apparent controversy is most likely owed to low levels of expression and to the lack of specific and sensitive antibodies, which reliably detect endogenous Zfyve26. It has been reported that in HeLa cells stably expressing ZFYVE26-GFP, GFP signals localized to puncta with a substantial overlap to Lamp1 or a lysotracker-positive compartment [15]. Upon transient overexpression of ZFYVE26-GFP in 3T3 cells or COS7 cells we only observed a co-localization with a minor population of Lamp1-positive structures, whereas the majority of ZFYVE26 co-localized with the endosomal marker EEA1. Though we cannot exclude that the protein is shifted to an EEA1-positive compartment upon overexpression, our localization is supported by our fractionation studies and is in good agreement with the fact that FYVE domains target proteins to phosphatidylinositol-3-phosphate-enriched membranes like early endosomes [9], [29]. Indeed, either disruption of the FYVE domain by site-directed mutagenesis or depletion of phosphatidylinositol-3-phosphate by wortmannin drastically altered the localization of ZFYVE26-GFP.
How can a predominantly endosomal localization of Zfyve26 be reconciled with aberrant endolysosomal material in aged Zfyve26 knockout mice? It has been shown that Zfyve26 co-immunoprecipitated with the AP5 components C20orf29 (AP5S1/σ5), KIAA0415 (AP5Z1/ζ), C14orf108 (AP5M1/μ5), and DKFZp761E198 (AP5B1/β5) [13], [14]. It thus seems likely that Zfyve26 plays some role in the largely unknown functions of AP5. While the cargoes of the AP5 complex have not yet been identified, it is conceivable that defects in AP5-mediated cargo sorting manifest in impairments in membrane compartments that a) normally represent the targets of cargo and lack this cargo in Zfyve26 loss-of-function situations or b) represent target compartments that are loaded with missorted cargo. The siRNAs directed against either C14orf108, the proposed AP5M1/μ5 subunit, or DKFZp761E198, the proposed AP5B1/β5 subunit, as well as against ZFYVE26 in HeLa cells resulted in clustered, mannose 6-phosphate receptor-positive puncta in the perinuclear region, membrane compartments that may represent multivesicular bodies and thus are part of the endolysosomal trafficking pathway [14]. Although knockdown of SPATACSIN or ZFYVE26 changed the localization/distribution of AP5 in HeLa cells [15], we did not observe any clear changes in the distribution of AP5Z1/ζ in MEFs or of AP5M1/μ5 in subcellular brain fractions. Our observation of Lamp1-positive deposits of electron-dense, autofluorescent lipopigments surrounded by membranes are well in line with defects in the trafficking from endosomes towards lysosomes. Our analysis of MEFs, which did not reveal a clear endolysosomal defect in the absence of Zfyve26, does not preclude a defect in neurons, as the effects may be cell type - or substrate-specific and/or time-dependent. The late onset neurodegeneration in Zfyve26 knockout mice may rather point to a minor defect in the endolysosomal sorting machinery, which accumulates over time. Indeed, the increase of the enzymatic activities of the lysosomal enzymes β-hexosaminidase and β-galactosidase in brain was only noted at 16 but not at 2 months of age. The increase in Cathepsin D, β-hexosaminidase, and β-galactosidase levels/activity is not necessarily in conflict with impaired lysosomal degradation. Whether this is rather caused by an altered composition or an increased size or number of lysosomes or both is still unclear.
Is it plausible that endosomal cargo sorting defects involving Zfyve26 and AP5 are the cellular basis for the progressing defects seen in aging Zfyve26 knockout mice and SPG15 patients? A strikingly similar pathology with the accumulation of large autofluorescent deposits and loss of Purkinje cells as well as long projection fibers within the spinal cord was reported for mice deficient for phosphatidylinositol 4-kinase 2α [30]. This kinase is predominantly located in the Golgi complex and endosomes and interacts with another adaptor complex, AP3, which targets cargo from endosomes to lysosomes [31]. Defects of the AP3 complex underlie a subtype of Hermansky-Pudlak syndrome, which is characterized by impaired function and/or biogenesis of lysosomes and lysosome-related organelles such as melanosomes and platelet dense granules. It thus results in oculocutaneous albinism, platelet storage pool deficiency, and ceroid lipofuscinosis [32]. Although the cargoes of the AP5 complex have not yet been identified it thus seems plausible that defects in endolysosomal sorting, which can impact on a variety of cellular functions including axonal transport or lysosomal function, result in axon degeneration and neuronal death. Indeed, defects in the AP4 complex, which is involved in transport between the trans-Golgi network and endosomes, cause neuroaxonal degeneration in SPG47, SPG50, SPG51, and SPG52 with early onset severe spasticity [33]. The long axonal projections of motoneurons and the uniquely complex dendritic arbors of Purkinje cells, respectively, may render these cells particularly vulnerable to defects of intracellular trafficking.
Materials and Methods
All animal experiments were approved by the Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz (TLLV) in Germany.
Targeted inactivation of the murine Zfyve26 gene
A clone isolated from a 129/SvJ mouse genomic λ library (Agilent Technologies, USA) was used to construct the targeting vector. A 13.4 kb fragment was cloned via NotI and BamHI into the pKO-V901 plasmid (Lexicon Genetics, USA) with a phosphoglycerate kinase (pgk) promoter–driven diphtheria toxin A cassette. A loxP site and an additional BamHI restriction site were placed at the EcoRV site into intron 14 and a pgk promoter–driven neomycin resistance cassette flanked by frt sites and an additional loxP site at the KpnI site into intron 15 (Fig. 2A). The construct was linearized with NotI and electroporated into R1 mouse embryonic stem (ES) cells. Neomycin-resistant clones were analyzed by Southern blot using BamHI and an external 782 bp probe (Mus musculus chr 12 : 80,383,750–80,384,531). Two correctly targeted ES cell clones were injected into C57BL/6 blastocysts to generate chimeras. Chimeric mice were mated to a cre-deleter mouse strain to remove exon 15 and the selection cassette [34]. Studies were performed in a mixed 129SvJ/C57BL/6 background in the F4 and F5 generation. For PCR genotyping on DNA isolated from tail biopsies, the primers f: 5′-CTTGTGTATTTTGCATAGGTGC-3′, r: 5′-TGACACTGAATGTTAAGA-3′, and del: 5′-TTCTGGAAGCGTCTGTAAAG-3′ were used in a single PCR mix. The primer pair f/r amplified a 187-bp wild-type allele, and the primer pair f/del a 250-bp KO allele. Mice were housed in a 12 h light/dark cycle and fed on a regular diet ad libitum.
Northern blot analysis
The Zfyve26 probe was cloned by PCR from a mouse brain cDNA using the forward primer 5′-AGGGAGTCTGAGTGCTG-3′ and the reverse primer 5′-GACTCTCAAGGCTGCTG-3′. The probe to detect Spatacsin transcripts was cloned with the forward primer 5′-GCAAACACTAACACACACTCCGCAGTGG-3′ and the reverse primer 5′-GCAACACCAGCACTAGATCCTGGC-3′. The Northern blot analysis was performed as described previously [35].
In situ-hybridization
in situ hybridizations were carried out on 20 µm sagittal cryosections of brain or transversal cryosections of spinal cord from 2-month-old wild-type mice using digoxigenin-labeled antisense and sense RNA probes as described [36]. The riboprobes covered either exon 16 (cDNA positions 2922–3683) or exons 22–25 (cDNA positions 4404–4847) of the Zfyve26 transcript (accession number: ENSMUST00000021547).
ZFYVE26-GFP overexpression
For overexpression of the ZFYVE26-GFP fusion protein (ZFYVE26-GFP), a human full length cDNA clone (DKFZp781H1112Q, RPDZ) was PCR-amplified and inserted via BglII and BamHI into the pEGFP-N1 vector (Clontech laboratories, USA). Mutations in the clone were corrected by PCR-based mutagenesis to obtain a cDNA corresponding to the human reference sequence ENSHU004078079. The point-mutation p.His1834Ala in ZFYVE26-GFP was inserted by PCR-based mutagenesis. The following primers were used for PCR mutagenesis: KpnI_f: 5′-GGTACCGGATGAGACTGAG-3′, H_in_A_f: 5′-CCATGTTTAACAGGCGTCATGCTTGTCGCCGCTGTGGCCGG-3′, H_in_A_r: 5′-CCGGCCACAGCGGCGACAAGCATGAGCCTGTTAAACATGG-3′, SspI_r: 5′-AATATTCAGCACATCTACCTTG-3′.
3T3-cells were maintained in DMEM (Invitrogen, Germany) supplemented with 10% fetal bovine serum, penicillin (100 UI/ml), and streptomycin (100 mg/ml). Before transfection cells were plated on coverslips and after 24 h transfected with 1 µg DNA/well in 24-well plates with lipofectamine 2000 reagent (Invitrogen, Germany) according to the instructions of the manufacturer. 24 to 48 h post-transfection cells were fixed in 4% para-formaldehyde at room temperature for 15 min and immunocytochemistry was performed as described in [37]. To inhibit phosphatidylinositol 3-kinase, cells were incubated with 100 nM wortmannin (Invitrogen, Germany) for 1 h. To quantify the degree of co-localization, at least 10 cells were imaged. The relative area of co-localization was determined by scatter plot analysis using the co-localization module of AxioVision (Release 4.8.2, Zeiss, Germany).
Antibodies
Anti-Zfyve26 antisera were raised by immunization of rabbits against either the N-terminal epitope TSSELSTSTSEGSLSA (residues 782–797 of mouse Zfyve26, NP_001008550.1) or the C-terminal epitope ENELVRSEFYYEQAPS (residues 1908–1923 of mouse Zfyve26, NP_001008550.1) and affinity-purified as described in [38]. The following antibodies were used for immunoblotting and immunofluorescence: rabbit anti-SPATACSIN, 1∶500, (Protein Tech, UK); mouse anti-M6PR and rabbit anti-EEA1, both 1∶500, (Abcam, UK); mouse anti-EEA1,1∶500, (BD Transduction laboratories, USA); mouse anti-NeuN and mouse anti-GFAP, both 1∶1,000, (Millipore, USA); mouse anti-γ-Adaptin and mouse anti-Calnexin, both 1∶1,000, (BD Biosciences, USA); rat anti-Lamp1 (clone CD107a), 1∶500, (BD Biosciences, USA); rat anti-Lamp1 (clone 1D4B), 1∶25, (Developmental Studies Hybridoma Bank, USA); rabbit anti-γ-Tubulin, 1∶2,000, (Sigma Aldrich, USA); mouse anti-Giantin, 1∶1,000, (Enzo Life Sciences, Germany); rabbit anti-phospho-Tau, 1∶500, (Biozol Diagnostika Vertrieb GmbH, Germany); mouse anti-microtubule-associated protein 2ab (MAP2ab), 1∶2,000, (Acris Antibodies GmbH, Germany); goat anti-CtsD (sc6486, Santa Cruz; USA, for Western studies); rabbit anti-CtsD, 1∶100 [39] (used for immunofluorescence studies); mouse anti-α-Tubulin (T9026, Sigma-Aldrich, USA); rabbit anti-M6PR (kind gift of K. von Figura).
Secondary antibodies for immunofluorescence: goat anti-rabbit, goat anti-mouse, or goat anti-rat coupled with Alexa 488 and Alexa 555, respectively, 1∶1,000, (Invitrogen, Germany); goat anti-rabbit coupled with Cy5, 1∶1,000, (Dianova, Germany); Goat anti-rat and anti-mouse Cy5, 1∶1,000, (Jackson ImmunoResearch Laboratories, USA). Secondary antibodies for Western blotting: goat anti-rabbit and goat anti-mouse, both 1∶4,000, (Amersham Bioscience, UK); goat anti-rat, 1∶2,000, (Santa Cruz, USA).
Immunoblotting analyses of cell and tissue lysates
Cells were harvested and lysed in 50 mM Tris/HCl pH 8, 1 mM EDTA, 0.5% NP40, 120 mM NaCl. Tissue lysates were prepared with the Ultra-Turrax T8 tissue homogenizer (IKA-WERKE, Germany) in homogenization buffer (300 mM Tris-HCl pH 8.8, 5 mM EDTA, 3 mM NaF, 10% Glycerol, 3% SDS, and complete protease inhibitor (Roche, Switzerland) as described [40]. Homogenates were centrifuged at 1,500 g to remove nuclei and insoluble debris. The supernatant was either denatured at room temperature for 15 min or/and at 95°C for 5 min in Laemmli sample buffer. After separation in a 6% SDS-polyacrylamide gel electrophoresis proteins were transferred onto nitrocellulose or PVDF membranes (Whatman, Germany). The Zfyve26 antibody directed against the C-terminus was used at a dilution of 1∶350 and the antibody directed against the N-terminus at a dilution of 1∶100. Both primary antibodies were detected with a horseradish peroxidase-conjugated secondary anti-rabbit antibody (Amersham Bioscience, UK) and the SuperSignal Western Blot Enhancer Kit (Thermo scientific, Germany).
Fractionation of brain material and density gradient analyses of isolated membranes
Membrane fractionations were done as described with slight modifications [41]. A brain from a 20-day-old mouse was homogenized in Buffer A (130 mM KCl, 25 mM Tris-HCl pH 7.4, 1 mM EGTA) including Complete protease inhibitor cocktail (Roche, Switzerland) and centrifuged at 1,000 g for 10 min. The supernatant (1000 g SN) was isolated and sucrose was added to a final concentration of 15%. Following centrifugation at 3,000 g for 10 min the resulting supernatant (3000 g SN) was diluted to a total volume of 4.5 ml using Buffer A containing 15% sucrose. A discontinuous sucrose gradient was generated by overlaying sucrose solutions in Buffer A: 3 ml 45%, 6 ml 30%, 4.5 ml of the diluted supernatant in 1 ml 5% sucrose and 2.5 ml 7.5%. The gradient was centrifuged at 100,000 g for 1 h. Light membranes were visible in the range of 15% and heavy membranes in the range of 35% sucrose. A discontinuous iodixanol gradient was generated by overlaying 2.2 ml of 15%, 12.5%, 10%, 7.5%, 5%, and 2.5% (v/v) iodixanol for light membrane fractionation and 3.25 ml of 25%, 23%, 21%, and 19% (v/v) iodixanol, all in Buffer A for heavy membrane fractionation. The light or the heavy membrane fraction were loaded at the bottom and centrifuged for 2 h at 110,000 g. 1.2 ml fractions were collected from the top, diluted to 8 ml with Buffer A and centrifuged at 130 000 g for 40 min. Pellets were resuspended in 50 µl SDS-sample buffer.
For crude fractionations 1/2 brain from a 16-month-old mouse was homogenized and centrifuged as described before. 1,000 g supernatants were diluted to 3 ml containing 15% sucrose. A discontinuous sucrose gradient with 3 ml 55%, 2 ml 45%, 4 ml 30%, 3 ml of the diluted supernatant in 15% sucrose, and 1 ml 7.5% sucrose was centrifuged at 100,000 g for 1 h. 13 fractions were collected, precipitated in 80% ethanol and pellets were resuspended in SDS-sample buffer and analyzed by SDS-PAGE and quantitative immunoblotting based on fluorescence using a LI-COR Odyssey detection system (LI-COR, Germany).
Cell culture
Mouse embryonic fibroblasts were prepared from E13.5 mouse pups as described in [42]. Spinal cord motoneurons were isolated from embryonic day 12.5 embryos as described previously in [43]. To determine motoneuron survival neurons were counted four hours after plating. This was repeated every day for five fields of view (1.16 mm2) with a phase-contrast microscope (Olympus, Germany). The number of initially counted cells was set 100% (day 0). The percentage of surviving cells was calculated for every day in culture. Axons of motoneurons were identified by positive anti-phospho-Tau staining, dendrites by positive anti-MAP2ab staining. The longest axonal extension was measured with the Neurolucida 8 software (MBF Bioscience, USA) after 4 days in culture and the number of axonal branches was counted. The results from at least four independent experiments were pooled.
Analysis of lysosomal function
Enzyme activity measurements, pulse chase and endocytosis experiments were performed as described in [44], [45].
Time-lapse imaging
Mitochondria were stained with Mito Tracker Green FM (final concentration 100 nM, Invitrogen, Germany) in living motoneurons after four days in culture for 30 min. After replacement of the medium, mitochondria were visualized with a fluorescence microscope (Axio Observer Z1, Zeiss, Germany) equipped with an incubation chamber (37°C, 5% CO2). Time-lapse images were acquired at a frequency of 15 Hz with an exposure time of 200 ms. For statistical analysis of mitochondrial movements ten independent experiments were performed.
Morris water maze
Spatial memory was assessed in a Morris water maze, consisting of circular tank (diameter 120 cm, depth 60 cm) filled with water at 25±1°C at a depth of 30 cm as described in [46] with some modifications. To escape from the water, the mice had to find a hidden platform (diameter 20 cm) submerged approximately 1 cm below the water surface. The platform was located at the center of one of the four quadrants of the pool. For efficient tracking of mice the water was colored white by the addition of 2 l milk. The maze was located in the middle of the room with prominent extra maze cues. Swim paths were recorded by a video camera. Latency, swim speed, path length, etc. were analyzed with the VideoMot2 software (TSE Systems, Germany). All mice were handled and habituated to the experimental situation 2 days before training. The mice were trained to find the hidden platform, which remained at a fixed location throughout testing. Each mouse received 2 blocks per day, each with 3 trials for 7 days, with an intertrial interval of approximately 60 s. Time intervals between blocks were 4 h. The mice were placed into the pool facing the side wall at one of the 8 start positions (N, W, S, E, NW, SW, NE, and SE; chosen randomly across trials) and allowed to swim until they found the platform. Any mouse that failed to find the platform within 1 min was guided and placed onto the platform and was then warmed for 45 s under an infrared lamp before commencing the next trial. 24 h after the last training day a probe trial was carried out. For this purpose, the platform was removed from the pool and the mice were allowed to swim freely for 60 s. The distance and percentage of the time spent in each quadrant was analyzed.
Quantitative gait analysis
Mice were trained to walk on a horizontal 20 cm elevated plastic beam (1,000 mm long, either 38 mm for cohorts at 8 and 12 months of age or 48 mm broad at 16 months of age) leading to their home cage (see movie S1 and movie S2). After the initial learning phase the foot-base-angle at toe-off positions of the hind-paws was measured using single video frames from recordings of beam walking mice [47]. To quantify ataxia, falls off the beam were counted.
Histological analysis
Hematoxylin and eosin (HE) stainings followed standard protocols (Carl Roth, Germany). Sudan black staining was performed as described in [48]. Immunohistochemical analysis was done on formalin fixed, free floating sections as described previously [37]. Nuclei were stained with Hoechst-33258 (1∶10,000, Molecular Probes, Germany). Purkinje cell loss and gliosis were analyzed on 40 µm sagittal brain sections using rabbit-anti-Calbindin and mouse anti-GFAP antibodies. Purkinje cell numbers were quantified on 8 µm HE stained paraffin sections on a Zeiss Axioskop 40 microscope. For statistical analysis the number of Purkinje cells per 1,000 µm distance along the Purkinje cell layer was counted from 3 different mice per genotype. For quantification of neurons in cortical layers NeuN immunohistochemistry was performed on 40 µm sagittal brain sections. Images of the motor cortex of sagittal sections of wild-type and Zfyve26 knockout brains were taken with a Leica TCS SP5 confocal scanning fluorescence microscope. Neurons were quantified with the cell counter plug in and the area measurement tool of ImageJ software (W. S. Rasband, National Institutes of Health, Bethesda, USA). Results are presented as mean ± SEM. “n” refers to the number of sections analyzed from 3 animals per genotype.
Spectral analysis
Fluorescence images were recorded on the stage of a confocal laser scanning microscope (LSM710, Zeiss, Germany). Autofluorescence and Cy5 fluorescence of the secondary antibodies were excited with the 488 nm line of an argon laser and the 633 nm line of a helium/neon laser. Fluorescence emitted from the sample was recorded with the spectral detector of the LSM710 in the wavelength range from 501–725 nm. Under these experimental conditions one can distinguish two autofluorescence components, one originating from the deposits and one from the background. The fluorescence signal recorded from the sample is a linear combination of these components and the Cy5 dye coupled to the secondary antibodies. Their contribution to the overall signal can be calculated by a linear unmixing algorithm, which optimizes the parameters of the relative contributions, so that the sum of the spectra of all components matches the recorded spectrum [17], [18]. To visualize the results the respective fluorescence contributions are encoded by false colors with the brightness being proportional to the intensity of the respective spectral components.
Ultrastructural analysis
For semi - and ultrathin sectioning, 4 animals per genotype were perfused with 50 ml fixative (4% paraformaldehyde, 1% glutaraldehyde). Brain and spinal cord were removed and post fixed overnight at 4°C. 150 µm sagittal and coronal sections of brain and spinal cord respectively were cut with a vibratome (Leica Microsystems, Germany) and processed as described in [49].
Statistics
For repeated experiments two-way ANOVA followed by Bonferroni post-hoc tests were used to compare between genotypes. For cortical morphology and quantitative western blot analysis Student's two-tailed t-test was used.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DepienneC, StevaninG, BriceA, DurrA (2007) Hereditary spastic paraplegias: an update. Curr Opin Neurol 20 : 674–680.
2. HardingAE (1983) Classification of the hereditary ataxias and paraplegias. Lancet 1 : 1151–1155.
3. ReidE (1999) The hereditary spastic paraplegias. J Neurol 246 : 995–1003.
4. BlackstoneC (2012) Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 35 : 25–47.
5. HaneinS, MartinE, BoukhrisA, ByrneP, GoizetC, et al. (2008) Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 82 : 992–1002.
6. SchuleR, SchlipfN, SynofzikM, KlebeS, KlimpeS, et al. (2009) Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 80 : 1402–1404.
7. SchuleR, ScholsL (2011) Genetics of hereditary spastic paraplegias. Semin Neurol 31 : 484–493.
8. LupasA (1996) Coiled coils: new structures and new functions. Trends Biochem Sci 21 (10) 375–82.
9. KutateladzeT, OverduinM (2001) Structural mechanism of endosome docking by the FYVE domain. Science 291 : 1793–1796.
10. SagonaAP, NezisIP, PedersenNM, LiestolK, PoultonJ, et al. (2010) PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol 12 : 362–371.
11. StevaninG, SantorelliFM, AzzedineH, CoutinhoP, ChomilierJ, et al. (2007) Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 39 : 366–372.
12. MartinE, YanicostasC, RastetterA, NainiSM, MaouedjA, et al. (2012) Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis 48 : 299–308.
13. SlabickiM, TheisM, KrastevDB, SamsonovS, MundwillerE, et al. (2010) A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol 8: e1000408.
14. HirstJ, BarlowLD, FranciscoGC, SahlenderDA, SeamanMN, et al. (2011) The fifth adaptor protein complex. PLOS Biol 9: e1001170.
15. HirstJ, BornerGH, EdgarJ, HeinMY, MannM, et al. (2013) Interaction Between AP-5 and the Hereditary Spastic Paraplegia Proteins SPG11 and SPG15. Mol Biol Cell 24 : 2558–69.
16. AndersonGW, GoebelHH, SimonatiA (2012) Human pathology in NCL. Biochim Biophys Acta 1832 (11) 1807–26.
17. DickinsonME, BearmanG, TilleS, LansfordR, FraserSE (2001) Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques 31 : 1272, 1274–1276, 1278.
18. ZimmermannT, RietdorfJ, PepperkokR (2003) Spectral imaging and its applications in live cell microscopy. FEBS Lett 546 : 87–92.
19. BoukhrisA, StevaninG, FekiI, DenoraP, ElleuchN, et al. (2009) Tunisian hereditary spastic paraplegias: clinical variability supported by genetic heterogeneity. Clin Genet 75 : 527–536.
20. FerreirinhaF, QuattriniA, PirozziM, ValsecchiV, DinaG, et al. (2004) Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest 113 : 231–242.
21. TarradeA, FassierC, CourageotS, CharvinD, VitteJ, et al. (2006) A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet 15 : 3544–3558.
22. BeetzC, KochN, KhundadzeM, ZimmerG, NietzscheS, et al. (2013) A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J Clin Invest 123 (10) 4273–4282.
23. FinkJK (2006) Hereditary spastic paraplegia. Curr Neurol Neurosci Rep 6 : 65–76.
24. EllederM (1978) A histochemical and ultrastructural study of stored material in neuronal ceroid lipofuscinosis. Virchows Arch B Cell Pathol 28 : 167–178.
25. TermanA, BrunkUT (2004) Lipofuscin. Int J Biochem Cell Biol 36 : 1400–1404.
26. PrasadVV, PullarkatRK (1996) Brain lysosomal hydrolases in neuronal ceroid-lipofuscinoses. Mol Chem Neuropathol 29 : 169–179.
27. BrunkUT, TermanA (2002) Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med 33 : 611–619.
28. MurmuRP, MartinE, RastetterA, EstevesT, MurielMP, et al. (2011) Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci 47 : 191–202.
29. StenmarkH, AaslandR, TohBH, D'ArrigoA (1996) Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem 271 : 24048–24054.
30. SimonsJP, Al-ShawiR, MinogueS, WaughMG, WiedemannC, et al. (2009) Loss of phosphatidylinositol 4-kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci U S A 106 : 11535–11539.
31. SalazarG, CraigeB, WainerBH, GuoJ, De CamilliP, et al. (2005) Phosphatidylinositol-4-kinase type II alpha is a component of adaptor protein-3-derived vesicles. Mol Biol Cell 16 : 3692–3704.
32. OhnoH (2006) Physiological roles of clathrin adaptor AP complexes: lessons from mutant animals. J Biochem 139 : 943–948.
33. HirstJ, IrvingC, BornerGH (2013) Adaptor Protein Complexes AP-4 and AP-5: New Players in Endosomal Trafficking and Progressive Spastic Paraplegia. Traffic 14 : 153–164.
34. SchwenkF, BaronU, RajewskyK (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23 : 5080–5081.
35. HenningsJC, PicardN, HuebnerAK, StauberT, MaierH, et al. (2012) A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubule. EMBO Mol Med 4 : 1057–1071.
36. ZimmerG, RudolphJ, LandmannJ, GerstmannK, SteineckeA, et al. (2011) Bidirectional ephrinB3/EphA4 signaling mediates the segregation of medial ganglionic eminence - and preoptic area-derived interneurons in the deep and superficial migratory stream. J Neurosci 31 : 18364–18380.
37. SinningA, LiebmannL, KougioumtzesA, WestermannM, BruehlC, et al. (2011) Synaptic glutamate release is modulated by the Na+-driven Cl−/HCO3 − exchanger Slc4a8. J Neurosci 31 : 7300–7311.
38. RustMB, AlperSL, RudhardY, ShmuklerBE, VicenteR, et al. (2007) Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest 117 : 1708–1717.
39. ClaussenM, KublerB, WendlandM, NeiferK, SchmidtB, et al. (1997) Proteolysis of insulin-like growth factors (IGF) and IGF binding proteins by cathepsin D. Endocrinology 138 : 3797–3803.
40. RustMB, FaulhaberJ, BudackMK, PfefferC, MaritzenT, et al. (2006) Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Circ Res 98 : 549–56.
41. RadhakrishnanA, GoldsteinJL, McDonaldJG, BrownMS (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab 8 : 512–521.
42. Joyner AL (1993) Gene targeting: a practical approach. IRL Press.
43. KarleKN, MöckelD, ReidE, SchölsL (2012) Neurogenetics 13 (2) 169–79.
44. MarschnerK, KollmannK, SchweizerM, BraulkeT, PohlS (2011) A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 333 : 87–90.
45. KollmannK, DammeM, MarkmannS, MorelleW, SchweizerM, et al. (2012) Lysosomal dysfunction causes neurodegeneration in mucolipidosis II ‘knock-in’ mice. Brain 135 : 2661–2675.
46. JacobsS, RuusuvuoriE, SipiläST, HaapanenA, DamkierHH, KurthI, et al. (2008) Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc Natl Acad Sci U S A 105 (1) 311–6.
47. IrintchevA, SimovaO, EberhardtKA, MorelliniF, SchachnerM (2005) Impacts of lesion severity and tyrosine kinase receptor B deficiency on functional outcome of femoral nerve injury assessed by a novel single-frame motion analysis in mice. The European journal of neuroscience 22 : 802–808.
48. SchnellSA, StainesWA, WessendorfMW (1999) Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem 47 : 719–730.
49. PoëtM, KornakU, SchweizerM, ZdebikAA, ScheelO, et al. (2006) Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci U S A
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- MAN1B1 Deficiency: An Unexpected CDG-II
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy