
A Reversible Histone H3 Acetylation Cooperates with Mismatch Repair and Replicative Polymerases in Maintaining Genome Stability
Mutations are a major driving force of evolution and genetic disease. In eukaryotes, mutations are produced in the chromatin environment, but the impact of chromatin on mutagenesis is poorly understood. Previous studies have determined that in yeast Saccharomyces cerevisiae, Rtt109-dependent acetylation of histone H3 on K56 is an abundant modification that is introduced in chromatin in S phase and removed by Hst3 and Hst4 in G2/M. We show here that the chromatin deacetylation on histone H3 K56 by Hst3 and Hst4 is required for the suppression of spontaneous gross chromosomal rearrangements, base substitutions, 1-bp insertions/deletions, and complex mutations. The rate of base substitutions in hst3Δ hst4Δ is similar to that in isogenic mismatch repair-deficient msh2Δ mutant. We also provide evidence that H3 K56 acetylation by Rtt109 is important for safeguarding DNA from small insertions/deletions and complex mutations. Furthermore, we reveal that both the deacetylation and acetylation on histone H3 K56 are involved in mutation avoidance mechanisms that cooperate with mismatch repair and the proofreading activities of replicative DNA polymerases in suppressing spontaneous mutagenesis. Our results suggest that cyclic acetylation and deacetylation of chromatin contribute to replication fidelity and play important roles in the protection of nuclear DNA from diverse spontaneous mutations.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003899
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003899
Summary
Mutations are a major driving force of evolution and genetic disease. In eukaryotes, mutations are produced in the chromatin environment, but the impact of chromatin on mutagenesis is poorly understood. Previous studies have determined that in yeast Saccharomyces cerevisiae, Rtt109-dependent acetylation of histone H3 on K56 is an abundant modification that is introduced in chromatin in S phase and removed by Hst3 and Hst4 in G2/M. We show here that the chromatin deacetylation on histone H3 K56 by Hst3 and Hst4 is required for the suppression of spontaneous gross chromosomal rearrangements, base substitutions, 1-bp insertions/deletions, and complex mutations. The rate of base substitutions in hst3Δ hst4Δ is similar to that in isogenic mismatch repair-deficient msh2Δ mutant. We also provide evidence that H3 K56 acetylation by Rtt109 is important for safeguarding DNA from small insertions/deletions and complex mutations. Furthermore, we reveal that both the deacetylation and acetylation on histone H3 K56 are involved in mutation avoidance mechanisms that cooperate with mismatch repair and the proofreading activities of replicative DNA polymerases in suppressing spontaneous mutagenesis. Our results suggest that cyclic acetylation and deacetylation of chromatin contribute to replication fidelity and play important roles in the protection of nuclear DNA from diverse spontaneous mutations.
Introduction
Mutations are the prerequisites for evolution and the humoral immune response. However, mutations are often detrimental due to their ability to trigger both inherited and sporadic diseases. Base substitutions, 1-bp deletions, and 1-bp insertions are the most common mutations [1], [2]. Cells can also acquire gross chromosomal rearrangements (GCRs) [3], [4], complex mutations [5], and other genetic alterations [1], [2], [6]. Though GCRs are relatively rare mutational events, they profoundly reshape genetic information. Mutations arise as a result of replication errors, defects in DNA repair, spontaneous and induced DNA damage, and several error-prone processes including somatic hypermutagenesis, mitotic gene conversion, and break-induced replication [2], [6]–[12]. DNA replication errors produce a large fraction of spontaneous mutations [7].
The bulk of nuclear DNA is replicated by the leading-strand polymerase ε and lagging-strand polymerase δ that both possess intrinsic 3′-5′ exonucleolytic activities [13], [14]. The suppression of DNA replication errors is in part achieved by the nucleotide selectivity at the active sites of replicative polymerases that permits DNA synthesis with an error rate of 10−4–10−5 [6]. The excision of incorrectly incorporated dNMPs by the 3′-5′ exonucleolytic activity of replicative polymerases further decreases the error rate ∼100-fold. In addition, mismatch repair (MMR) promotes high-fidelity DNA replication by correcting replication errors which escaped the proofreading activities of replicative polymerases. MMR is a multifunctional process, but correction of DNA replication errors is its primary function [2], [11], [15]–[21]. Eukaryotic MMR is initiated by the binding of MutSα (MSH2-MSH6 heterodimer) or MutSβ (MSH2-MSH3 heterodimer) to a mispair. After detecting a mismatch, MutSα or MutSβ activates the endonuclease activity of MutLα (MLH1-PMS2 in humans and Mlh1-Pms1 in S. cerevisiae) in the presence of ATP, a strand break, and PCNA loaded by RFC [22]–[25]. A MutLα incision 5′ to the mismatch initiates the downstream events leading to the correction of the mismatch [26], [27]. MMR improves fidelity of DNA replication 10–104-fold depending on the sequence context. Thus, replicative polymerases and MMR are the major factors in high-fidelity DNA replication [28]–[31].
Several reversible histone modifications have been implicated in DNA replication, repair, and damage response [32], [33]. Histone H3 K56 acetylation is one such modification located in the αN-helix that is adjacent to the histone fold domain [34], [35]. When histone H3 acetylated on K56 (H3K56ac) is part of a nucleosome, the acetylation is near the entry and exit sites of DNA and appears to loosen the histone-DNA contacts [34]. Nearly all newly synthesized yeast H3 histones are acetylated on K56 [36] by the histone acetyltransferase Rtt109 and histone chaperone Asf1 in S phase [37]–[40]. The loss of yeast H3K56ac enhances the sensitivity of cells to several DNA damaging drugs [35], [40]–[42] and destabilizes stalled replication forks [43]. During DNA damage response, yeast H3K56ac is required for both restoration of chromatin on repaired DNA and subsequent recovery of the cells from the DNA damage checkpoint [41]. H3K56ac has been identified in human cells where it is also involved in DNA damage response [44], [45].
The NAD-dependent histone deacetylases Hst3 and Hst4 erase H3K56ac marks from the newly generated chromatin in G2/M [36], [46], [47]. Like H3 K56 acetylation, H3 K56 deacetylation by Hst3 and Hst4 is important for DNA damage response. In the presence of DNA damage in G2/M in wild-type strains, H3 K56 deacetylation is delayed to allow DNA repair to take place [35]. Furthermore, it is known that H3 K56 acetylation and deacetylation are critical for selecting sister chromatid as the template for repair of replication-born double strand breaks by homologous recombination (HR) [48]. About 92% of chromatin histone H3 molecules are continuously acetylated on K56 residues in hst3Δ hst4Δ strains [36]. Strains lacking both HST3 and HST4 display spontaneous DNA damage, a strong sensitivity to genotoxic agents, a five-fold increase in mitotic homologous recombination, and an elevated level of chromosome loss in mitosis [36], [47], [49], [50]. Hst3 and Hst4 are members of the conserved sirtuin family also containing Hst1 and Hst2 [49], [51]. The targets of Hst1 and Hst2 enzymes are not well defined. A recent study reported that Hst1 is important for histone H3 K4 deacetylation in euchromatin [52].
Nuclear DNA is part of chromatin, but little is known about the relationship between chromatin and mutation avoidance. Previous studies have demonstrated that the yeast chromatin factors Caf1, Asf1, Hst3, and Rtt109 are involved in the suppression of GCRs [39], [53]–[55]. Furthermore, human CAF-1 has been shown to interact functionally and physically with the mismatch recognition factor MutSα and modulate MMR in cell-free extracts and reconstituted systems [56], [57]. A recent report has described that a depletion of the histone methyltransferase SETD2 triggers microsattelite instability and an increased mutation frequency at HPRT [58]. Because microsattelite instability is a hallmark of MMR defects and the MSH6 subunit of MutSα recognizes H3K36me3, these findings suggest that SET2D-dependent H3K36me3 is required for the action of human MMR in vivo [58].
In this work, we analyzed the impacts of both H3 K56 deacetylation and acetylation on spontaneous mutagenesis in S. cerevisiae. We found that H3 K56 deacetylation by the combined action of Hst3 and Hst4 plays a major role in the defense against GCRs, base substitutions, 1-bp insertions/deletions, and complex mutations. Our analysis also showed that in addition to being part of the protection from GCRs [54], H3 K56 acetylation is involved in the prevention of small insertions/deletions and complex mutations. Furthermore, our results revealed that both the acetylation and deacetylation of H3 K56 are important for genetic stabilization mechanisms that act in concert with MMR and the proofreading activities of replicative DNA polymerases to suppress spontaneous mutagenesis.
Results
Spontaneous mutagenesis in strains deficient in H3 K56 deacetylation
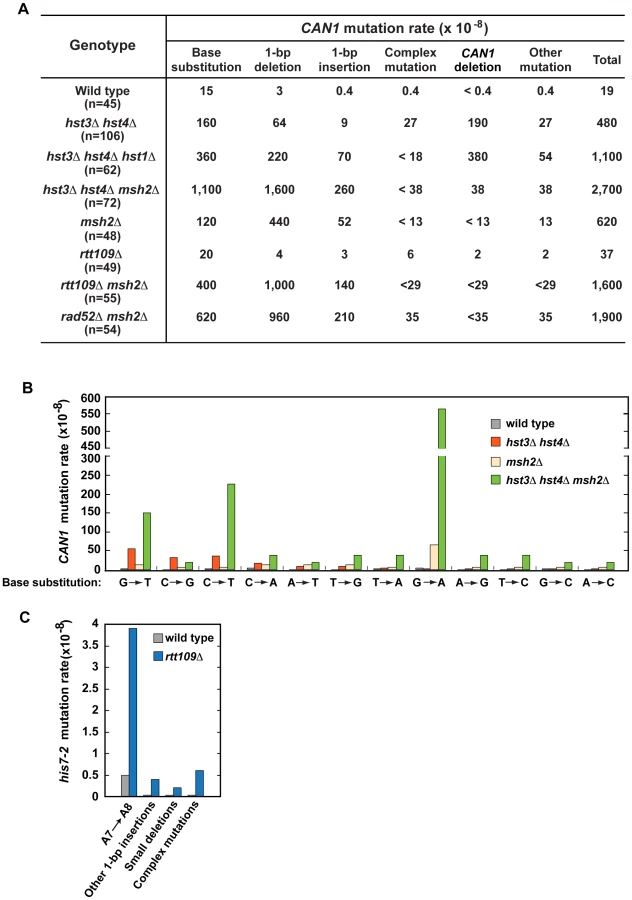
We started this work to investigate whether chromatin is involved in the defense against spontaneous point mutations in the haploid yeast S. cerevisiae. Many of our experiments relied on CAN1 and his7-2 reporters for scoring mutations. CAN1 is a counter-selectable marker that allows the selection of any mutation that inactivates the gene including base substitutions, small insertions/deletions and complex mutations. In addition, CAN1 can be inactivated by GCRs involving the 43-kb CAN1-containing region of chromosome V [3]. The his7-2 reporter permits the selection of net +1 frameshift mutations causing a reversion to HIS7 [59]. As shown in Table 1, analysis of several histone deacetylase and acetyltransferase mutants revealed that the CAN1 and his7-2 mutation rates for three different hst3Δ hst4Δ strains are about 25 times as high as those for isogenic wild-type strains. However, deletion of HST3 or HST4 alone causes little or no mutator phenotype (Table 1). We also established that the CAN1 mutation rates in hst3Δ hst4Δ are very similar to those in the MMR-deficient msh2Δ and mlh1Δ strains (Figure 1C). Collectively, these data demonstrated that the loss of HST3 and HST4 strongly promotes spontaneous mutagenesis.


Hst3 and Hst4 remove acetylations on H3 K56 residues that are introduced by Rtt109 [36]–[40], [46]. No other enzymatic activity has been assigned to Hst3 and Hst4. Based on this information, we hypothesized that Hst3 and Hst4 participate in the suppression of spontaneous mutations (Table 1) by deacetylating chromatin histones H3 on K56. If this hypothesis is correct, the loss of H3K56ac by deletion of RTT109 or introduction of H3K56R should make the H3 K56 deacetylation activities of Hst3 and Hst4 unnecessary, and therefore suppress the mutator phenotype of hst3Δ hst4Δ. (H3K56R variant mimics histone H3 that is not acetylated on lysine 56 [35], [60].) However, if H3 K56 deacetylation by Hst3 and Hst4 is not involved in the protection of yeast genome from spontaneous mutations, the loss of H3K56ac should not affect the mutator phenotype of hst3Δ hst4Δ. We found that deletion of RTT109 or introduction of H3K56R suppresses the mutator phenotype of hst3Δ hst4Δ to the level observed in rtt109Δ and H3K56R (Table 1). We concluded from these data that H3 K56 deacetylation by Hst3 and Hst4 is required for the prevention of spontaneous mutations.
H3K56ac is weakly mimicked by H3K56Q [35], [46], [60], [61]. Consistent with H3K56Q being a weak mimic of H3K56ac [46], [60], [61], we observed that the CAN1 and his7-2 mutation rates for H3K56Q are increased, but 6 and 2 times lower, respectively, than those for hst3Δ hst4Δ (Table 1). Furthermore, we found that the mutation rates for H3K56Q and hst3Δ hst4Δ H3K56Q do not differ from each other (Table 1). Therefore, these data further support the conclusion that H3 K56 deacetylation by Hst3 and Hst4 is required for mutation avoidance.
Nicotinamide (NAM) is a potent inhibitor of Hst3, Hst4, and other NAD-dependent sirtuins [36], [50], [62]. Yeast strains grown in 25-mM NAM-containing media accumulate an abnormally high level of H3K56ac [36]. We studied whether the presence of NAM in the culture medium promotes spontaneous mutagenesis of several yeast strains. We found that exposure to 25-mM or 50-mM NAM increases the mutation rates in wild type (Figures 1A and 1B). For example, the CAN1 mutation rate for wild type treated with 50-mM NAM increases 30-fold compared to that for untreated wild type. Importantly, our control experiments established that exposing H3K56R and rtt109Δ to 25-mM or 50-mM NAM has no effect on their mutation rates (Figures 1A and 1B). Together, these results provided independent evidence that H3 K56 deacetylation is important for the suppression of spontaneous mutagenesis.
We also found that the CAN1 and his7-2 mutation rates in the hst3Δ hst4Δ strain grown in the presence of 25-mM NAM are twice and five times higher, respectively, than those in untreated strain (Figures 1A and 1B). This finding suggested that an NAD-dependent histone deacetylase activity participates in the defense against spontaneous mutations in hst3Δ hst4Δ.
Hst1 and Hst2 are homologous to Hst3 and Hst4, but their biological functions remain enigmatic [36], [49]. In light of our evidence that the mutation rates in hst3Δ hst4Δ are increased in the presence of 25-mM NAM (Figures 1A and 1B), we sought to determine whether Hst1 and Hst2 are involved in the suppression of spontaneous mutations. We found that the CAN1 and his7-2 mutation rates in the hst1Δ, hst2Δ, hst1Δ hst3Δ, hst1Δ hst2Δ hst3Δ, and hst1Δ hst2Δ hst4Δ strains are not significantly different from those in wild type (Table 1). Furthermore, deletion of HST2 in the hst3Δ hst4Δ and hst3Δ hst4Δ hst1Δ strains does not increase spontaneous mutagenesis above the existing levels. However, the mutation rates in hst3Δ hst4Δ hst1Δ are twice higher than those in hst3Δ hst4Δ (Table 1). Together, these findings suggested that Hst1, but not Hst2, contributes to maintaining genome integrity in strains lacking Hst3 and Hst4.
Spontaneous mutagenesis in strains deficient in H3 K56 acetylation
The histone acetyltransferase Rtt109 produces H3K56ac in the presence of the histone chaperone Asf1 [37]–[40]. We inquired whether H3 K56 acetylation plays a role in mutation avoidance. We determined that deletion of RTT109 causes 9 - and 2-fold increases of the his7-2 and CAN1 mutation rates, respectively (Table 2). The mutation rates for asf1Δ and H3K56R are nearly identical to those for rtt109Δ (Table 2). Importantly, we found that there is epistasis between H3K56R and rtt109Δ for CAN1 and his7-2 mutations (Table 2). The simplest interpretation of these results is that H3 K56 acetylation by Rtt109 is involved in a mutation avoidance mechanism that suppresses spontaneous mutations in his7-2 and CAN1.

We considered the possibility that the absence of H3K56ac causes a defect in replication-coupled nucleosome assembly, which in turn increases spontaneous mutagenesis. The current view suggests that normal replication-coupled chromatin assembly in yeast depends on histone chaperones Caf1 (Cac1-Cac2-Cac3 heterotrimer) and Rtt106 [33]. However, as shown in Table S1, deletions of the replication histone chaperone genes CAC2 and RTT106 have nearly no effect on the CAN1 and his7-2 mutation rates. These results suggested that the increased mutagenesis in strains lacking H3K56ac is not caused by defects in the Caf1 - and Rtt106-dependent chromatin assembly.
H3K56ac is required for conferring cellular resistance to several DNA-damaging drugs [35]. In this pathway, H3K56ac acts through the ubiquitin ligase containing Rtt101, Mms1, and Mms22 subunits [63], [64]. Our data revealed that deletion of RTT101, MMS1, or MMS22 causes an ∼7-fold increase in his7-2 frameshifts (Table S2). Furthermore, we established that rtt101Δ and rtt109Δ are epistatic for his7-2 frameshifts (Table S2). These results suggested that the Rtt101 cullin-containing ubiquitin ligase is part of an H3 K56 acetylation-dependent mutation avoidance mechanism.
H3K56ac is involved in the regulation of budding yeast transcription [65]. In one mechanism of transcriptional regulation, the presence of H3K56ac leads to the SWR-C-dependent removal of the histone variant H2A.Z from promoter-proximal nucleosomes [65], [66]. In strains deficient in H2A.Z, transcription of ∼320 genes is upregulated while transcription of ∼480 genes is repressed [65]. To test whether the H3K56ac-dependent transcription regulation plays a role in the control of spontaneous mutagenesis, we measured CAN1 and his7-2 mutation rates in htz1Δ and swr1Δ strains. (HTZ1 is the only gene for the histone variant H2A.Z and SWR1 encodes the catalytic subunit of the SWR-C chromatin remodeling complex.) As shown in Table 2, the CAN1 and his7-2 mutation rates in the htz1Δ and swr1Δ strains are nearly identical to those in wild type. These findings indicated that the defects in the SWR-C - and H2A.Z-dependent transcription regulation do not increase the levels of can1 and HIS7 mutations in strains proficient in both the acetylation and deacetylation of H3 K56.
The deacetylation and acetylation of histone H3 K56 are important for mutation avoidance mechanisms that cooperate with MMR and the proofreading activities of replicative DNA polymerases δ and ε
Analysis of genetic interactions has been critical for understanding the functions of numerous proteins involved in mutation avoidance. Previous studies have defined the existence of multiplicative, synergistic, and additive relationships between mutants that inactivate different mutation avoidance mechanisms [29], [67]–[71]. In a synergistic relationship, the relative mutation rate for a double mutant is greater than the sum of those for the single mutants [29]. A multiplicative relationship is a form of synergistic relationship in which the relative mutation rate in a double mutant is equal to the product of those in the single mutants [29]. The presence of a synergistic or multiplicative relationship indicates that one of the mutants is deficient in one mechanism and the other mutant in a different mechanism, and that the two mechanisms act in concert to suppress the same pool of DNA lesions [29]. On the other hand, the existence of an additive relationship indicates that either mechanism suppresses a different pool of DNA lesions [29]. In an additive relationship, the relative mutation rate for a double mutant is equal to the sum of those for the single mutants [29], [71].
In hst3Δ hst4Δ strains, nearly all H3 histones are acetylated on K56 at replication forks [36], [47]. We thought that the presence of excess H3K56ac might interfere with high-fidelity DNA replication. Therefore, we decided to test whether histone H3 K56 deacetylation by Hst3 and Hst4 contributes to DNA replication fidelity. In these experiments we used four replication fidelity mutants: msh2Δ and mlh1Δ completely inactivate MMR [59], [72], pol2-4 disables the proofreading activity of DNA polymerase ε [73], and pol3-5DV eliminates the proofreading activity of DNA polymerase δ [74]. Based on the results of the previous research [29], [67]–[71] described above, we predicted that if a histone H3 K56 deacetylation-dependent mutation avoidance mechanism cooperates with MMR and the proofreading activities of replicative polymerases in promoting replication fidelity, each of triple mutant combinations containing hst3Δ hst4Δ and one of the replication fidelity mutants (msh2Δ, mlh1Δ, pol2-4, or pol3-5DV) should display synergistic or multiplicative, but not additive, increases in the relative CAN1 and his7-2 mutation rates. We found that the hst3Δ hst4Δ msh2Δ, hst3Δ hst4Δ mlh1Δ, hst3Δ hst4Δ pol2-4, and hst3Δ hst4Δ pol3-5DV triple mutants indeed show synergistic increases in the relative CAN1 and his7-2 mutation rates (Figure 1C and Table S3). In addition, weak synergies were observed when hst3Δ, but not hst4Δ, was combined with msh2Δ, mlh1Δ, pol2-4, or pol3-5DV (Table S3). Taken together, these findings suggested that an H3 K56 deacetylation-dependent mutation avoidance mechanism act in concert with MMR and the proofreading activities of replicative polymerases δ and ε to maintain high-fidelity DNA replication.
Because replication-coupled nucleosome assembly incorporates H3K56ac in chromatin in S phase [35], we tested whether this modification is important for maintaining high-fidelity DNA replication. We found a multiplicative increase in the relative CAN1 mutation rate when an H3 K56 acetylation mutant (rtt109Δ, H3K56R, or asf1Δ) was combined with a replication fidelity mutant (msh2Δ, pol2-4, or pol3-5DV) (Table 2). Furthermore, we established that all these double mutant combinations display synergistic increases in his7-2 mutation rates (Table 2). Collectively, these data suggested that an H3K56ac-dependent mutation avoidance mechanism cooperates with MMR and the proofreading activities of DNA polymerases to promote replication fidelity.
In addition to H3 K56, Rtt109 acetylates other targets [39], [75], [76]. Analysis of data in Table 2 indicated that the synergies between rtt109Δ and replication fidelity mutants for his7-2 mutations are often weaker than those between asf1Δ or H3K56R and msh2Δ, pol2-4, or pol3-5DV. Therefore, acetylation of a different target by Rtt109 may compromise replication fidelity.
Mutations formed in H3 K56 deacetylation - and H3 K56 acetylation-deficient strains
To characterize spontaneous mutagenesis caused by the deficiency in H3 K56 deacetylation (Figure 1 and Table 1), we determined mutations that occurred within the 1.77-kb CAN1 ORF in the wild-type and hst3Δ hst4Δ strains by PCRs and DNA sequencing (Figures 2, S1, and S2). Consistent with a previous report [77], we observed that in the wild-type strain 79% of can1 mutations are base substitutions (Figure 2A). Genetic alterations detected in the hst3Δ hst4Δ strain include base substitutions, 1-bp deletions, 1-bp insertions, complex mutations, and deletions of CAN1 gene (Figures 2A and S2). Of those, deletions of CAN1 gene are the most common mutations generated at a rate of 190×10−8. This unexpected finding suggested that strains defective in H3 K56 deacetylaton are very susceptible to GCRs and we confirmed this idea in experiments described in the next subsection. We also found that base substitutions in the hst3Δ hst4Δ strain accumulate at a high rate of 160×10−8. Strikingly, the rate of base substitutions in hst3Δ hst4Δ is comparable with that in MMR-deficient msh2Δ. This finding suggested that H3 K56 deacetylation is a major player in the protection of S. cerevisiae from base substitutions. The most common base substitution in the spectrum of hst3Δ hst4Δ is a G→T transversion produced at a rate of 50×10−8 (Figures 2B and S2B). Analysis of the spectrum also suggested that C→G transversions and C→T transitions are formed at high rates in the H3 K56 deacetylation-deficient strain. Among other mutations detected in hst3Δ hst4Δ are six medium-size deletions ranging from 40-bp to 1,036-bp. Examination of the end points of the deletions revealed that five out of the six deletions occurred between perfect or nearly perfect direct repeats (Figure S2D).
Deletion of HST1 in hst3Δ hst4Δ promotes spontaneous mutagenesis (Table 1). To obtain additional insight into the interaction between hst1Δ and hst3Δ hst4Δ, we determined can1 mutation spectrum of hst3Δ hst4Δ hst1Δ. Analysis of the can1 mutation spectrum showed that the rates of base substitutions, 1-bp deletions, 1-bp insertions, and deletions of CAN1 gene for hst3Δ hst4Δ hst1Δ are 2–8 times higher than those for hst3Δ hst4Δ (Figures 2A). We also found that the rate of base substitutions for hst3Δ hst4Δ hst1Δ exceeds that for msh2Δ by 3-fold. Complex mutations comprising 2 or more mutations within an ∼10-bp DNA are a signature of the action of DNA polymerase ζ [5]. Surprisingly, the mutation spectrum of hst3Δ hst4Δ hst1Δ does not contain even a single complex mutation whereas six complex mutations are present in the spectrum of hst3Δ hst4Δ (Figures 2A and S2C). Collectively, these findings suggested that deletion of HST1 in hst3Δ hst4Δ significantly affects the dynamics of DNA metabolism.
To characterize the synergy between hst3Δ hst4Δ and msh2Δ (Figure 1C and Table S3), we determined can1 mutation spectra of the msh2Δ and hst3Δ hst4Δ msh2Δ strains. As expected from the results of an earlier work [72], 71% and 19% of mutations in the msh2Δ spectrum are 1-bp deletions and base substitutions, respectively (Figure 2A). Analysis of the data indicated that the rate of CAN1 gene deletions in hst3Δ hst4Δ msh2Δ is 5 times lower than that in hst3Δ hst4Δ (Figure 2A). This result provided us with the first clue that MMR might be involved in the formation of a large fraction of CAN1 deletions in H3 K56 deacetylation-defective strains. The can1 mutation spectrum of hst3Δ hst4Δ msh2Δ is dominated by base substitutions and 1-bp deletions accumulating at the rates of 1,100×10−8 and 1,600×10−8, respectively (Figure 2A). Among different base substitutions observed in the spectrum of hst3Δ hst4Δ msh2Δ, G→A changes are the most frequent (Figures 2B and S2B). Further analysis of the data revealed that there is a synergistic relationship [29] between hst3Δ hst4Δ and msh2Δ for base substitutions, 1-bp deletions, and 1-bp insertions (Figures S2A and S2B). For example, the relative rate of G→A substitutions for hst3Δ hst4Δ msh2Δ is 8 times as high as the sum of those for msh2Δ and hst3Δ hst4Δ (Figure S2B). Taken together, these findings established that H3 K56 deacetylation cooperates with MMR to prevent base substitutions, 1-bp deletions, and 1-bp insertions.
To better understand the H3 K56 acetylation-dependent suppression of spontaneous mutations, we determined spectra of mutations of the rtt109Δ and rtt109Δ msh2Δ strains (Figure 2A, 2C, and S2A). Compared to wild type, rtt109Δ displays higher rates of 1-bp insertions, complex mutations, and deletions of CAN1 (Figure 2A). The most common mutation in the his7-2 reporter of the rtt109Δ mutant was an A insertion that extended the A7 into an A8 run (Figure 2C). In addition, the HIS7 spectrum contains other net 1-bp insertions, small deletions, and complex mutations consisting of a 1-bp insertion and an adjacent base substitution. Noticeably, the rate of complex mutations in his7-2 for rtt109Δ is 20 times as high as that for wild type. Collectively, these results demonstrated that H3 K56 acetylation is important for the protection from 1-bp insertions, small deletions, and complex mutations. Comparison of can1 mutation spectra of the rtt109Δ, msh2Δ, and rtt109Δ msh2Δ strains revealed a synergy between rtt109Δ and msh2Δ for both base substitutions and 1-bp insertions/deletions (Figure S2A). Therefore, these data established that an H3 K56 acetylation-dependent mutation avoidance mechanism acts synergistically with MMR to prevent 1-bp deletions, base substitutions, and 1-bp insertions.
GCRs in hst3Δ hst4Δ strains
The genomic DNAs of 40% of our can1 hst3Δ hst4Δ isolates did not support PCR amplification of can1, but templated the expected POL2 PCR product (Figure S1). This finding implied that the hst3Δ hst4Δ mutant loses all or part of CAN1 due to GCRs (Figure 2A). To further investigate this phenomenon, we carried out experiments that took advantage of contour-clamped homogenous electric field (CHEF) electrophoresis coupled with Southern blot hybridization. The data revealed the presence of a rearranged chromosome V in can1 hst3Δ hst4Δ isolates that did not support PCR-based amplification of can1 (Figure 3A). Some of the isolates appear to carry fusions of the chromosome V arm with a different chromosome (Figure 3A, lanes 2, 4, and 6), while the other isolates contain deletions within chromosome V (Figure 3A, lanes 7–16). Such chromosomal rearrangements have been detected in previous studies [78], [79].

To provide further evidence that the defect in H3 K56 deacetylation triggers GCRs, we measured the rate of GCRs in the wild-type and hst3Δ hst4Δ strains using an approach developed by Richard Kolodner and coworkers [4]. URA3 was inserted 2.1-kb telomeric to CAN1 and the simultaneous loss of the two markers occurring as a result of a GCR was measured (Figures 3B and 3C). The rate of GCRs in the hst3Δ hst4Δ strain is 15,600-fold as high as that in wild type (Figure 3C). This finding demonstrated that the lack of H3 K56 deacetylation causes a dramatic increase in the rate of GCRs. Combining hst3Δ hst4Δ with pol3-5DV does not significantly change the rate of GCRs. Strikingly, the hst3Δ hst4Δ msh2Δ and hst3Δ hst4Δ mlh1Δ strains display GCR rates that are 15 times lower than that in isogenic hst3Δ hst4Δ (Figure 3C). Furthermore, we found that deletion of MSH3 or MSH6 in the hst3Δ hst4Δ mutant decreases the rate of GCRs (Figure 3C). These results suggested that the formation of the majority of GCRs in hst3Δ hst4Δ strains involves MMR action dependent on both MutSα and MutSβ. A model shown in Figure 3D outlines a possible mechanism of this phenomenon and is described in the Discussion section.
Analysis of genetic interactions involving H3 K56 deacetylation and H3 K56 acetylation mutants
Next, we investigated whether several DNA repair proteins contribute to the high mutation rates in the hst3Δ hst4Δ strains (Table 3). As described above, the spectrum of hst3Δ hst4Δ contains complex mutations (Figures 2A and S2C). Rev3 is the catalytic subunit of DNA polymerase ζ [80] that produces complex and other mutations during replication of damaged and undamaged DNA and during double-strand break repair [71], [81]. The CAN1 and his7-2 mutation rates for rev3Δ hst3Δ hst4Δ are nearly identical to those for hst3Δ hst4Δ. Therefore, these results implied that DNA polymerase ζ is not responsible for the majority of mutations occurring in the H3 K56 deacetylation-deficient strains.

Deletion of RTT101 or CTF18 suppresses a strong defect of hst3Δ hst4Δ strains for growth at 37°C [47], [82]. Rtt101 is required for the progression of replication forks through pause sites and damaged DNA template [83], and is part of the H3K56ac-dependent resistance to genotoxic stress [82]. Ctf18 is the largest subunit of the Ctf18-RFC complex, which is essential for sister chromatid cohesion [84] and unloads PCNA from DNA [85]. We analyzed the involvement of both RTT101 and CTF18 in the formation of spontaneous mutations in hst3Δ hst4Δ. As shown in Table 3, the CAN1 and his7-2 mutation rates in rtt101Δ hst3Δ hst4Δ are 12 and 6 times lower, respectively, than those in hst3Δ hst4Δ. This finding implicated Rtt101 in the formation of the majority of can1 and HIS7 mutations in H3 K56 deacetylation-deficient strains. Furthermore, we found that the mutation rates in ctf18Δ hst3Δ hst4Δ are lower than those in hst3Δ hst4Δ. This result indicated that Ctf18-RFC is involved in promoting spontaneous mutagenesis in H3 K56 deacetylation-deficient strains.
The presence of complex mutations in the mutation spectra of rtt109Δ (Figures 2A and 2C) suggested that DNA polymerase ζ might contribute to spontaneous mutagenesis in the H3 K56 acetylation-deficient strains. We determined that deletion of REV3 in rtt109Δ completely suppresses the CAN1 mutation rate and decreases the his7-2 mutation rate two-fold (Table 3). These results support the idea that DNA polymerase ζ is involved in the formation of mutations in H3 K56 acetylation-deficient strains.
Rad52 and Rad51 are key components of HR [12]. To characterize the genetic interactions between H3 K56 acetylation and HR, we measured the CAN1 and his7-2 mutation rates in the rad51Δ, rad51Δ rtt109Δ, rad52Δ, and rad52Δ rtt109Δ strains (Table 3). Unfortunately, the relative CAN1 mutation rates in the single and double mutants do not allow us to distinguish between epistasis and additivity in the genetic interactions of rtt109Δ with the HR alleles (Table 3). However, we found that rtt109Δ displays epistatic relationships with rad51Δ and rad52Δ for his7-2 mutations (Table 3). This finding suggested that the recombination proteins and H3K56 acetylation act in the same pathway to promote the integrity of replication fidelity.
Because our results provided evidence that H3K56 acetylation acts synergistically with MMR (Table 2) and epistatically with HR (Table 3) to control spontaneous mutagenesis, we hypothesized that HR might contribute to fidelity of DNA replication. To test this hypothesis, we studied the genetic interactions between rad52Δ and msh2Δ (Figures 4A and 4B). We found the presence of a synergistic relationship between rad52Δ and msh2Δ for both CAN1 and his7-2 mutations. Rev3 produces the majority of mutations in rad52 strains by acting on ssDNA generated by the resection of double-strand breaks [86]. We established that in the rev3Δ background, the relationship between rad52Δ and msh2Δ is nearly multiplicative for both CAN1 and his7-2 mutations (Figures 4A and 4B). Next, we compared the can1 mutation spectra of the rad52Δ msh2Δ and msh2Δ strains (Figure 2A). The mutation spectrum of rad52Δ msh2Δ is similar to that of msh2Δ, but the rates of the most common classes of mutations (1-bp deletions, base substitutions, and 1-bp deletions) for msh2Δ are 2–3 times lower than those for rad52Δ msh2Δ. Taken together, these results suggested that Rad52-dependent HR contributes to fidelity of DNA replication.

Discussion
Chromatin controls many critical aspects of metabolism in eukaryotes. Besides being a major regulator of transcription, chromatin profoundly affects DNA damage response, replication, and repair [32], [33], [87]. However, our knowledge about the relationship between chromatin and spontaneous mutagenesis is very limited. Previous research has identified that yeast Asf1, Caf1, Hst3, and Rtt109-dependent H3 K56 acetylation are involved in the control of GCRs [39], [53]–[55]. Additionally, a recent study reported that SET2D-dependent H3K36me3 regulates the mismatch correction function of human MMR [58]. Up to date, no information has been available about the involvement of either histone acetylation or deacetylation in the protection from point and complex mutations. In yeast S. cerevisiae, H3K56ac is an abundant posttranslational modification introduced in and removed from chromatin in a cell cycle-dependent manner [34]–[36], [46]. In this work, we investigated the impact of both the deacetylation and acetylation of H3 K56 on spontaneous mutagenesis in S. cerevisiae. We demonstrated that H3 K56 deacetylation by Hst3 and Hst4 plays a critical role in the suppression of GCRs, base substitutions, small insertions/deletions, and complex mutations (Figures 2, 3, and 5). Remarkably, a strain deficient in Hst3 - and Hst4-dependent H3 K56 deacetylation forms GCRs at a rate that is 15,600-fold as high as that in isogenic wild type (Figure 3C). Furthermore, we showed that the rates of base substitutions in the hst3Δ hst4Δ and msh2Δ strains are similar to one another (Figure 2A). This finding suggests that H3 K56 deacetylation is as important for the prevention of base substitutions as MMR. We also showed that H3 K56 acetylation by Rtt109 and Asf1 is involved in the protection of DNA from 1-bp insertions, small deletions, and complex mutations (Figures 2A and 2C); however the effects of H3 K56 acetylation are weaker than those of H3 K56 deacetylation. Therefore, our findings indicate that in addition to controlling gene transcription and GCRs [39], [53]–[55], [87], histone acetylation and deacetylation are required for the defense against point and complex mutations.

This study was greatly facilitated by the availability of the H3K56R and H3K56Q alleles [35], [46], [60] (Figure 1 and Tables 1 and 2). In our experiments, H3K56R mimicked well H3 unacetylated on K56, but H3K56Q behaved as a weak mimic of H3K56ac. The latter conclusion is based on the observation that the mutation rates for hst3Δ hst4Δ exceeded those for H3K56Q and hst3Δ hst4Δ H3K56Q by 2–6 fold (Table 1). Previous studies have also found that H3K56Q mimics weakly H3K56ac [46], [60], [61].
Exposure of the hst3Δ hst4Δ strain to 25–50-mM NAM increases the mutation rates 2–5-fold (Figures 1A and 1B). This finding suggested that another NAD-dependent histone deacetylase is involved in mutation avoidance. Consistent with this, we found that the CAN1 and his7-2 mutation rates in hst1Δ hst3Δ hst4Δ are twice higher than those in hst3Δ hst4Δ (Table 1). Given that 8% of histone H3 is still unacetylated on K56 in hst3Δ hst4Δ strains [36] and that the CAN1 and his7-2 mutation rates for hst1Δ H3K56Q and H3K56Q do not differ from each other (Table 1), we hypothesize that Hst1 is involved in the suppression of spontaneous mutagenesis in hst3Δ hst4Δ cells by weakly deacetylating H3 on K56. Alternatively, Hst1 may promote genetic stability by acting on a different target. Since the removal of euchromatic H3K4ac mainly depends on Hst1 [52], H3K4ac may be this target.
Mutagens, in general, produce a lesion by directly acting upon DNA, which is later converted into a mutation. One of the few deviations from this rule is the demonstration that cadmium cations trigger genetic instability in yeast strains by inhibiting an enzymatic system, MMR [88]. We showed that exposure of yeast strains to 50-mM NAM, a specific inhibitor of the NAD-dependent histone deacetylases [62], produces a strong 30-fold increase in the CAN1 mutation rate (Figure 1A). This effect depends on the presence of both H3 K56 and RTT109. To the best of our knowledge, these data have provided the first example of a small molecule that inhibits chromatin-modifying enzymes and by doing so strongly promotes spontaneous mutagenesis.
MMR and the proofreading activities of DNA polymerases δ and ε are critical for maintaining high-fidelity DNA replication [2], [18]. We found the presence of synergistic increases in CAN1 and his7-2 mutation rates when hst3Δ hst4Δ is combined with msh2Δ, mlh1Δ, pol2-4, or pol3-5DV (Figures 1C, 2A, and 2B and Table S3). Furthermore, we established the existence of a synergy between hst3Δ hst4Δ and msh2Δ for base substitutions, 1-bp insertions, and 1-bp deletions (Figure 2A, 2B, and S2A). It is also evident that the relationships of rtt109Δ, asf1Δ, and H3K56R with msh2Δ, pol2-4, and pol3-5DV are synergistic for his7-2 frameshifts and multiplicative for CAN1 mutations (Table 2). The presence of synergistic and multiplicative relationships supports the view that both the deacetylation and acetylation of H3 K56 are involved in mutation avoidance pathways that act in concert with MMR and the proofreading activities of the replicative polymerases to promote DNA replication fidelity. The absolute CAN1 mutation rates for hst3Δ hst4Δ msh2Δ (Table S3) and msh2Δ rtt109Δ (Table 2) are 4.2 times and 7.6 times lower, respectively, than that for the pol2-4 msh2Δ mutant [69]. Therefore, this comparison suggests that the contributions of the acetylation and deacetylation of H3 K56 to replication fidelity are not as strong as that of the proofreading activity of DNA polymerase ε.
GCRs have been implicated in triggering many different cancers [9]. S. cerevisiae has been instrumental for dissecting the mechanisms of GCRs [4], [78], [89], [90]. A study that used a URA3-CAN1 cassette containing the two genes 7.5-kb apart from each other described that deletion of HST3 or RTT109 increases the rate of GCRs four-fold [55]. To analyze GCRs, we utilized a URA3-CAN1 cassette in which the distance between URA3 and CAN1 is 2.1-kb (Figures 3B and 3C). We determined that the deletions of HST3 and RTT109 cause 45 - and 6-fold increases of the GCR rate, respectively (Figure 3C). Surprisingly, the rate of GCRs in hst3Δ hst4Δ exceeds those in the corresponding single mutants and rtt109Δ by at least 350-fold. Therefore, our findings (Figure 3C) are in good accord with and extend the previous observations that identified that Hst3 and Rtt109-dependent H3 K56 acetylation play roles in the control of GCRs [39], [53], [89]. In addition, our data (Figure 3C) support the view that Hst3 is the principal enzyme for H3 K56 deacetylation [36], [46].
Msh2 and Mlh1 are the key components of yeast MMR [2], [15]. Msh2 is a subunit of the mismatch recognition factors MutSα and MutSβ, whereas Mlh1 forms MutLα endonuclease by dimerizing with Pms1. Strikingly, deletion of MSH2 or MLH1 in hst3Δ hst4Δ reduces the rate of GCRs by 15-fold (Figure 3C). On the other hand, the rate of GCRs in the hst3Δ hst4Δ pol3-5DV mutant is nearly identical to that in hst3Δ hst4Δ. Therefore, these data demonstrate that MMR, but not the proofreading activity of DNA polymerase δ, is required for the generation of the majority of GCRs in the H3 K56 deacetylation-defective strains. We infer from these results that histone H3 K56 deacetylation is necessary to suppress malfunction of MMR. It is possible that in addition to promoting GCRs, MMR malfunction may result in the formation of some point mutations in hst3Δ hst4Δ (Figures 2A and 5). We speculate that MMR malfunction triggered by a defective environment may be responsible for the formation of a subset of cancer-initiating GCRs and point mutations. It has been known that MMR initiates several neurodegenerative diseases by destabilizing a number of DNA triplet repeats [2]. Thus, the idea that MMR can cause pathogenic consequences has already gained significant experimental support. We also analyzed the importance of the Msh6 subunit of MutSα and the Msh3 subunit of MutSβ for GCR formation (Figure 3C). The results of this analysis suggested that both MutSα and MutSβ contribute to the high rate of GCRs in hst3Δ hst4Δ, but the impact of the latter complex is somewhat weaker compared to that of the former.
How does MMR contribute to the formation of GCRs in hst3Δ hst4Δ cells? Our data permit us to suggest a speculative model shown in Figure 3D. It is known that in the absence of nucleosomes or concomitant nucleosome assembly the MutSα-dependent endonuclease activity of MutLα causes excessive degradation of mismatch-containing DNA in cell-free extracts and defined systems [22], [23], [56]. Therefore, we hypothesize that excessive and persistent nicking of DNA by MutLα may occur in the presence of the defective H3 K56 deacetylation. Such strand breaks can be converted into double-strand breaks in the next round of replication. If DNA flanking an end of one double-strand break carries a sequence that is a direct repeat of DNA flanking an end of another double-strand break, the MutSβ-dependent single strand annealing (SSA) mechanism [91]–[93] can join these two ends producing a GCR. Previous studies already demonstrated the importance of the MutSβ-dependent SSA mechanism for the repair of double-strand breaks flanked by direct repeats [91]–[93]. In this mechanism, MutSβ stabilizes the annealed DNA ends permitting the Rad1-Rad10 nuclease to cleave nonhomologous DNA tails [94]. The involvement of MutSβ and SSA, which is a major mechanism for repairing double-strand breaks flanked by direct repeats [95], in the formation of GCRs in hst3Δ hst4Δ cells is consistent with the following findings. First, the loss of MutSβ in hst3Δ hst4Δ decreases the GCR rate four-fold (Figure 3C). Second, five out of six identified medium-size deletions in CAN1 of the hst3Δ hst4Δ mutant were between direct repeats (Figure S2D).
In addition, a different mechanism may lead to GCRs in the hst3Δ hst4Δ strains. In this mechanism, H3 K56 hyperacetylation, MutSα or MutSβ, and a mismatch activate MutLα endonuclease to initiate the excision of the mismatch on opposite strands. Such aberrant excision may produce a double-strand break. When two double-strand breaks arise in the same hst3Δ hst4Δ cell, they may be repaired by the SSA mechanism causing a GCR. This mechanism is somewhat related to the one that has been proposed to explain the mismatch repair-dependent killing of E. coli dam recA mutants [96].
Though there are strong synergistic relationships between the H3 K56 acetylation mutants (rtt109Δ, asf1Δ, and H3K56R) and the replication fidelity defects (msh2Δ, pol2-4, and pol3-5DV) for CAN1 mutations, the double mutants show weaker synergistic increases in his7-2 mutations (Table 2). Furthermore, hst3Δ hst4Δ displays weak synergistic relationships with the MMR-deficient and proofreading mutants for CAN1 and his7-2 mutations (Figure 1C and Table S3). These findings suggest that a large fraction of mutations in both hst3Δ hst4Δ and rtt109Δ strains is produced from DNA lesions/mismatches formed outside S phase. In wild-type strains, H3K56ac appears in S phase and is removed in G2/M [36], [46]. Wild-type, hst3Δ, and hst4Δ strains do not have H3K56ac in G1, unlike hst3Δ hst4Δ [36]. Therefore, it is likely that a significant fraction of pre-mutagenic lesions/mismatches in hst3Δ hst4Δ is formed in G1 as a result of the presence of H3K56ac. Compared to wild type, rtt109Δ does not have H3K56ac in S phase and a part of G2/M. Hence, it is possible that S phase-independent pre-mutagenic lesions/mismatches in rtt109Δ arise in G2/M as a consequence of the lack of H3K56ac. In addition, the indicated variations in the presence or absence of H3K56ac in the different stages of the cell cycle provide a good explanation of why hst3Δ hst4Δ and rtt109Δ impact spontaneous mutagenesis differently (Tables 1–3 and Figures 2A and 3C).
What are the mechanisms that could be responsible for the generation of pre-mutagenic lesions in hst3Δ hst4Δ during G1 and in rtt109Δ during G2/M? Studies of gene transcription have identified many factors that recognize/read the presence or absence of histone modifications including histone acetylations [87], [97]. In addition, some factors read a specific combination of modifications [87]. After forming a complex with a modified/unmodified residue(s), the factor alters transcription of the affected gene. Thus, it is plausible that factors that read the absence or presence of H3K56ac alone or in combination with different modifications in the different stages of the cell cycle change transcription, DNA repair, and/or other mechanisms in a way that results in spontaneous mutagenesis. For example, transcription-dependent variations in the levels of some DNA repair proteins triggered by the defects in the deacetylation or acetylation of H3 K56 may shift the dynamics of DNA metabolism towards increased formation of spontaneous mutations. Consistent with this, it is known that H3K56ac is involved in several mechanisms that regulate gene transcription in yeast [34], [37], [65], and that transcription of ∼370 genes is deregulated in H3K56Q cells [65].
One of the mechanisms of transcriptional regulation that involves H3K56ac uses this modification to facilitate SWR-C-dependent removal of the H2A.Z variant from promoter-proximal nucleosomes [65]. Transcription of ∼900 genes is upregulated or downregulated in mutants lacking H2A.Z [65]. We tested whether defects in this H3K56ac-dependent transcriptional regulation affect spontaneous mutagenesis. However, we found that strains lacking H2A.Z or Swr1 do not have increased levels of CAN1 and his7-2 mutations (Table 2). Thus, the H3K56ac-dependent transcriptional regulation does not contribute to spontaneous mutagenesis in strains with the intact control of both H3 K56 acetylation and H3 K56 deacetylation. Nevertheless, it is still possible that this mechanism of transcriptional regulation contributes to the formation of mutations in strains that are deficient in the acetylation or deacetylation of H3 K56. Furthermore, the defects in the acetylation and deacetylation of H3 K56 may promote the formation of spontaneous mutations via a different mechanism of transcriptional regulation.
The abundant and persistent H3K56ac in hst3Δ hst4Δ mutants impairs DNA replication [47]. Replication forks in hst3Δ hst4Δ are able to adapt to the high level of H3K56ac when RFC1 is overexpressed or CTF18 is deleted [47]. The overexpression of RFC1 or deletion of CTF18 also suppresses the temperature-sensitive phenotype of the cells. Additionally, we observed that deletion of CTF18 in hst3Δ hst4Δ strongly reduces the mutation rates (Table 3). Because RFC loads PCNA onto DNA and CTF18-RFC unloads the clamp from DNA [85], these results suggest that a higher concentration of PCNA at replication forks allows the cells to adapt to the abundant and persistent H3K56ac. Intriguingly, PCNA is also required for MMR [22], [98], [99]. Thus, we speculate that inadequate concentrations of PCNA at replication forks in hst3Δ hst4Δ strains may impair both the replicative proofreading and MMR and this compromises replication fidelity. Nevertheless, it is also feasible that factors that recognize unacetylated H3 K56 and promote mutation avoidance cannot be recruited to replication forks in hst3Δ hst4Δ cells. Alternatively, the presence of the excessive H3K56ac may cause recruitment of mutagenic factors to the replication forks.
What is the mechanism that causes spontaneous mutagenesis in S phase in rtt109Δ cells? Our results suggest that H3K56ac is involved in a yet unknown HR mechanism that promotes replication fidelity (Figure 4 and Tables 2 and 3). The progression of replication forks is often impeded by spontaneous DNA damage. Therefore, it is possible that H3K56ac is important for an error-free bypass of spontaneous lesions by the HR machinery during DNA replication. In this mechanism, the presence of H3K56ac may be necessary for recruiting an interacting complex that promotes efficient chromatin remodeling around the lesions and by doing so facilitates an error-free bypass. Understanding the mechanisms that depend on the acetylation and deacetylation of H3 K56 to prevent spontaneous mutagenesis will require further experimentation.
In summary, our findings revealed that the cell cycle-regulated acetylation and deacetylation of chromatin on H3 K56 are critical for suppressing spontaneous mutagenesis (Figure 5). The acetylation and deacetylation of H3 K56 are involved in mutation avoidance mechanisms that act in concert with MMR and replicative polymerases to maintain genome stability. The lack of H3K56ac appears to compromise an HR mechanism that promotes replication fidelity. Defective H3 K56 deacetylation causes spontaneous mutagenesis involving Rtt101 and Ctf18, and results in the formation of MMR-dependent GCRs.
Materials and Methods
Strains
The S. cerevisiae wild-type strains used in this work are E134 (MATα ade5-1 lys2::InsE-A14 trp1-289 his7-2 leu2-3,112 ura3-52) [68], E35 (MATα ade5-1 lys2::InsE-A8 trp1-289 his7-2 leu2-3,112 ura3-52) [68], BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0), and 1B-D770 (MATa ade5-1 lys2::Tn5-13 trp1-289 his7-2 leu2-3,112 ura3-4) [59]. Strain SY579 and plasmids pPK588 and pPK589 have previously been described [60], [100]. If not indicated, the mutant strains are derivatives of E134. All strains used in this work are listed in Table S4.
To create gene replacements, disruption cassettes with homologous or heterologous markers [101] were amplified in PCRs and introduced into yeast cells by the lithium acetate/PEG-based transformation method [102]. Yeast genomic DNAs were isolated from recombinant isolates with MasterPure Yeast DNA purification kits (Epicentre) and all gene replacements were verified by PCR. The pol2-4 [67] and pol3-5DV [74] alleles were introduced by the integration-excision method. To analyze GCRs, S. cerevisiae URA3 gene amplified from the pFL34 plasmid with primers (5′ - ATACATGCACATATAGCTACTACATAGTCAAGAACATATCATAACATTTGTCTGGCTTTTCAATTCATC-3′) and (5′ - GTCGGTAGAGCCAGCATCAGATGCAAAGCCATGCAAAGACTGATATAAAGACTGTTATACAGATCTGAGCTTTTTCTTTCC-3′) was inserted at position 29617 of chromosome V, which is 2,077 bp telomeric to CAN1 and 3′ adjacent to SIT1.
Measurement of spontaneous mutation rates
Spontaneous mutation rates were measured using fluctuation analysis carried out according to a previously described procedure [59]. On average 15 cultures (no less than 9 cultures), started from single colonies of two-four freshly prepared independent isolates of the same genotype, were used to determine spontaneous mutation rate for this genotype. The cultures were grown to saturation in 3–50 ml YPD medium (1% yeast extract, 2% bacto-peptone, 2% dextrose) supplemented with 60 mg/L adenine and 60 mg/L uracil at 30°C for 20–48 h. When indicated, nicotinamide was added to the supplemented YPD medium to the final concentration of 25 mM or 50 mM. Each saturated culture was plated, after dilution, on a synthetic complete (SC) medium for scoring the total number of cells. The cultures were also plated on SC medium lacking histidine for scoring His+ revertants, SC medium lacking arginine and supplemented with 60 mg/L L-canavanine for scoring can1 mutants, and/or SC medium lacking arginine and supplemented with 60 mg/L L-canavanine and 1 g/L 5-FOA for scoring GCRs. The plates were incubated for 3–5 days at 30°C, and the colonies were counted.
The CAN1 and his7-2 mutation rates were calculated from the total numbers of cells and mutants in the cultures with the Drake formula and are presented as median values with 95% confidence intervals [7], [59]. Where indicated, the significance of observed differences in the CAN1 and his7-2 mutation rates was analyzed with Mann-Whitney U two-tailed test (GraphPad Prism 6 software), where the null hypothesis is that there is no difference between the two data sets. The rates of GCRs were calculated with the Ma-Sandri-Sarkar maximum likelihood method [103], [104] using the web tool FALCOR at http://www.keshavsingh.org/protocols/FALCOR.html [105], [106].
Analysis of mutation spectra
Mutation spectra in CAN1 gene were determined essentially as described [71]. Patches were started from single colonies on YPD plates and then replica plated on SC plates supplemented with 60 mg/L L-canavanine and lacking arginine. A single CanR clone from each patch was randomly selected, purified on the selective medium, and then propagated by patching on a YPD plate. Genomic DNAs were isolated from the patched cultures with a MasterPure Yeast DNA purification kit (Epicentre). 2,057-bp fragments containing the entire length of can1 ORF were amplified with primers 1 (5′-GCAGAAAGAAGAGTGGTTGCGAAC-3′) and 2 (5′-GAGAATGCGAAATGGCGTGGAAATG-3′) in PCR reactions. The amplified fragments were purified with a PCR purification kit (Qiagen) and sequenced such that the entire DNA sequence of the mutant ORF in each clone was determined.
To generate HIS7 mutation spectra for the wild-type and rtt109Δ strains, 1.4-ml or 2.8-ml saturated cultures started from single colonies were concentrated and plated on SC medium lacking histidine. One His+ clone from each plate was randomly selected and processed as above. 2005-bp fragments spanning HIS7 ORF were PCR-amplified with primers 3 (5′-CTCCACGGCTAATTAGGTGATCATG-3′) and 4 (5′-CCTACTGACACCACCAATAATACAAC-3′). The PCR fragments were purified as described above and part of HIS7 ORF, corresponding to chromosome II coordinates 716234 – 715434, was sequenced. his7-2 reverts to HIS7 by acquiring a +1-net frameshift in a 51-bp region (chromosome II coordinates 716023 - 715973) containing an A7 run [59].
CHEF gel electrophoresis and Southern hybridization
Yeast cells were embedded into 0.8% agarose plugs at a concentration of 6×108 cells/ml, and chromosomal DNA was separated by a contour-clamped homogeneous electric field (CHEF) gel electrophoresis in a 1.2% agarose gel/0.5×TBE for 40 hours at 6 V/cm and at 14°C, using the CHEF Mapper XA system (Bio-Rad). The included angle was 120 degrees. The initial and final switch times were 36.63 sec and 2 min 6.67 sec, respectively. The separated yeast chromosomal DNAs were transferred onto a nylon membrane and probed with a 32P-labeled MET6-specific probe. (The probe was generated by a random prime labeling of a MET6 PCR fragment amplified with primers 5′-GACGCCATCAAGGGCTTGCCAG-3′ and 5′-CGTTAGCTTCTAGGGCAGCAGC-3′.) The indirectly labeled yeast chromosomal DNAs were visualized with a Kodak BioMax film.
Supporting Information
Zdroje
1. KunkelTA (2004) DNA replication fidelity. J Biol Chem 279 : 16895–16898.
2. IyerRR, PluciennikA, BurdettV, ModrichPL (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106 : 302–323.
3. ChenC, UmezuK, KolodnerRD (1998) Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell 2 : 9–22.
4. ChenC, KolodnerRD (1999) Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet 23 : 81–85.
5. HarfeBD, Jinks-RobertsonS (2000) DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol Cell 6 : 1491–1499.
6. KunkelTA, BebenekK (2000) DNA replication fidelity. Annu Rev Biochem 69 : 497–529.
7. DrakeJW (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci USA 88 : 7160–7164.
8. GeacintovNE, SwenbergCE (1991) Chemical, molecular biology, and genetic techniques for correlating DNA base damage induced by ionizing radiation with biological end points. Basic Life Sci 58 : 453–473 discussion 473–454.
9. KolodnerRD, PutnamCD, MyungK (2002) Maintenance of genome stability in Saccharomyces cerevisiae. Science 297 : 552–557.
10. MalkovaA, HaberJE (2012) Mutations arising during repair of chromosome breaks. Annu Rev Genet 46 : 455–473.
11. BoiteuxS, Jinks-RobertsonS (2013) DNA Repair Mechanisms and the Bypass of DNA Damage in Saccharomyces cerevisiae. Genetics 193 : 1025–1064.
12. San FilippoJ, SungP, KleinH (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 : 229–257.
13. PursellZF, IsozI, LundstromEB, JohanssonE, KunkelTA (2007) Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science 317 : 127–130.
14. Nick McElhinnySA, GordeninDA, StithCM, BurgersPM, KunkelTA (2008) Division of labor at the eukaryotic replication fork. Mol Cell 30 : 137–144.
15. KolodnerRD, MarsischkyGT (1999) Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 9 : 89–96.
16. HarfeBD, Jinks-RobertsonS (2000) DNA Mismatch Repair and Genetic Instability. Annu Rev Genet 34 : 359–399.
17. SurteesJA, ArguesoJL, AlaniE (2004) Mismatch repair proteins: key regulators of genetic recombination. Cytogenet Genome Res 107 : 146–159.
18. KunkelTA, ErieDA (2005) DNA Mismatch Repair. Annu Rev Biochem 74 : 681–710.
19. ModrichP (2006) Mechanisms in eukaryotic mismatch repair. J Biol Chem 281 : 30305–30309.
20. LiGM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18 : 85–98.
21. Pena-DiazJ, JiricnyJ (2012) Mammalian mismatch repair: error-free or error-prone? Trends Biochem Sci 37 : 206–214.
22. KadyrovFA, DzantievL, ConstantinN, ModrichP (2006) Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126 : 297–308.
23. KadyrovFA, HolmesSF, AranaME, LukianovaOA, O'DonnellM, et al. (2007) Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem 282 : 37181–37190.
24. PluciennikA, DzantievL, IyerRR, ConstantinN, KadyrovFA, et al. (2010) PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci US A 107 : 16066–16071.
25. IyerRR, PluciennikA, GenschelJ, TsaiMS, BeeseLS, et al. (2010) MutLalpha and proliferating cell nuclear antigen share binding sites on MutSbeta. J Biol Chem 285 : 11730–11739.
26. ConstantinN, DzantievL, KadyrovFA, ModrichP (2005) Human mismatch repair: Reconstitution of a nick-directed bidirectional reaction. J Biol Chem 280 : 39752–39761.
27. KadyrovFA, GenschelJ, FangY, PenlandE, EdelmannW, et al. (2009) A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci USA 106 : 8495–8500.
28. StrandM, ProllaTA, LiskayRM, PetesTD (1993) Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365 : 274–276.
29. MorrisonA, JohnsonAL, JohnstonLH, SuginoA (1993) Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J 12 : 1467–1473.
30. ParsonsR, LiGM, LongleyMJ, FangWH, PapadopoulosN, et al. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell 75 : 1227–1236.
31. AlbertsonTM, OgawaM, BugniJM, HaysLE, ChenY, et al. (2009) DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci U S A 106 : 17101–17104.
32. GrothA, RochaW, VerreaultA, AlmouzniG (2007) Chromatin challenges during DNA replication and repair. Cell 128 : 721–733.
33. RansomM, DenneheyBK, TylerJK (2010) Chaperoning histones during DNA replication and repair. Cell 140 : 183–195.
34. XuF, ZhangK, GrunsteinM (2005) Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121 : 375–385.
35. MasumotoH, HawkeD, KobayashiR, VerreaultA (2005) A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature 436 : 294–298.
36. CelicI, MasumotoH, GriffithWP, MeluhP, CotterRJ, et al. (2006) The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 16 : 1280–1289.
37. SchneiderJ, BajwaP, JohnsonFC, BhaumikSR, ShilatifardA (2006) Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem 281 : 37270–37274.
38. HanJ, ZhouH, HorazdovskyB, ZhangK, XuRM, et al. (2007) Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315 : 653–655.
39. DriscollR, HudsonA, JacksonSP (2007) Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science 315 : 649–652.
40. TsubotaT, BerndsenCE, ErkmannJA, SmithCL, YangL, et al. (2007) Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell 25 : 703–712.
41. ChenCC, CarsonJJ, FeserJ, TamburiniB, ZabaronickS, et al. (2008) Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134 : 231–243.
42. HylandEM, CosgroveMS, MolinaH, WangD, PandeyA, et al. (2005) Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol 25 : 10060–10070.
43. FrancoAA, LamWM, BurgersPM, KaufmanPD (2005) Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev 19 : 1365–1375.
44. YuanJ, PuM, ZhangZ, LouZ (2009) Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 8 : 1747–1753.
45. TjeertesJV, MillerKM, JacksonSP (2009) Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 28 : 1878–1889.
46. MaasNL, MillerKM, DeFazioLG, ToczyskiDP (2006) Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 23 : 109–119.
47. CelicI, VerreaultA, BoekeJD (2008) Histone H3 K56 hyperacetylation perturbs replisomes and causes DNA damage. Genetics 179 : 1769–1784.
48. Munoz-GalvanS, JimenoS, RothsteinR, AguileraA (2013) Histone H3K56 acetylation, Rad52, and non-DNA repair factors control double-strand break repair choice with the sister chromatid. PLoS Genet 9: e1003237.
49. BrachmannCB, ShermanJM, DevineSE, CameronEE, PillusL, et al. (1995) The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev 9 : 2888–2902.
50. HachinoheM, HanaokaF, MasumotoH (2011) Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae. Genes Cells 16 : 467–477.
51. HaigisMC, SinclairDA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5 : 253–295.
52. GuillemetteB, DrogarisP, LinHH, ArmstrongH, Hiragami-HamadaK, et al. (2011) H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet 7: e1001354.
53. MyungK, PennaneachV, KatsES, KolodnerRD (2003) Saccharomyces cerevisiae chromatin-assembly factors that act during DNA replication function in the maintenance of genome stability. Proc Natl Acad Sci USA 100 : 6640–6645.
54. ChanJE, KolodnerRD (2011) Rapid analysis of Saccharomyces cerevisiae genome rearrangements by multiplex ligation-dependent probe amplification. PLoS Genet 8: e1002539.
55. PutnamCD, Allen-SolteroSR, MartinezSL, ChanJE, HayesTK, et al. (2012) Bioinformatic identification of genes suppressing genome instability. Proc Natl Acad Sci U S A 109: E3251–3259.
56. KadyrovaLY, Rodriges BlankoE, KadyrovFA (2011) CAF-I-dependent control of degradation of the discontinuous strands during mismatch repair. Proc Natl Acad Sci U S A 108 : 2753–2758.
57. SchopfB, BregenhornS, QuivyJP, KadyrovFA, AlmouzniG, et al. (2012) Interplay between mismatch repair and chromatin assembly. Proc Natl Acad Sci U S A 109 : 1895–1900.
58. LiF, MaoG, TongD, HuangJ, GuL, et al. (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153 : 590–600.
59. ShcherbakovaPV, KunkelTA (1999) Mutator phenotypes conferred by MLH1 overexpression and by heterozygosity for mlh1 mutations. Mol Cell Biol 19 : 3177–3183.
60. RechtJ, TsubotaT, TannyJC, DiazRL, BergerJM, et al. (2006) Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci U S A 103 : 6988–6993.
61. ErkmannJA, KaufmanPD (2009) A negatively charged residue in place of histone H3K56 supports chromatin assembly factor association but not genotoxic stress resistance. DNA Repair (Amst) 8 : 1371–1379.
62. BittermanKJ, AndersonRM, CohenHY, Latorre-EstevesM, SinclairDA (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 277 : 45099–45107.
63. MichelJJ, McCarvilleJF, XiongY (2003) A role for Saccharomyces cerevisiae Cul8 ubiquitin ligase in proper anaphase progression. J Biol Chem 278 : 22828–22837.
64. KroganNJ, CagneyG, YuH, ZhongG, GuoX, et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440 : 637–643.
65. WatanabeS, Radman-LivajaM, RandoOJ, PetersonCL (2013) A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science 340 : 195–199.
66. DionMF, KaplanT, KimM, BuratowskiS, FriedmanN, et al. (2007) Dynamics of replication-independent histone turnover in budding yeast. Science 315 : 1405–1408.
67. MorrisonA, SuginoA (1994) The 3′-->5′ exonucleases of both DNA polymerases delta and epsilon participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol Gen Genet 242 : 289–296.
68. TranHT, KeenJD, KrickerM, ResnickMA, GordeninDA (1997) Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol 17 : 2859–2865.
69. TranHT, GordeninDA, ResnickMA (1999) The 3′-->5′ exonucleases of DNA polymerases delta and epsilon and the 5′-->3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol 19 : 2000–2007.
70. KirchnerJM, TranH, ResnickMA (2000) A DNA polymerase epsilon mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast. Genetics 155 : 1623–1632.
71. NorthamMR, RobinsonHA, KochenovaOV, ShcherbakovaPV (2010) Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 184 : 27–42.
72. MarsischkyGT, FilosiN, KaneMF, KolodnerR (1996) Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10 : 407–420.
73. MorrisonA, BellJB, KunkelTA, SuginoA (1991) Eukaryotic DNA polymerase amino acid sequence required for 3′----5′ exonuclease activity. Proc NatlAcad Sci U S A 88 : 9473–9477.
74. JinYH, ObertR, BurgersPM, KunkelTA, ResnickMA, et al. (2001) The 3′-->5′ exonuclease of DNA polymerase delta can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc Natl Acad Sci U S A 98 : 5122–5127.
75. FillinghamJ, RechtJ, SilvaAC, SuterB, EmiliA, et al. (2008) Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 28 : 4342–4353.
76. BerndsenCE, TsubotaT, LindnerSE, LeeS, HoltonJM, et al. (2008) Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat Struct Mol Biol 15 : 948–956.
77. KimN, HuangSN, WilliamsJS, LiYC, ClarkAB, et al. (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332 : 1561–1564.
78. MyungK, ChenC, KolodnerRD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411 : 1073–1076.
79. MyungK, DattaA, KolodnerRD (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104 : 397–408.
80. MorrisonA, ChristensenRB, AlleyJ, BeckAK, BernstineEG, et al. (1989) REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J Bacteriol 171 : 5659–5667.
81. HolbeckSL, StrathernJN (1997) A role for REV3 in mutagenesis during double-strand break repair in Saccharomyces cerevisiae. Genetics 147 : 1017–1024.
82. CollinsSR, MillerKM, MaasNL, RoguevA, FillinghamJ, et al. (2007) Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 : 806–810.
83. LukeB, VersiniG, JaquenoudM, ZaidiIW, KurzT, et al. (2006) The cullin Rtt101p promotes replication fork progression through damaged DNA and natural pause sites. Curr Biol 16 : 786–792.
84. HannaJS, KrollES, LundbladV, SpencerFA (2001) Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol 21 : 3144–3158.
85. BylundGO, BurgersPM (2005) Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol 25 : 5445–5455.
86. RocheH, GietzRD, KunzBA (1995) Specificities of the Saccharomyces cerevisiae rad6, rad18, and rad52 mutators exhibit different degrees of dependence on the REV3 gene product, a putative nonessential DNA polymerase. Genetics 140 : 443–456.
87. ShahbazianMD, GrunsteinM (2007) Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76 : 75–100.
88. JinYH, ClarkAB, SlebosRJ, Al-RefaiH, TaylorJA, et al. (2003) Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet 34 : 326–329.
89. PutnamCD, HayesTK, KolodnerRD (2009) Specific pathways prevent duplication-mediated genome rearrangements. Nature 460 : 984–989.
90. LobachevKS, GordeninDA, ResnickMA (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108 : 183–193.
91. SaparbaevM, PrakashL, PrakashS (1996) Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1-RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 142 : 727–736.
92. SugawaraN, PaquesF, ColaiacovoM, HaberJE (1997) Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci U S A 94 : 9214–9219.
93. StudamireB, PriceG, SugawaraN, HaberJE, AlaniE (1999) Separation-of-function mutations in Saccharomyces cerevisiae MSH2 that confer mismatch repair defects but do not affect nonhomologous-tail removal during recombination. Mol Cell Biol 19 : 7558–7567.
94. LyndakerAM, AlaniE (2009) A tale of tails: insights into the coordination of 3′ end processing during homologous recombination. Bioessays 31 : 315–321.
95. HaberJE (1995) In vivo biochemistry: physical monitoring of recombination induced by site-specific endonucleases. Bioessays 17 : 609–620.
96. McGrawBR, MarinusMG (1980) Isolation and characterization of dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol Gen Genet 178 : 309–315.
97. YunM, WuJ, WorkmanJL, LiB (2011) Readers of histone modifications. Cell Res 21 : 564–578.
98. UmarA, BuermeyerAB, SimonJA, ThomasDC, ClarkAB, et al. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 87 : 65–73.
99. DzantievL, ConstantinN, GenschelJ, IyerRR, BurgersPM, et al. (2004) A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell 15 : 31–41.
100. SuttonA, BucariaJ, OsleyMA, SternglanzR (2001) Yeast ASF1 protein is required for cell cycle regulation of histone gene transcription. Genetics 158 : 587–596.
101. GueldenerU, HeinischJ, KoehlerGJ, VossD, HegemannJH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23.
102. GietzRD, WoodsRA (2002) Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350 : 87–96.
103. MaWT, GV SandriGV, SarkarS (1992) Analysis of the Luria-Delbrück distribution using discrete convolution powers. J Appl Prob 29 : 255–267.
104. AsterisG, SarkarS (1996) Bayesian procedures for the estimation of mutation rates from fluctuation experiments. Genetics 142 : 313–326.
105. HallBM, MaCX, LiangP, SinghKK (2009) Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 25 : 1564–1565.
106. RoscheWA, FosterPL (2000) Determining mutation rates in bacterial populations. Methods 20 : 4–17.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy