
The Rad4 ATR-Activation Domain Functions in G1/S Phase in a Chromatin-Dependent Manner
DNA damage checkpoint activation can be subdivided in two steps:
initial activation and signal amplification. The events distinguishing these two phases and their genetic determinants remain obscure. TopBP1, a mediator protein containing multiple BRCT domains, binds to and activates the ATR/ATRIP complex through its ATR-Activation Domain (AAD). We show that Schizosaccharomyces pombe Rad4TopBP1 AAD–defective strains are DNA damage sensitive during G1/S-phase, but not during G2. Using lacO-LacI tethering, we developed a DNA damage–independent assay for checkpoint activation that is Rad4TopBP1 AAD–dependent. In this assay, checkpoint activation requires histone H2A phosphorylation, the interaction between TopBP1 and the 9-1-1 complex, and is mediated by the phospho-binding activity of Crb253BP1. Consistent with a model where Rad4TopBP1 AAD–dependent checkpoint activation is ssDNA/RPA–independent and functions to amplify otherwise weak checkpoint signals, we demonstrate that the Rad4TopBP1 AAD is important for Chk1 phosphorylation when resection is limited in G2 by ablation of the resecting nuclease, Exo1. We also show that the Rad4TopBP1 AAD acts additively with a Rad9 AAD in G1/S phase but not G2. We propose that AAD–dependent Rad3ATR checkpoint amplification is particularly important when DNA resection is limiting. In S. pombe, this manifests in G1/S phase and relies on protein–chromatin interactions.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002801
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002801
Summary
DNA damage checkpoint activation can be subdivided in two steps:
initial activation and signal amplification. The events distinguishing these two phases and their genetic determinants remain obscure. TopBP1, a mediator protein containing multiple BRCT domains, binds to and activates the ATR/ATRIP complex through its ATR-Activation Domain (AAD). We show that Schizosaccharomyces pombe Rad4TopBP1 AAD–defective strains are DNA damage sensitive during G1/S-phase, but not during G2. Using lacO-LacI tethering, we developed a DNA damage–independent assay for checkpoint activation that is Rad4TopBP1 AAD–dependent. In this assay, checkpoint activation requires histone H2A phosphorylation, the interaction between TopBP1 and the 9-1-1 complex, and is mediated by the phospho-binding activity of Crb253BP1. Consistent with a model where Rad4TopBP1 AAD–dependent checkpoint activation is ssDNA/RPA–independent and functions to amplify otherwise weak checkpoint signals, we demonstrate that the Rad4TopBP1 AAD is important for Chk1 phosphorylation when resection is limited in G2 by ablation of the resecting nuclease, Exo1. We also show that the Rad4TopBP1 AAD acts additively with a Rad9 AAD in G1/S phase but not G2. We propose that AAD–dependent Rad3ATR checkpoint amplification is particularly important when DNA resection is limiting. In S. pombe, this manifests in G1/S phase and relies on protein–chromatin interactions.
Introduction
The DNA damage checkpoint is an elaborate signal transduction pathway that monitors the integrity of the DNA, prevents cell cycle progression and promotes appropriate DNA metabolism [1] reviewed in [2]. The DNA damage sensors associated with checkpoint activation define two separate DNA structure-dependent signal transduction cascades. Each pathway engages a phospho-inositol-3 kinase-like protein kinase (PIKK); either the Ataxia Telangiectasia Mutated (ATM) or the Ataxia Telangiectasia and Rad3 related (ATR) kinase [3]. ATM detects DNA double strand breaks (DSBs) by interaction with the Mre11-Rad50-Nbs1 repair complex, while ATR primarily senses single stranded-DNA (ss-DNA) through interactions with RPA. Both ATM and ATR are conserved in the model organisms S. pombe and S. cerevisiae.
For ATR to recognise a DNA lesion, single-stranded DNA (ssDNA) needs to be formed - for example by DNA repair-dependent DNA processing [4] or following the replication machinery encountering the unrepaired lesion [5]. Once ssDNA is generated, it is immediately coated by replication protein A (RPA) (Reviewed in: [6]). Multiple ATR molecules are initially recruited to ssDNA regions via ATRs obligate binding partner, ATRIP, which binds directly to RPA [7], [8]. ATR-ATRIP recruitment to ssDNA-RPA is necessary for “basal” ATR activation, but is insufficient for full checkpoint activation: co-recruitment of a second complex consisting of three PCNA-like proteins, Rad9, Hus1 and Rad1 (known as the 9-1-1 clamp) is also necessary. 9-1-1 is loaded in parallel to ATR recruitment at 5′ ssDNA/dsDNA junctions by the checkpoint clamp loader Rad17-RFC[2–5] [9], [10], [11]. (Figure 1A).

When ATR-ATRIP is first loaded at the site of ssDNA, its “basal” kinase activity promotes phosphorylation of its immediate neighbours, including ATRIP [12], [13], an in trans phosphorylation of a residue within ATR itself, T1989 [14], and the subunits of the 9-1-1 clamp [15], [16]. Dependent on the concomitant recruitment of 9-1-1, a further protein, TopBP1, is recruited [17]. TopBP1 is recruited via an interaction between its BRCT (1+2) domains and a constitutive phosphosphorylation on the C-terminus of Rad9 [18], [19]. Similarly in both yeast systems, Saccharomyces cerevisiae and Schizosaccharomyces pombe, the TopBP1 homologs, Dpb11 and Rad4 respectively, are recruited by the phosphorylation of the C-terminus of Rad9Ddc1 creating a binding site for a pair of BRCT domains (Figure 1A). In S. pombe, the C-terminal phosphorylations occur on Rad9 at residues T412 and S423 [20]. This subsequently recruits Rad4TopBP1 via interaction with BRCT pair (3+4). However, unlike in mammalian cells, T412 and S423 in S. pombe are directly targeted by Rad3ATR in response to its ssDNA/RPA binding and concomitant 9-1-1 loading [16], [20], [21]. Despite these differences, in both S. pombe [20] and mammalian cells [14], Rad4TopBP1 recruitment promotes the formation of a Rad3ATR/9-1-1/Rad4TopBP1 complex (Figure 1A). However, the mode of interaction of Rad3ATR and Rad4TopBP1 within this complex has not been defined in S. pombe.
Mammalian TopBP1 can directly activate ATR-ATRIP both in vitro in the absence of ssDNA/RPA and when over-expressed in cells. TopBP1-dependent ATR activation requires an ATR activation domain (AAD) situated between the 6th and 7th BRCT domains [17] and mutation of a conserved aromatic residue within this unstructured region, W1147, prevents this mode of ATR activation. The AAD contacts a region within the C-terminus of ATR, between the kinase and FATC domains [22], which has been termed the PIKK Regulatory Domain (PRD). Mutation of a conserved PRD residue, K2598, similarly abolishes TopBP1-dependent ATR activation. In both mammalian cells and Xenopus extracts the interaction between TopBP1 and ATR-ATRIP appears to be essential for checkpoint activation in response to replication stress [17], [22], although the initial in trans phosphorylation of ATR on T1989, reportedly essential for full ATR activation, is TopBP1-independent [14].
In the budding yeast model system, the intrinsically disordered C-terminal extension of the TopBP1 homolog, Dpb11TopBP1, contains an AAD which interacts with and activates Mec1ATR via a pair of aromatic residues, W700 and Y735 [22], [23], [24], [25]. Interestingly, in S. cerevisiae, at least two distinct Mec1ATR activation domains have been identified: in addition to the Dbp11TopBP1 AAD, the C-terminal tail of Ddc1Rad9 (S. cerevisiae homolog of the 9-1-1 subunit Rad9) contains an AAD that can directly stimulate Mec1ATR activity in vitro and contributes to checkpoint activation in vivo [11], [26]. The key residues in Ddc1Rad9 required for Mec1ATR activation are W352 and W544. W352 resides on the surface of the PCNA-like domain, while W544 lies within the intrinsically disordered C-terminus. In vivo the Ddc1Rad9 AAD is essential for Mec1ATR activation when S. cerevisiae cells are in G1 [24], [26], while the Ddc1Rad9 AAD acts redundantly with the C-terminal AAD of Dpb11TopBP1 during checkpoint activation in G2. It is proposed that, at least in S. cerevisiae, a minimum of one other protein contains an equivalent AAD (Reviewed in [27]).
We have previously shown that S. pombe Rad4TopBP1 is not required for the Rad3ATR-dependent and DNA damage-dependent phosphorylation of Rad26ATRIP or the 9-1-1 clamp subunits [16], [20], demonstrating that Rad3ATR is active at sites of DNA damage in the absence of activation by the Rad4TopBP1 AAD. However, the presence of Rad4TopBP1 is clearly required to form a robust Rad3ATR/9-1-1/Rad4TopBP1 complex [20], to recruit the Crb253BP1 mediator protein [28], [29] and for Rad3ATR to phosphorylate downstream substrates such as Chk1-S345 [30] and promote robust checkpoint activation.
To further explore the role of Rad4TopBP1 in checkpoint activation in S. pombe we identified and characterised the Rad4TopBP1 AAD. We show that Rad4TopBP1 can interact with Rad3ATR via its AAD and that the AAD contributes to Rad3ATR activation in vivo. We observe that the biological function of the Rad4TopBP1 AAD is most important in G1/S phase, when resection is limited, and that reducing DSB resection in G2 following ionising radiation results in compromised Chk1 phosphorylation in the absence of Rad4TopBP1 AAD function. In order to separate out and study Rad4TopBP1 AAD-dependent Rad3ATR activation we developed a Rad4TopBP1 AAD-dependent lacO-LacI checkpoint activation system for S. pombe and used this to show that Rad4TopBP1 AAD-dependent Rad3ATR activation is also dependent on histone H2A phosphorylation. Consistent with a role for this chromatin modification, mutations in Crb2 that interfere with phospho-binding by Crb2 also decrease Rad4TopBP1 AAD-dependent checkpoint activation. Thus, the Rad4TopBP1 AAD-dependent Rad3ATR activation pathway is chromatin dependent, implying a role in checkpoint amplification and maintenance.
Results
In S. cerevisiae, two aromatic residues, W700 and Y735 were identified as critical for Dbp11TopBP1 AAD activity [24], [25]. We similarly created an alignment of Rad4TopBP1 with the C-terminal tails of a variety of TopBP1 homologs and identified a short sequence encompassing Y599 in S. pombe (Figure 1B, 1C), which we subsequently confirmed as defining the AAD activity of Rad4TopBP1 (see below). To characterise the function of the Rad4TopBP1 AAD, we separately mutated the conserved aromatic amino acid to create strain rad4-Y599R or deleted the minimal conserved motif to create rad4-Δ[595–601].
To establish if the Rad4TopBP1 AAD interacts with Rad3ATR we expressed and purified recombinant Rad4[288–648] (that includes BRCT (3+4) and the unstructured C-terminal tail) as a fusion with GST. Constructs containing a mutation in either the AAD (Y599R), deletion of residues 643–645 encompassing a putative cyclin binding motif (RxL), or mutation of a CDK phosphorylation site (S641A) were similarly purified. Wild-type and all the mutated recombinant proteins, when incubated with yeast extracts, bound to and co-purified the control interacting partner of Rad4TopPB1, Cdc13CyclinB (unpublished data). Wild-type Rad4-GST also co-purified Rad3ATR-Myc from extracts prepared from 3myc-rad3 cells (Figure 1D). In contrast, the amount of Rad3ATR-Myc pulled-down with recombinant AAD-mutated Rad4TopBP1-GST was reproducibly reduced (n = 4). Neither Rad3ATR-myc nor Cdc13CyclinB were pulled-down with a GST-only control. We conclude that, in vitro, this residue of Rad4TopBP1 is part of a Rad4TopBP1 interaction domain for Rad3ATR, consistent with it having the properties of an AAD. We also note that the interaction with Cdc13CyclinB is not abolished by loss of the RxL motif and that mutation of S641 affects the Rad4TopBP1:Rad3ATR interaction in vitro (Figure 1D, lane 4).
rad4-AAD mutants show checkpoint defects associated with S phase
Both rad4-Y599R and rad4-Δ[595–601] displayed normal cell cycle progression (data not shown), indicating that, as is observed in S. cerevisiae [24], the Rad4TopBP1 AAD is not required for unperturbed DNA replication. In response to UV, MMS and HU treatment, rad4-Y599R and rad4-Δ[595–601] cells showed intermediate sensitivity when compared to rad4+ and checkpoint defective rad3Δ (Figure 1E and Figure S1A). To establish if the sensitivity to DNA damage correlated with a defective G2 DNA damage checkpoint, we monitored cell cycle progression after cells were synchronised in G2 and UV-irradiated. Following exposure to 50 Jm-2 (Figure 2A), rad4-Y599R cells displayed premature release from cell cycle arrest (∼20 min earlier than rad4+ after 50 Jm-2). We next monitored the sensitivity and checkpoint response to ionising radiation. rad4-Y599R mutant cells displayed only very mild sensitivity to IR (Figure 2B) and the checkpoint was mildly extended (Figure 2C). We do not know the reason for the slight extension of the G2 delay: no obvious increase in numbers or duration of Rad22Rad52 foci were observed, indicating no significant delay to DSB repair (Figure S1B).

We next examined Chk1 phosphorylation status in rad4+ and rad4-Y599R cells as a surrogate for checkpoint activation (Figure 2D). In response to 200 Jm-2 UV irradiation, asynchronously growing rad4-Y599R cells displayed reduced Chk1 phosphorylation when compared to rad4+ cells, consistent with the partial checkpoint defect observed. Conversely, in response to IR, no significant difference is seen between rad4-Y599R and rad4+. An upstream target of Rad3ATR is the C-terminus of histone H2A [31], [32]. To establish if the UV-specific defect in Rad3ATR-dependent phosphorylation is specific to Chk1, we monitored γH2A formation following either UV or IR treatment (Figure 2E). As was seen for Chk1 phosphorylation, a significant decrease in γH2A is observed in rad4-Y599R cells following UV but not IR treatment when compared to rad4+ cells.
The pattern of DNA damage sensitivity seen for rad4-Y599R cells is consistent with a specific sensitivity within S phase. >70% of fission yeast cells in an asynchronous culture are in G2 and mitosis is followed rapidly by S phase: G1 is extremely short. In response to IR, the G2 DNA damage checkpoint is robustly activated and DNA repair completed before cells pass through mitosis and into S phase [33]. Thus, following IR, relatively few cells replicate damaged DNA. Conversely, the G2 checkpoint is not robustly activated following UV [34] and the majority of UV-irradiated cells pass through mitosis and enter S phase with damaged DNA. To monitor S phase-specific events, we thus examined γH2A induction in cells treated with either hydroxyurea (HU), an inhibitor of ribonucleotide reductase, or Camptothecin (CPT), an inhibitor of topisomerase I (Figure 2F). Consistent with both agents manifesting cytotoxicity in S phase, γH2A levels were significantly reduced when comparing rad4-Y599R with rad4+ cells. Finally, since replication of UV damaged DNA induces Cds1Chk2 activity [35], we monitored the kinase activity of immuno-precipitated Cds1Chk2 following UV irradiation of rad4-Y599R and rad4+ cells (Figure 2G). Cds1Chk2 activation was reproducibly lower for rad4-Y599R, indicating an impaired S phase checkpoint activation (n = 3).
The rad4-AAD mutants are sensitive to DNA damage in S-phase
If the rad4-Y599R mutant is deficient in activation of Rad3ATR in S phase, we would anticipate increased sensitivity to IR within S phase when compared to rad4+ cells. To test this possibility, rad4-Y599R mutant and rad4+ cells where either synchronised in G2 cells using cdc25-22 or in G1 using a cdc10-m17. Following the block, cells were released by reducing the temperature and cell cycle progression was monitored by FACS analysis (Figure 3A, 3B). Cells were irradiated with 50 Gy IR at the times indicated. rad4-Y599R cells showed significant increased sensitivity when compared to rad4+ when irradiated in S phase, but not when irradiated in G2, when S phase is complete (i.e. see Figure 3B). In an equivalent cdc25-22 block and release experiment, we monitored Chk1 phosphorylation and γH2A induction (Figure 3C). Unlike when asynchronous rad4-Y599R cells are irradiated (>70% of such cells are in G2), when rad4-Y599R cells were irradiated in early-mid S phase, Chk1 phosphorylation was moderately reduced for the first 40 minutes after irradiation and γH2A levels were similarly decreased when compared to rad4+. Interestingly, following progression through S phase and into G2 (150 minute time point), Chk1 phosphorylation levels increased significantly in rad4-Y599R cells, although the same was not seen for γH2A levels.

To determine that the use of cdc25-22 synchronisation was not generating an artefact (Cdc25 is an activator of Cdc2-Cyclin B, which itself is required for normal DNA damage responses in G2 [36]), we used the alternative method of synchronisation where cells were arrested in G1 using cdc10-M17 and released directly into S phase (Figure 3D). Unlike IR treatment of asynchronous cultures where equivalent levels of γH2A were observed (Async IR), treatment of rad4-Y599R cells at 30, 60 or 90 minutes after release from arrest resulted in decreased γH2A levels and Chk1 phosphorylation when compared to rad4+ control cells.
LacO array-dependent checkpoint activation in S. pombe requires the Rad4TopBP1 AAD
In S. cerevisiae, co-localisation of two or more checkpoint proteins to arrays of lacO repeats bypasses the requirement for DNA damage in Mec1-mediated checkpoint activation [37]. To establish the role of the Rad4TopBP1 AAD in a what has previously been characterised as an RPA-ssDNA independent system, rad4TopBP1, rad9 and rad3ATR were each fused to a construct encoding GFP, the E. coli lac-repressor (LacI) and a nuclear localization signal (NLS); GFP/LN (Figure 4A). The resulting plasmids express the fusion construct under the control of a thiamine-repressible (nmt41) promoter. We established that each of the fusion constructs were functional by expressing them individually in the corresponding null mutants. Each was able to suppress the DNA damage sensitivity (and for rad4TopBP1, the thermosensitivity) of the appropriate mutant, although for rad4-GFP/LN genotoxin resistance was not restored to wild-type levels (Figure S1C–S1E). When expressed in cells harbouring 256 repeats of the lac operator sequence (lacO) integrated at the ura4+ locus, each fusion protein formed a single nuclear focus. No foci were detected in cells devoid of lacO arrays (Figure 4B).
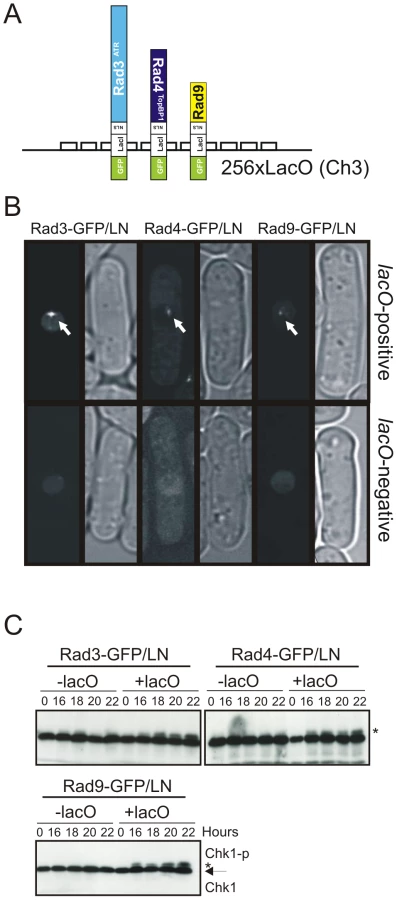
We used Chk1 phosphorylation as a readout for DNA damage checkpoint activation (Figure 4C). Following thiamine removal (induction takes between 12 and 16 hours [38]), Chk1 became phosphorylated in lacO containing cells, but not in lacO-negative control cells, when either Rad3ATR, Rad4TopBP1 or Rad9 LacI fusion proteins were expressed. Similar results were obtained when each pair-wise combinations of two fusion proteins were expressed (Figure 5B). In S. pombe, DNA damage checkpoint activation results in cell cycle arrest and cell elongation. Elongated cells were observed upon expression of single fusion proteins (data not shown), confirming checkpoint activation. From these data we conclude that, in S. pombe, as in mammals [39] tethering of any of these single checkpoint proteins to a lacO array is sufficient to activate the DNA damage checkpoint and that, in contrast to the analogous experiments reported for S. cerevisiae, forced co-localisation of two checkpoint proteins is not required [37].

To establish if the Rad4TopBP1 AAD is involved in this damage-independent mode of checkpoint activation, we tested if Rad3ATR tethering could result in Chk1 phosphorylation in a rad4-Y599R mutant background (Figure 5A). While Rad3-GFP/LN expression resulted in induced Chk1 phosphorylation in rad4+ cells, Rad3-GFP/LN expression did not increase Chk1 phosphorylation in rad4-Y599R cells, demonstrating a role for the Rad4TopBP1 AAD. Next we established if expression and tethering of the AAD-defective Rad4-Y599R protein to lacO arrays was able to activate the checkpoint (Figure 5B). No induction of Chk1 phosphorylation was observed. Furthermore, while co-expression and tethering Rad3ATR and Rad9, or of Rad3ATR and Rad4TopBP1 resulted in checkpoint activation (Figure 5B), we observed that co-expression of Rad3ATR with Rad4TopBP1-Y599R mutant protein did not result in Chk1 phosphorylation. This data suggests that the AAD-defective mutant protein can act as a dominant negative, at least in this specific situation, preventing the endogenous wild-type Rad4TopBP1 from functioning with the tethered Rad3ATR to activate the checkpoint. It also supports the idea that the Rad4TopBP1 AAD domain is required for the activation of Rad3ATR and not simply recruiting it.
H2A phosphorylation is required for Rad4TopBP1 AAD–dependent checkpoint activation
While Rad3ATR kinase activity is essential for Chk1 phosphorylation in response to DNA damage [40], it also depends on the 9-1-1 clamp, the Rad17 clamp loader and the Crb253BP1 mediator. To characterise the dependencies for lacO-dependent checkpoint activation we examined which checkpoint genes were required for Chk1 phosphorylation during Rad3ATR tethering (Figure 5C). Rad3ATR-GFP/LN was expressed in lacO-positive strains deleted for rad1, rad9 (encoding 9-1-1 components), rad17 (clamp loader) and crb2. Each was required for Chk1 phosphorylation. Thus, Rad3ATR tethering is not sufficient for checkpoint activation: the clamp loader, the 9-1-1 clamp complex and the Crb2 mediator are all required and this artificial checkpoint activation system does not bypass the usual requirements. However, Brc1, the proposed MDC1/PTIP ortholog is not required for Chk1 phosphorylation in this system (Figure S1F)
In both S. pombe and S. cerevisiae recruitment of the 53BP1 ortholog (Crb2 and Rad9 respectively) to chromatin in response to IR requires prior phosphorylation of histone H2A [32], [41], [42]. In addition to H2A phosphorylation, recruitment also requires the largely constitutive methylation of a further histone residue, H3K79 in S. cerevisiae or H4K20 in S. pombe. These modifications are effected by distinct methylransferases in the two yeasts: Dot1 methylates H3K79 in S. cerevisiae [43] while Set9 methylates H3K20 in S. pombe [44], [45]. In S. pombe it has been demonstrated that the C-terminal BRCT domains of Crb253BP1 binds directly to γH2A [42] while the Tudor domain binds directly to di-methylated H3K20 [46]. Both interactions are required for Crb253BP1 chromatin association and show an epistatic relationship [45].
Using our Chk1 phosphorylation assay in response to Rad3ATR tethering, we tested two strains harbouring charge reversal mutations of residues within the phospho-acceptor site of the C-terminal Crb253BP1 BRCT domains, crb2-K617E and crb2-K619E (Figure 5C) that disrupt the interaction with γH2A [42]. Chk1 phosphorylation was reduced in both mutants. We next established if checkpoint activation by Rad3ATR tethering was affected in cells containing mutants in the two H2A genes that replace the phosphorylated residue with alanine, hta1-S129A hta2-S128A [32]. Chk1 phosphorylation was not observed in this background (Figure 5D), indicating that the Rad3ATR tethering-dependent and Rad4TopBP1 AAD-dependent checkpoint activation acts in the context of chromatin modification.
Rad4TopBP1 tethering bypasses the requirement for Rad9 C-terminal phosphorylation
Upon activation of the DNA damage checkpoint in S. pombe, Rad3ATR phosphorylates the Rad9 C-terminus on T412 and this is required to recruit Rad4TopBP1 [20]. Recruitment of Rad4TopBP1 allows subsequent recruitment of Crb253BP1 and consequent Chk1 activation [29]. A similar requirement for TopBP1 recruitment via Rad9 C-terminal phosphorylation is also evident in S. cerevisiae and mammalian cells [19], [47], [48]. As expected, expression of Rad3ATR-LacI in cells harbouring a rad9-T412A mutation did not result in Chk1 phosphorylation (Figure 5E) implying that lacO-recruited Rad3ATR must phosphorylate endogenous Rad9 to promote Rad4TopBP1 recruitment to activate the checkpoint.
We reasoned that the requirement for Rad9-T412 phosphorylation during activation by Rad3ATR tethering may solely be to bring the Rad4TopBP1 AAD into proximity of Rad3ATR. In this case, we should be able to bypass the requirement for Rad9-T412 phosphorylation specifically for checkpoint activation by Rad3ATR tethering by recruiting both Rad3ATR and Rad4TopBP1 at the same time. Indeed, Chk1 phosphorylation was restored when we co-expressed Rad3ATR-LacI and Rad4TopBP1-LacI in a rad9-T412A mutant background (Figure 5E). Since checkpoint activation by co-expression of Rad3ATR-LacI and Rad4TopBP1-LacI remains lacO dependent (Figure S1G), these data suggest that Rad4TopBP1 AAD can activate the Rad3ATR-dependent checkpoint cascade in the absence of the recruitment activity of the Rad9 C-terminal tail.
The Rad4TopBP1 AAD is important when resection is limiting
We have shown that the Rad4TopBP1 AAD functions to protect cells from insult during S phase, but is not required for G2 checkpoint activation after IR. Further, we demonstrated that when rad4-Y599R (AAD-defective) mutant cells were synchronised in S phase, phosphorylation of both Chk1 and H2A in response to IR treatment was reduced when compared to rad4+ cells. The increase in Cdc2-Cdc13ClyclinB (CDK) activity as cells progress from S phase into G2 [49] is known to establish conditions conducive to HR by regulating factors required for DNA resection [36], [50], [51]. A consequence of this is that, in response to IR treatment but not in response to UV treatment, ssDNA RPA is predicted to be more prevalent in G2 cells when compared to G1/S phase cells. We thus predicted that reducing resection rates associated with IR treatment in G2 cells would create a dependency for full Chk1 phosphorylation on the Rad4TopBP1 AAD and thus that Chk1 phosphorylation would be reduced in rad4-Y599R strains compared to rad4+ strains in response to an equal dose of IR.
To test this prediction we examined the induction of Chk1 phosphorylation in response to 100 Gy IR in exo1Δ rad4+ and exo1Δ rad4-Y599R cells (Figure 6). First we established that, when exo1 was deleted, Rad11RPA foci were reduced in number, consistent with the expectationt hat resection is decreased in this background (Figure 6A). In contrast to rad4+ cells, where exo1 deletion did not reduce Chk1 phosphorylation levels, Chk1 phosphorylation was reduced to approximately 50% when exo1 was deleted in AAD-defective cells. Previous work in both budding and fission yeasts has indicated that, in the absence of resection, Chk1 phosphorylation can occur through an alternative double strand break end-dependent Tel1ATM pathway, as opposed to the canonical resection and ssDNA/RPA-dependent Rad3ATR pathway [52]. Such a response could potentially mask some aspect of defects seen in the exo1Δ background. Thus, we first confirmed that loss of tel1 alone does not influence Chk1 phosphorylation in our assay (Figure S1H) and then concomitantly deleted Tel1ATM in both rad4+ and rad4-Y599R strains (Figure 6B, 6C). In the tel1Δ exo1Δ background, Chk1 phosphorylation was decreased by approximately 50% for both rad4+ and rad4-Y599R when compared to the exo1Δ alone background. These data are consistent with a general increase in Tel1-dependent checkpoint signalling when resection is reduced by exo1 deletion. In the background of the rad4-Y599R mutation, this is superimposed on a decrease in Rad3ATR-dependent signalling caused by the reduced resection.

The Rad4TopBP1 and Rad9 AADs co-operate in G1/S
A second ATR activation domain has recently been identified in the S. cerevisiae Ddc1Rad9 C-terminal tail [26]. Mutations in this domain define a function in Mec1ATR activation during G1, complementary to the function of the Dpb11TopBP1 AAD in promoting robust checkpoint activation in G2 in this organism [24]. Sequence alignments show that the two key Ddc1Rad9 AAD aromatic residues are conserved in S. pombe as Y271 (equating to W352Sc within the PCNA-like domain) and W348 (equating to W544Sc in the intrinsically disordered C-terminal tail) ([26] and Figure 7A, 7B).We thus created an rad9-AAD mutant by mutating both aromatic residues to alanine.

Analysis of the resulting rad9-AAD strain demonstrates no clear sensitivity to DNA damaging agents that create problems during S phase, including CPT, MMS and UV (Figure 7C) or in G2 to IR. However, some increased sensitivity is evident to CPT and MMS when the Rad4TopBP1 AAD mutant is present in the same strain. We next assayed the ability of rad9-AAD mutants to activate Chk1 in response to either IR or UV treatment. Consistent with the lack of sensitivity, the level of Chk1 phosphorylation after IR was not reduced (Figure 7D), either in rad9-AAD mutant alone or in the rad9-AAD rad4-Y599R double mutant when compared to the rad4-Y599R single. In response to UV treatment, there was again no decrease observed for the single rad9-AAD mutant, but a further and reproducible decrease was seen for the rad9-AAD rad4-Y559R double mutant when compared to rad4-Y599R alone (Figure 7E). Thus, the putative Rad9-AAD domain in S. pombe plays, at most, only a minor role in activating Rad3ATR in response to DNA damage and this is only revealed in the absence of the Rad4TopBP1 AAD.
Discussion
Understanding the mechanism of ATR activation is an important facet of gaining insight into how cells respond to unwanted DNA structures, itself a key aspect in maintaining genomic integrity. TopBP1 was initially implicated in the ATR-dependent checkpoint in fission yeast and later this was extended to higher eukaryotes [53]. TopBP1 is a multi-BRCT-domain containing protein that acts to scaffold proteins during both the initiation of DNA replication and in response to DNA damage, a function dependent of the phospho-binding ability of the BRCT-domain pairs within TopBP1 [25]. In addition to scaffolding phospho-proteins, TopBP1 was shown to be able to directly activate ATR in Xenopus and human cells through a small domain of TopBP1 which is not part of any BRCT pair [17]. The ATR activating domain is sufficient, both in vitro and in vivo, to activate ATR - although it is not always necessary for ATR activation and the pathway in which this TopBP1 AAD domain functions is yet to be fully understood. It has recently been shown in S. cerevisiae that the AAD of the TopBP1 homolog (Dpb11) is also able to activate the ATR homolg (Mec1). However, the Dpb11 AAD plays a relatively minor role in checkpoint activation which is specific to G2 phase [24]. In S. cerevisiae, a second ATR activation domain within the C-terminal tail of the 9-1-1 subunit, Ddc1Rad9 acts to help activate ATR in G1 and G2 and it is only when the function of this AAD is ablated a role for the Dpb11 AAD becomes apparent [26]. However, loss of both domains does not prevent checkpoint activation entirely, suggesting other AADs or modes of activation. Conversely, in the Xenopus system, ATR activation via the TopBP1 AAD is evident in S phase.
Here we show that, in S. pombe, the activation of the ATR homolog (Rad3) by a Rad4TopBP1 AAD is conserved. We demonstrate that the Rad4TopBP1 AAD makes a contribution to checkpoint activation and that this is specific to G1/S phase and is not evident in G2. Note that log phase S. pombe spend little, if any, time in G1 and thus, while we can arrest cells before the onset of replication with cell cycle mutants, we cannot make a clear physiological distinction between G1 and S phase. We go on to demonstrate that, when DNA resection was limited in G2 by ablation of the Exo1 nuclease, checkpoint activation in response to DNA damage during G2 becomes partially dependent on the Rad4TopBP1 AAD, mimicking what we observed in G1/S cells. This leads us to propose that there is a threshold of ssDNA required for activation of the DNA damage checkpoint and that the Rad4TopBP1 AAD serves to amplify checkpoint signals when ssDNA is limiting.
We next used a genetic system to separate Rad3ATR activation from the production of DNA damage and therefore ssDNA, thus allowing us to assess the pathway of Rad3ATR activation dependent on the Rad4TopBP1 AAD. In this system, specific checkpoint proteins are recruited to a defined chromatin locus through dsDNA:protein binding [37], [39]. Interestingly, recruitment of any one of the three checkpoint proteins (Rad3ATR, Rad4TopBP1 and Rad9) tested was sufficient to generate a checkpoint response and these responses followed the expected dependencies. This suggests that the recruitment of multiple copies of a single checkpoint protein results in the formation of active checkpoint complexes that utilise the endogenous proteins. Using this system, we observed that the ability of the Rad4TopBP1 AAD to activate Rad3ATR is fully dependent on phosphorylation of H2A (γH2A) and requires the ability of Crb2 to bind γH2A. This leads us to conclude that, in the absence of ssDNA, the ATR activation domain of Rad4TopBP1 is particularly important for Rad3ATR activation and acts in a chromatin:protein interaction dependent manner. Taking these data together with the requirement of the Rad4TopBP1 AAD to amplify checkpoint signals in either G1/S or G2 when resection was limited, we propose that Rad4TopBP1 acts to amplify the checkpoint in a chromatin-dependent manner when single-stranded DNA levels are limiting. We can therefore hypothesise that there is a threshold level for the amount of active Rad3ATR required for a full checkpoint response. When ss-DNA is limited, such as in S-phase, the chromatin-dependent Rad4TopBP1 AAD-dependent pathway for Rad3ATR activation becomes important to amplify the levels of activated Rad3ATR to obtain a full checkpoint response (Figure 8).

In addition to analysing the Rad4TopBP1 AAD, we also created a mutant predicted to disable the Rad9 equivalent of the S. cerevisiae Ddc1Rad9 AAD and analysed the effect of this mutant in checkpoint activation. Unlike in S. cerevisiae, we observed no significant effect on DNA damage-induced checkpoint activation either in G1/S phase or G2. Although when combined with a Rad4TopBP1 AAD mutant, an additive effect to S-phase but not G2 DNA damage can be seen. This suggests that the Rad9 AAD acts in a separate but redundant pathway for Rad3ATR activation in G1/S with the Rad4TopBP1 AAD. It appears that, during evolution, the mechanism of activating the ATR pathway has diverged significantly with the roles of different ATR activating domains being of more or less importance in different organisms.
It will be interesting to establish if the ATR activating domain of TopBP1 in metazoan systems is particularly important in the context of low levels of ssDNA and whether its function is dependent on γH2AX, especially as a 53BP1(Crb2) and TopBP1 pathway for checkpoint activation in G1 has been previously reported in the mammalian system [54]. The differences in the dependencies of the specific ATR activators in different cell cycle phases between S. pombe and S. cerevisiae is not surprising as the checkpoint mechanism between these organisms has diverged. For example, in S. cerevisiae, the S phase checkpoint is activated independently of the 9-1-1 complex, whereas in S. pombe and mammalian cells ATR activation appears to be largely - if not entirely - dependent on 9-1-1 loading. Such distinctions are likely a result of evolutionary adaptation to the different cell cycle profiles of the two yeasts and it is interesting to note that significant evolutionary plasticity surrounds the interface between TopBP1 and the checkpoint apparatus. These distinctions will have to be considered when extrapolating mechanistic data from yeast to human systems. None the less, we believe that our findings shed light on the role of TopBP1 AAD in DNA damage responses and offer useful insights into metazoan mechanisms of DNA damage signalling.
Materials and Methods
S. pombe strain construction and biological methods
Standard S. pombe protocols were carried out as previously described [55]. rad4 and rad9 mutant strains were created using PCR site directed mutagenesis and integrated at their endogenous locus using Cre recombinase-mediated cassette exchange [56] In brief, this system uses a “base strain” which is engineered so that the gene of interest is either replaced with the ura4 marker (i.e. rad9), or in the case of essential genes (i.e. rad4) has the marker integrated immediately after the stop codon. In both cases the gene/marker and loci's promoter region are flanked by loxP and loxM sites. These two variant lox sites are incompatible with each other. The marker (and, for essential genes, the actual gene also) is then replaced by transforming in either the wild type (as a control: rad+) or the various mutated copies on a plasmid. These are flanked by the equivalent loxP and loxM sites and the plasmid expresses Cre recombinase, which results in loxP:loxP and loxM:loxM recombination. For cdc10-M17 synchronisation cells were grown to log phase at the permissive temperature (25°C) and shifted to the restrictive temperature of 36°C for 3.5 hours. Cells were then either irradiated with the indicated dose of gamma irradiation at 36°C and released at 25°C, or directly released at 25°C and irradiated at the given time points after release. cdc25-22 block and release [57] and lactose gradient synchronisation [12] were performed as described previously. For FACS analysis cells were resuspended in 50 mM tri-sodium citrate, 1 mg/ml final concentration RNAseA [Sigma], stained with 5 µg/ml Propidium iodide [Sigma] and analysed on FacsCalibur [Becton Dickinson].
Imaging
For live cell imaging concentrated culture was mounted onto a 2.5% agar patch in standard YE medim [Microworks] and imaged on a Deltavsion Microscope. Septation index was counted as previously described [12]. lacO::NAT chk1-HA strains were created by inserting the 10 Kb lacO repeats into the PUC19 plasmid containing the NAT marker and homology to ura4. This was integrated into the genomic ura4 locus. The appropriate strains were transformed [58] with pRep41-GFP-LacI-NLS (GFP/LN) into which either rad3 or rad9 had been cloned in frame for N-terminal tagging or rad4 cloned in frame for C-terminal tagging. Transformants were grown and expression of the fusion protein induced by the removal of thiamine. All lacO repeats were checked by Southern hybridisation.
Biochemical assays
Protein extracts for western were prepared by TCA (trichloro-acetic acid) extraction from 1×108 cells and resuspended in SDS sample buffer [59]. Crude extracts for affinity analysis were prepared by mechanical disruption in liquid nitrogen. Antibodies used: α-HA [Santa cruz] 1∶2500, α-Myc [Santa cruz] 1∶2000, α-GFP [Roche] 1∶2500, α-H2ApS129 [Abcam] 1∶2500 or 1∶1000, α-Tubulin [Sigma] 1∶5000, α-Cdc2 Sc-53 [Santa cruz] 1∶2500. α-Cdc13 [Jacky Hayles] 1∶500. α-Cds1 1∶5000 [35]. The secondary antibodies used were Hrp rabbit α mouse [Dako] 1∶2500 or Hrp swine α Rabbit [Dako]1∶2500. Chk1-HA phosphorylation was quantified as a percentage of total signal minus back ground on a ImageQuant LAS 4000 [GE Healthcare]. Cds1 kinase assay was carried out as described [35].
Supporting Information
{kind=link}
Zdroje
1. WeinertTAHartwellLH 1988 The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241 317 322
2. CicciaAElledgeSJ 2010 The DNA damage response: making it safe to play with knives. Molecular cell 40 179 204
3. BakkenistCJKastanMB 2004 Initiating cellular stress responses. Cell 118 9 17
4. MimitouEPSymingtonLS 2008 Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455 770 774
5. ByunTSPacekMYeeMCWalterJCCimprichKA 2005 Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes & development 19 1040 1052
6. FanningEKlimovichVNagerAR 2006 A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic acids research 34 4126 4137
7. ZouLElledgeSJ 2003 Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300 1542 1548
8. BallHLEhrhardtMRMordesDAGlickGGChazinWJ 2007 Function of a conserved checkpoint recruitment domain in ATRIP proteins. Molecular and cellular biology 27 3367 3377
9. BermudezVPLindsey-BoltzLACesareAJManiwaYGriffithJD 2003 Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proceedings of the National Academy of Sciences of the United States of America 100 1633 1638
10. EllisonVStillmanB 2003 Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 1 e33 doi:10.1371/journal.pbio.0000033
11. MajkaJBinzSKWoldMSBurgersPM 2006 Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. The Journal of biological chemistry 281 27855 27861
12. EdwardsRJBentleyNJCarrAM 1999 A Rad3-Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nature cell biology 1 393 398
13. CortezDGuntukuSQinJElledgeSJ 2001 ATR and ATRIP: partners in checkpoint signaling. Science 294 1713 1716
14. LiuSShiotaniBLahiriMMarechalATseA 2011 ATR autophosphorylation as a molecular switch for checkpoint activation. Molecular cell 43 192 202
15. KostrubCFKnudsenKSubramaniSEnochT 1998 Hus1p, a conserved fission yeast checkpoint protein, interacts with Rad1p and is phosphorylated in response to DNA damage. The EMBO journal 17 2055 2066
16. HarrisSKemplenCCaspariTChanCLindsayHD 2003 Delineating the position of rad4+/cut5+ within the DNA-structure checkpoint pathways in Schizosaccharomyces pombe. Journal of cell science 116 3519 3529
17. KumagaiALeeJYooHYDunphyWG 2006 TopBP1 activates the ATR-ATRIP complex. Cell 124 943 955
18. St OngeRPBesleyBDPelleyJLDaveyS 2003 A role for the phosphorylation of hRad9 in checkpoint signaling. The Journal of biological chemistry 278 26620 26628
19. LeeJKumagaiADunphyWG 2007 The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. The Journal of biological chemistry 282 28036 28044
20. FuruyaKPoiteleaMGuoLCaspariTCarrAM 2004 Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes & development 18 1154 1164
21. TaricaniLWangTS 2006 Rad4TopBP1, a scaffold protein, plays separate roles in DNA damage and replication checkpoints and DNA replication. Molecular biology of the cell 17 3456 3468
22. MordesDAGlickGGZhaoRCortezD 2008 TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes & development 22 1478 1489
23. Navadgi-PatilVMBurgersPM 2008 Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. The Journal of biological chemistry 283 35853 35859
24. Navadgi-PatilVMKumarSBurgersPM 2011 The unstructured C-terminal tail of yeast Dpb11 (human TopBP1) protein is dispensable for DNA replication and the S phase checkpoint but required for the G2/M checkpoint. The Journal of biological chemistry 286 40999 41007
25. PfanderBDiffleyJF 2011 Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. The EMBO journal 30 4897 4907
26. Navadgi-PatilVMBurgersPM 2009 The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Molecular cell 36 743 753
27. Navadgi-PatilVMBurgersPM 2011 Cell-cycle-specific activators of the Mec1/ATR checkpoint kinase. Biochemical Society transactions 39 600 605
28. SakaYEsashiFMatsusakaTMochidaSYanagidaM 1997 Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes & development 11 3387 3400
29. MochidaSEsashiFAonoNTamaiKO'ConnellMJ 2004 Regulation of checkpoint kinases through dynamic interaction with Crb2. The EMBO journal 23 418 428
30. Lopez-GironaATanakaKChenXBBaberBAMcGowanCH 2001 Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proceedings of the National Academy of Sciences of the United States of America 98 11289 11294
31. DownsJALowndesNFJacksonSP 2000 A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408 1001 1004
32. NakamuraTMDuLLRedonCRussellP 2004 Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Molecular and cellular biology 24 6215 6230
33. ChristensenPUBentleyNJMartinhoRGNielsenOCarrAM 2000 Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proceedings of the National Academy of Sciences of the United States of America 97 2579 2584
34. CallegariAJKellyTJ 2006 UV irradiation induces a postreplication DNA damage checkpoint. Proceedings of the National Academy of Sciences of the United States of America 103 15877 15882
35. LindsayHDGriffithsDJEdwardsRJChristensenPUMurrayJM 1998 S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes & development 12 382 395
36. CaspariTMurrayJMCarrAM 2002 Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes & development 16 1195 1208
37. BonillaCYMeloJAToczyskiDP 2008 Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Molecular cell 30 267 276
38. MaundrellK 1993 Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123 127 130
39. Lindsey-BoltzLASancarA 2011 Tethering DNA damage checkpoint mediator proteins topoisomerase IIbeta-binding protein 1 (TopBP1) and Claspin to DNA activates ataxia-telangiectasia mutated and RAD3-related (ATR) phosphorylation of checkpoint kinase 1 (Chk1). The Journal of biological chemistry 286 19229 19236
40. WalworthNCBernardsR 1996 rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271 353 356
41. HammetAMagillCHeierhorstJJacksonSP 2007 Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO reports 8 851 857
42. KilkennyMLDoreASRoeSMNestorasKHoJC 2008 Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes & development 22 2034 2047
43. GiannattasioMLazzaroFPlevaniPMuzi-FalconiM 2005 The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. The Journal of biological chemistry 280 9879 9886
44. SandersSLPortosoMMataJBahlerJAllshireRC 2004 Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119 603 614
45. DuLLNakamuraTMRussellP 2006 Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes & development 20 1583 1596
46. GreesonNTSenguptaRAridaARJenuweinTSandersSL 2008 Di-methyl H4 lysine 20 targets the checkpoint protein Crb2 to sites of DNA damage. The Journal of biological chemistry 283 33168 33174
47. DelacroixSWagnerJMKobayashiMYamamotoKKarnitzLM 2007 The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes & development 21 1472 1477
48. PudduFGranataMDi NolaLBalestriniAPiergiovanniG 2008 Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Molecular and cellular biology 28 4782 4793
49. NurseP 2000 A long twentieth century of the cell cycle and beyond. Cell 100 71 78
50. IraGPellicioliABalijjaAWangXFioraniS 2004 DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 1011 1017
51. HuertasPCortes-LedesmaFSartoriAAAguileraAJacksonSP 2008 CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455 689 692
52. LimboOPorter-GoffMERhindNRussellP 2011 Mre11 nuclease activity and Ctp1 regulate Chk1 activation by Rad3ATR and Tel1ATM checkpoint kinases at double-strand breaks. Molecular and cellular biology 31 573 583
53. GarciaVFuruyaKCarrAM 2005 Identification and functional analysis of TopBP1 and its homologs. DNA repair 4 1227 1239
54. CescuttiRNegriniSKohzakiMHalazonetisTD 2010 TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. The EMBO journal 29 3723 3732
55. MorenoSKlarANurseP 1991 Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods in enzymology 194 795 823
56. WatsonATGarciaVBoneNCarrAMArmstrongJ 2008 Gene tagging and gene replacement using recombinase-mediated cassette exchange in Schizosaccharomyces pombe. Gene 407 63 74
57. FuruyaKMiyabeITsutsuiYPaderiFKakushoN 2010 DDK phosphorylates checkpoint clamp component Rad9 and promotes its release from damaged chromatin. Molecular cell 40 606 618
58. SugaMHatakeyamaT 2005 A rapid and simple procedure for high-efficiency lithium acetate transformation of cryopreserved Schizosaccharomyces pombe cells. Yeast 22 799 804
59. CaspariTDahlenMKanter-SmolerGLindsayHDHofmannK 2000 Characterization of Schizosaccharomyces pombe Hus1: a PCNA-related protein that associates with Rad1 and Rad9. Molecular and cellular biology 20 1254 1262
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy