
Natural Genetic Variation of Integrin Alpha L () Modulates Ischemic Brain Injury in Stroke
During ischemic stroke, occlusion of the cerebrovasculature causes neuronal cell death (infarction), but naturally occurring genetic factors modulating infarction have been difficult to identify in human populations. In a surgically induced mouse model of ischemic stroke, we have previously mapped Civq1 to distal chromosome 7 as a quantitative trait locus determining infarct volume. In this study, genome-wide association mapping using 32 inbred mouse strains and an additional linkage scan for infarct volume confirmed that the size of the infarct is determined by ancestral alleles of the causative gene(s). The genetically isolated Civq1 locus in reciprocal recombinant congenic mice refined the critical interval and demonstrated that infarct size is determined by both vascular (collateral vessel anatomy) and non-vascular (neuroprotection) effects. Through the use of interval-specific SNP haplotype analysis, we further refined the Civq1 locus and identified integrin alpha L (Itgal) as one of the causative genes for Civq1. Itgal is the only gene that exhibits both strain-specific amino acid substitutions and expression differences. Coding SNPs, a 5-bp insertion in exon 30b, and increased mRNA and protein expression of a splice variant of the gene (Itgal-003, ENSMUST00000120857), all segregate with infarct volume. Mice lacking Itgal show increased neuronal cell death in both ex vivo brain slice and in vivo focal cerebral ischemia. Our data demonstrate that sequence variation in Itgal modulates ischemic brain injury, and that infarct volume is determined by both vascular and non-vascular mechanisms.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003807
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003807
Summary
During ischemic stroke, occlusion of the cerebrovasculature causes neuronal cell death (infarction), but naturally occurring genetic factors modulating infarction have been difficult to identify in human populations. In a surgically induced mouse model of ischemic stroke, we have previously mapped Civq1 to distal chromosome 7 as a quantitative trait locus determining infarct volume. In this study, genome-wide association mapping using 32 inbred mouse strains and an additional linkage scan for infarct volume confirmed that the size of the infarct is determined by ancestral alleles of the causative gene(s). The genetically isolated Civq1 locus in reciprocal recombinant congenic mice refined the critical interval and demonstrated that infarct size is determined by both vascular (collateral vessel anatomy) and non-vascular (neuroprotection) effects. Through the use of interval-specific SNP haplotype analysis, we further refined the Civq1 locus and identified integrin alpha L (Itgal) as one of the causative genes for Civq1. Itgal is the only gene that exhibits both strain-specific amino acid substitutions and expression differences. Coding SNPs, a 5-bp insertion in exon 30b, and increased mRNA and protein expression of a splice variant of the gene (Itgal-003, ENSMUST00000120857), all segregate with infarct volume. Mice lacking Itgal show increased neuronal cell death in both ex vivo brain slice and in vivo focal cerebral ischemia. Our data demonstrate that sequence variation in Itgal modulates ischemic brain injury, and that infarct volume is determined by both vascular and non-vascular mechanisms.
Introduction
Stroke is the second leading cause of death and the most common cause of acquired adult disability worldwide [1], [2]. Ischemic stroke is caused by disrupted blood flow within the territory of an occluded blood vessel that results in death of brain cells (infarct). The severity of cerebral infarction primarily depends on the re-perfusion and response of the blood vessels, but neuronal cell death is also determined by intrinsic molecular cascades including excitotoxicity, oxidative stress, apoptosis, and inflammation [3]. More recently, emerging data suggest that the dynamic interaction between vascular cells, glia, neurons and associated tissue matrix proteins – the neurovascular unit – plays a crucial role in the pathogenesis of ischemic brain injury [4].
Although genome-wide association studies have made some progress in the identification of stroke susceptibility genes [5], the genetic determinants for stroke outcome have yet to be fully explained. Because variation in the anatomic location of the occluded artery, the extent and duration of occlusion, time until treatment, and other contributing factors cannot be controlled in patients, very few genetic factors have been identified that contribute to the severity of brain damage in human ischemic stroke. By contrast, infarct volume in murine models of focal cerebral ischemia (stroke) has been shown to vary widely among inbred strains, suggesting strong genetic control [6]–[8]. In a mouse model of ischemic stroke, we have previously demonstrated that infarct volume varies up to 30-fold in 16 common inbred mouse strains. Using a forward genetic mapping analysis in an F2 intercross between C57BL/6J (B6 hereafter) and BALB/cByJ (BALB/c) strains, we have identified three distinct quantitative trait loci (QTLs) that modulate the volume of cerebral infarction. In particular, a single locus mapping to distal chromosome 7, Civq1 (cerebral infarct volume QTL1), accounts for the major portion of variation (56%) in infarct volume [8]. In the present study, through the use of multiple QTL mapping analyses, generation of sub-congenic mouse lines, genome-wide association across inbred strains, and ancestral SNP haplotype analyses, we have identified that genetic variation in integrin alpha L (Itgal) modulates ischemic brain injury in mice.
Results
Characterization of cerebral infarct volumes in 32 different inbred mouse strains
Using 16 common inbred mouse strains, we have previously demonstrated that infarct volume after permanent distal middle cerebral artery occlusion (MCAO) is under strong genetic control, and had mapped Civq1 on chromosome 7 as a major genetic determinant of infarct volume [8]. To further explore the naturally occurring genetic variation in infarct volume, and provide additional statistical power and map resolution, we determined infarct volumes on additional 16 inbred strains, representing the priority strains for the Mouse Phenome Project (http://phenome.jax.org). We excluded wild-derived strains to avoid spurious false positive association [9]–[11]. The ischemic damage was localized exclusively in the frontal and parietal cortex and infarct volumes were highly reproducible among individual animals of the same inbred strain. Similar to our previous report [8], we observed large variability in infarct volumes among strains (Figure 1A,B). Strains I/LnJ, LP/J, C3H/HeJ, AKR/J, A/J, BALB/c, SWR/J (SWR), BUB/BnJ, MRL/MpJ, and C58/J exhibited marked sensitivity to ischemic injury and developed large infarct volumes (>17.0 mm3). By contrast, strains C57BLKS/J, FVB/NJ (FVB), C57BR/cdJ, CBA/J, NON/LtJ, DBA/2J, NZL/LtJ, RIIIS/J, 129X1/SvJ, NOD/ShiLtJ, SJL/J, DBA/1J, C57L/J, B6, and 129S1/SvImJ were relatively resistant to cerebral ischemia, showing infarct volume smaller than 5 mm3 (Figure 1B). Mean infarct volume ranged from 0.9 to 43.0 mm3 between the strain pairs at the phenotypic extremes (C57BLKS/J vs. C58/J), representing a 47-fold difference in the trait.

Genome-wide association study remapped and reduced Civq1
From these combined data on 32 total strains (255 total mice), we preformed genome-wide associations for this trait. To correct for population structure and genetic relatedness among inbred strains of mice, we employed the Efficient Mixed Model Association (EMMA) [12]. The association scan was carried out using the 4 million high-density SNP panel using the EMMA server (http://mouse.cs.ucla.edu/emmaserver). We identified a significant region of association (Chr7 : 132.35–134.81 Mb) that co-localized within the 95% confidence interval of Civq1 that we previously mapped in linkage analyses (Figure 2A) [8]. A total of 69 SNPs across the ∼2.5 Mb region reached the statistically significant threshold (P<10−5) for cerebral infarct volume and the most significant association without any missing alleles was at a SNP (rs32965660, P = 1.25×10−6) at 132,390,712 bp on the chromosome.
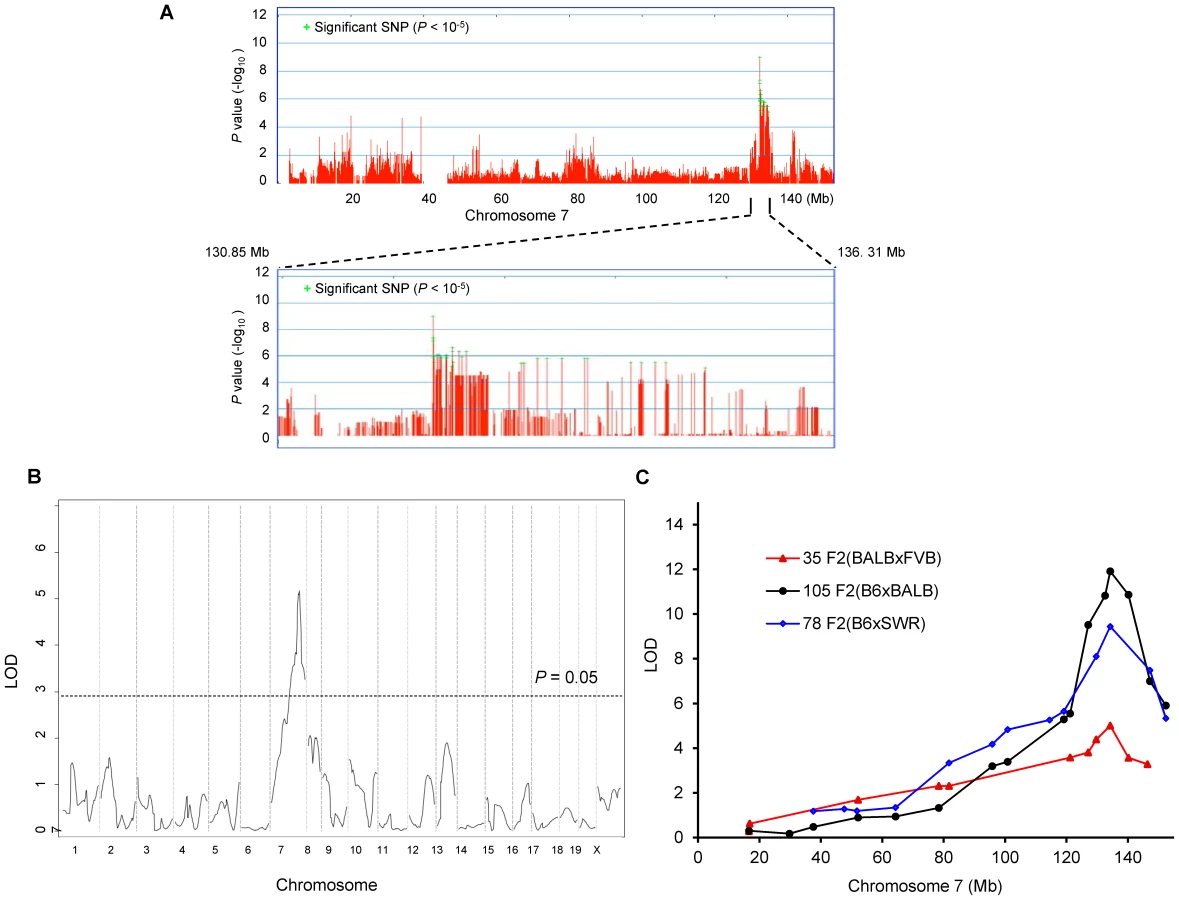
Our EMMA associated region for Civq1 on chromosome 7 covers approximately 2.5 Mb (132.35–134.81 Mb). A previous report calculated a median distance of 3 Mb between the actual causal variant and the closest marker in EMMA analysis [11], so we expanded our candidate interval for an additional 1.5 Mb region flanking either side of the associated SNPs. There are 124 known genes in the expanded associated region (130.85–136.31 Mb) on chromosome 7 (Figure 2A). Because of lack of precision in genome-wide association studies due to incomplete understanding of linkage disequilibrium in the mouse genome, statistical power is highly dependent on the number and genetic relatedness of the inbred mouse strains used [13], [14]. A previous study suggests that for a trait with a genetic effect contributing in the range of 30% to the total variance (Civq1 = 56%), 30 strains or more are required for acceptable power [15]. This suggests that our analysis is sufficiently powered, and that the causative gene for the Civq1 locus is located within the expanded 5.5 Mb region, and most likely, in the 2.5 Mb region, reduced by our EMMA analysis using 32 inbred strains.
Civq1 is remapped in an intercross between FVB and BALB/c strains
Since we previously identified Civq1 in two different genetic crosses (B6×BALB/c and B6×SWR), as well as in the Chromosome Substitution Strain 7 (CSS7) where A/J chromosome 7 was introgressed into the B6 background, and each cross includes B6 as one of the parental strains [8], it is possible that the sequence variant underlying Civq1 is unique to B6, occurring in this strain only after it was separated from its last common ancestor with the other strains [16]–[19]. To determine whether allelic variation at Civq1 is unique to the B6 strain or instead due to a sequence variant mapping within an ancestral murine haplotype block [20], we performed an additional intercross between the large infarct strain, BALB/c, and a different small infarct strain FVB; two strains which exhibit a 10-fold difference in infarct volume (Figure 1B). By substituting FVB for B6 as the “small infarct” strain in this new cross, we could effectively determine whether FVB and B6 share the “protective” allele at Civq1. Because our goal was to determine whether we would remap Civq1 in this new cross, and to date, Civq1 had shown effect sizes in excess of 50% in other crosses, we surmised that if Civq1 was responsible for the difference between these two parental strains, the locus could be identified with a minimum number of F2 animals. Even with only 35 F2 (FVB×BALB/c) mice, we identified a statistically significant locus (LOD = 5.2) that mapped to the identical position (peak LOD at rs13479513) on chromosome 7 as that of Civq1 (Figure 2B). Interestingly, the locus identified in this F2 (FVB× BALB/c) cross exhibits a large effect size (∼85%), even stronger than observed in our original two crosses (56–57%). In this cross, Civq1 accounts for nearly all of the phenotypic difference in infarct volume observed between FVB and BALB/c strains (Figure S1), and this may explain the highly significant LOD score obtained with only 35 F2 progeny. These combined data further validate the importance of Civq1 in the determination of infarct volume across common inbred mouse strains (Figure 2C). More importantly, these data strongly suggest that the sequence variant underlying Civq1 is located within an ancestral haplotype block that has been inherited across multiple inbred mouse strains, rather than being unique to strain B6. This further supports the use of ancestral haplotype association mapping approaches to fine map the Civq locus, towards the identification of the causative gene variant(s).
Congenic mouse lines retaining the infarct volume phenotype reduce the critical QTL interval and display vascular and non-vascular effects on stroke outcome
To validate the phenotypic effects of the isolated Civq1 locus, and to narrow down the critical region of the QTL, we created recombinant congenic mouse lines (C.B6-Civq1) carrying different segments of the Civq1 region from B6 introgressed into the BALB/c background. Congenic Line 1 contains approximately 22.6 Mb of the B6 region of Civq1, and Line 3 is a fully nested sub-congenic line containing a smaller region completely contained within the larger congenic region (C.B6-Civq1-1 : 119.3–141.9 Mb and C.B6-Civq1-3 : 126.2–135.8 Mb). We also attempted to generate a reciprocal congenic line (B6.C-Civq1) containing the Civq1 region from BALB/c on the B6 background (Figure 3A), but for unknown reasons, the reciprocal congenic line was embryonic lethal when crossed to homozygosity (0 homozygotes out of 52 progeny from a heterozygous congenic cross). Nonetheless, since both Civq1 exhibit heterozygous effects in the mapping crosses, we retained the heterozygous congenic line (B6.C-Civq1(Het)) for analysis (retaining approximately 16 Mb of BALB/c genomic from D7Mit238 to rs32420445).

We first analyzed infarct volume of the congenic lines. Both C.B6-Civq1-1 and -3 lines showed significantly reduced (∼30%) volume of cerebral infarction compared to control BALB/c mice (Figure 3B,C). There was no significant difference in infarct volume between the two lines carrying the larger or fully nested, smaller, segment of B6 Civq1 region, providing evidence that the 9.6 Mb region (126.2–135.8 Mb) located between the markers D7Mit238 and rs45999701 defines the critical interval for Civq1 (Figure 3A).
In a B6×BALB/c intercross, a locus modulating the number of pial collateral arteries (Collateral artery number QTL, Canq1) [21] was mapped that appears to coincide with our infarct volume locus, Civq1. This suggested that allelic variation in the same gene(s) might modulate the phenotypes of both infarct volume and collateral vessel formation in the brain. We next determined the collateral artery phenotype of these same congenic lines, measuring pial collateral arteries connecting between MCA and ACA (Figure 3D). Consistent with previous reports [21], BALB/c mice have on average less than one collateral artery per cerebral hemisphere, compared to an average of 10 in B6 mice. Both Lines 1 and 3 of the (C.B6-Civq1) congenic lines showed an approximately 50% increase in the number of collateral arteries when compared to control background BALB/c mice, and similar to the infarct volume data, there was no difference between C.B6-Civq1-1 and -3 lines (Figure 3E). Surprisingly, although the heterozygous reciprocal congenic mice (B6.C-Civq1(Het)) showed no difference in pial collateral number when compared with B6 controls (Figure 3E), the infarct volume of the heterozygous congenic mice was significantly increased when compared to B6 mice (Figure 3C). Infarct volume of the congenic mice (B6.C-Civq1(Het)) was ∼3-fold larger than that of B6 mice (14.7 mm3 vs. 4.5 mm3).
To examine whether the Civq1 locus confers a collateral-independent, tissue-intrinsic effect on cerebral infarction, we performed the widely used brain slice-based assay where transient oxygen-glucose deprivation (OGD) is used to induce neuronal cell death. Neuronal degeneration was measured via biolistic transfection of the vital fluorescent reporter, YFP, which creates a dispersed ‘sentinel’ population of cortical pyramidal neurons that can be used to quantify neuronal vitality and numbers [22], [23]. We first examined neuronal cell death for parental B6 and BALB/c strains. Subjecting YFP-transfected coronal brain slices to transient OGD resulted in the degeneration and clearance of a large proportion of cortical pyramidal neurons by 24 hr post OGD treatment. Intriguingly, similar to the sensitivity to focal cerebral ischemia, cell viability in YFP-transfected brain slices in B6 mice was significantly higher than that in BALB/c mice (Figure 3F,G). Based on this finding, we next determined the phenotype of the C.B6-Civq1-3 congenic mice. The congenic mice displayed a significantly increased number of YFP-positive live cortical neurons in OGD-treated brain slices when compared with control BALB/c mice (40%; Figure 3F,G). There was no difference in YFP transfection efficiency and viability in non OGD-treated brain slices between these mouse strains (Figure S2). In support of differential resistance to OGD in brain tissues between these strains, western blot analysis showed that the level of cleaved Caspase-3 was significantly reduced in lysates of brain slices from C.B6-Civq1-3 mice compared to control BALB/c mice after OGD treatment (Figure 3S). Taken together, these results show that the B6 allele(s) of at least one of the causal gene(s) underlying Civq1 provides non-vascular, tissue-intrinsic resistance to ischemic brain injury. More importantly, although the sum of genetic evidence to date suggested that Civq1, regulating infarct volume, and Canq1, controlling collateral artery density, are mapped to the identical genomic region, the data from the congenic strain (B6.C-Civq1(Het)) and these ex vivo, OGD experiments using brain slice explants that lack functioning vasculature suggest that the Civq1 locus may be more complex than Canq1, containing at least one gene variant that modulates ischemic brain injury independent of collateral artery density.
Interval-specific ancestral SNP haplotype analysis and fine-mapping of Civq1 toward candidate gene identification
The congenic line (C.B6-Civq1-3) reduces the critical QTL interval to a 9.6 Mb interval between D7Mit238 at 126.2 Mb and rs45999701 at 135.8 Mb, but this region still harbors over 200 genes. Although genome-wide association analysis can be employed to significantly reduce a QTL interval for candidate gene identification [24], the phenotype-associated EMMA interval in this region of chromosome 7 encompasses approximately 2.5 Mb, consisting of a genomic region of unusually high gene density, harboring more than 100 potential candidate genes. To further dissect the interval, we compared ancestral SNP haplotype patterns across the inbred mouse lineages, specifically focusing on those strains for which we had generated independent genetic mapping information [25]–[27]. Interval-specific SNP haplotype block analysis can reduce confidence intervals by identifying high-priority regions within a QTL interval that are likely to harbor the causal polymorphism [27], [28]. Because Civq1 was identified in three different genetic crosses (B6×BALB/c, B6×SWR, and FVB×BALB/c) and mapped more broadly to chromosome 7 using the CSS (B6×A/J) series, allelic variation at Civq1 is most likely harbored by a gene that maps within an ancestral haplotype block that is shared between BALB/c, A/J, and SWR (large infarct volumes), but that is different from B6 and FVB strains (small infarct volumes). As illustrated in Figure 4, defining a haplotype block to be three or more adjacent consecutive shared SNP alleles [25], we identified all SNP haplotype blocks throughout the 3.3 Mb critical region of Civq1 (132.5–135.8 Mb), a region consistent with each of the 95% confidence intervals of the 4 independent linkage peaks for Civq1. Only 4 genes (4933440M02Rik, Fam57b (1500016O10Rik), Qprt, and Itgal) fall within haplotype blocks matching the phenotype pattern of the mapping strains (Figure 4).

Itgal harbors non-synonymous coding SNPs and is differentially expressed between mouse strains
To identify the causal gene for Civq1, we first sought the presence of non-synonymous coding SNPs in these genes. Re-sequencing of the 4 candidate genes identified non-synonymous amino acid substitutions in Qprt and Itgal. Qprt encoding quinolinate phosphoribosyltransferase harbors two coding SNPs (E205K and D253N) that segregate with the infarct volume phenotype (Figure S4). These changes occur at residues that are not well conserved between mammalian species and that are predicted to be functionally benign by in silico amino acid substitution analysis in the three different databases, PolyPhen (http://genetics.bwh.harvard.edu/pph/), PMut (http://mmb2.pcb.ub.es:8080/PMut/), and Panther (http://www.pantherdb.org/) [29]. Itgal (CD11a) encodes an α subunit of β2-integrin Lymphocyte Function associated Antigen-1 (LFA-1) that mediates adhesion and migration of leukocytes. The gene harbors two coding SNPs that create W972R and P978L polymorphisms located in the calf-2 extracellular domain of the protein. Strains B6 and FVB that exhibit small infarcts, encode the W972 and P978 isoform, whereas BALB/c, A/J, and SWR strains that exhibit large infarcts, encode the R972 and L978 isoform. Interestingly, despite a lack of conservation at these residues across other species, the W972R change is predicted to be deleterious to the protein in the three in silico databases. No coding changes were identified in the two uncharacterized genes, 4933440M02Rik and Fam57b.
Next, to identify genes that show different levels of mRNA between the mapping strains, a surrogate measure of the effects of “regulatory” sequence variants (broadly defined), we performed quantitative real time PCR (qRT-PCR) for the 4 candidate genes. In adult brain cortex, only a single gene, Itgal, shows a strain-specific expression difference; an 8-fold higher transcript level in B6 cortex than seen in cortex from the large infarct strains (BALB/c, CSS7, and SWR) (Figure 5A). Since the causative variant for the infarct phenotype would need to reside within the mapped Civq1 interval, we performed allele-specific gene expression analysis to determine whether the observed expression difference is due to cis-acting variation [30]. Similar to the qRT-PCR results, allele-specific gene expression analysis confirmed that the level of B6 Itgal transcript is approximately 6-fold higher than BALB/c transcript in the adult cortex in F1 (B6×BALB/c) animals (Figure 5B), providing further evidence that regulatory genetic variation in cis causes the difference in Itgal mRNA abundance in the brain. These differences at the mRNA level were also seen at the protein level, as detected by flow cytometry of CD45-positive cells isolated from adult brain. We found that the level of ITGAL protein is significantly higher in B6 than in BALB/c mice (Figure 5C). Because the Civq1 locus contains at least one gene that modulates infarct volume via effects on collateral artery formation [21], we also examined the mRNA level for each of the 4 candidate genes in postnatal day 1 (P1) cortex (Table S1), consistent with the time of development of these vessels [31]. Itgal did not display allele-specific differential gene expression (Figure S5), consistent with a collateral-independent effect on infarct volume. We also noted that neither Fam57b nor Qprt showed allele-specific expression, and we were unable to detect 4933440M02Rik in either P1 or adult cortices. These other genes are therefore also unlikely to play a role in infarction via effects on collateral vessel anatomy.

Increased mRNA level of a splice variant of Itgal corresponds with allelic variation between mouse strains
While performing the complete re-sequencing of all of the coding exons (including at least 50 bp of flanking intron) for the 4 candidate genes, we found that the large infarct strains, BALB/c, SWR, and A/J, harbor a complex rearrangement in the distal region of the Itgal gene; a ∼150 bp deletion in intron 29 and multiple insertions and deletions (indels) in the coding and 3′-UTR of exon 30b of an alternative splice form of Itgal (Itgal-003, ENSMUST00000120857) (Figure 6A,B). Further sequencing of cDNA of the Itgal-003 transcript identified a 5-bp insertion in the coding region of exon 30b in BALB/c, SWR, and A/J strains causing a frameshift in the encoded protein, resulting in novel amino acid sequence and a shorter cytoplasmic tail of the protein, as compared to strains B6 and FVB (Figure 6C).

We also found that the mRNA level of Itgal-003 markedly differed between the 5 mapping strains in the P1 cortex. The mRNA level of this splice variant was substantially higher (>11-fold) in the large infarct strains (BALB/c, SWR, and CSS7) than that of the small infarct strains, B6 and FVB mice (Figure 6D). Using the more accurate allele-specific expression analysis, the level of BALB/c Itgal-003 transcript is approximately 20-fold higher than B6 transcript in P1 cerebral cortex in F1 (B6×BALB/c) animals (Figure 6E), indicating that sequence variation in cis leads to increased levels of the Itgal-003 transcript in the BALB/c strain. Since the interplay between neurons, endothelial cells, and glial cells plays a crucial role in the early development of the neurovascular unit, and thus, the pathogenesis of cerebral ischemia [4], we investigated the mRNA profile of Itgal-003 in both CD146 (LSEC)-positive endothelial cells and CD11b-positive brain macrophages isolated from F1 embryos (E18.5). As illustrated in Figure 6E, BALB/c-specific Itgal-003 transcript is expressed 43 times and 25 times higher in endothelial cells and macrophages, respectively, than the B6-specific transcript in the allele-specific expression analysis. Interestingly, the Itgal-003 transcript was not detected in the adult cerebral cortex by RT-PCR, suggesting that this splice variant is acting primarily or exclusively during brain development. To date, 5 Itgal splice isoforms have been identified in mice (Figure S6), so we determined the relative allele-specific expression of all the other Itgal splice isoforms. No difference was found in P1 and adult cerebral cortices for the other isoforms (data not shown). To further determine whether the strain specific level of Itgal-003 transcript is also reflected at the level of the protein, we generated an Itgal-003-specific antibody against the unique cytoplasmic tail peptide to assay protein expression (Figure 6C). Consistent with the mRNA level, western blot analysis of P1 cerebral cortex demonstrated markedly increased protein level in strain BALB/c compared to strains B6 and FVB (Figure 6F).
Itgal is neuroprotective for ischemic brain damage
To determine whether Itgal is involved in ischemic brain injury in vivo, we next examined the phenotype of Itgal knockout mice [32]. A recent study reported that there was no difference in collateral vessel number or in infarct volume between Itgal knockout and control B6 mice [33]. However, despite also finding no difference in number of collateral arteries (Figure 7A), we observed that infarct volume in Itgal knockout mice (n = 30) of B6 background (17 generations backcrossed into B6) was ∼3-fold larger than that observed in B6 mice (15.5 mm3 vs. 4.7 mm3) (Figure 7B,C). We believe that this difference in infarct volume results from the use of different surgical techniques. When performing permanent distal MCAO, care has to be taken to position the occlusion proximal to the lenticulo-striate branches [34], [35]. If the artery is occluded more distally, smaller and more variable infarcts are produced. In support of this explanation for the difference between the studies, they report a difference in infarct volume of ∼3-fold between B6 and BALB/c strains [33], whereas we routinely observe that infarct volume in BALB/c mice is ∼7-fold larger than that of B6 mice (4.7 mm3 vs. 34.2 mm3) (Figure 1B).

Since the Civq1 locus retaining infarct volume phenotype displays both vascular and non-vascular neuroprotective effects on ischemic tissue injury, we hypothesized that the increased infarct volume in Itgal knockout mice might be related to neuroprotection after focal cerebral ischemia. To examine this further, we determine the level of OGD-induced neuronal cell death in brain slices from Itgal knockout mice, again counting YFP-transfected cortical pyramidal neurons in brain slices. As a control, there was no difference in YFP-transfection efficiency and viability in non OGD-treated brain slices between B6 and Itgal knockout mice (Figure S2). Consistent with the infarct volume data, Itgal knockout mice showed an approximately 50% increase in neuronal cell death after transient OGD, compared to control B6 mice (Figure 7D,E). After OGD treatment, the level of cleaved Caspase-3 was also significantly increased in lysates of brain slices from Itgal knockout mice compared to B6 mice (Figure S3). To further investigate whether reduced levels of Itgal modulate ischemic brain injury, and particularly whether these effects were collateral vessel-independent, we employed an ex vivo model of cerebral ischemia, using siRNA to knockdown Itgal expression in cortical brain slices (where collateral circulation is no longer relevant). Consistent with the increased neuronal cell death observed in Itgal knockout mice, after transient oxygen deprivation, Itgal siRNA-treated brain slices from B6 mice show markedly increased levels of cleaved Caspase-3, a marker of neuronal cell death (Figure 8). These results indicate that Itgal plays a protective role in ischemic brain damage, independent of tissue reperfusion through the collateral vessels.

It should be noted that the Itgal knockout mouse line used in our study was generated by replacing the exons 1 and 2 with a Neo-cassette [32] but more recent data shows that there are 5 known alternative splicing transcripts of the gene, including transcripts that do not include these two exons (Figure S6). Thus, we examined whether this Itgal knockout mouse line generates null alleles for all of the splice variants of the gene. We found that a splicing variant that uses a different initiation site (Itgal-004, ENSMUST00000118405) was detected in P1 and adult cortices by RT-PCR (Figure S6), suggesting that the phenotypes we observed in the knockout mice represent only partial knockout of the entire complement of Itgal gene transcripts/functions. Thus, this well-established Itgal knockout line may retain residual or additional isoform protein functions, and in the sense of total Itgal gene function(s), thus represents a hypomorphic allele.
The Itgal knockout allele was generated using 129Sv (Stevens) genomic DNA and a 129S7 (129S7/SvEvBrd-Hprtb-m2) ES cell line. Thus, it is formally possible that the some of the differences in infarct volume that are seen in the Itgal knockout were contributed by 129 alleles flanking the deleted Itgal locus. Although the original strains used in the knockout construction are not widely available, we have determined infarct volume and collateral artery number for the related 129S1/SvImJ strain which is derived from the original 129/Sv strain [36]. Both infarct volume and collateral artery number of 129S1/SvImJ mice are not significantly different from those of B6 mice (Figure S7). Thus, we conclude that the effects seen with the Itgal KO mice are primarily due to the loss of the Itgal gene, and not linked (129Sv or 129S7) polymorphisms, although we cannot rule out modest bystander effects.
Discussion
Although several approaches have been proposed to reduce ischemic brain damage, including reperfusion, neuroprotection, and neuronal regeneration, for the vast majority of stroke patients, current therapies are limited. We reasoned that given the multiplicity of mechanisms causing cell death in stroke, approaches that augment endogenous protective pathways might be more likely to lead to success. Thus, we have attempted to identify novel genetic factors modulating stroke outcomes by exploiting naturally occurring endogenous genetic variation determining ischemic brain injury. In crosses between inbred mouse strains that exhibit large differences in infarct volumes, we previously identified a QTL (Civq1) mapping to distal chromosome 7 that determines more than 50% of the variation observed between the strains. In this study, we present evidence that Itgal is one of the genes underlying the complex Civq1 locus.
Despite the almost routine detection of QTLs for important disease traits in both rodents and humans, identification of causal genes underlying QTLs remains a major obstacle, in large part due to the large confidence intervals for the typical QTL, often covering hundreds of candidate genes [26]. To narrow the Civq1 interval we employed three different methods that capitalize on the structure of the mouse genome: (1) generation of interval-specific congenic lines, (2) genome wide association (EMMA) analysis across more than 30 inbred mouse strains, and (3) interval-specific SNP haplotype analysis using the 5 inbred strains from our experimental crosses. The latter approach was most effective at reducing the list of candidate genes in the Civq1 interval to only 4 genes that clearly fall within shared SNP haplotype blocks. Of these 4 genes, Itgal was the only gene that harbored non-synonymous coding SNPs and exhibited altered mRNA abundance, with both molecular phenotypes co-segregating with the infarct volume phenotypes in the 5 strains used in QTL mapping. We also found that allelic variation in an alternative splicing variant of Itgal (Itgal-003) resulted not only in differential transcript abundance, but also in a truncated cytoplasmic tail of the protein, consisting mostly of a novel amino acid sequence.
Itgal encodes the α subunit of LFA-1(αLβ2) integrin, which is highly expressed in microglia, spleen, bone marrow, and most immune cell populations. The binding of intracellular proteins to the cytoplasmic tail of Itgal is essential to the activation of LFA-1 integrin. The canonical splice isoform of Itgal (Itgal-002, ENSMUST00000117762) contains this important functional domain, conserved among all integrin family members. Mutation of the cytoplasmic tail of Itgal (Ital-002) has been shown to inhibit its interactions with intracellular proteins, destabilize integrin conformation, and disconnect to cytoskeleton [37], [38]. By contrast, the function of the unique, truncated sequence of the cytoplasmic tail found in Itgal-003 remains unknown. The increased expression of the Itgal-003 in large infarct strains may interfere with the functions of the well-studied, reference isoform of Itgal, or with other α subunits (αD, αM, and αX) that can bind to the β2 subunit, resulting in inhibition of cell adhesion and migration during the development and/or tissue injury. In addition to these differences in the cytoplasmic tail of Itgal-003, the inbred mouse strains show two amino acid substitutions (W972R and P978L) in the calf-2 domain that segregate with the infarct volume phenotypic difference. These two coding changes in ITGAL fall in relatively poorly conserved residues of the protein and the 972R BALB/c allele is shared with other species including cow, sheep, and opossum. However, the lack of conservation at these residues itself does not allow us conclude that these changes are inconsequential because non-synonymous coding SNPs causing risk for complex genetic traits tend not to fall within highly conserved residues/regions of proteins [39]. In point of fact, W972R is predicted to be deleterious by multiple in silico amino acid substitution databases. Although the exact role of the calf-2 domain is not fully understood, a point mutation in the calf-2 domain of αvβ3 integrin in Glazmann thrombasthenia patients disrupts the normal contacts between α and β subunits, resulting in impaired cellular transport from the endoplasmic reticulum [40]. Thus, the two amino acid substitutions in the calf-2 domain of ITGAL may have an effect on α-β formation and stabilization of LFA-1 integrin. Taken together, natural DNA sequence variation in the mapping strains in Itgal generates both qualitative (a strain-specific splice variant and two amino acid coding changes) and quantitative (overall transcript level) changes in the transcript and encoded protein. We do not know exactly which of these changes is most relevant to the infarct volume phenotype but we surmise that it is likely to be a combination of some or all of these. Importantly, we have shown that a congenic animal that retains all of these strain-specific differences in Itgal variation from B6 shows a similar phenotype to B6 mice, whereas the Itgal gene knockout appears similar to BALB/c strain. In addition, siRNA knockdown of Itgal in an ex vivo cerebral ischemia model using cortical brain slices from B6 mice also increases the extent of neuronal cell death. These combined data suggest that in comparison to the B6 Itgal allele, the net effect of the BALB/c allele is at least a partial loss of function.
The identification of Civq1 has raised the question of the role of the causative gene(s) in pathophysiology of ischemic stroke. A previous study suggested that the differential ischemic outcomes between inbred mouse strains is related to intrinsic differences in ischemic tolerance or protection pathways in neural tissue [7]. More recently, the extent of pial collateral circulation in the brain has been shown to be inversely correlated with infarct volume data between 15 inbred strains [41]. The authors also identified a QTL (Canq1) for collateral vessel number mapping to the identical genomic position as Civq1, thereby proposing that the causative gene controlling collateral circulation might also determine the differential infarct volume [21]. Surprisingly, we have observed that the genetically isolated Civq1 locus retaining infarct volume phenotype displays both vascular (collateral circulation) and non-vascular (neuroprotection) effects in the modulation of ischemic brain damage after MCAO. These observations are in accord with our previous study that this locus displays a non-vascular protective effect on ischemic insult in skeletal muscle [42]. In a mouse model of hind-limb ischemia, we previously mapped a strong genetic locus determining limb necrosis and recovery of perfusion (Lsq1) at the identical genomic position as Civq1 on chromosome 7 [43]. Using an in vitro model of hypoxia and nutrient deprivation, where collateral circulation and indeed all circulation is absent, we have found that isolated primary myocytes from BALB/c are more sensitive to hypoxia and nutrient deprivation than B6 myocytes, recapitulating the strain-specific response to hind-limb ischemia. More importantly, muscle cells from the congenic mice (C.B6-Civq1-3) are protected from this same in vitro hypoxic insult, indicating that the B6 allele(s) of the causative gene(s) plays an important role in survival of muscle cells, independent of any vascular contribution to ischemia [42]. Therefore, given the physiological similarities between the two ischemic models and the identical map position, we propose that the same causative gene(s) underlying Civq1/Lsq1 determines ischemic tissue damage in multiple tissues, possibly through the same physiological mechanism.
A number of studies have demonstrated that microglia play an important protective role in ischemic brain damage through microglial migration to the site of injury [44], [45], which is controlled by the Itgal protein [46], [47]. Microglial cells with down-regulated Itgal expression fail to protect neurons after OGD in cultured hippocampal brain slices, suggesting that the migration and adhesion of microglial cells regulated by Itgal is important for the beneficial effect of microglia in stroke [47]. These published data further support Itgal as one of the genes underlying Civq1.
However, Itgal also shows deleterious effects in stroke, as Itgal is also involved in inflammatory injury after cerebral ischemia. After the ischemic insult, the brain is invaded by blood-circulating leukocytes, and LFA-1 (containing Itgal) regulates the interaction between circulating leukocytes and endothelial cells. Deficiency of Itgal shows a protective effect on ischemia-reperfusion injury using a transient MCAO model [48]. However, it should be stressed that permanent (our study) and transient MCAO [48] models exhibit different pathophysiologies [49]. Large numbers of circulating blood cells enter the brain at time points later than 24 hr after the ischemic insults [50], [51], but we measure infarct volume at 24 hr after MCAO. Thus, microglia, expressing Itgal, may play their critical, protective role in the early stages of stroke.
Thus far, we have emphasized vascular-independent functions of Itgal in the modulation of infarct volume. But it is clear that the Civq1 locus also contains genetic determinants that modulate infarct volume via changes in collateral vessel anatomy. The identity and nature of these genes and gene variants remains to be determined. Recent studies have demonstrated that microglia regulate vascular anastomosis and increase vascular complexity by assisting endothelial tip cell fusion during brain development [52]. LFA-1 (containing Itgal) modulates adhesion of monocyte to collateral endothelium involved in arteriogenesis [53], [54]. Given the important role of microglia and Itgal in these processes related to collateral vessel development, the question remains then why the Itgal knockout mouse line used in this study did not exhibit a collateral vessel phenotype. Because an engineered knockout allele is rarely equivalent to a naturally-occurring variant allele at a QTL [14] and an Itgal splice isoform was in fact detected in these Itgal “knockout” mice, we cannot exclude the possibility that collateral vessel development and neuroprotection are genetically regulated by different alleles or splice isoforms of Itgal.
Alternatively, it is quite likely that more than one gene underlies the complex Civq1 locus. One or more genes may modulate infarct volume by their effect on neuroprotection or inflammatory physiology, and another gene or genes may modulate infarct volume by regulating collateral artery formation. Recent successes in QTL gene identification support this conjecture. Multiple, physically linked, smaller effect genes often contribute to the overall effect of robust, large effect QTLs [14], [55]–[58]. In this light, we note that a number of gene products mapping within the Civq1 interval have well-defined roles in the immune response (cytokine receptors, integrins), tissue remodeling (MMP21, ADAM12), or metabolism (cytochrome c oxidase), each of which could be involved in the overall response to ischemia. Generation of sub-congenic strains that further divide the Civq1 locus may help identify these genes.
Although the Itgal haplotype across 32 inbred strains generally correlates with infarct volume, the correlation falls off for several outlier strains. For example, despite that fact that the NOD strain shares the BALB/c haplotype for the Itgal locus, this strain exhibits a small infarct volume and its mRNA expression level is significantly higher than those of the large infarct strains (Figure S8). Similarly, strain C3H shares the B6 haplotype including a high mRNA expression level, the W972 and P978 SNPs, and lacks the genomic deletion, but the C3H infarct volume is larger than most of the small infarct strains. These data suggests for certain outlier strains, loci other than the otherwise large-effect Civq1 locus can cause profound phenotypic effects. In line with this hypothesis, we have identified a novel genetic locus mapping to mouse chromosome 8 in an intercross between B6 and one of these outlier strains, C3H [59]. Overall, these results are consistent with what we have shown from our previous mapping data, namely, that genetic variation in Itgal is not solely causative for the differential phenotype. Even within the Civq1 locus, there are additional genes for determining infarct volume (ie, a gene(s) regulating collateral vessel density). Nonetheless, our data using congenic and Itgal knockout mice show that at least one of the causative gene(s) underlying Civq1 functions in the survival of brain tissue independent of a vascular contribution to the ischemic response. Taken together, these data show that the extent of the collateral circulation will not be the sole mechanism underlying Civq1/Lsq1, and possibly for other loci as well.
In summary, by showing evidence of its role in regulation of ischemic brain injury in a mouse model of stroke, we have identified Itgal as one of possibly many quantitative trait genes for Civq1. Natural DNA sequence variation in Itgal generates qualitative and quantitative change of the transcript and the encoded protein, and a knockout allele, even while retaining one of the newly described splice isoforms, shows a robust effect on infarct volume in vivo and in vitro. Future studies using cell type-specific knockout mice will help dissect cellular and molecular mechanisms of Itgal in both vascular and non-vascular contributions to ischemic brain injury. Ultimately, this work will provide insight into the endogenous protective pathways involved in the pathophysiology of ischemic tissue damage, and in the long-term, provide novel targets for potential therapeutic intervention of ischemic stroke.
Materials and Methods
Ethics statement
All experiments were performed under protocols approved by the Animal Care and Use Committee of Duke University.
Animals
All inbred strains and Itgal knockout mice (B6.129S7-Itgaltm1Bll/J) were obtained from the Jackson Laboratory (Bar Harbor, Me) either directly or bred locally from breeding pairs of each strain. Mice were age-matched (12±1 week) for all experiments.
Permanent distal MCAO stroke model
Focal cerebral ischemia was induced by direct occlusion of the distal MCA as described previously [8]. Briefly, mice were anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg), and the right MCA was exposed by a 0.5-cm vertical skin incision midway between the right eye and ear. After the temporalis muscle was split, a 2-mm burr hole was drilled at the junction of the zygomatic arch and the squamous bone. While visualizing with a stereomicroscope, the right MCA was cauterized using an electrocauterizer (Fine Science Tools). The cauterized MCA segment was then transected with micro scissors to verify that the occlusion was complete. The surgical site was closed with 6-0 sterile nylon sutures, and 0.25% bupivicaine was applied. Animals were maintained at 37°C during and after surgery until fully recovered from anesthesia, when they were returned to their cages and allowed free access to food and water. All mice were housed in an air-ventilated room with ambient temperature maintained at 25±0.5°C.
Measurement of infarct volume
Twenty-four hours after surgery, the animal was euthanized and the brain was removed, chilled at −80°C for 3 min to slightly harden the surface and sliced into 1-mm coronal sections using a brain matrix. Slices were stained with 2% TTC (2,3,5-Triphenyl-tetrazolium chloride) as previously described [8]. Infarct volumes were calculated by measuring infarct areas on the separate slices, multiplying areas by slice thickness, and summing all slices; this “indirect” morphometric method corrects for edematous swelling.
Collateral artery measurement
Under ketamine (100 mg/kg) and xylazine (5 mg/kg), the left ventricle of the heart was cannulated. The right atrium of the heart was incised to allow for venous outflow and the circulation was cleared and maximally dilated with heparin (50 µg/ml), adenosine (1 mg/mL) and papaverine (40 µg/mL) in phosphate-buffered saline (PBS). Immediately after the PBS infusion, the skull and dura were carefully removed and blue polyurethane (PU4ii, Vasqtec) with a viscosity sufficient to restrict capillary transit (1∶1 resin∶methylethyl ketone) was injected. Formalin (10% in PBS) was applied topically to the cortex, and the polyurethane was allowed to cure for 20 minutes. After post-fixation in 10% formalin overnight, the pial circulation was imaged (Leica MZ16FA). Analysis was confined the measurement of collaterals between the MCA and ACA trees [41].
OGD of brain slices
Brain slice isolation and OGD experiments were performed as previously described [22], [23]. B6, BALB/c, C.B6-Civq1-3, and Itgal KO mice were euthanized at postnatal day 10. Each brain was cut into 250 µm coronal slices on a Vibratome (Vibratome) in chilled culture medium containing 15% heat-inactivated horse serum, 10 mM KCl, 10 mM HEPES, 100 U/ml penicillin/streptomycin, 1 mM MEM sodium pyruvate, and 1 mM L-glutamine in Neurobasal A supplemented with NMDA inhibitor (1 µM MK-801). Brains were divided into “hemi-coronal” slices. For OGD, slices were suspended at 34°C for 5.5 min in glucose-free, N2-bubbled artificial CSF containing 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 24 mM D-glucose, and 10 mM HEPES. Control and OGD-treated brain slices were plated onto a 12-well plate with solid culture medium made by the addition of 0.5% agarose. After explanting the brain slices, plates were placed for recovery at 37°C for 30 min in a humidified incubator under 5% CO2. Gold particle-coated plasmids containing Yellow Fluorescent Protein (YFP) were introduced into the brain slices by biolistic transfection using a Helios Gene Gun (Bio-Rad, Herculues). Slice cultures were maintained at 37°C for 24 hr in humidified incubator under 5% CO2.
Genotyping
Single nucleotide polymorphism (SNP) genotyping was performed using the GoldenGate genome-wide mouse 377 SNP panel (Illumina). Genomic positions of genetic markers (NBCI Build37/mm9) were retrieved from the UCSC genome browser (http://www.genome.ucsc.edu/) and converted to cM using the mouse map converter (http://cgd.jax.org/mousemapconverter). Additional SNP (rs13479513, rs13468481, rs45999701, rs32420445, rs37240399 and rs6216320) and microsatellite (D7Mit281, D7Mit238, and D7Mit68) markers were used for fine-mapping of the locus. The distal boundary of the critical interval (9.6 Mb) of C.B6-Civq1-3 was determined by locations of maximal breakpoints for the genotyped markers. The critical interval was homozygous for BALB/c alleles at 126.2 Mb (D7Mit238) and at 135.82 Mb (rs45999701).
Linkage analysis
Genome-wide scans were plotted using the J/QTL mapping program, version 1.2.1 (http://research.jax.org/faculty/churchill/). Suggestive (P = 0.63) and significant (P = 0.05) thresholds were established empirically for each phenotypic trait by 1,000 permutation tests using all informative markers. The percentage of total trait variance attributable to each locus was determined using the Fit-QTL function provided within the J/QTL software.
Genome-wide association EMMA analysis
Genome-wide association mapping for infarct volumes for the 32 strains was performed with EMMA using the UCLA web-based server (http://mouse.cs.ucla.edu/emmaserver) [12]. Analysis of individual phenotypic data was performed with SNP panels consisting of 4 million SNPs [11]. We determined confidence intervals by expanding the interval around the peak SNP to include all neighboring SNPs surpassing the significance threshold (P = 10−5). For single SNP associations, the QTL confidence interval was set at 3 Mb (1.5 Mb kb on either side of the peak SNP). SNP-associated P values were transformed with −log10 (P value) for graphing association scores.
Interval-specific SNP haplotype analysis
For the 9.6 Mb interval on chromosome 7, SNP data were obtained from the Mouse Phenome Database (http://phenome.jax.org/), the Wellcome Trust Sanger Institute Mouse Genome Browser (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl), and the Center for Genome Dynamics (http://cgd.jax.org). Physical map position was based on the genomic sequence from the NCBI Build 37/mm9. Haplotype blocks were defined as three or more adjacent informative SNPs shared between the large infarct strains (BALB/c, A/J and SWR/J) which differed from the haplotype for the small infarct strains (B6 and FVB) [25].
Generation and purification of the anti-Itgal-003 antibody
The cytoplasmic tail peptide (GQRRDIGMDQEERAGPGRL) of Itgal-003 was synthesized and then used to generate an Itgal-003-specific polyclonal antibody (Bethyl Laboratories). The rabbit antiserum was affinity purified and tested for immunoreactivity for Itgal-003 by immunoblotting.
Isolation of embryonic macrophages and endothelial cells
Each E18.5 embryonic brain was cut into small pieces, incubated in DMEM containing 10% fetal bovine serum and Collagenase type IV (0.2 mg/ml, Sigma) for 30 min at 37°C and then passed through a 19G syringe to obtain a homogeneous cell suspension. Cells were washed with PBS supplemented with 0.5 mM EDTA and 0.5% BSA and incubated in 10 µl anti-CD131 (Endothelial cells) or anti-CD11b (Macrophages) antibody-conjugated magnetic beads (Miltenyi Biotech) for 15 min on ice. These cells were applied to MACS MS separation columns on magnetic stands and washed with 1.5 ml PBS supplemented with 0.5 mM EDTA and 0.5% BSA. The column was removed from the magnetic stand and magnetically labeled cells were isolated by flushing out fractions. Isolated cells were used to extract total RNA using Trizol (Invitrogen).
SYBR_quantitative RT-PCR
Quantification of RT-PCR products were measured by examining the increase in fluorescence that was induced by SYBR green binding to dsDNA (Applied Biosystems). The reaction was analyzed on an ABI 7700 Sequence Detection system using the following conditions: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. All samples were run in triplicate and additional assays for endogenous controls (Gapdh and/or Hprt1) were performed to control for input cDNA template quantity. Relative quantification was determined for each sample by calculating the mean Ct value using the 2−ΔΔCt method. Sequence detection software (SDS version 2.1.1, Applied Biosystems) was used for this analysis.
Allele-specific SNapShot gene expression analysis
This approach requires at least one SNP in the transcript to distinguish the alleles of the two strains. PCR was performed using an appropriate dilution of cDNA generated from the cerebral cortex of F1 (B6×BALB) animals. Amplicons containing coding SNPs were amplified by conventional PCR, and 15 ul of PCR products were treated with 1 U exonuclease I (New England Biolabs) and 5 U of Shrimp Alkaline Phosphatase (SAP) (Promega). Purified PCR products were used in combination with a conventional primer designed to sit at the nucleotide to the immediate 5′ position of a coding SNPs in the transcript. Cycling conditions were as follows: 40 cycles of 95C° for 10 s, 50C° for 5 s, and 60C° for 30 s. The primer is then labeled with a fluorescently tagged dideoxynucleotide through a single base pair extension (SNaPshot kit from ABI). These products were treated with 1 U of SAP prior to running on an ABI 3130 sequencer, and peak heights were determined using Gene Mapper software (ABI). To determine conditions under which the SNaPshot assays were quantitative, genomic DNA from the F1 animals was amplified and also analyzed by SNaPshot using the same extension. The expression ratio of each transcript allele was normalized to the ratio of the two alleles in the F1 genomic DNA.
Itgal siRNA knock-down experiments
To reduce Itgal mRNA expression by RNA interference, the Itgal-specific siRNA was purchased from Thermo Scientific and used as recommended. For the ex vivo stroke experiments, siRNA was delivered to the cortical brain slices before the oxygen deprivation. Briefly, after explanting brain slices from B6 mice (age P10), plates were placed for recovery at 37°C for 30 min in a humidified incubator under 5% CO2. 5 µM of non-target pooled (D-001910-10-05) or Itgal-specific pooled (E-046772-00-0005) siRNAs were introduced onto the brain slices and the slices were maintained at 37°C in a humidified incubator under 5% CO2. Forty eight hours after introducing the siRNA, brain slices were deprived of oxygen using glucose-free, N2-bubbled artificial CSF. The slices were then incubated for an additional 24 hr.
Flow cytometry
Mice were perfused with PBS and brains were dissected out and weighed. Brains were teased apart and digested for 1 hour at 37C with 2 mg/mL collagenase A (Roche) and 0.25 mg/mL DNase I (Roche). Cells were strained and centrifuged in a 30% over 70% Percoll (Invitrogen) in PBS density gradient. Cells from the interface were isolated and red blood cells were lysed with ACK lysis buffer. Cells were counted and stained with the following antibodies: CD11a-PE or IgG2a-PE (eBioscience); CD11c-PE-Cy5.5 (eBioscience); CD45-PE-Cy7 (eBioscience); Ly6G-AF700 (eBioscience); IA-IE-Qdot655 (eBioscience); and CD11b-BV780 (Biolegend). Flow cytometry was run on a LSR-II in the Duke Human Vaccine Institute Flow Research Facility. Analysis was done with FlowJo (Treestar).
Western blot analysis
Following anesthesia, the whole brains were isolated from mice, and were homogenized in cold lysis buffer (50 mM Tris/HCl [pH 7.4], 1 mM EGTA, 1 mM DTT) containing a protease and phosphatase inhibitor cocktail (Thermo Scientific). After low speed centrifugation (1,000×g, 5 min), supernatants (30 ug) were then electrophoresed in a 4–12% polyacrylamide gel and electro-blotted for 2 hr on PVDF (Polyvinylidine Fluoride) membranes at room temperature. The blot was incubated with the polyclonal anti-Itgal-003 (1∶1,000) or anti-alpha-Tubulin (1∶5,000) primary antibodies overnight at 4C°. To detect the level of apoptosis, six brain slices from each group were collected after OGD treatment. Anti cleaved-Caspase3 antibody (1∶3,000, Cell Signaling Tech) was applied to detect level of apoptotic cell death. The protein bands were visualized using the chemiluminescence reaction (ECL detection kit).
Statistical analysis
Results were represented as the mean±SEM. Statistical analysis of infarct volume, number of primary branch from MCA as well as collateral vessel number were performed using 1-way ANOVA or non-parametric Kruskal–Wallis test. For qRT-PCR and SNapShot allele-specific expression analyses, student's t-test was used to determine if the fold change was significantly different.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. StrongK, MathersC, BonitaR (2007) Preventing stroke: saving lives around the world. Lancet Neurol 6 : 182–187.
2. DichgansM (2007) Genetics of ischaemic stroke. Lancet Neurol 6 : 149–161.
3. HossmannKA (2006) Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol 26 : 1057–1083.
4. MoskowitzMA, LoEH, IadecolaC (2010) The science of stroke: mechanisms in search of treatments. Neuron 67 : 181–198.
5. BevanS, TraylorM, Adib-SamiiP, MalikR, PaulNL, et al. (2012) Genetic Heritability of Ischemic Stroke and the Contribution of Previously Reported Candidate Gene and Genomewide Associations. Stroke 43 : 3161–7.
6. BaroneFC, KnudsenDJ, NelsonAH, FeuersteinGZ, WilletteRN (1993) Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab 13 : 683–692.
7. MajidA, HeYY, GiddayJM, KaplanSS, GonzalesER, et al. (2000) Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 31 : 2707–2714.
8. KeumS, MarchukDA (2009) A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circ Cardiovasc Genet 2 : 591–598.
9. PayseurBA, PlaceM (2007) Prospects for association mapping in classical inbred mouse strains. Genetics 175 : 1999–2008.
10. MhyreTR, CheslerEJ, ThiruchelvamM, LunguC, Cory-SlechtaDA, et al. (2005) Heritability, correlations and in silico mapping of locomotor behavior and neurochemistry in inbred strains of mice. Genes Brain Behav 4 : 209–228.
11. KirbyA, KangHM, WadeCM, CotsapasC, KostemE, et al. (2010) Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics 185 : 1081–1095.
12. KangHM, ZaitlenNA, WadeCM, KirbyA, HeckermanD, et al. (2008) Efficient control of population structure in model organism association mapping. Genetics 178 : 1709–1723.
13. Burgess-HerbertSL, TsaihSW, StylianouIM, WalshK, CoxAJ, et al. (2009) An experimental assessment of in silico haplotype association mapping in laboratory mice. BMC Genet 10 : 81.
14. FlintJ, EskinE (2012) Genome-wide association studies in mice. Nat Rev Genet 13 : 807–817.
15. CervinoAC, DarvasiA, FallahiM, MaderCC, TsinoremasNF (2007) An integrated in silico gene mapping strategy in inbred mice. Genetics 175 : 321–333.
16. YalcinB, WongK, AgamA, GoodsonM, KeaneTM, et al. (2011) Sequence-based characterization of structural variation in the mouse genome. Nature 477 : 326–329.
17. YangH, WangJR, DidionJP, BuusRJ, BellTA, et al. (2011) Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet 43 : 648–655.
18. FrazerKA, EskinE, KangHM, BogueMA, HindsDA, et al. (2007) A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature 448 : 1050–1053.
19. KeaneTM, GoodstadtL, DanecekP, WhiteMA, WongK, et al. (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477 : 289–294.
20. WadeCM, KulbokasEJ3rd, KirbyAW, ZodyMC, MullikinJC, et al. (2002) The mosaic structure of variation in the laboratory mouse genome. Nature 420 : 574–578.
21. WangS, ZhangH, DaiX, SealockR, FaberJE (2010) Genetic architecture underlying variation in extent and remodeling of the collateral circulation. Circ Res 107 : 558–568.
22. WangJK, PortburyS, ThomasMB, BarneyS, RiccaDJ, et al. (2006) Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc Natl Acad Sci U S A 103 : 10461–10466.
23. DunnDE, HeDN, YangP, JohansenM, NewmanRA, et al. (2011) In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J Neurochem 119 : 805–814.
24. PletcherMT, McClurgP, BatalovS, SuAI, BarnesSW, et al. (2004) Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol 2: e393.
25. Burgess-HerbertSL, CoxA, TsaihSW, PaigenB (2008) Practical applications of the bioinformatics toolbox for narrowing quantitative trait loci. Genetics 180 : 2227–2235.
26. FlintJ, ValdarW, ShifmanS, MottR (2005) Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet 6 : 271–286.
27. WangX, KorstanjeR, HigginsD, PaigenB (2004) Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res 14 : 1767–1772.
28. ParkYG, CliffordR, BuetowKH, HunterKW (2003) Multiple cross and inbred strain haplotype mapping of complex-trait candidate genes. Genome Res 13 : 118–121.
29. AdzhubeiIA, SchmidtS, PeshkinL, RamenskyVE, GerasimovaA, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7 : 248–249.
30. CowlesCR, HirschhornJN, AltshulerD, LanderES (2002) Detection of regulatory variation in mouse genes. Nat Genet 32 : 432–437.
31. ChalothornD, FaberJE (2010) Formation and maturation of the native cerebral collateral circulation. J Mol Cell Cardiol 49 : 251–259.
32. DingZM, BabenseeJE, SimonSI, LuH, PerrardJL, et al. (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 163 : 5029–5038.
33. WangS, ZhangH, WiltshireT, SealockR, FaberJE (2012) Genetic dissection of the Canq1 locus governing variation in extent of the collateral circulation. PLoS One 7: e31910.
34. HossmannKA (2008) Cerebral ischemia: models, methods and outcomes. Neuropharmacology 55 : 257–270.
35. CarmichaelST (2005) Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx 2 : 396–409.
36. AdamsDJ, QuailMA, CoxT, van der WeydenL, GorickBD, et al. (2005) A genome-wide, end-sequenced 129Sv BAC library resource for targeting vector construction. Genomics 86 : 753–758.
37. LuCF, SpringerTA (1997) The alpha subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J Immunol 159 : 268–278.
38. LuoBH, CarmanCV, SpringerTA (2007) Structural basis of integrin regulation and signaling. Annu Rev Immunol 25 : 619–647.
39. ThomasPD, KejariwalA (2004) Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A 101 : 15398–15403.
40. RosenbergN, YatuvR, SobolevV, PeretzH, ZivelinA, et al. (2003) Major mutations in calf-1 and calf-2 domains of glycoprotein IIb in patients with Glanzmann thrombasthenia enable GPIIb/IIIa complex formation, but impair its transport from the endoplasmic reticulum to the Golgi apparatus. Blood 101 : 4808–4815.
41. ZhangH, PrabhakarP, SealockR, FaberJE (2010) Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab 30 : 923–934.
42. McClungJM, McCordTJ, KeumS, JohnsonS, AnnexBH, et al. (2012) Skeletal muscle-specific genetic determinants contribute to the differential strain-dependent effects of hindlimb ischemia in mice. Am J Pathol 180 : 2156–2169.
43. DokunAO, KeumS, HazarikaS, LiY, LamonteGM, et al. (2008) A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation 117 : 1207–1215.
44. LambertsenKL, ClausenBH, BabcockAA, GregersenR, FengerC, et al. (2009) Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci 29 : 1319–1330.
45. IadecolaC, AnratherJ (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17 : 796–808.
46. UllrichO, DiestelA, EyupogluIY, NitschR (2001) Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol 3 : 1035–1042.
47. NeumannJ, GunzerM, GutzeitHO, UllrichO, ReymannKG, et al. (2006) Microglia provide neuroprotection after ischemia. FASEB J 20 : 714–716.
48. ArumugamTV, SalterJW, ChidlowJH, BallantyneCM, KevilCG, et al. (2004) Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol 287: H2555–2560.
49. HossmannKA (2012) The two pathophysiologies of focal brain ischemia: implications for translational stroke research. J Cereb Blood Flow Metab 32 : 1310–1316.
50. SchillingM, BesselmannM, LeonhardC, MuellerM, RingelsteinEB, et al. (2003) Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 183 : 25–33.
51. GelderblomM, LeypoldtF, SteinbachK, BehrensD, ChoeCU, et al. (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40 : 1849–1857.
52. FantinA, VieiraJM, GestriG, DentiL, SchwarzQ, et al. (2010) Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116 : 829–840.
53. HeilM, ClaussM, SuzukiK, BuschmannIR, WilluweitA, et al. (2000) Vascular endothelial growth factor (VEGF) stimulates monocyte migration through endothelial monolayers via increased integrin expression. Eur J Cell Biol 79 : 850–857.
54. HeilM, SchaperW (2004) Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ Res 95 : 449–458.
55. FlintJ (2011) Mapping quantitative traits and strategies to find quantitative trait genes. Methods 53 : 163–174.
56. LegareME, BartlettFS2nd, FrankelWN (2000) A major effect QTL determined by multiple genes in epileptic EL mice. Genome Res 10 : 42–48.
57. AlemayehuA, BreenL, KrenovaD, PrintzMP (2002) Reciprocal rat chromosome 2 congenic strains reveal contrasting blood pressure and heart rate QTL. Physiol Genomics 10 : 199–210.
58. FrantzS, ClemitsonJR, BihoreauMT, GauguierD, SamaniNJ (2001) Genetic dissection of region around the Sa gene on rat chromosome 1: evidence for multiple loci affecting blood pressure. Hypertension 38 : 216–221.
59. ChuPL, KeumS, MarchukDA (2013) A novel genetic locus modulates infarct volume independent of the extent of collateral circulation. Physiol Genomics 45 : 751–63.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy