
Rad52 Sumoylation Prevents the Toxicity of Unproductive Rad51 Filaments Independently of the Anti-Recombinase Srs2
The budding yeast Srs2 is the archetype of helicases that regulate several aspects of homologous recombination (HR) to maintain genomic stability. Srs2 inhibits HR at replication forks and prevents high frequencies of crossing-over. Additionally, sensitivity to DNA damage and synthetic lethality with replication and recombination mutants are phenotypes that can only be attributed to another role of Srs2: the elimination of lethal intermediates formed by recombination proteins. To shed light on these intermediates, we searched for mutations that bypass the requirement of Srs2 in DNA repair without affecting HR. Remarkably, we isolated rad52-L264P, a novel allele of RAD52, a gene that encodes one of the most central recombination proteins in yeast. This mutation suppresses a broad spectrum of srs2Δ phenotypes in haploid cells, such as UV and γ-ray sensitivities as well as synthetic lethality with replication and recombination mutants, while it does not significantly affect Rad52 functions in HR and DNA repair. Extensive analysis of the genetic interactions between rad52-L264P and srs2Δ shows that rad52-L264P bypasses the requirement for Srs2 specifically for the prevention of toxic Rad51 filaments. Conversely, this Rad52 mutant cannot restore viability of srs2Δ cells that accumulate intertwined recombination intermediates which are normally processed by Srs2 post-synaptic functions. The avoidance of toxic Rad51 filaments by Rad52-L264P can be explained by a modification of its Rad51 filament mediator activity, as indicated by Chromatin immunoprecipitation and biochemical analysis. Remarkably, sensitivity to DNA damage of srs2Δ cells can also be overcome by stimulating Rad52 sumoylation through overexpression of the sumo-ligase SIZ2, or by replacing Rad52 by a Rad52-SUMO fusion protein. We propose that, like the rad52-L264P mutation, sumoylation modifies Rad52 activity thereby changing the properties of Rad51 filaments. This conclusion is strengthened by the finding that Rad52 is often associated with complete Rad51 filaments in vitro.
Published in the journal:
. PLoS Genet 9(10): e32767. doi:10.1371/journal.pgen.1003833
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003833
Summary
The budding yeast Srs2 is the archetype of helicases that regulate several aspects of homologous recombination (HR) to maintain genomic stability. Srs2 inhibits HR at replication forks and prevents high frequencies of crossing-over. Additionally, sensitivity to DNA damage and synthetic lethality with replication and recombination mutants are phenotypes that can only be attributed to another role of Srs2: the elimination of lethal intermediates formed by recombination proteins. To shed light on these intermediates, we searched for mutations that bypass the requirement of Srs2 in DNA repair without affecting HR. Remarkably, we isolated rad52-L264P, a novel allele of RAD52, a gene that encodes one of the most central recombination proteins in yeast. This mutation suppresses a broad spectrum of srs2Δ phenotypes in haploid cells, such as UV and γ-ray sensitivities as well as synthetic lethality with replication and recombination mutants, while it does not significantly affect Rad52 functions in HR and DNA repair. Extensive analysis of the genetic interactions between rad52-L264P and srs2Δ shows that rad52-L264P bypasses the requirement for Srs2 specifically for the prevention of toxic Rad51 filaments. Conversely, this Rad52 mutant cannot restore viability of srs2Δ cells that accumulate intertwined recombination intermediates which are normally processed by Srs2 post-synaptic functions. The avoidance of toxic Rad51 filaments by Rad52-L264P can be explained by a modification of its Rad51 filament mediator activity, as indicated by Chromatin immunoprecipitation and biochemical analysis. Remarkably, sensitivity to DNA damage of srs2Δ cells can also be overcome by stimulating Rad52 sumoylation through overexpression of the sumo-ligase SIZ2, or by replacing Rad52 by a Rad52-SUMO fusion protein. We propose that, like the rad52-L264P mutation, sumoylation modifies Rad52 activity thereby changing the properties of Rad51 filaments. This conclusion is strengthened by the finding that Rad52 is often associated with complete Rad51 filaments in vitro.
Introduction
Homologous recombination (HR) is fundamental for the repair of DNA double-strand breaks (DSBs). It is also involved in the error-free fill-in of single-strand gaps generated by replication fork stalling or incomplete DNA repair. Defects in HR are associated with many cancers, both hereditary and sporadic [1], which underlines the essential nature of this process. The mechanisms and proteins involved in HR have been well conserved throughout evolution and much of our knowledge on HR comes from studies conducted with the yeast Saccharomyces cerevisiae (S. cerevisiae) (reviewed in [2], [3]). HR involves the interaction of a 3′-single stranded DNA (ssDNA) end with a homologous double-strand DNA (dsDNA) molecule, which is used as a template for DNA synthesis. In eukaryotes, the recombinase Rad51 forms a nucleoprotein filament on the ssDNA which undergoes synapsis and strand invasion of the homologous duplex DNA to form a stable D-loop (reviewed in [4]). However, the presence of replication protein A (RPA) previously bound to ssDNA prevents Rad51-mediated strand exchange in vitro. This inhibition is overcome by the addition of Rad52 or the Rad55-Rad57 heterodimer (the Rad51 paralogs of S. cerevisiae), defining these proteins as Rad51 filament mediators (reviewed in [4]).
Rad52 exhibits the greatest Rad51 filament mediator activity in S. cerevisiae. Via its interaction with both RPA and Rad51, it stimulates the removal of RPA and recruits Rad51 to DNA (reviewed in [4]). This mediator function is highlighted by the severe phenotypes caused by null mutations of the RAD52 gene: γ-ray sensitivity and highly reduced levels of both mitotic and meiotic HR (reviewed in [3]). The Rad52 protein also has the capacity to anneal homologous ssDNA in vitro [5]. This activity is involved in Rad51-independent single-strand annealing (SSA) [6] and possibly in the capture of the second end of a DSB to generate double Holliday junction (dHJ) intermediates [7]–[9].
The S. cerevisiae Rad52 protein is subject to post-translational modifications but it is unclear how these modulate Rad52 activities. Rad52 is constitutively phosphorylated throughout the cell cycle on some serine and/or threonine residues and additional phosphorylations are induced specifically in S phase [10]. The phosphorylated residues have not yet been identified. Rad52 also undergoes sumoylation at lysines 10, 11 and 220 after exposure to DNA-damaging agents that induce DSBs. This modification depends on the SUMO-conjugating enzyme Ubc9 and on the SUMO-ligase Siz2 [11]. ssDNA accumulation is necessary for sumoylation of Rad52 [11], [12]. It has been reported that loss of Rad52 sumoylation decreases protein stability without significantly affecting HR levels or recruitment of Rad52 to DNA damage [11], [13]. Sumoylation appears to facilitate the exclusion of Rad52 recombination foci from the nucleolus to maintain a low level of recombinational repair at the ribosomal gene locus [14]. Recently, it was shown that Rad52, RPA and Rad59 are modified by a sumoylation wave leading to simultaneous multisite modification. Catalyzed by a DNA-bound SUMO ligase, this wave stabilizes physical interactions between the proteins [15]. However, Rad52 sumoylation might also restrict Rad51 filament formation through the SUMO-targeted Cdc48 segregase that can curb Rad52-Rad51 physical interaction and displace these proteins from DNA [16]. How exactly phosphorylation and sumoylation of Rad52 affect Rad51 filament formation remains to be determined.
Contrasting with its primordial role in DNA repair, HR may lead to potentially lethal intermediates. This was first revealed by the study of the Srs2 helicase, a major actor in the avoidance of such intermediates (reviewed in [17]). UV sensitivity of srs2Δ cells is suppressed by the ablation of the Rad51 protein [18], which suggests that Srs2 is required for the elimination of toxic UV-induced recombination structures. Furthermore, it was shown that leaky alleles of RAD51 or RAD52, which form abortive recombination intermediates, trigger Srs2 activity [19], [20]. Finally, negative interactions between srs2Δ and genes involved in DNA replication or recombination, such as sgs1Δ, rad54Δ or mrc1Δ, are rescued by mutations preventing or altering HR (rad51Δ, rad52Δ, rad55Δ and rad57Δ) 21–23. Thus, it was concluded that ablation of these genes can also induce the formation of lethal recombination intermediates normally eliminated by Srs2.
All these studies indicate that a key role of Srs2 is to avoid the formation of lethal structures induced by HR. Several studies point out that Rad51 filaments on ssDNA could be the lethal structures eliminated by Srs2. First, in vitro experiments show that Srs2 can disrupt Rad51 filaments thanks to its translocase activity [24], [25]. Second, srs2Δ strains show a three - to four-fold increase in the number of budded cells that contain a Rad51 or Rad54 focus compared with wild-type (WT) cells [26]. Finally, it has also been reported that some of the srs2Δ phenotypes, like the co-lethality with rad54Δ or the high sensitivity to a persistent DSB, depend at least partially on the DNA damage checkpoint [27], [28]. Unproductive Rad51 filaments could induce this persistent checkpoint response.
Srs2 is also necessary to complete DSB repair by HR in haploid cells, when the homologous sequence is located on another chromosome. In this context, the low viability of srs2Δ cells is associated with a strong increase in the level of crossing-over (CO) associated with gene conversion [29], [30]. This suggests that Srs2 avoids the formation of CO by promoting synthesis-dependent strand annealing [31]. This is supported by recent in vivo work showing that Srs2 promotes the formation of non-crossing over products mostly through its helicase activity [32], possibly by dismantling nicked HJs. Additionally, the increased sensitivity to UV and γ-ray irradiation of srs2Δ homozygous diploid cells compared with haploids was proposed to be related to the resolution of specific interactions between homologous chromosomes, probably related to HR [33]. Altogether, these data suggest that lethal recombination structures eliminated by Srs2 could also be intertwined recombination intermediates.
To gain insight into the nature of lethal recombination intermediates eliminated by Srs2, we searched for mutants that suppress the sensitivity of srs2Δ cells to the radiomimetic DNA alkylating agent methyl methanesulfonate (MMS). This screen was designed to select against mutations in genes that are essential for HR because they generally confer high sensitivity to this drug. Yet we found an allele of RAD52 (rad52-L264P) that can completely suppress sensitivity to MMS in srs2Δ cells. Our extensive analysis indicates that rad52-L264P suppresses defects in srs2Δ cells associated with presynaptic Rad51 filaments rather than with the resolution of recombination intermediates. This suppression is related to a modulation of Rad52 mediator activity. We also show that, like rad52-L264P, Rad52 sumoylation leads to the suppression of srs2Δ cells defects, suggesting that sumoylation modulates Rad52 mediator activity.
Results
A novel allele of RAD52 bypasses the requirement of SRS2 for resistance to DNA damage
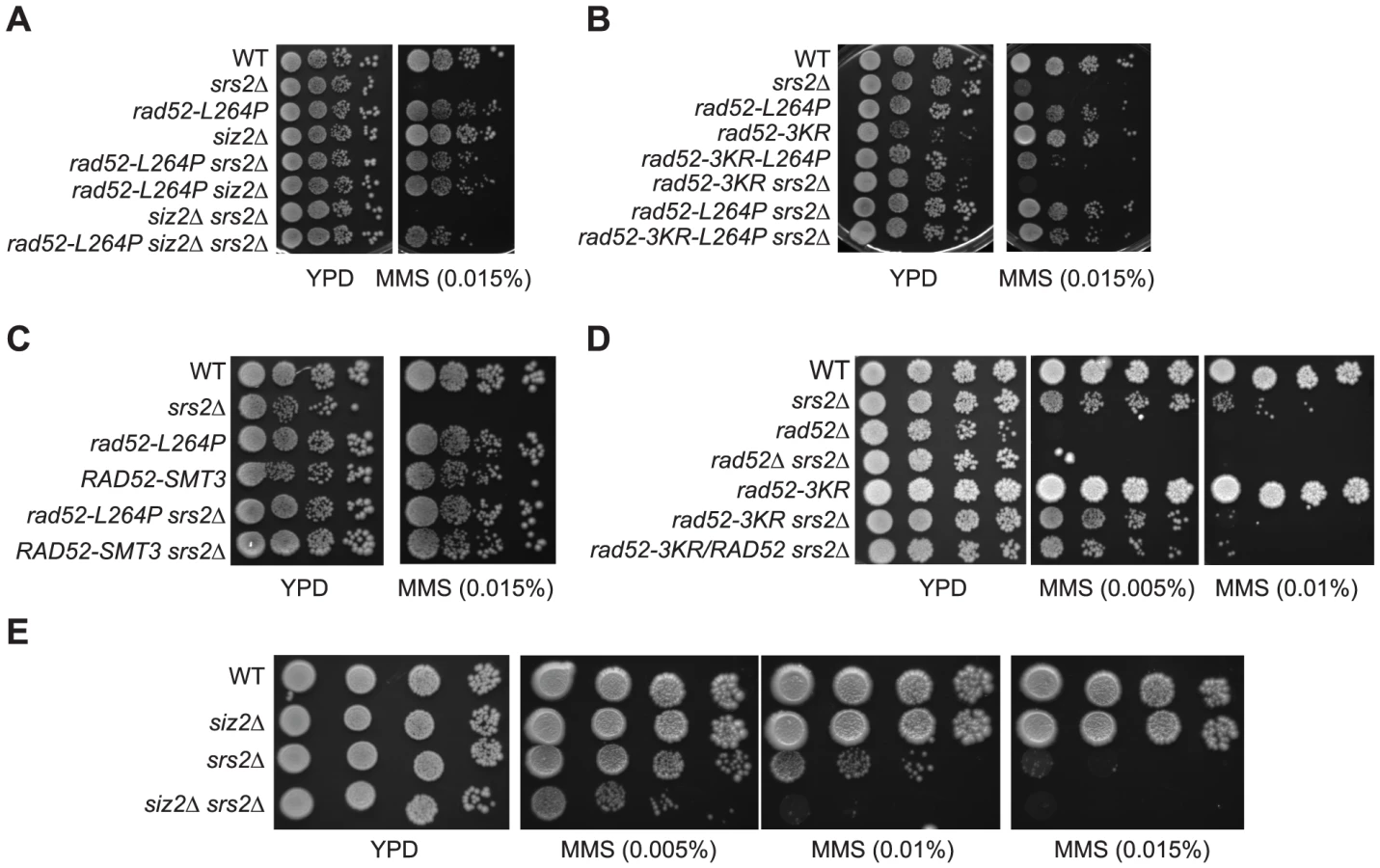
To characterize factors involved in the formation of toxic recombination intermediates eliminated by Srs2, we selected mutants that suppress the MMS sensitivity of srs2Δ haploid cells by plating them on rich medium containing 0.015% MMS. Several well-grown colonies were isolated and backcross analyses showed that the suppressor mutations responsible for MMS resistance were all monogenic. Genetic analyses showed that they affected different genes. We genetically mapped one of these mutations at 25 cM from the PIF1 gene. Around this position, we found RAD52 to be a good candidate since it was involved in DNA repair. The sequence of the RAD52 gene showed a single T to C transversion at position 790 of the open reading frame (ORF), leading to a change of leucine 264 to a proline. To confirm that this mutation is solely responsible for the phenotype, directed mutagenesis was used to create an integrative plasmid which was introduced by gene replacement in a new srs2Δ strain using the pop-in pop-out technique. MMS sensitivity suppression was equivalent to that observed in the original suppressed strain (Figure 1A).

Rad52 is composed of a highly conserved N-terminus that forms ring structures [34], [35]. Outside the N-terminus, the protein is less conserved. Sequence alignments show that L264 belongs to a stretch of well-conserved amino acids (positions 261–283) in Hemiascomycetes, located outside the highly conserved N-terminus (Figure 1B). L264 is located ten residues upstream of the QDDD motif essential for RPA binding [36], (Figure 1B). Note that while rad52-L264P is only slightly sensitive to MMS (Figure 1A), rad52-Q275A/D276A/D277A/D278A is a null allele [36].
We wondered if the change to a proline and not the loss of the leucine itself was significant for the suppression phenotype. Therefore, we replaced the leucine with an alanine by directed mutagenesis. We found that rad52-L264A suppresses the MMS sensitivity of srs2Δ cells, but not as well as rad52-L264P (Figure 1A). Therefore, even if changing the leucine to a proline has a more radical effect, probably because it affects the domain more extensively, it is the leucine ablation that confers the suppression phenotype.
rad52-L264P cells are proficient in HR
To characterize the effect of the mutation on Rad52 activities, we constructed a rad52-L264P single mutant strain. rad52-L264P is not sensitive to incubation at 16°C or 37°C (Figure S1A) and, unlike the deletion of RAD52, it does not affect the rate of spontaneous mutagenesis in the CAN1 gene (Figure S1B). The growth rate is also unchanged compared with WT (90 minutes), while the deletion of RAD52 leads to a significant increase of the doubling time (155 minutes). Surprisingly, cells carrying this mutation are strongly resistant to MMS, while rad52Δ makes cells extremely sensitive (Figures 1A and 9D). rad52-L264P cell resistance to γ-ray and to UV is also comparable to that of WT cells (Figure 2A and B). UV-induced recombination between his7-1 and his7-2 heteroalleles in rad52-L264P homozygous diploid cells is indistinguishable from that of WT cells (Figure 2D), suggesting that gene conversion is not affected by this mutation. Altogether, these data show that rad52-L264P does not substantially affect the activity of the protein.

UV irradiation and γ-ray sensitivities as well as the UV-induced hyper-recombination phenotype of srs2Δ are suppressed by rad52-L264P
rad52-L264P completely suppresses srs2Δ haploid cell growth defect on MMS plates (Figure 1A). It also suppresses srs2Δ γ-ray and UV sensitivities (Figure 2A and B). UV induces mostly single strand gaps, while γ-ray and MMS produce also DSBs. Therefore, the deadly recombination intermediates induced by both kinds of lesions in srs2Δ cells are not toxic or are not formed in the rad52-L264P srs2Δ background.
rad52-L264P also completely suppresses the previously described UV-induced srs2Δ hyper-recombination phenotype [33], [37]. Frequencies of UV-induced recombination in diploid cells measured between his7-1 and his7-2 heteroalleles are the same in rad52-L264P srs2Δ and in WT cells (Figure 2D).
Interestingly, the UV sensitivity and the hyper-recombination phenotype of srs2Δ/srs2Δ diploid cells were only partially suppressed when rad52-L264P and RAD52 alleles were co-expressed in comparison with homozygous rad52-L264P diploids (Figure 2E). Therefore, the WT and rad52-L264P alleles of RAD52 are co-dominant, which implies that rad52-L264P cannot be a simple hypomorphic allele. The Rad52-L264P protein mediates the formation of enough functional Rad51 filaments for HR and DNA repair to occur without leading to accumulation of deadly recombination intermediates in srs2Δ cells.
rad52-L264P suppresses srs2Δ synthetic sickness or lethality with DNA repair and replication mutants
Deleting numerous genes involved in DNA replication or recombination can also induce the formation of lethal recombination intermediates in the absence of Srs2. The deletion of those genes reveals negative interactions with srs2Δ in a RAD51-dependent manner. Some of these genes, like RAD50 and RAD54, are involved in the normal maturation process of recombination intermediates [22], [27], [38]. Another set of genes is involved in DNA replication. Among them are RRM3 [39], MRC1 and CTF18 [23]. The srs2Δ mutation is also synthetically lethal with sgs1Δ, but Sgs1 is involved in recombination and potentially in replication. Therefore, it is not clear which function of Sgs1 is required to avoid srs2Δ death [40]. We wondered if some of these negative interactions would be suppressed by rad52-L264P. A rad52-L264P srs2Δ strain was crossed with strains containing deletions of the genes interacting negatively with srs2Δ (Figure 3A). Interestingly, tetrad analysis showed that all the negative interactions we tested were suppressed by rad52-L264P. Triple mutant strains' doubling times ranged from 92 to 147 min (Table S1), indicating that even if barriers to replication might persist in some background, the suppression is rather strong. We conclude from these experiments that there is a feature common to toxic recombination intermediates eliminated by Srs2 in the different recombination and replication mutants as well as in cells exposed to DNA-damaging agents. Therefore, the toxicity of these intermediates disappears in rad52-L264P srs2Δ cells or, alternatively, the intermediates themselves are not formed.

rad52-L264P suppresses srs2Δ defects associated with presynaptic Rad51 nucleoprotein filament formation
The defects associated with srs2Δ in haploid cells can be suppressed by rad52-L264P. However, resistance to γ-ray and UV irradiation is only partially restored in srs2Δ rad52-L264P homozygous diploids (Figure 2C and D). It has been proposed that the higher sensitivity of srs2Δ homozygous diploids compared with haploids is related to the resolution of specific interactions between homologous chromosomes, probably related to HR [33]. Thus, rad52-L264P would not suppress srs2Δ deficiency in the resolution of recombination intermediates involving homologous chromosomes. This would also mean that the important role of Srs2 in DNA damage resistance in haploid cells would not be related to the resolution of recombination intermediates. We propose rather that the defect in Srs2 observed in haploid cells is related to the formation of Rad51 filaments that are toxic because they do not achieve strand invasion (because they are deficient or because a homologous sequence is not available to perform strand invasion) and cannot be removed from ssDNA. These filaments can be defined as toxic presynaptic Rad51 filaments. Conversely, the fraction of srs2Δ sensitivity that cannot be suppressed by rad52-L264P in diploid cells may be related to a “postsynaptic” role of Srs2. To test this hypothesis, we took advantage of two recombination systems allowing the repair of a single DSB created by a galactose-inducible HO endonuclease. Both systems require Srs2 to survive the formation of the DSB. The first one involves SSA between direct repeats located 25 kb apart (Figure 4A) and the other one uses ectopic gene conversion to repair the DSB (Figure 4D) [28], [29]. Srs2 is required in SSA to remove Rad51 accumulating on ssDNA generated from DSB processing [41], while during ectopic gene conversion srs2Δ cells fail to properly resolve recombination intermediates [29]. According to our hypothesis, rad52-L264P should only suppress the poor viability of srs2Δ cells after HO induction in the SSA system. This is exactly what we observed: rad52-L264P suppressed srs2Δ low cell viability strongly in the SSA system (Figure 4B), but only marginally in the gene conversion ectopic system (Figure 4E). Monitoring of DSB repair in both systems by Southern blot analysis showed that rad52-L264P restored a normal level of SSA product formation in srs2Δ cells (Figure 4A). However, the gene conversion products in the ectopic system did not accumulate as much as in WT cells (Figure 4D). Note that the survival rates after DSB induction in the SSA and the ectopic systems were largely unchanged in rad52-L264P cells in comparison to WT cells (Figure 4B and E). Moreover, DSB repair analysis by Southern blotting showed that the kinetics of repair in both systems were unaffected by rad52-L264P (Figure 4A and D).

We conclude that the rad52-L264P mutation only bypasses srs2Δ defects associated with presynaptic Rad51 nucleoprotein filament formation. This finding implies that rad52-L264P cannot suppress the high level of CO observed in srs2Δ cells either [29], [30]. Southern blot quantification (Figure 4D) confirmed the increased amount of CO in srs2Δ cells (18%) compared with WT cells (5%). As expected, the amount of CO in rad52-L264P srs2Δ cells was still very high (13%). To confirm this result, we used the arg4 ectopic recombination system described by Robert et al. [30]. We found CO in 64% of ARG+ recombinants in rad52-L264P srs2Δ cells in this background, a value comparable to the 60% measured in srs2Δ cells (Escartin F, De Cian A, Coïc E, Gilquin B, Le Cam E, Veaute X, unpublished data). Altogether, these results show that the genetic interactions between srs2Δ and rad52-L264P are related to Rad51 filament formation and not to postsynaptic resolution of intertwined recombination intermediates.
Persistence of DNA repair checkpoint induced by DSB formation in srs2Δ cells is suppressed by rad52-L264P
Elimination of the checkpoint response in the mec1Δ sml1Δ derivative of the srs2Δ strain suppresses, like rad52-L264P, the poor viability related to the formation of the DSB in the SSA system [28]. As a consequence, we wondered if the Rad52-L264P mutant protein could somehow lower the persistence of the checkpoint related to the maintenance of extensive ssDNA formed around the DSB before repair could take place. The analysis of Rad53 by western blot (Figure 4C) shows that it is similarly phosphorylated two hours after HO induction in both rad52-L264P and WT strains. We confirmed that this modification disappears after 12 hours in WT and rad52-L264P cells and persists in srs2Δ cells even 24 hours after DSB formation. It was previously shown that the persistence of the checkpoint activation in srs2Δ cells is dependent on Rad51 [28] because the nucleofilament protects ssDNA from degradation [41]. rad52-L264P suppresses the checkpoint activation in srs2Δ cells as shown by the similarity of the Rad53 phosphorylation kinetics in rad52-L264P srs2Δ compared with WT. This implies that the Rad51 filaments formed by the mutated Rad52 protein allow the checkpoint to be turned off, even in the absence of Srs2. This result strengthens our conclusion concerning the involvement of rad52-L264P in the suppression of srs2Δ defects in presynaptic Rad51 filament management and suggests that Rad52-L264P-mediated Rad51 filaments are different from Rad52-mediated filaments.
During ectopic gene conversion, the srs2Δ mutant defect is associated with a slower disappearance of Rad53 phosphorylation. The retarded bands decrease in intensity after 8 hours in WT cells compared with 12 hours in srs2Δ cells (Figure 4F). This slower recovery is not suppressed by rad52-L264P, confirming the absence of suppression of srs2Δ defects in this system.
rad52-L264P suppresses srs2Δ low cell viability in the SSA system through subtle changes in Rad52 mediator activity
We carried out chromatin immunoprecipitation (ChIP) experiments to study the recruitment of RPA, Rad52 and Rad51 to ssDNA in order to monitor Rad51 filament formation in cells that express Rad52-FLAG or Rad52-L264P-FLAG. We used the SSA assay described above because DSB repair requires the formation of long ssDNA tails [28], [29]. This allowed us to follow Rad51 filament formation on ssDNA for a long period of time. Quantitative PCR was carried out using primer sets that amplify DNA at 0.6 kb and 7.6 kb upstream of the DSB site during a time-course experiment to follow DSB induction. We found an increase in the relative enrichment of RPA, Rad52-FLAG and Rad51 at the site of DSB formation compared to the uncut control ARG5,6 locus, indicative of the formation of ssDNA and subsequently of Rad51 filaments (Figure 5). The increase of RPA binding was higher in Rad52-L264P-FLAG than in Rad52-FLAG expressing cells at positions 0.6 kb and 7.6 kb upstream of the DSB site. The highest RPA increase in Rad52-L264P-FLAG expressing cells (2-fold in comparison to what is detected in Rad52-FLAG expressing cells) was observed four hours after HO induction and was associated with a decrease in Rad52-L264P-FLAG and Rad51 binding at the same time point (2 - and 3-fold, respectively, in comparison to Rad52-expressing cells, Figure 5). However, Rad51 binding was still 30-fold higher than the enrichment value observed at the ARG5,6 locus without DSB. The lower Rad51 recruitment in Rad52-L264P-FLAG expressing cells might be caused by a reduced mediator activity of this Rad52 mutant. Alternatively, it might be the result of a modification of the Rad51 filament properties. For example, their average length could be shorter in Rad52-L264P-FLAG than in Rad52-FLAG expressing cells.

Srs2 removes Rad51 filaments differently according to the distance from the 3′-end of ssDNA tails
We also observed that the recruitment of RPA, Rad52 and Rad51 was lower at position 7.6 kb than at position 0.6 kb in Rad52-FLAG expressing cells (Figure 5). This could be linked to a limited amount of proteins to cover long ssDNA tails. However, western blot analysis of the cell extracts used for ChIP showed that the protein level of RPA and Rad52-FLAG remained unchanged after DSB formation (Figure S3), whereas the pool of Rad51 increased. Consequently, the lower recruitment of Rad51 at 7.6 kb could be related to a specific activity of Srs2 at this position. Indeed, we found that Rad51 recruitment is of the same order of magnitude at the 7.6 kb and 0.6 kb positions in srs2Δ cells. The finding that Rad51 binding at 0.6 kb was not significantly affected in srs2Δ cells in comparison to WT cells indicates that Srs2 does not influence significantly Rad51 recruitment at position 0.6 kb, while it is very active in eliminating Rad51 filaments at position 7.6 kb. This observation suggests that Rad51 filaments forming far away from the DSB site might be responsible for the death of srs2Δ cells upon DSB formation. Finally, reduced Rad51 binding at 7.6 kb was observed also in rad52-L264P cells and was confirmed in rad52-L264P srs2Δ cells as well. This lower recruitment (or the modification of Rad51 filaments properties) can explain the resistance of such cells to DNA damage.
rad52-L264P does not affect the interaction with RPA, Rad51 and Rad59
We then asked whether the lower recruitment of Rad52-L264P and Rad51 in rad52-L264P cells observed by ChIP could be related to a modification of the interaction of RPA or Rad51 with Rad52-L264P. Indeed, L264 is located just upstream of the QDDD motif essential for RPA binding [36] (Figure 1B). Therefore, it was possible that the rad52-L264P mutation affects RPA binding [42], [43]. We evaluated Rad52-RPA interaction by co-immunoprecipitation experiments. Rad52 was immunoprecipitated with a polyclonal antibody and the precipitated fraction was analyzed on western blots with a polyclonal antibody directed against the RPA complex (Figure 6A). We first observed that the rad52-L264P mutation did not substantially affect the binding to RPA, which was expected for a mutant protein still able to manage DNA repair by HR. However, to make sure that there was no difference between the WT and the mutant protein in binding to RPA, we added increasing salt concentrations to the cell extracts to test the robustness of this interaction. As shown in Figure 6A, the amount of RPA co-immunoprecipitated was equivalent in extracts expressing the WT or the mutant protein. In both cases, the interaction was abolished at 500 mM NaCl. In the same experiment, we looked at the interaction between the mutated Rad52-L264P protein and Rad59 [44] and found no major differences between the WT and the mutant protein at 150 mM NaCl (Figure S2). Since Rad59 interaction is not crucial for Rad52/Rad51-dependent recombination [45], we did not investigate further the effect of rad52-L264P on this interaction. We also measured the effect of rad52-L264P on Rad51 binding [44]. For that purpose, we used Rad52-FLAG and Rad52-L264P-FLAG tagged proteins and found that the mutation did not affect this interaction (Figure 6B). Similarly to what is observed for RPA, the destabilization of the interaction with Rad51 by increasing NaCl concentrations was comparable when using WT Rad52 or Rad52-L264P.

Purified Rad52-L264P is more efficient in Rad51 nucleoprotein filament formation than the WT protein
To characterize the biochemical properties of Rad52-L264P we purified recombinant Rad52-L264P and Rad52 from Escherichia coli (Figure 7A). Using electrophoretic mobility shift assays (Figure 7B), we found that Rad52 binding to ssDNA was not significantly affected by the mutation. Next, we investigated whether the L264P mutation affected Rad52 annealing activity by incubating Rad52-L264P and WT Rad52 with complementary ssDNA strands and monitoring the formation of dsDNA. Again, no significant difference was observed (Figure 7C). This was further confirmed in a reaction where ssDNA was coated first with RPA to reflect the in vivo conditions (Figure 7D). We also observed that Rad52-L264P annealing activity was sensitive to RPA, as previously reported for WT Rad52 [7]. Finally, Rad52-L264P and Rad52 annealing activity were similarly affected by Rad51 filaments and free Rad51 proteins [7] (Figure S4A–C).

To determine whether the L264P mutation has an impact on Rad52 recombination mediator activity, we used a well-established DNA strand exchange system that involves plasmid DNA substrates (Figure 7E). In this system, RPA that is pre-bound to ssDNA partially inhibits strand exchange by Rad51. The addition of Rad52 together with Rad51 increases the formation of products through Rad52 interaction with RPA (reviewed in [4]). We found that Rad52-L264P was approximately 8-fold more efficient than WT Rad52 in catalyzing DNA strand exchange.
As this finding indicates that the mediator activity of Rad52-L264P is modified, we studied more precisely the effect of the L264P mutation on the formation of Rad51 filaments and their stability by using electrophoretic analysis of glutaraldehyde-fixed Cy5-labeled DNA-protein complexes. First, we optimized RPA, Rad51 and Rad52 stoichiometry at 60 mM NaCl to obtain the best Rad52 antagonism of the inhibitory effect of pre-bound RPA on Rad51 nucleoprotein filament formation (Figure S5). In these conditions, Rad52-L264P showed a slightly increased mediator activity in comparison to WT Rad52 (Figure 7F). Challenging Rad51 filament formation with increasing salt concentrations showed that the mediator activity of both WT and mutant Rad52 was very sensitive to salt. Indeed, the addition of only 60 mM NaCl to the reaction (120 mM in total) was sufficient to reduce Rad51 filament formation from 30 to 10% of the protein/DNA complexes. Quantification of Rad51 filament formation at increasing NaCl concentrations confirmed that Rad52-L264P was slightly more efficient than WT Rad52 (the difference was significant at 100 and 120 mM NaCl). However, the salt titration midpoint was the same for both WT and mutant protein (around 90 mM), again indicating that the difference in the mediator activity between Rad52-L264P and Rad52 is minimal. Western blot analysis using an anti-Rad51 polyclonal antibody confirmed the slightly higher mediator activity of Rad52-L264P. Electron microscopy (EM) analysis of the resulting protein-ssDNA complexes confirmed the sensitivity of the reaction to NaCl concentration and the higher mediator activity of Rad52-L264P (Figure 7G). We also measured the stability of the nucleoprotein filaments formed by Rad52 or Rad52-L264P by adding increasing concentrations of NaCl after Rad51 filament formation (Figure S5B) and found no difference. The salt-titration mid-point was around 400 mM.
In addition, western blot analysis using an anti-Rad52 polyclonal antibody revealed that Rad52 remained associated with complete Rad51 filaments (Figures 7F and S5). This association was quite unstable because the addition of 60 mM NaCl after complete formation of filaments resulted in Rad52 dissociation from the complex (Figure S5C). EM analysis of the nucleoprotein complexes formed with Rad52 (at 60 mM NaCl) showed that 55% of complete Rad51 filaments remained associated with the mediator protein (Figures 7F and S5). Rad52 was localized mostly at the end of filaments (75%), but in many cases multiple Rad52 spots were distributed all along the filament. In absence of RPA pre-bound to ssDNA, Rad52 was rarely associated with Rad51 filaments (Figure S5A). Thus, Rad52 spot formation might be dependent on the previous binding of RPA to ssDNA. Rad52 might remain associated with residual RPA bound to DNA between Rad51 molecules. Alternatively, as only a few complete Rad51 filaments are formed in the absence of RPA, Rad52 association might be restricted to complete Rad51 filaments. Our results also show that Rad52-L264P association with Rad51 filaments was increased (74% of complete Rad51 filaments compared with 55% for WT Rad52, Figure 7G and S5), while the proportion of Rad52-L264P spots at the end of filaments was comparable (78%). This increased association of Rad52-L264P together with its higher mediator activity might modify qualitatively Rad51 filament properties and cause the suppression of srs2Δ phenotypes in vivo.
SIZ2 overexpression suppresses the MMS sensitivity of srs2Δ cells via Rad52 sumoylation
In parallel to the search for point mutations suppressing the MMS sensitivity of srs2Δ mutants, we also screened a multi-copy genomic library for high-dosage suppressors. After a second round of selection, plasmids bearing a suppressor were sequenced. Strikingly, we isolated a plasmid carrying the SIZ2 gene (Figure 8A). Siz2 is the only SUMO ligase involved in Rad52 sumoylation after the formation of chemically induced DNA damage (like MMS) [11]. Consequently, we speculated that the stimulation of Rad52 sumoylation was responsible for the suppression conferred by SIZ2 overexpression since our study of rad52-L264P shows that subtle changes in Rad52 activity can bypass the requirement for Srs2 in haploid cells. Indeed, we found that the suppression of the MMS sensitivity of srs2Δ conferred by SIZ2 overexpression is no longer observed in cells bearing a sumoylation-deficient rad52-3KR allele, where the three SUMO acceptor sites, lysines 10, 11 and 220 are replaced by arginines [11] (Figure 8A). As a control, we checked that the rad52-3KR allele was not sensitive to MMS (Figure 9B and D), as previously described [11]. Therefore, the suppression of the MMS sensitivity of srs2Δ cells by the overexpression of SIZ2 occurs through the sumoylation of Rad52.

To check that SIZ2 overexpression truly stimulates Rad52 sumoylation, extracts from cells overexpressing SIZ2 and His-tagged SUMO were subjected to Ni-NTA pull-down experiments under denaturing conditions (Figure 8B). The amount of Rad52 sumoylation was compared to that of cells expressing SIZ2 under normal physiological conditions. Because the FLAG tag we used to detect Rad52 contains a poly-histidine chain, the protein was also retained on the Ni-NTA beads, allowing us to quantify the amount of sumoylated protein relative to the total amount of Rad52. Overexpression of SIZ2 increases the amount of sumoylated Rad52 3-fold compared with its basal expression. This result strengthens our genetic experiments establishing that an increase in the pool of sumoylated Rad52 allows Srs2 activity to be bypassed.
Rad52-L264P behaves as Rad52-SUMO
Since Rad52 sumoylation leads to the suppression of MMS sensitivity of srs2Δ cells, we wondered whether the suppression by rad52-L264P was dependent on an increase in the pool of sumoylated Rad52 induced by the mutation. As shown in Figure 8B, rad52-L264P does not increase Rad52 sumoylation. We also found that the suppression conferred by rad52-L264P was not dependent on SIZ2 since the deletion of this gene in rad52-L264P srs2Δ cells did not affect MMS resistance (Figure 9A). Additionally, a rad52-L264P allele that cannot be sumoylated (rad52-3KR-L264P) still suppresses the MMS sensitivity of srs2Δ mutants (Figure 9B). Altogether, these results clearly show that Siz2-mediated Rad52-L264P sumoylation is not required to bypass Srs2. We also checked if the Rad52-L264P protein was sumoylated by another SUMO ligase. We found that Rad52-L264P MMS-induced sumoylation was still dependent on Siz2 (Figure S6). To reconcile our findings that srs2Δ MMS sensitivity can be suppressed on the one hand by Rad52 sumoylation and on the other hand by rad52-L264P, even if sumoylation cannot occur, we propose that Rad52-L264P behaves as if it is constitutively sumoylated, even when it is not physically modified. This would mean that the mutation has the same effects on Rad52 activities, as does sumoylation. Consequently, cells that constitutively express only sumoylated Rad52 should suppress the MMS sensitivity of srs2Δ cells as strongly as rad52-L264P.
To test this hypothesis, we fused the SMT3 gene, coding for the SUMO modifier, to the 3′ end of the endogenous RAD52 gene, to generate cells expressing only a Rad52 protein bearing a C-terminal SUMO fusion. The part of SMT3 coding for the last three amino acids and the two glycines required for conjugation [46] was removed to avoid subsequent conjugation of SUMO with other proteins. The resulting strain displays only mild MMS sensitivity (Figure 9C), showing that the addition of SUMO does not substantially affect Rad52 activity in DNA repair. srs2Δ cells bearing the RAD52-SMT3 fusion allele were as MMS-resistant as those carrying rad52-L264P. Therefore, in cells that produce only Rad52-SUMO fusion proteins, Srs2 becomes incidental to MMS resistance. We also monitored the effect of the fusion protein on survival of srs2Δ cells in both SSA and MAT ectopic recombination systems (Figure 4B and E). As for Rad52-L264P, the expression of the Rad52-SUMO protein instead of Rad52 can alleviate the strong lethality associated with srs2Δ in the SSA system but not in the ectopic system. Additionally, ChIP analysis showed that Rad52-SUMO behaves as Rad52-L264P. Using FLAG-tagged proteins, we also observed a 2-fold decrease in Rad52-SUMO recruitment to ssDNA compared with Rad52 (Figure 5). This is associated with a 2-fold decrease in Rad51 binding, as we observed for Rad52-L264P. These results reinforce the idea that Rad52 sumoylation affects Rad52 activities in the same way as rad52-L264P.
Since Rad52 sumoylation can bypass the requirement for Srs2 in the avoidance of toxic Rad51 filaments, it would be expected that the expression of the Rad52-3KR protein, which cannot be sumoylated, would lead to a negative interaction with srs2Δ. This is exactly what we observed (Figure 9D). This negative effect is also observed in a siz2Δ srs2Δ double mutant (Figure 9E). It is interesting to note that the negative effect of rad52-3KR in a srs2Δ background is dominant to the RAD52 allele. Indeed, the introduction of the RAD52 allele in a rad52-3KR srs2Δ strain does not increase MMS resistance (Figure 9D). We suggest that Rad52-3KR mediates toxic Rad51 filaments that can be disassembled only by Srs2.
It has been reported that a plasmid carrying rad52-3KR is able to suppress (albeit weakly) rrm3Δ srs2Δ synthetic lethality and sgs1Δ srs2Δ growth defect [11]. However, we found that rad52-L264P suppresses these negative interactions (Figure 3A), while this allele codes for a protein behaving as if it were sumoylated. We confirmed our conclusion by tetrad analysis showing that a single integrated copy of rad52-3KR, or siz2Δ, cannot rescue rrm3Δ srs2Δ and sgs1Δ srs2Δ negative interactions (Figure 3B).
Finally, it has been reported that the Rad52-3KR protein is more prone to proteasome degradation than the WT protein, mostly in cells lacking Srs2 or Rrm3 helicases [11]. Since rad52-L264P makes the protein behave as if it were constitutively sumoylated, it was possible that it could suppress the increased instability of Rad52-3KR. Expression shut-off experiments were performed with different mutation of Rad52 in srs2Δ mutants. We found that the Rad52-3KR-L264P protein was not more stable than Rad52-3KR (Figure S7). It is possible that the decreased stability of Rad52-3KR is related to the substitution of the three targeted lysines and not to the inability of the protein to be sumoylated.
Discussion
A novel allele of RAD52 circumvents the requirement for Srs2 activity in DNA repair
We found a novel allele, rad52-L264P, which suppresses the requirement for Srs2 activity in DNA damage tolerance. A profusion of RAD52 mutants displaying a separation of function has been studied, revealing the multifunctional nature of the protein. Some of them were affected in DSB repair activity but not in spontaneous recombination [34], [47]. Others were differentially affected in the mediator and the ssDNA annealing activity of Rad52 [48]–[50]. Some of these mutants, which were shown to affect Rad52 mediator activity, display a partial resistance to DNA damage [49], [50], but their viability is still reduced by several orders of magnitude. Lastly, some have been shown to alter the choice of the donor template during spontaneous HR [51]. rad52-L264P contrasts with all these mutations because it does not confer any of the rad52Δ null mutation-associated phenotypes (defect in vegetative growth, increased spontaneous mutagenesis, very high MMS and γ-ray sensitivities, large decrease in DSB repair by SSA and gene conversion). In addition, it is not a hypomorphic allele since it is co-dominant when it is combined with the WT allele.
Several alleles of RAD52 that partially suppressed the sensitivity of srs2Δ cells to DNA damage have been previously described [20]. They were all highly defective in DNA repair and HR, but not as much as the null allele. The viability of cells carrying these alleles is reduced 10 - to 20-fold at low doses of MMS, and HR is reduced 10-fold. They were all dominant-negative and, unlike what we observed for rad52-L264P, they have to be combined with the WT allele or overexpressed to suppress only partially the MMS or UV sensitivities of srs2Δ mutants. In addition, none of these RAD52 mutants suppressed both phenotypes, while this is the case for rad52-L264P. It was proposed that these rad52 alleles are able to rescue srs2Δ mutants partially by reducing HR efficiency. Some of these mutations code for C-terminal truncations unable to bind Rad51. Therefore, they could bind ssDNA without forming Rad51 filaments. Others were N-terminal truncations, probably impaired in DNA binding, which suggests that their overexpression resulted in depletion of the protomer pool of Rad51. It has been proposed that both kinds of mutations result in a large decrease in Rad51 filament formation, resulting in a limited suppression of srs2Δ MMS or UV sensitivities. However, rad52-L264P suppresses many defects of srs2Δ haploid cells without displaying any rad52Δ phenotype. Thus, in contrast with previously described alleles, rad52-L264P allows an extensive genetic and molecular investigation of the toxic recombination intermediates formed by Rad52 and eliminated by Srs2 in WT cells.
rad52-L264P specifically bypasses the role of Srs2 in the removal of toxic Rad51 filaments
We found that rad52-L264P cannot overcome the function of Srs2 in the resolution of postsynaptic recombination intermediates, but only those associated with the removal of presynaptic Rad51 filaments. It was reported previously that the Srs2 anti-recombination function in removing toxic Rad51 filaments is genetically separable from its role in promoting the resolution of postsynaptic recombination intermediates, which depends exclusively on Srs2 Cdk1-dependent phosphorylation [52]. Our results are in agreement with this finding. rad52-L264P does not suppress the increased sensitivity to UV and γ-ray irradiation of srs2Δ/srs2Δ diploid cells. It is also unable to suppress the strong lethality associated with ectopic HR in srs2Δ cells. Additionally, it cannot suppress the high rate of CO found in these cells. We propose that these defects are related to the resolution of HR intermediates between homologous or ectopic chromosomes that absolutely requires the helicase activity of Srs2 (Figure 10A). Additionally, our results support the recent finding that the post-synaptic role of Srs2 is to dismantle HJs through its helicase activity [32] rather than by displacing Rad51. However, rad52-L264P perfectly rescues the lethality associated with DSB repair by SSA between distant repeats. In this assay, a Rad51-dependent strand invasion step is not involved in repair [28]. Therefore, Srs2 is required to remove Rad51 filaments forming on ssDNA around the initial DSB to allow proper repair. By extension, our findings suggest that Rad51 filaments are also responsible for srs2Δ defects after γ-ray and UV irradiation and in cells bearing mutations in genes that impair replication or recombination (Figure 10A).

The nature of toxic Rad51 filaments eliminated by Srs2
In all the situations where the requirement for Srs2 can be bypassed by rad52-L264P, we propose that the formation of extensive ssDNA may potentially lead to the establishment of Rad51 filaments that cannot recombine (Figure 10A). These “unproductive” filaments would be a signal to trigger Srs2 translocase activity in WT cells. In the SSA assay, ChIP experiments showed that Srs2 is more active in removing Rad51 at 7.6 kb than at 0.6 kb from the DSB site, thus indicating how Srs2 might interfere with such unproductive filaments in WT cells. Indeed, Srs2 job might be to remove extended rather than short Rad51 filaments located near the 3′-end of the ssDNA tail. The length of 5′-ssDNA resection is in part related to strand invasion. Indeed, it is well documented that the formation of a DSB in a unique sequence results in extended resection [53]. Therefore, the formation of extended Rad51 filaments, which is a potential mark of strand invasion failure, would trigger Srs2 activity. The formation of filaments near the ssDNA end might be strictly controlled, while the lower recruitment of Rad52 at more distant sites might affect the quality of the filaments, triggering Srs2 activity. This would allow DSB repair by SSA to occur when HR is inefficient. In srs2Δ cells, the persistence of extended, toxic Rad51 filaments would lead to DSB repair towards a dead-end.
Likewise, synthetic lethality of srs2Δ with replication mutants, like mrc1Δ, would be related to unproductive Rad51 filament formation. In mrc1Δ cells, uncoupling between DNA synthesis and DNA unwinding by the MCM helicases [54] leads to the formation of complementary ssDNAs on each newly replicated sister chromatid. In the absence of Srs2, Rad51 filaments formed on these ssDNAs would not be productive because of the lack of a dsDNA template on each sister chromatid. Such filaments could impede replication fork restart. Considering that Srs2 is more active on Rad51 filaments not associated with a 3′-end, we suggest that Srs2 might specifically recognize and eliminate Rad51 filaments formed on parental ssDNA at the replication fork because they are not associated with such an end.
The precise nature of toxic recombination intermediates in rad50Δ, rad54Δ or sgs1Δ has yet to be clarified. Rad50 has an essential role in sister-chromatid recombination (reviewed in [55]) and Rad54 is necessary to initiate DNA synthesis after the formation of the D-loop [56], to extend this structure [57] and to remove the dsDNA template nucleosomes [58]. Sgs1 is probably involved in both replication and recombination mechanisms. Sgs1 could help replication progression and could prevent the formation of ssDNA on which Rad51 filaments could nucleate in srs2Δ cells [40]. Furthermore, several lines of evidence suggest that Sgs1 is also involved in the dissolution of dHJ [59], [60] and it was proposed that in sgs1Δ cells Srs2 could prevent the formation of these structures [40]. Therefore, in all these backgrounds, toxic recombination intermediates accumulating in the absence of Srs2 could be presynaptic Rad51 filaments or unprocessed postsynaptic recombination intermediates. However, since rad52-L264P can suppress rad50Δ, rad54Δ and sgs1Δ synthetic growth defect or lethality with srs2Δ, it seems more relevant that these negative interactions are the consequence of the accumulation of ineffective Rad51 filaments.
Finally, γ-ray and UV irradiation would sensitize srs2Δ cells because of the formation of unproductive Rad51 filaments on extensive ssDNA. These would accumulate because of DNA damage-induced fork stalling or unfulfilled DNA repair.
In contrast, rad52-L264P cannot suppress the increased sensitivity to UV and γ-ray irradiation of srs2Δ/srs2Δ diploid cells compared with srs2Δ haploids. Nor can it suppress the inability to repair a DSB by ectopic recombination nor the high level of CO associated with gene conversion of srs2Δ haploid cells. We propose that these defects are related to the resolution of HR intermediates between homologous or ectopic chromosomes which absolutely requires the helicase activity of Srs2 (Figure 10A).
How does Rad52-L264P bypass Srs2?
In vitro experiments showed that Rad52-L264P mediator and strand exchange activities are stronger than those of WT Rad52. This intriguing gain of function of Rad52-L264P is not correlated with any modification of its binding to RPA or Rad51 in vivo. Thus, further biochemical analyses of Rad52-L264P are required to provide valuable insights on the mechanism by which Rad52 mediates Rad51 filament formation. In vivo, other Rad51 filament mediators, which might act with Rad52 as a complex, could be affected by the rad52-L264P mutation. This would explain the reduction in Rad51 binding associated with increased RPA recruitment, measured by ChIP, in rad52-L264P cells, while, in vitro, Rad52-L264P is a better mediator than the WT protein. The evidence that Rad52-L264P restricts the formation of (or shortens) Rad51 filaments without affecting HR efficiency could explain how this mutation can bypass Srs2, not only in SSA, but also after irradiation or in mutants that are synthetically lethal with srs2Δ.
Alternatively, Rad52-L264P could change substantially the properties of Rad51 filaments. Western blot analysis and EM images of Rad51 filaments nucleated in vitro show that Rad52 remains associated with complete Rad51 filaments. This association is weak because Rad52 is released by the addition of only 60 mM NaCl, but it could be stabilized in vivo by chaperone proteins. The presence of Rad52 within the Rad51 filament might have consequences on homology search and strand invasion. Indeed, Rad52-L264P association with Rad51 filaments is increased in comparison to WT Rad52, a result that can be correlated with the more efficient strand exchange activity observed with the mutant protein. Rad52-L264P might also affect Rad51 filament properties in a way that would suppress their potential toxicity in srs2Δ cells. For example, such filaments would not prevent the restart of stalled replication forks, thereby bypassing the need for Srs2.
Sumoylation affects Rad52 in the same way as the rad52-L264P mutation
We also found that Rad52-SUMO fusion protein and Rad52-L264P show a similar ability to avoid or restrict the formation of toxic Rad51 filaments. It was previously reported that Rad52 is sumoylated simultaneously with RPA and Rad59 following treatment with a high dose of MMS [15]. This sumoylation wave might stabilize complexes engaged on their substrates rather than to promote specificity. However, recent work showed that Cdc48 interacts with sumoylated Rad52 and consequently dissociates it from DNA [16]. This interaction might be part of another Rad51 filament formation regulation process, acting in parallel with Srs2 activity. Here, we demonstrate that increasing the pool of sumoylated Rad52 suppresses srs2Δ deficiencies in haploid cells. We thus propose that Rad52 sumoylation might modulate its mediator activity and/or change the properties of the formed Rad51 filaments, maybe through Cdc48 activity, in the same way as Rad52-L264P (Figure 10B).
It seems likely that sumoylation of Rad52 is a conserved process because mono - and disumoylation of human Rad52 were also observed in HEK293T cells [11]. It would be interesting to know if this modification induces changes in Rad51 filament properties in mammals as it does in yeast. In this case, it might be important to explore the genetic interactions between Rad52 sumoylation and the newly characterized anti-recombinase PARI [61], which could be the mammalian Srs2 ortholog. Lastly, it is tempting to compare the potential differences between Rad52 un-sumoylated and sumoylated proteins in yeast and those of Rad52 and BRCA2 in metazoans. Recently, it was shown in human cells that these two proteins co-exist as mediators of the Rad51 filaments [62]. Even if the roles of Rad52 in recombination in yeast and metazoans are certainly different, it is possible that in both cases different Rad51 filaments are nucleated.
Materials and Methods
Plasmids
For integration into the yeast genome, the rad52-L264P allele was cloned into the Yiplac211 integrative plasmid. rad52-L264P was PCR amplified from genomic DNA with primers containing restriction sites suitable for cloning. The digested PCR product was ligated into the EcoRI and HindIII sites of Yiplac211 to give YipLac211-rad52-L264P. YipLac211-rad52L264A was made by directed mutagenesis from YipLac211-rad52-L264P (Phusion Site-Directed Mutagenesis kit, Finnzymes) with primers changing the CCC codon coding for P264 to a GCC codon coding for A264. The rad52-L264P mutation was introduced in the same way into pYI211::Kan-rad52-K10,11,220R (D2535, provided by S. Jentsch) with primers changing the CTC codon coding for L264 to a CCC codon coding for P264. Yep181-CUP-His7-Smt3 [63] was used to overexpress His7-Smt3 in cells in order to immunoprecipitate SUMO-conjugated proteins. To create a fusion between RAD52 and SMT3, the SMT3 ORF was fused to the 3′ intergenic sequences of RAD52 in a vector bearing the NATMX cassette coding for the resistance to clo-NAT (pAG25, [64]). First, the SMT3 ORF was PCR amplified from FF18733 genomic DNA with primers suitable for cloning. The last three amino acids and the two glycines required for conjugation [46] were removed to avoid subsequent interaction of SUMO with other proteins. The natural SMT3 stop codon was conserved. The PCR product was ligated into the HindIII and BamHI sites of pAG25 resulting in pAG25-SMT3. This construct created a NheI restriction site just 5 bp before the BamHI sites. The RAD52 3′ intergenic sequence was then ligated into the NheI and BamHI sites of this plasmid, generating pEC54. pSIZ2 is a subclone of a plasmid suppressing the MMS sensitivity of srs2Δ cells isolated from an overexpression library built for this study.
Yeast strains
Strains used in this study are listed in Table S2. Experiments were mostly conducted in the FF18733 background. Diploid cells used in survival and recombination assays were the result of crosses between two different cell backgrounds: FF18733 and FF18985 in order to monitor HR between the his7-1 and his7-2 alleles. All the deletion mutants were constructed by the one-step gene disruption method [65]. Multiple mutant strains were derived from meiotic segregants from FF18733 or FF18985 isogenic diploids. Mutations rad52-L264P and rad52-L264A were introduced into yeast cells with the pop-in pop-out technique using the integrative plasmids Yiplac211-rad52-L264P and Yiplac211-rad52-L264A. The non-sumoylable rad52-3KR and rad52-3KR-L264P alleles were targeted at the URA3 locus by transformation of the integrative plasmids pYI211::Kan-rad52-K10,11,220R (D2535 [11]) and pYI211::Kan-rad52-K10,11,220R-L264P. The SMT3 gene, coding for the SUMO radical, was fused in vivo to the 3′ end of the RAD52 gene. The insert of pEC54, containing the SMT3 ORF and a NATMX resistance cassette, was PCR amplified with primers designed to introduce SMT3 in phase with RAD52, without affecting the intergenic sequences surrounding RAD52. Transformants were selected on clo-NAT containing medium and checked by colony PCR. The production of the fusion protein was checked on western blot (Figure S8). The strain bearing the Rad59-9xMYC fusion protein was made using the method described in [66] in the FF18733 background. The Rad52-FLAG strains were constructed as previously described in the same background [67].
Sequence alignment
Homologous sequences of S. cerevisiae Rad52 were retrieved using PSI-Blast searches against the nr database [68], [69]. A multiple sequence alignment of the full-length sequences of these homologs was obtained using Muscle software [70]. However, within the C-terminal disordered tail the algorithm did not satisfactorily align the small linear motifs surrounding L264 and the alignment had to be manually refined. Propensities to adopt secondary structures were estimated using PsiPred restricting the alignments to subsets of species such as those from the Hemiascomycetes group [71]. The final alignment was represented using Jalview [72].
Irradiation and induced recombination
UV irradiation was performed using a 264 nm source delivering 1 J/m2/s. γ-ray irradiation was performed using a 137Cs source at a dose of 50 Gy/min. Cells growing exponentially were plated at appropriate dilutions on rich medium (YPD) and synthetic plates. Survival was determined as the number of cell-forming colonies on YPD at a given dose divided by the number of non-irradiated colonies. We determined HR frequencies by dividing the number of recombinant colonies growing on selective medium by the number of unselected colonies subjected to the same dose of irradiation. The values obtained after irradiation were corrected by subtracting the number of spontaneous recombinants present on the non-irradiated plates.
Measurement of spontaneous mutation rates
Spontaneous formation of canavanine-resistant colonies was quantified by a fluctuation test based on a minimum of 27 independent cultures of each strain, initiated from approximately 200 cells and grown to saturation [73].
Survival following DSB formation
Cells were grown overnight in liquid culture containing lactate before plating. Survival following HO-induced DSB was measured as the number of cells growing on galactose-containing medium divided by the number of colonies growing on YPD. The results shown are the average of at least 3 independent experiments.
Physical analysis of HO-induced SSA and ectopic gene conversion
Cells were grown in YPD until late exponential phase. Cells were then used to inoculate 400 ml of YPLactate. Cultures were grown to a concentration of 5 to 10×106 cells/ml. A 50 ml sample was removed for the 0 hour time-point and then galactose was added to a final concentration of 2%. Incubation was continued and 50 ml samples were removed at given times. Cells were harvested by centrifugation and washed with water. Cell pellets were then frozen at −20°C. DNA was extracted from the thawed cell pellets and digested accordingly. DNA fragments were separated by electrophoresis on 0.8% agarose gels, transferred to nylon membranes and hybridized with a suitable radioactive probe (Ready-prime II, GE Health Care). Blots were analyzed by using a Typhoon 9600 phosphorimager (GE Health Care) and quantified with ImageQuant Software. The amount of product in the ectopic gene conversion system was measured at 10 hours to avoid the over-estimation of srs2Δ cells that had completed repair. The checkpoint is turned off after the completion of repair in this system and cells resume growth. This repeatedly distorts the quantification at 24 hours. However, in the SSA system, we quantified the amount of repair at 24 hours because the reaction was far from complete at 10 hours (the first products appear at 6 hours) and the persistence of the checkpoint impedes srs2Δ cell growth.
Measurement of CO rate in the arg4 ectopic recombination system
We used the system described in [30]. However, we used a colony-PCR assay to detect the CO among ARG4 recombinant colonies (Escartin F, De Cian A, Coïc E, Gilquin B, Le Cam E, Veaute X, unpublished data). Briefly, we used primers allowing the discrimination of the parental configurations of the arg4 locus on chromosome VIII from the reciprocal translocation produced by the CO associated with gene conversion.
Western blot analysis of Rad53 phosphorylation
Cells were harvested during the time-course experiments previously described for the physical analysis of HO-induced SSA and ectopic gene conversion. Protein extracts were prepared by trichloroacetic acid precipitation. Proteins were separated on 10% SDS-PAGE with an acrylamide/bisacrylamide ratio of 30∶0.4, for 2 hours at 150 V and transferred to nitrocellulose membrane. Membrane was incubated overnight with a goat polyclonal antibody raised against the C terminus sequence of Rad53 (Santa Cruz Biotechnology, yC-19) at a 1/1300 dilution in PBS, 0.1% Tween, 5% milk (w/v); then incubated for 1 hour with secondary horseradish peroxidase-conjugated anti-goat antibody (Santa Cruz Biotechnology, Sc-2020) at a 1/5000 dilution in the same buffer. The blot was revealed by chemiluminescence (ECL Plus, GE Healthcare).
ChIP experiments and quantitative PCR analyses
Samples were collected during the same time-course experiment performed to monitor the physical analysis of HO-induced SSA (see below). ChIP was carried out as previously described with minor modifications [74]. Samples were incubated with 2 µg of rabbit anti-RPA polyclonal antibody (a gift from V. Géli), of mouse anti-FLAG monoclonal antibody (Sigma) or of rabbit anti-Rad51 polyclonal antibody (Santa Cruz Biotechnology). 50 µl of Magnetic Dynabeads Protein A (Invitrogen) was added to each sample when treated with rabbit antibodies and 50 µl of Magnetic Dynabeads Pan mouse otherwise. After washes, elution of the proteins and reversal of crosslink, samples were treated with proteinase K followed by purification of the DNA with QIAquick PCR purification kit (Qiagen). Quantitative PCR reactions of 180 bp fragments at 0.6 kb or 7.6 kb proximal to the DSB site and at the ARG5,6 locus were performed using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) on an Eppendorf Realplex system.
Co-immunoprecipitation
Yeast cells were grown in YPD medium to a concentration of 2.5×106 cells/ml. Cells were harvested and washed twice with PBS. Extracts were prepared as previously described [75] without DNAse treatment. The whole cell extract (1 mg) was incubated for 1 hour at 4°C, either with a rabbit anti-Rad52 polyclonal antibody (a gift from S. Jentsch's lab), or with 1 µg of a rabbit anti-Rad51 polyclonal antibody (Santa Cruz Biotechnology). Then, 50 µl of Dynabeads coupled to Protein A (Invitrogen) was added, and the incubation was continued for another hour. The immunoprecipitates were washed twice with 1 ml of lysis buffer and resuspended in 30 µl of Laemmli buffer. The eluted proteins were analyzed by western blot. Proteins were separated on 10% SDS-PAGE and transferred to Hybond-C super membrane (Amersham Biosciences). Proteins were detected with rabbit anti-Rad52 polyclonal antibody (1/2000), rabbit anti-RPA polyclonal antibody (a gift from V. Géli, 1/2500), mouse anti-MYC monoclonal antibody (Sigma, 1/1000), mouse anti-FLAG monoclonal antibody (Sigma, 1/10000) and rabbit anti-Rad51 polyclonal antibody (1/2000). Blots were then incubated with a secondary antibody: horseradish peroxidase-conjugated goat anti-mouse antibody or horseradish-peroxidase-conjugated goat anti-rabbit antibody (GE Healthcare 1/10000). Protein-antibody complexes were visualized by enhanced chemiluminescence using the GE Healthcare ECL Plus system.
Protein purification
RPA was purified from the protease-deficient yeast stain BJ5496 (ura3-52, trp1, leu2Δ1, his3Δ200, pep4::HIS3, prbΔ1.6R, can1). Cells were transformed with three plasmids containing the RFA1, RFA2, or RFA3 ORF under the control of a GAL promoter (a gift from R. Kolodner). The RPA heterotrimer was purified as described [76]. Rad51 was overexpressed in E. coli BL21 (DE3) pLysS cells transformed with the pEZ5139 plasmid (provided by S. Kowalczykowski) and then purified as described previously [77]. Rad52 and Rad52-L264P were purified from BRL (DE3) pLysS cells transformed with the pET15b-Rad52 or pET15b-Rad52-L264P plasmid. Cells were grown in 8-liters of LB broth containing 100 µg/ml ampicillin at 37°C until A600 = 0.8. Protein expression was induced by addition of 0.1 mM IPTG followed by incubation at 30°C for 3 h. Cell lysis was carried out in 50 mM MES (pH 6.5), 450 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol, 1 mM AEBSF, 10 mM Benzamidine and 2 mM Pepstatin by sonication. Proteins were purified as described previously [78] until the hydroxyapatite column step. Fractions containing Rad52 or Rad52-L264P were pooled and precipitated with 0.45 g/ml ammonium sulfate. Pellets were suspended in 20 mM Tris HCl pH 7.5, 1 M NaCl, 1 mM DTT, 1 mM EDTA and 10% glycerol and then loaded onto Superdex 200 columns (24 ml). Peak fractions were diluted 20 times to obtain a final concentration of 50 mM NaCl and then loaded onto Resource S columns (1 ml). Fractions containing purified Rad52 or Rad52-L264P were pooled, diluted to a final concentration of 200 mM NaCl and finally concentrated using Amicon Ultra 3000 ultrafiltration devices (Millipore). Rad52 and Rad52-L264P concentrations were determined using an extinction coefficient of 2.43×104 at 280 nm.
Electrophoretic mobility shift assay
Increasing amounts of Rad52 or Rad52-L264P (Figure 7B) were incubated with 0.27 µM 5′ end-Cy5-labeled XV2 oligonucleotide (5′-TGG GTG AAC CTG CAG GTG GGC AAA GAT GTC CTA GCA ATG TAA TCG TCA AGC TTT ATG CCG TT-3′) in buffer E (10 mM Tris-HCl pH 8, 5 mM MgCl2, 100 mM NaCl) at 37°C for 10 min. Complexes were separated on 8% native polyacrylamide gels.
DNA annealing
Primary DNA annealing reactions (Figure 7C) were carried out using the same Cy5-labeled XV2 oligonucleotide as for the electrophoretic mobility shift assay and a reverse-complement oligonucleotide (XV98). Each primer (340 nM nucleotides) was resuspended in buffer E and then mixed (time 0). The annealing reaction was started by adding different concentrations of Rad52 or Rad52-L264P (final volume: 50 µl) and incubation at 25°C. An aliquot of 9 µl was collected every two minutes, transferred into 6 µl of stop buffer (20 µM unlabeled XV2, 0.5% SDS, 0.5 mg/ml Proteinase K) and incubated at 25°C for another 5 min. The effect of RPA and Rad51 on the reaction (Figure 7D and S4) was investigated at 30°C with primers 25 and 26 and the buffers previously described in [7]. RPA or Rad51 (or only storage buffer for control reactions) were incubated with the individual primers for 5 min before mixing the nucleoprotein complexes to start the reaction. All samples were separated on 8% native TBE polyacrylamide gels. Fluorescent signals were revealed with a Typhoon 9400 scanner and quantified with ImageQuant (Molecular Dynamics).
DNA strand exchange reaction
33 µM (nucleotides) viral (+) strand of φX174 DNA were coated first with 1.1 µM RPA by incubation in SEB buffer (42 mM MOPS pH 7.2, 3 mM Mg acetate, 1 mM DTT, 20 mM NaCl, 25 µg/ml BSA and 2.5 mM ATP) in a final volume of 12.5 µl at 37°C for 5 min. Rad51 filament formation was initiated by adding 5.5 µM Rad51 and different amounts of Rad52 or Rad52-L264P (Figure 7D), or storage buffers as controls. Reactions were incubated at 37°C for 15 min. The addition of 33 µM (nucleotides) of PstI-linearized φX174 dsDNA and 4 mM spermidine initiated the strand exchange reaction. After incubation at 37°C for 90 min, samples were deproteinized by addition of 2 µl of 10 mg/ml Proteinase K, 5% SDS solution at 37°C for 10 min and analyzed by electrophoresis (0.8% agarose gels in 1× TAE buffer). Gels were stained with ethidium bromide and protein bands quantified with ImageQuant (Molecular Dynamics).
Purification of the 400 nt-long Cy5-labeled ssDNA fragment used for the salt titration analysis
A 5′-biotinylated 400 bp dsDNA fragment was prepared by PCR using the pBR322 plasmid as template. PCR products were loaded onto HiTrap Streptavidin HP columns (GE Healthcare). The non-biotinylated Cy5-labeled strand was purified by elution with 60 mM NaOH.
Salt titration of protein-DNA complexes
Salt titration of protein-DNA complex formation was performed by first incubating 82.5 nM RPA (1/30 nt) with 2.5 µM ssDNA (5′ end-Cy5-labeled 400 nt-long fragment) in SEB buffer in a final volume reaction of 10 µl at 37°C for 5 min. Increasing concentrations of NaCl were added (Figure 7E), followed by addition of 0.83 µM Rad51 (1/3 nt) and 90.5 nM Rad52 or Rad52-L264P (1/27 nt). After 15 min incubation at 37°C, reactions were stopped with 0.25% glutaraldehyde. 4 µl of 40% sucrose was added to facilitate loading on agarose gel. Nucleoprotein electrophoresis was carried out using 0.5% agarose gels in 1X TAE buffer for 1.5 hours at 150 mA. Fluorescent signals were revealed with a Typhoon 9400 scanners and quantified with ImageQuant (Molecular Dynamics). Western blot analysis was performed after washing the gels twice with transfer buffer (25 mM Tris-HCl, 0.2 M glycine, 0.015% SDS) for 20 min. Proteins were then transferred to PVDF membranes with a semi-dried blotter (Biorad) at 0.8 mA/cm2 for 1.25 hours. Membranes were saturated with PBS, 0.1% Tween, 5% milk for 1 hour. Hybridizations with anti-Rad51, anti-Rad52 or anti-RPA antibodies were performed as described for co-immunoprecipitation experiments. Salt titrations of the protein-DNA complex stability were performed as above, but by first incubating proteins and ssDNA in the presence of 60 mM NaCl at 37°C for 15 min to allow the formation of protein-DNA complexes. Additional NaCl was then added to the indicated final concentrations (Figure S5) and the protein-DNA complexes were incubated at 37°C for another 30 min. Reactions were fixed with 0.25% glutaraldehyde. Nucleoprotein gel electrophoresis and western blot analysis were performed as before.
Electron microscopy analysis
For transmission electron microscopy studies, a fraction of the complex formation reactions was handled as previously described [31]. Positive staining images were taken in order to monitor filament dynamics and formation, whereas negative staining images were taken to obtain structural information on the position of Rad52 (along or at the end of the filaments).
Western blot analysis of Rad52 sumoylation
Rad52 sumoylation was induced in exponential phase (5×106 cells/ml) by the addition of 0.3% of MMS for 3 hours at 30°C. Rad52 proteins were detected by western blotting as described [11] using a rabbit anti-Rad52 polyclonal antibody at 1/2000 dilution (from S. Jentsch's lab).
Ni-NTA pull-down of sumoylated Rad52
Rad52-FLAG cells over-expressing His7-SMT3 were collected in exponential phase (2.5×106 cells/ml). Lysates and Ni-NTA pull-Down of sumoylated proteins were carried out according to [63]. Rad52-FLAG was detected with a mouse anti-FLAG monoclonal antibody at 1/10000 dilution (Sigma) on western blots.
Cycloheximide expression shut-off experiment
Strains were grown to 2.5×106 cells/ml. Expression was shut-off by addition of cycloheximide to a final concentration of 50 µg/ml. For each time-point, 2 ml samples of yeast cells were harvested and protein extracts were prepared [79]. Rad52-FLAG was detected on western blots with a mouse anti-FLAG monoclonal antibody at 1/10000 dilution (Sigma).
Supporting Information
{kind=link}
Zdroje
1. CerbinskaiteA, MukhopadhyayA, PlummerER, CurtinNJ, EdmondsonRJ (2011) Defective homologous recombination in human cancers. Cancer Treat Rev 38 : 89–100.
2. PâquesF, HaberJE (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63 : 349–404.
3. KroghBO, SymingtonLS (2004) Recombination proteins in yeast. Annu Rev Genet 38 : 233–271.
4. San FilippoJ, SungP, KleinH (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 : 229–257.
5. MortensenUH, BendixenC, SunjevaricI, RothsteinR (1996) DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A 93 : 10729–10734.
6. SugawaraN, HaberJE (1992) Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol 12 : 563–575.
7. WuY, KantakeN, SugiyamaT, KowalczykowskiSC (2008) Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem 283 : 14883–14892.
8. LaoJP, OhSD, ShinoharaM, ShinoharaA, HunterN (2008) Rad52 promotes postinvasion steps of meiotic double-strand-break repair. Mol Cell 29 : 517–524.
9. ShiI, HallwylSC, SeongC, MortensenU, RothsteinR, et al. (2009) Role of the Rad52 amino-terminal DNA binding activity in DNA strand capture in homologous recombination. J Biol Chem 284 : 33275–33284.
10. Antunez de MayoloA, LisbyM, ErdenizN, ThyboT, MortensenUH, et al. (2006) Multiple start codons and phosphorylation result in discrete Rad52 protein species. Nucleic Acids Res 34 : 2587–2597.
11. SacherM, PfanderB, HoegeC, JentschS (2006) Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol 8 : 1284–1290.
12. AltmannovaV, Eckert-BouletN, ArnericM, KolesarP, ChaloupkovaR, et al. (2010) Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res 38 : 4708–4721.
13. OhuchiT, SekiM, BranzeiD, MaedaD, UiA, et al. (2008) Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair (Amst) 7 : 879–889.
14. Torres-RosellJ, SunjevaricI, De PiccoliG, SacherM, Eckert-BouletN, et al. (2007) The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9 : 923–931.
15. PsakhyeI, JentschS (2012) Protein Group Modification and Synergy in the SUMO Pathway as Exemplified in DNA Repair. Cell 151 : 807–820.
16. BerginkS, AmmonT, KernM, SchermellehL, LeonhardtH, et al. (2013) Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat Cell Biol 15 : 526–532.
17. SymingtonLS, HeyerWD (2006) Some disassembly required: role of DNA translocases in the disruption of recombination intermediates and dead-end complexes. Genes Dev 20 : 2479–2486.
18. AboussekhraA, ChanetR, AdjiriA, FabreF (1992) Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol 12 : 3224–3234.
19. ChanetR, HeudeM, AdjiriA, MaloiselL, FabreF (1996) Semidominant mutations in the yeast Rad51 protein and their relationships with the Srs2 helicase. Mol Cell Biol 16 : 4782–4789.
20. MilneGT, HoT, WeaverDT (1995) Modulation of Saccharomyces cerevisiae DNA double-strand break repair by SRS2 and RAD51. Genetics 139 : 1189–1199.
21. GangloffS, SoustelleC, FabreF (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet 25 : 192–194.
22. PalladinoF, KleinHL (1992) Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics 132 : 23–37.
23. XuH, BooneC, KleinHL (2004) Mrc1 is required for sister chromatid cohesion to aid in recombination repair of spontaneous damage. Mol Cell Biol 24 : 7082–7090.
24. VeauteX, JeussetJ, SoustelleC, KowalczykowskiSC, Le CamE, et al. (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423 : 309–312.
25. KrejciL, Van KomenS, LiY, VillemainJ, ReddyMS, et al. (2003) DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423 : 305–309.
26. BurgessRC, LisbyM, AltmannovaV, KrejciL, SungP, et al. (2009) Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. J Cell Biol 185 : 969–981.
27. KleinHL (2001) Mutations in recombinational repair and in checkpoint control genes suppress the lethal combination of srs2Δ with other DNA repair genes in Saccharomyces cerevisiae. Genetics 157 : 557–565.
28. VazeMB, PellicioliA, LeeSE, IraG, LiberiG, et al. (2002) Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 : 373–385.
29. IraG, MalkovaA, LiberiG, FoianiM, HaberJE (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 : 401–411.
30. RobertT, DervinsD, FabreF, GangloffS (2006) Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25 : 2837–2846.
31. DupaigneP, Le BretonC, FabreF, GangloffS, Le CamE, et al. (2008) The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell 29 : 243–254.
32. MitchelK, LehnerK, Jinks-RobertsonS (2013) Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9: e1003340.
33. AboussekhraA, ChanetR, ZgagaZ, Cassier-ChauvatC, HeudeM, et al. (1989) RADH, a gene of Saccharomyces cerevisiae encoding a putative DNA helicase involved in DNA repair. Characteristics of radH mutants and sequence of the gene. Nucleic Acids Res 17 : 7211–7219.
34. MortensenUH, ErdenizN, FengQ, RothsteinR (2002) A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics 161 : 549–562.
35. ShinoharaA, ShinoharaM, OhtaT, MatsudaS, OgawaT (1998) Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 3 : 145–156.
36. PlateI, HallwylSC, ShiI, KrejciL, MullerC, et al. (2008) Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. J Biol Chem 283 : 29077–29085.
37. RongL, PalladinoF, AguileraA, KleinHL (1991) The hyper-gene conversion hpr5-1 mutation of Saccharomyces cerevisiae is an allele of the SRS2/RADH gene. Genetics 127 : 75–85.
38. SchildD (1995) Suppression of a new allele of the yeast RAD52 gene by overexpression of RAD51, mutations in srs2 and ccr4, or mating-type heterozygosity. Genetics 140 : 115–127.
39. SchmidtKH, KolodnerRD (2006) Suppression of spontaneous genome rearrangements in yeast DNA helicase mutants. Proc Natl Acad Sci U S A 103 : 18196–18201.
40. FabreF, ChanA, HeyerWD, GangloffS (2002) Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A 99 : 16887–16892.
41. YeungM, DurocherD (2011) Srs2 enables checkpoint recovery by promoting disassembly of DNA damage foci from chromatin. DNA Repair (Amst) 10 : 1213–1222.
42. SugiyamaT, NewJH, KowalczykowskiSC (1998) DNA annealing by Rad52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci U S A 95 : 6049–6054.
43. SongB, SungP (2000) Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J Biol Chem 275 : 15895–15904.
44. DavisAP, SymingtonLS (2001) The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single-strand annealing. Genetics 159 : 515–525.
45. Cortes-LedesmaF, MalagonF, AguileraA (2004) A novel yeast mutation, rad52-L89F, causes a specific defect in Rad51-independent recombination that correlates with a reduced ability of Rad52-L89F to interact with Rad59. Genetics 168 : 553–557.
46. JohnsonES (2004) Protein modification by SUMO. Annu Rev Biochem 73 : 355–382.
47. LettierG, FengQ, de MayoloAA, ErdenizN, ReidRJ, et al. (2006) The role of DNA double-strand breaks in spontaneous homologous recombination in S. cerevisiae. PLoS Genet 2: e194.
48. BaiY, DavisAP, SymingtonLS (1999) A novel allele of RAD52 that causes severe DNA repair and recombination deficiencies only in the absence of RAD51 or RAD59. Genetics 153 : 1117–1130.
49. Boundy-MillsKL, LivingstonDM (1993) A Saccharomyces cerevisiae RAD52 allele expressing a C-terminal truncation protein: activities and intragenic complementation of missense mutations. Genetics 133 : 39–49.
50. KrejciL, SongB, BussenW, RothsteinR, MortensenUH, et al. (2002) Interaction with Rad51 is indispensable for recombination mediator function of Rad52. J Biol Chem 277 : 40132–40141.
51. de MayoloAA, SunjevaricI, ReidR, MortensenUH, RothsteinR, et al. (2010) The rad52-Y66A allele alters the choice of donor template during spontaneous chromosomal recombination. DNA Repair (Amst) 9 : 23–32.
52. SaponaroM, CallahanD, ZhengX, KrejciL, HaberJE, et al. (2010) Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet 6: e1000858.
53. LeeSE, MooreJK, HolmesA, UmezuK, KolodnerRD, et al. (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94 : 399–409.
54. KatouY, KanohY, BandoM, NoguchiH, TanakaH, et al. (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424 : 1078–1083.
55. SymingtonLS (2002) Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66 : 630–70.
56. LiX, HeyerWD (2009) Rad54 controls access to the invading 3′-OH end after Rad51-mediated DNA strand invasion in homologous recombination in Saccharomyces cerevisiae. Nucleic Acids Res 37 : 638–646.
57. RossiMJ, MazinAV (2008) Rad51 protein stimulates the branch migration activity of Rad54 protein. J Biol Chem 283 : 24698–24706.
58. SinhaM, PetersonCL (2008) A Rad51 presynaptic filament is sufficient to capture nucleosomal homology during recombinational repair of a DNA double-strand break. Mol Cell 30 : 803–810.
59. CejkaP, KowalczykowskiSC (2010) The full-length Saccharomyces cerevisiae Sgs1 protein is a vigorous DNA helicase that preferentially unwinds holliday junctions. J Biol Chem 285 : 8290–8301.
60. MankouriHW, AshtonTM, HicksonID (2011) Holliday junction-containing DNA structures persist in cells lacking Sgs1 or Top3 following exposure to DNA damage. Proc Natl Acad Sci U S A 108 : 4944–4949.
61. MoldovanGL, DejsuphongD, PetalcorinMI, HofmannK, TakedaS, et al. (2011) Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI. Mol Cell 45 : 75–86.
62. FengZ, ScottSP, BussenW, SharmaGG, GuoG, et al. (2011) Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A 108 : 686–691.
63. UlrichHD, DaviesAA (2009) In vivo detection and characterization of sumoylation targets in Saccharomyces cerevisiae. Methods Mol Biol 497 : 81–103.
64. GoldsteinAL, McCuskerJH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15 : 1541–1553.
65. ItoH, FukudaY, MurataK, KimuraA (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153 : 163–168.
66. LongtineMS, McKenzieAr, DemariniDJ, ShahNG, WachA, et al. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 : 953–961.
67. De AntoniA, GallwitzD (2000) A novel multi-purpose cassette for repeated integrative epitope tagging of genes in Saccharomyces cerevisiae. Gene 246 : 179–185.
68. AltschulSF, MaddenTL, SchafferAA, ZhangJ, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402.
69. SchafferAA, AravindL, MaddenTL, ShavirinS, SpougeJL, et al. (2001) Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 29 : 2994–3005.
70. EdgarRC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 : 1792–1797.
71. JonesDT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292 : 195–202.
72. WaterhouseAM, ProcterJB, MartinDM, ClampM, BartonGJ (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 : 1189–1191.
73. FosterPL (2006) Methods for determining spontaneous mutation rates. Methods Enzymol 409 : 195–213.
74. SugawaraN, HaberJE (2006) Repair of DNA double strand breaks: in vivo biochemistry. Methods Enzymol 408 : 416–429.
75. Strahl-BolsingerS, HechtA, LuoK, GrunsteinM (1997) Sir2 and Sir4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11 : 83–93.
76. KantakeN, SugiyamaT, KolodnerRD, KowalczykowskiSC (2003) The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J Biol Chem 278 : 23410–23417.
77. ZaitsevaEM, ZaitsevEN, KowalczykowskiSC (1999) The DNA binding properties of Saccharomyces cerevisiae Rad51 protein. J Biol Chem 274 : 2907–2915.
78. NewJH, SugiyamaT, ZaitsevaE, KowalczykowskiSC (1998) Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 391 : 407–410.
79. BelleA, TanayA, BitinckaL, ShamirR, O'SheaEK (2006) Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci U S A 103 : 13004–13009.
80. SungP (1997) Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem 272 : 28194–28197.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A GDF5 Point Mutation Strikes Twice - Causing BDA1 and SYNS2
- Dominant Mutations in Identify the Mlh1-Pms1 Endonuclease Active Site and an Exonuclease 1-Independent Mismatch Repair Pathway
- Eleven Candidate Susceptibility Genes for Common Familial Colorectal Cancer
- The Histone H3 K27 Methyltransferase KMT6 Regulates Development and Expression of Secondary Metabolite Gene Clusters
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy