
A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
Growth and Differentiation Factor 5 (GDF5) is a secreted growth factor that belongs to the Bone Morphogenetic Protein (BMP) family and plays a pivotal role during limb development. GDF5 is a susceptibility gene for osteoarthritis (OA) and mutations in GDF5 are associated with a wide variety of skeletal malformations ranging from complex syndromes such as acromesomelic chondrodysplasias to isolated forms of brachydactylies or multiple synostoses syndrome 2 (SYNS2). Here, we report on a family with an autosomal dominant inherited combination of SYNS2 and additional brachydactyly type A1 (BDA1) caused by a single point mutation in GDF5 (p.W414R). Functional studies, including chondrogenesis assays with primary mesenchymal cells, luciferase reporter gene assays and Surface Plasmon Resonance analysis, of the GDF5W414R variant in comparison to other GDF5 mutations associated with isolated BDA1 (p.R399C) or SYNS2 (p.E491K) revealed a dual pathomechanism characterized by a gain - and loss-of-function at the same time. On the one hand insensitivity to the main GDF5 antagonist NOGGIN (NOG) leads to a GDF5 gain of function and subsequent SYNS2 phenotype. Whereas on the other hand, a reduced signaling activity, specifically via the BMP receptor type IA (BMPR1A), is likely responsible for the BDA1 phenotype. These results demonstrate that one mutation in the overlapping interface of antagonist and receptor binding site in GDF5 can lead to a GDF5 variant with pathophysiological relevance for both, BDA1 and SYNS2 development. Consequently, our study assembles another part of the molecular puzzle of how loss and gain of function mutations in GDF5 affect bone development in hands and feet resulting in specific types of brachydactyly and SYNS2. These novel insights into the biology of GDF5 might also provide further clues on the pathophysiology of OA.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003846
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003846
Summary
Growth and Differentiation Factor 5 (GDF5) is a secreted growth factor that belongs to the Bone Morphogenetic Protein (BMP) family and plays a pivotal role during limb development. GDF5 is a susceptibility gene for osteoarthritis (OA) and mutations in GDF5 are associated with a wide variety of skeletal malformations ranging from complex syndromes such as acromesomelic chondrodysplasias to isolated forms of brachydactylies or multiple synostoses syndrome 2 (SYNS2). Here, we report on a family with an autosomal dominant inherited combination of SYNS2 and additional brachydactyly type A1 (BDA1) caused by a single point mutation in GDF5 (p.W414R). Functional studies, including chondrogenesis assays with primary mesenchymal cells, luciferase reporter gene assays and Surface Plasmon Resonance analysis, of the GDF5W414R variant in comparison to other GDF5 mutations associated with isolated BDA1 (p.R399C) or SYNS2 (p.E491K) revealed a dual pathomechanism characterized by a gain - and loss-of-function at the same time. On the one hand insensitivity to the main GDF5 antagonist NOGGIN (NOG) leads to a GDF5 gain of function and subsequent SYNS2 phenotype. Whereas on the other hand, a reduced signaling activity, specifically via the BMP receptor type IA (BMPR1A), is likely responsible for the BDA1 phenotype. These results demonstrate that one mutation in the overlapping interface of antagonist and receptor binding site in GDF5 can lead to a GDF5 variant with pathophysiological relevance for both, BDA1 and SYNS2 development. Consequently, our study assembles another part of the molecular puzzle of how loss and gain of function mutations in GDF5 affect bone development in hands and feet resulting in specific types of brachydactyly and SYNS2. These novel insights into the biology of GDF5 might also provide further clues on the pathophysiology of OA.
Introduction
Growth and Differentiation Factor 5 (GDF5), which is also known as Cartilage-Derived Morphogenetic Protein 1 (CDMP1) belongs to the Transforming Growth Factor Beta superfamily (TGFB) and the subordinated group of Bone Morphogenetic Proteins (BMPs) [1]. GDF5 has a fundamental role during limb development, where it controls the size of the initial cartilaginous condensations as well as the process of joint development [2]–[4]. As a positive key regulator of early chondrogenesis, dimeric GDF5 initiates signaling by interacting preferably with two distinct BMP type I receptors, BMPR1A and BMPR1B, whereas binding via BMPR1B is favored over BMPR1A [5], [6]. Upon receptor phosphorylation, intracellular SMAD transducer proteins are activated in order to regulate target gene transcription [7], [8]. GDF5 activity is counteracted by BMP antagonists such as NOGGIN (NOG), which mask the receptor binding sites of GDF5 by a direct protein-protein interaction, thereby impeding receptor binding of the ligand and thus signaling [9], [10].
Alterations in GDF5 signaling due to specific point mutations have been associated with various diseases affecting bone and cartilage development [2], [11], [12]. Activating mutations in GDF5 lead to a gain of function phenotype, resulting in increased chondrogenic activity as described for proximal symphalangism (SYM1, MIM #185800) and the multiple synostoses syndrome 2 (SYNS2; MIM #610017) [13]–[18]. The SYM1 phenotype is characterized by ankylosis of the proximal interphalangeal joints as well as fusion of carpal and tarsal bones. Additional symphalangism in the elbow and knee joint caused by GDF5 mutations is a hallmark of the SYNS2 phenotype. In contrast to activating GDF5 mutations, loss of function mutations result in hypoplastic or absent skeletal elements as described for the molecular disease family of brachydactylies. Depending on the affected phalanges, five different types of brachydactylies are categorized (A–E) including three subgroups (A1–A3) [11]. So far, mutations in GDF5 have been linked to isolated traits of BDA1 (MIM #112500), BDA2 (MIM #112600) and BDC (MIM 113100) [16], [18]–[21]. Extreme shortening of digits and limbs are caused by homozygous loss-of-function mutations in GDF5, which are associated with different types of acromesomelic chondrodysplasia (Grebe MIM #200700, Hunter Thompson MIM #201250, Du Pan MIM#228900) [22].
Here we describe a family carrying a mutation in the mature domain of GDF5, p.W414R, showing combined clinical features of BDA1 and SYNS2. In this work we unravel the unique pathomechanism behind GDF5W414R and thus demonstrate how one mutation in GDF5 confers a gain - and loss-of-function phenotype simultaneously.
Results
GDF5W414R results in SYNS2 and BDA1
We report on a family of Mexican descent with an autosomal dominant form of SYNS2 with additional BDA1 (Figure 1A). Sequencing of the GDF5 gene revealed a c.T1240C mutation (p.W414R) in three affected individuals from three generations. A mutation in NOG, a candidate gene for SYNS1, was excluded. The affected individuals are presented with multiple synostoses including proximal and distal symphalangism, metacarpophalangeal synostosis, and synostosis of carpal and tarsal bones as well as BDA1 with severe hypoplasia and even aplasia of the middle phalanges (Figure 1B and Table 1). Additional symptoms such as hearing impairment or short stature were not observed.


GDF5W414R is positioned within the NOG and BMPRI binding interface
The three mutations of interest (GDF5W414R, GDF5R399C, GDF5E491K) were highlighted in the GDF5 structure model (Figure 2A). GDF5W414R is positioned within the long loop of finger 1, whereas GDF5E491K is located within the second finger of the GDF5 dimer. GDF5R399C is located at the N-terminal end, right in front of the β1 sheet of the first finger [23]. As shown in Figure 2B, all mutated sites in GDF5 are conserved among different species (human, mouse, chicken). Based on the crystal structures of the BMP7:NOG, BMP2:BMPR1A and GDF5:BMPR1B complexes, we predicted residues of GDF5 that are involved in binding to NOG or to the BMP type I receptors (Figure 2B) [5], [24]–[26]. Both SYNS2 associated variants, GDF5W414R and GDF5E491K, are located within the NOG interaction site. Contrary, GDF5R399C, which is linked to an isolated BDA1 phenotype, is positioned outside of the NOG binding interface. Since all three mutations might also interfere with BMP type I receptor recruitment, we analyzed the interactions of the three mutations (GDF5W414R, GDF5R399C, GDF5E491K) to NOG and to BMPR1A and BMPR1B.

GDF5W414R is insensitive to inhibition by NOG
NOG, the main regulator of GDF5 activity, was initially identified to be mutated in patients with SYNS1 [27]. As GDF5W414R is associated with the SYNS2 phenotype and furthermore located within the critical NOG binding site, we examined the signaling potency of the GDF5 mutations compared to wild type GDF5 in the absence and presence of NOG. We performed in vitro chondrogenesis assays and used the respective chicken GDF5 constructs to infect chicken limb bud micromass cultures with and without NOG. Similar expression levels of wild type and mutant GDF5 were confirmed by Western blot (Figure S1, Text S1). As a chondrogenic marker, the extracellular matrix (ECM) produced by the limb bud cells was stained with Alcian blue (Figure 3).

In the absence of NOG, quantification of Alcian blue revealed a strong induction of early chondrogenesis for wild type GDF5 and GDF5W414R as well as for the BDA1 causing variant GDF5R399C and the SYM1/SYNS2-associated variant GDF5E491K. However, co-infection of NOG suppressed chondrogenesis effectively in wild type GDF5 expressing cells, while GDF5W414R infected cells displayed a clear insensitivity towards NOG. NOG-resistance was also found for the GDF5E491K variant. In contrast, cartilage formation was strongly inhibited in micromass cells expressing GDF5R399C.
The reduced sensitivity of GDF5W414R to NOG was also detected in Biacore measurements. In contrast to the high binding affinity of wild type GDF5 to NOG (apparent KD: ∼2 nM), GDF5W414R showed a markedly reduced (∼12 fold) binding to NOG (apparent KD: ∼25.5 nM) (Table 2).

GDF5W414R shows specific loss of BMPR1A signaling
As GDF5W414R is located in the overlapping interface of the high affinity BMP type I receptors and NOG, we analyzed subsequently signaling activities of wild type GDF5 and GDF5W414R after co-expression of either one of the type I receptors, Bmpr1a or Bmpr1b. Signaling activities were determined in NIH/3T3 cells using a Smad Binding Element (SBE) luciferase reporter gene assay (Figure 4A–C).
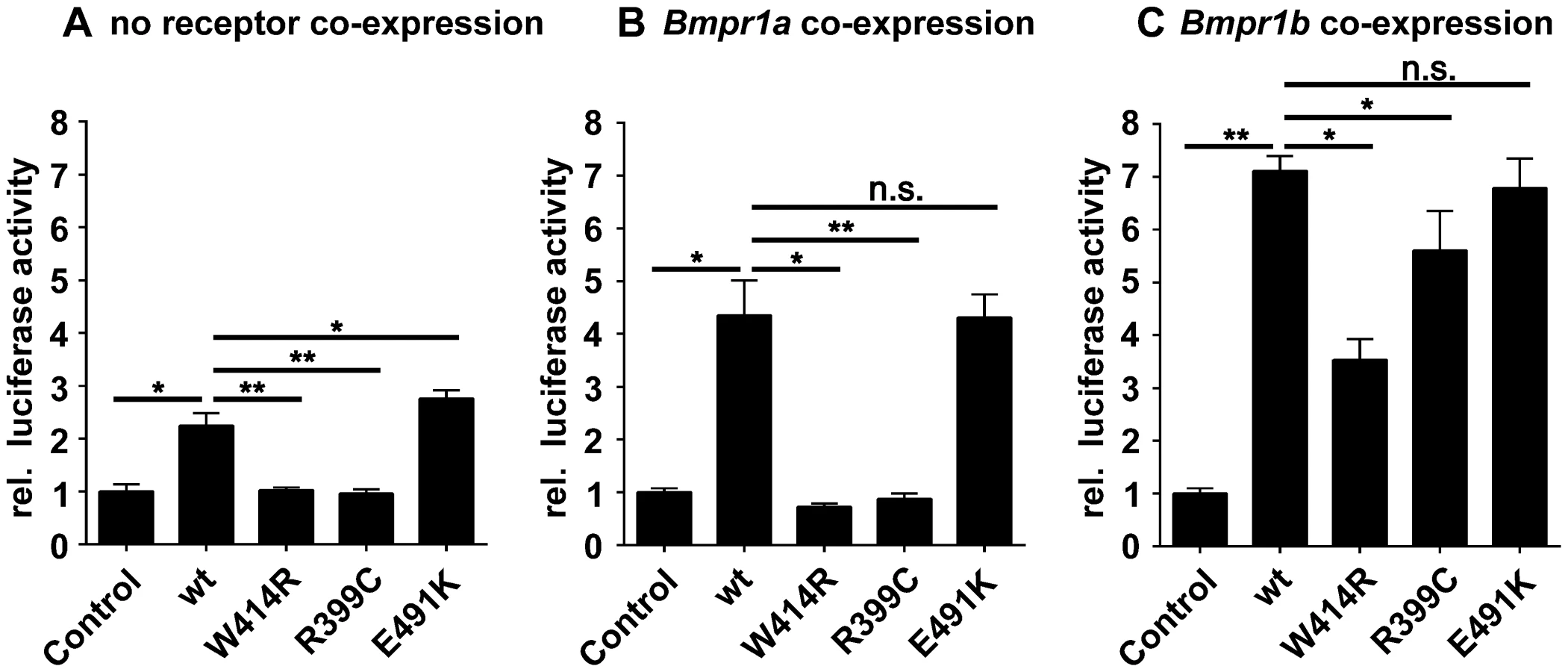
Overexpression of wild type GDF5 in combination with either one of the two type I receptors, Bmpr1a and Bmpr1b, resulted in a strong induction of luciferase activity. As expected from our Biacore data, wild type GDF5-induced signaling via Bmpr1b was stronger compared to signaling mediated via Bmpr1a (Figure 4B–C; Table 2). In case of GDF5W414R, no reporter gene activity was observed when Bmpr1a was additionally transfected (Figure 4B). However, co-transfection of Bmpr1b led to a clear induction of the SBE reporter, even though to a slightly lesser extent compared to wild type GDF5 (Figure 4C). For the BDA1 associated variant GDF5R399C, we revealed the same signaling pattern in our luciferase assay as for GDF5W414R, which leads to the assumption that the pathomechanism of BDA1 is presumably connected with an alteration of the GDF5:BMPR1A binding interaction. In contrast, the SYM1/SYNS2 causing GDF5 variant GDF5E491K promotes GDF5 signaling via Bmpr1a and Bmpr1b to a similar extent when compared to wild type GDF5.
Biacore analysis supported the findings from our cell based assays since GDF5W414R showed a clear deviation from the wild type GDF5 receptor binding pattern. We could demonstrate a 7-fold lower affinity of GDF5W414R to BMPR1A (apparent KD: ∼124 nM) compared to wild type GDF5 (apparent KD: ∼17 nM) and only a 3-fold lower affinity for BMPR1B (apparent KD: ∼3,3) compared to wild type GDF5 (apparent KD: ∼1,1 nM) (Table 2).
In summary, Biacore analysis and in vitro overexpression studies indicate a functional link between the phenotypic features of BDA1 and an impaired BMPR1A signaling of BDA1 associated GDF5 variants.
Activity of GDF5W414R is reduced in the absence of Bmpr1b
In order to confirm the previous hypothesis, that GDF5W414R is not able to transduce signaling via BMPR1A, we conducted an in vitro chondrogenesis assay using primary mesenchymal cells derived from Bmpr1b null mice (Figure 5A–C). Assuming that in wild type cells GDF5 signaling is mediated via Bmpr1a and Bmpr1b, a Bmpr1b knock-out would lead to a situation where solely Bmpr1a transmits GDF5-specific signals. Hence, we hypothesized that GDF5W414R would not able to stimulate chondrogenic differentiation in cells lacking Bmpr1b, due to its insufficiency in binding to Bmpr1a.

As anticipated, heterozygous and homozygous Bmpr1b cells resulted in decreased chondrogenic activity for both, wild type GDF5 as well as GDF5W414R. However, wild type GDF5 stimulation led to an induction of chondrogenesis even in the complete absence of Bmpr1b, whereas GDF5W414R stimulated cells displayed a complete loss of chondrogenic activity, indicating that a loss of binding of GDF5 to BMPR1A represents the centerpiece of the BDA1 pathomechanism.
Gdf5 expression co-localizes with Nog and Bmpr1b during limb development
To reconstruct the progress of GDF5 dependent limb development we analyzed the gene expression of Gdf5 and its main antagonist Nog as well as its BMP type I receptors Bmpr1a and Bmpr1b in mice limb buds at stages E11.5 to E13.5, which represent critical phases of limb development.
At stage E11.5, Gdf5 is expressed in the anterior part of the limb bud (Figure 6A/E). Here, expression signals for Nog and Bmpr1b partly co-localize with Gdf5 in the distal region (Figure 6B/F and D/H). Additionally, Bmpr1b and Nog show signals in the area of the later developing shoulder and Nog in the elbow joint as well. In contrast, Bmpr1a expression concentrates in the surrounding epithelium and underlying mesenchyme but sparing the central mesenchyme (Figure 6C/G). At stage E12.5 the expression pattern becomes more defined for Gdf5, Nog and Bmpr1b in the digital rays (Figure 6A′/E′, 6B′/F′ and 6D′/H′) and for Bmpr1a in direct proximity in the inter-phalangeal regions and the surrounding epithelium (Figure 6C′/G′). From stage E13.5 onwards, Gdf5 expression concentrates in the joint interzones (Figure 6A″/E′), flanked by Nog and Bmpr1b expression (Figure 6B′/F′ and 6D′/H′). Apart from the distal tips, Bmpr1a is still expressed in the surrounding limb epithelium and interdigital mesenchyme.

The expression pattern analysis shows a co-localization for Gdf5, Nog and Bmpr1b and in case of Bmpr1a, expression in direct proximity to Gdf5.
Discussion
Here we describe a novel GDF5 Trp to Arg transition (p.W414R) in patients with multiple synostoses syndrome 2 (SYNS2), including proximal and distal symphalangism, metacarpophalangeal synostosis, and synostosis of carpal and tarsal bones as well as BDA1 with severe hypoplasia and even aplasia of the middle phalanges. We identified that BDA1 and SYNS2 caused by GDF5W414R are due to two independent molecular mechanisms involving specifically the BMP receptor BMPR1A and the BMP antagonist NOG, respectively.
Interestingly, mutations in NOG as well as in GDF5 can lead to similar phenotypic characteristics of SYM1 and SYNS1/2 [15], [17], [27]. However, joint fusions caused by NOG mutations often affect the ossicles leading to hearing impairment, whereas mutations in GDF5 including GDF5W414R spare this feature [27]–[29]. Regarding the literature, SYM1 - and SYNS2-associated mutations in GDF5 like GDF5N445T and GDF5S475N were shown to destabilize the GDF5/NOG interaction thus leading to a severe insensitivity towards the antagonist, also called NOG resistance [15], [17]. Therefore, we likewise analyzed GDF5W414R concerning its interaction with NOG. W414 is located within the putative NOG binding interface, which was predicted based on the published superimposed GDF5:NOG complex [15], [30]. Interaction analyses of GDF5W414R using chondrogenic differentiation assays together with Biacore binding studies revealed a NOG resistance as molecular cause of the joint fusion phenotype similar to GDF5N445T and GDF5S475N [15], [17]. In addition, we identified NOG insensitivity as the effect of the SYM1 associated GDF5E491K mutation [14]. Hence, in case of SYM1 and SYNS2, an impaired GDF5/NOG interaction interferes with the negative feedback loop by which GDF5 is antagonized and thus balanced within the fine-tuned signaling network. Consequently, GDF5 variants associated with joint fusions exert an enhanced chondrogenic activity and can be referred to as gain of function mutations (Figure 7) [11]. The tight connection between GDF5 and NOG and their major importance for the development of joints become further visible as the results of our expression analyses of the developing mouse limb show overlapping temporal and spatial expression patterns of Gdf5 and Nog.

To elucidate the underlying molecular mechanism by which GDF5W414R causes joint fusions in combination with brachydactyly, we further analyzed how the mutation interferes with its cognate transmembrane BMP type I receptors. Situated in the long loop of finger 1 between the ß-sheets ß1/2 and ß3/ß4, W414 is positioned outside of the wrist epitope, which is mainly responsible for binding the BMP type I receptors BMPR1A and BMPR1B [5], [6], [23]. On the basis of the GDF5:BMPR1B crystal structure and the modeled GDF5:BMPR1A interaction, the contact of both BMP type I receptors with W414 was confirmed [5], [23]. As suggested, a transition of hydrophobic Trp to hydrophilic Arg at this highly conserved position results in impaired BMP type I receptor activation as shown in reporter gene assays and Biacore binding studies. Most strikingly, GDF5W414R displayed a complete loss of BMPR1A activation, whereas signaling via BMPR1B was only moderately decreased. Possibly, a mutation interfering with the BMP type 1 receptor binding has in general a more drastic effect for the BMPR1A than for the BMPR1B, because the interaction with BMPR1A is per se lower. The remaining signaling activity of GDF5W414R via BMPR1B seems to be sufficient to preserve its biological functionality as seen in our chondrogenic differentiation assays. A recently published GDF5 mutation (GDF5R399C) is likewise reported to cause BDA1. However, in this French Canadian family BDA1 occurs as an isolated trait in contrast to the phenotype of GDF5W414R, which is combined with features of synostoses [19]. GDF5R399C is located at the N-terminus of the mature GDF5, right in front of the first β-sheet of finger 1, and is predicted to interfere with both BMP type I receptors [23]. Accordingly, we revealed a BMP type I receptor activation pattern similar to that of GDF5W414R indicating that the disruption of BMPR1A signaling is a hallmark of the BDA1 pathomechanism (Figure 7).
There are two studies which deal explicit with the analyses of specific functions of BMPR1A and BMPR1B, one was done in chicken and the other one was done in mice [31], [32]. In the chicken study the expression patterns of both receptors were distinct during limb development. BMPR1B was strongly expressed in precartilaginous condensations, whereas BMPR1A was reported to be expressed throughout the limb mesenchyme. To rule out a functional difference between BMPR1A and BMPR1B, the authors overexpressed either constitutive active (c.a.) or dominant negative (d.n.) variants of BMPR1A and BMPR1B in vivo in the chicken limb bud or in vitro in chicken micromass cultures [32]. Interestingly, BMPR1B turned out to be necessary for early steps of cartilage formation, whereas BMPR1A was shown to elicit an important function in prehypertrophic chondrocytes. Expression of c.a. BMPR1A led to a delay of chondrogenic differentiation; similar to the phenotype caused by overexpression of IHH [33]. Another study indicated that the IHH-regulated process of chondrogenic differentiation indeed requires BMP signaling [34]. As both signals, IHH and BMP, are required for maintaining a normal proliferation rate and regular differentiation of chondrocytes, these findings could explain how BDA1 can be caused by mutations in IHH or the BMP pathway. A few years later a study was undertaken in mice, where authors concluded that Bmpr1a and Bmpr1b have mostly redundant functions in chondrogenesis [31]. This statement was made due to the observation that single knock outs of either Bmpr1a or Bmpr1b showed only subtle skeletal phenotypes, whereas the double knock out displayed a very strong phenotype with a nearly absent endochondral skeleton. Nevertheless, the phenotypes of each knock out are very distinct, for example Bmpr1b null mice displayed defects in the appendicular skeleton, whereas Bmpr1a cKO shows a generalized chondrodysplasia and the more severe phenotype seen in the double knock out could also be explained by an additive or synergistic effect. Therefore we suggest that both receptors have unique functions and loss of binding of the GDF5 mutants to one of the two receptors cannot be compensated by the other receptor.
Loss of GDF5 receptor binding in general plays a central role within the molecular disease family of brachydactylies. For example, the GDF5 variant GDF5L441P causes BDA2 due to an impaired BMPR1B binding [11], [16]. Vice versa, specific mutations in BMPR1B are associated with BDA2 [35]. Compared to BDA1, where all middle phalanges are affected and distal symphalangism can occur, BDA2 is characterized by short or absent middle phalanges only of the second and sometimes fifth finger. BDC comprises features of BDA1 and BDA2 and primarily affects the middle phalanges of the second, third and fifth fingers and the first metacarpal bone. Interestingly, the molecular reason for BDC is functional haploinsufficiency of GDF5 [21]. The implication of GDF5 in chondrogenesis and joint formation can finally be highlighted in connection with osteoarthritis (OA; MIM #165720), the most common form of late-onset destruction of articular cartilage in synovial joints nowadays [36]. Among various genetic loci, GDF5 has been discovered as the most consistent and robust risk factor of OA, whereby decreased Gdf5 mRNA levels have been found to account for a murine OA-like phenotype [36]–[38]. Furthermore Masuya et al. identified a Trp to Arg transition in a large ENU mutagenesis screen, which was described to impair joint formation and thereby cause OA [39]. This mutation (p.W408R) is the mouse homologue to the GDF5W414R mutation we described in this work. Therefore, understanding the biology of GDF5W414R might also give insights into the pathophysiology of OA.
In summary, we revealed that GDF5W414R, in contrast to wild type GDF5, loses the BMPR1A signaling route and at the same time increases the alternative signaling via BMPR1B in the presence of NOG. Therefore, the reduced sensitivity of W414R to Noggin and its reduced interaction with BMPR1A do not actually “neutralize” each other, but lead to a misbalance of BMPR1A and BMPR1B signaling. Hence, our study assembles another part of the molecular puzzle how loss and gain of function mutations in GDF5 affect bone development in hands and feet and result in specific types of brachydactyly and SYNS2.
Materials and Methods
Clinical investigation and molecular analysis
All clinical investigations have been performed according to Declaration of Helsinki principles. The study was approved by the local institutional review board “Ethikkommission der Charité - Universitätsmedizin Berlin”. Informed consent for genetic testing was obtained from the patient or their legal guardians respectively. Genomic DNA of affected family members were extracted from peripheral blood samples by standard methods. The coding regions of NOG and GDF5 as well as the flanking intronic sequences were amplified by standard PCR protocols. The primer sequences and PCR conditions for the molecular testing were previously described [20], [30]. PCR products were analyzed on 2% agarose gels. Sequencing was done using the ABI Prism BigDye Terminator Sequencing Kit (Applied Biosystems) with PCR primers used as sequencing primers. Products were evaluated on an automated capillary sequencer (Applied Biosystems).
Chicken micromass cultures
Cloning of the coding sequences of chicken GDF5 and NOG into RCAS(BP)A or RCAS(BP)B, respectively was previously described [15]. Mutations (GDF5W414R, GDF5R399C, GDF5E491K) were introduced into the coding sequence of chicken GDF5 in pSLAX13 by in vitro mutagenesis. Primer sequences are available in the supplement (Table S1). Production of viral supernatant in DF1 cells and concentration of viral particles was performed as described previously [40]. Fertilized chicken eggs were obtained from VALO BioMedia GmbH (Osterholz-Scharmbeck, Germany) and incubated at 38°C in a humidified egg incubator for 4.5 days. Micromass cultures were plated in a drop containing 2×105 cells. Infection was performed with concentrated viral supernatants: RCASBP(A) containing cDNA encoding chicken wild type GDF5 and the GDF5 mutants GDF5W414R, GDF5R399C, and GDF5E491K with a titer of 1×107 plaque forming units (PFU)/ml. RCASBP(B) containing the cDNA encoding chicken wild type NOG was applied with a titer of 2,5×106 PFU/ml. Culture medium containing DMEM-F12 (Biochrom), 10% FBS (Biochrom), 0,2% chicken serum (Sigma), 2 mM L-Gln (Lonza), 100 U/ml penicillin, and 100 µg/ml streptomycin (Lonza) was replaced every 2 days. For each condition, three replicates were performed in parallel. Quantification of Alcian blue dye was performed at 595 nm after extraction with Guanidin-HCl.
Recombinant proteins
Recombinant human (rh) GDF5 and its variant rhGDF5W414R were dissolved in 10 mM HCl and provided by Biopharm GmbH.
BIAcore binding assay
The BIA2000 system (Biacore) was used to analyze the binding affinities of recombinant human GDF5 and its variant GDF5W414R to immobilized NOG and ectodomains of BMPR1A, BMPR1B and BMPR2, as previously described [16].
Luciferase activity assay
Coding sequences of human GDF5 and mouse Bmpr1a and Bmpr1b were cloned into pSLAX13. Mutations (GDF5W414R, GDF5R399C, GDF5E491K) were introduced into the coding sequence of human GDF5 in pSLAX13 by in vitro mutagenesis. Primer sequences are available in the supplement (Table S1). Inserts were subcloned into pCS2+ via ClaI.
Luciferase reporter gene assays were performed using the murine fibroblast cell line NIH/3T3 (ATCC) which was maintained in DMEM high glucose (Lonza) with 10% FCS (Biochrom), 2 mM L-Gln (Lonza), 100 U/ml penicillin, and 100 µg/ml streptomycin (Lonza). Prior to transfection, cells were seeded in a 96-well plate at a density of 1×104 cells per well. BMP receptors and GDF5 constructs were transfected for 40 hours together with the Smad Binding Element luciferase construct SBE-pGL3 [41] and the normalization vector pRLTk (Promega) using Lipofectamine 2000 (Invitrogen). Luciferase activity was determined as described previously [42].
Mouse micromass cultures
Limb mesenchymal cells were isolated from stage E13.5 embryos resulting from matings of C57BL/6, Bmpr1btm1kml heterozygous or homozygous knock-out mice on a C57BL/6 background [43]. Mouse embryos were genotyped using primers for Bmpr1b and neomycin (Table S2), if applicable embryos were pooled according to their phenotypes. Isolation of mouse micromass cells was performed as described for chicken micromass cultures with minor modifications. For mouse micromass cultures no additional chicken serum was used. After 24 h mouse micromass cultures were stimulated with 5 nM of recombinant human wild type GDF5 and GDF5W414R.
Whole mount in situ hybridization
C57BL/6 mouse embryos were harvested at stages E11.5–13.5 and fixed in 4% PFA. Whole mount in situ hybridization was performed as previously described [44].
DIG-labeled RNA antisense-probes were generated by in vitro transcription using the coding sequences of mouse Bmpr1a, Bmpr1b and Nog as a template. The probe for mouse Gdf5 was previously published [45]. Signal detection was performed with BMPurple (Roche). 3D imaging of labeled limbs was done by optical projection tomography (OPT) scans as previously described [46].
Statistics
Statistical analyses were performed using a two-tailed Student's T-test. Results are presented as mean ± SEM. P values of less than 0.05 were considered significant.
Supporting Information
{kind=link}
Zdroje
1. ChangSC, HoangB, ThomasJT, VukicevicS, LuytenFP, et al. (1994) Cartilage-derived morphogenetic proteins. New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J Biol Chem 269 : 28227–28234.
2. StrickerS, MundlosS (2011) Mechanisms of Digit Formation: Human Malformation Syndromes Tell the Story. Developmental Dynamics 240 : 990–1004.
3. BuxtonP, EdwardsC, ArcherCW, Francis-WestP (2001) Growth/differentiation factor-5 (GDF-5) and skeletal development. J Bone Joint Surg Am 83-A Suppl 1: S23–30.
4. StormEE, HuynhTV, CopelandNG, JenkinsNA, KingsleyDM, et al. (1994) Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 368 : 639–643.
5. KotzschA, NickelJ, SeherA, SebaldW, MullerTD (2009) Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for GDF-5-type I receptor specificity. EMBO J 28 : 937–947.
6. MuellerTD, NickelJ (2012) Promiscuity and specificity in BMP receptor activation. FEBS Lett 586 : 1846–1859.
7. NoheA, KeatingE, KnausP, PetersenNO (2004) Signal transduction of bone morphogenetic protein receptors. Cell Signal 16 : 291–299.
8. SchmiererB, HillCS (2007) TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8 : 970–982.
9. MiyazonoK, KamiyaY, MorikawaM (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147 : 35–51.
10. BragdonB, MoseychukO, SaldanhaS, KingD, JulianJ, et al. (2011) Bone morphogenetic proteins: a critical review. Cell Signal 23 : 609–620.
11. MundlosS (2009) The brachydactylies: a molecular disease family. Clin Genet 76 : 123–136.
12. LoriesRJ, LuytenFP (2005) Bone morphogenetic protein signaling in joint homeostasis and disease. Cytokine Growth Factor Rev 16 : 287–298.
13. DawsonK, SeemanP, SebaldE, KingL, EdwardsM, et al. (2006) GDF5 is a second locus for multiple-synostosis syndrome. Am J Hum Genet 78 : 708–712.
14. WangX, XiaoF, YangQ, LiangB, TangZ, et al. (2006) A novel mutation in GDF5 causes autosomal dominant symphalangism in two Chinese families. Am J Med Genet A 140A: 1846–1853.
15. SeemannP, BrehmA, KonigJ, ReissnerC, StrickerS, et al. (2009) Mutations in GDF5 reveal a key residue mediating BMP inhibition by NOGGIN. PLoS Genet 5: e1000747.
16. SeemannP, SchwappacherR, KjaerKW, KrakowD, LehmannK, et al. (2005) Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J Clin Invest 115 : 2373–2381.
17. SchwaerzerGK, HiepenC, SchreweH, NickelJ, PloegerF, et al. (2012) New insights into the molecular mechanism of multiple synostoses syndrome (SYNS): mutation within the GDF5 knuckle epitope causes noggin-resistance. J Bone Miner Res 27 : 429–442.
18. PlogerF, SeemannP, Schmidt-von KeglerM, LehmannK, SeidelJ, et al. (2008) Brachydactyly type A2 associated with a defect in proGDF5 processing. Hum Mol Genet 17 : 1222–1233.
19. ByrnesAM, RacachoL, NikkelSM, XiaoF, MacDonaldH, et al. (2010) Mutations in GDF5 presenting as semidominant brachydactyly A1. Hum Mutat 31 : 1155–1162.
20. SchwabeGC, TurkmenS, LeschikG, PalanduzS, StoverB, et al. (2004) Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1. Am J Med Genet A 124A: 356–363.
21. EvermanDB, BartelsCF, YangY, YanamandraN, GoodmanFR, et al. (2002) The mutational spectrum of brachydactyly type C. Am J Med Genet 112 : 291–296.
22. ThomasJT, KilpatrickMW, LinK, ErlacherL, LembessisP, et al. (1997) Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet 17 : 58–64.
23. NickelJ, KotzschA, SebaldW, MuellerTD (2005) A single residue of GDF-5 defines binding specificity to BMP receptor IB. J Mol Biol 349 : 933–947.
24. KellerS, NickelJ, ZhangJL, SebaldW, MuellerTD (2004) Molecular recognition of BMP-2 and BMP receptor IA. Nat Struct Mol Biol 11 : 481–488.
25. KirschT, SebaldW, DreyerMK (2000) Crystal structure of the BMP-2-BRIA ectodomain complex. Nat Struct Biol 7 : 492–496.
26. GroppeJ, GreenwaldJ, WiaterE, Rodriguez-LeonJ, EconomidesAN, et al. (2002) Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 420 : 636–642.
27. GongY, KrakowD, MarcelinoJ, WilkinD, ChitayatD, et al. (1999) Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat Genet 21 : 302–304.
28. BrownDJ, KimTB, PettyEM, DownsCA, MartinDM, et al. (2002) Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the gene encoding noggin. Am J Hum Genet 71 : 618–624.
29. ManginoM, FlexE, DigilioMC, GiannottiA, DallapiccolaB (2002) Identification of a novel NOG gene mutation (P35S) in an Italian family with symphalangism. Hum Mutat 19 : 308.
30. LehmannK, SeemannP, SilanF, GoeckeTO, IrgangS, et al. (2007) A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am J Hum Genet 81 : 388–396.
31. YoonBS, OvchinnikovDA, YoshiiI, MishinaY, BehringerRR, et al. (2005) Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci U S A 102 : 5062–5067.
32. ZouH, WieserR, MassagueJ, NiswanderL (1997) Distinct roles of type I bone morphogenetic protein receptors in the formation and differentiation of cartilage. Genes Dev 11 : 2191–2203.
33. VortkampA, LeeK, LanskeB, SegreGV, KronenbergHM, et al. (1996) Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273 : 613–622.
34. MininaE, WenzelHM, KreschelC, KarpS, GaffieldW, et al. (2001) BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 128 : 4523–4534.
35. LehmannK, SeemannP, StrickerS, SammarM, MeyerB, et al. (2003) Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc Natl Acad Sci U S A 100 : 12277–12282.
36. ReynardLN, LoughlinJ (2012) Genetics and epigenetics of osteoarthritis. Maturitas 71 : 200–204.
37. LiuJ, CaiW, ZhangH, HeC, DengL (2013) Rs143383 in the Growth Differentiation Factor 5 (GDF5) Gene Significantly Associated with Osteoarthritis (OA)-A Comprehensive Meta-analysis. Int J Med Sci 10 : 312–319.
38. DaansM, LuytenFP, LoriesRJ (2011) GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann Rheum Dis 70 : 208–213.
39. MasuyaH, NishidaK, FuruichiT, TokiH, NishimuraG, et al. (2007) A novel dominant-negative mutation in Gdf5 generated by ENU mutagenesis impairs joint formation and causes osteoarthritis in mice. Hum Mol Genet 16 : 2366–2375.
40. MorganBA, FeketeDM (1996) Manipulating gene expression with replication-competent retroviruses. Methods Cell Biol 51 : 185–218.
41. JonkLJ, ItohS, HeldinCH, ten DijkeP, KruijerW (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273 : 21145–21152.
42. HampfM, GossenM (2006) A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal Biochem 356 : 94–99.
43. YiSE, DaluiskiA, PedersonR, RosenV, LyonsKM (2000) The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 127 : 621–630.
44. PryceBA, BrentAE, MurchisonND, TabinCJ, SchweitzerR (2007) Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Dev Dyn 236 : 1677–1682.
45. SharpeJ, AhlgrenU, PerryP, HillB, RossA, et al. (2002) Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science 296 : 541–545.
46. QuintanaL, SharpeJ (2011) Preparation of mouse embryos for optical projection tomography imaging. Cold Spring Harb Protoc 2011 : 664–669.
47. RobinsonPN, KohlerS, BauerS, SeelowD, HornD, et al. (2008) The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet 83 : 610–615.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy