
Control of CpG and Non-CpG DNA Methylation by DNA Methyltransferases
The enzymatic control of the setting and maintenance of symmetric and non-symmetric DNA methylation patterns in a particular genome context is not well understood. Here, we describe a comprehensive analysis of DNA methylation patterns generated by high resolution sequencing of hairpin-bisulfite amplicons of selected single copy genes and repetitive elements (LINE1, B1, IAP-LTR-retrotransposons, and major satellites). The analysis unambiguously identifies a substantial amount of regional incomplete methylation maintenance, i.e. hemimethylated CpG positions, with variant degrees among cell types. Moreover, non-CpG cytosine methylation is confined to ESCs and exclusively catalysed by Dnmt3a and Dnmt3b. This sequence position–, cell type–, and region-dependent non-CpG methylation is strongly linked to neighboring CpG methylation and requires the presence of Dnmt3L. The generation of a comprehensive data set of 146,000 CpG dyads was used to apply and develop parameter estimated hidden Markov models (HMM) to calculate the relative contribution of DNA methyltransferases (Dnmts) for de novo and maintenance DNA methylation. The comparative modelling included wild-type ESCs and mutant ESCs deficient for Dnmt1, Dnmt3a, Dnmt3b, or Dnmt3a/3b, respectively. The HMM analysis identifies a considerable de novo methylation activity for Dnmt1 at certain repetitive elements and single copy sequences. Dnmt3a and Dnmt3b contribute de novo function. However, both enzymes are also essential to maintain symmetrical CpG methylation at distinct repetitive and single copy sequences in ESCs.
Published in the journal:
. PLoS Genet 8(6): e32767. doi:10.1371/journal.pgen.1002750
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002750
Summary
The enzymatic control of the setting and maintenance of symmetric and non-symmetric DNA methylation patterns in a particular genome context is not well understood. Here, we describe a comprehensive analysis of DNA methylation patterns generated by high resolution sequencing of hairpin-bisulfite amplicons of selected single copy genes and repetitive elements (LINE1, B1, IAP-LTR-retrotransposons, and major satellites). The analysis unambiguously identifies a substantial amount of regional incomplete methylation maintenance, i.e. hemimethylated CpG positions, with variant degrees among cell types. Moreover, non-CpG cytosine methylation is confined to ESCs and exclusively catalysed by Dnmt3a and Dnmt3b. This sequence position–, cell type–, and region-dependent non-CpG methylation is strongly linked to neighboring CpG methylation and requires the presence of Dnmt3L. The generation of a comprehensive data set of 146,000 CpG dyads was used to apply and develop parameter estimated hidden Markov models (HMM) to calculate the relative contribution of DNA methyltransferases (Dnmts) for de novo and maintenance DNA methylation. The comparative modelling included wild-type ESCs and mutant ESCs deficient for Dnmt1, Dnmt3a, Dnmt3b, or Dnmt3a/3b, respectively. The HMM analysis identifies a considerable de novo methylation activity for Dnmt1 at certain repetitive elements and single copy sequences. Dnmt3a and Dnmt3b contribute de novo function. However, both enzymes are also essential to maintain symmetrical CpG methylation at distinct repetitive and single copy sequences in ESCs.
Introduction
DNA methylation at the C-5 positions of cytosine (5mC) is a key epigenetic modification in mammals essential for normal development [1], [2]. Cytosine methylation is predominantly found in CpG dinucleotide context and about 70 to 80% of all CpGs are methylated. These methylated CpGs are usually located in CpG poor regions and often in repetitive sequences [3]–[5]. About 40% of the genome consists of repetitive elements. Four main groups of repetitive elements can be discriminated: long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long-terminal repeat (LTR) retrotransposons and (peri-) centromeric satellites. For these elements, the maintenance of DNA methylation during development and aging is important for transcriptional silencing and genome stability [6], [7].
The establishment and maintenance of methylation patterns at palindromic CpG sequences (CpG dyads) is performed by three catalytically active DNA methyltransferases (Dnmts). In vitro experiments suggest that Dnmt1 prefers hemimethylated CpG (hemi-mCpG) dyads and maintains the methylation pattern on the newly synthesized strand after replication (maintenance methylation). In vitro, Dnmt1 shows a low activity on unmethylated CpG dyads. Dnmt3a and Dnmt3b methylate DNA de novo, independent of the methylation status of the complementary CpG position [8], [9]. In vitro analyses furthermore suggest that methylation by Dnmt3b and the maintenance function of Dnmt1 mostly occur in a processive manner, whereas Dnmt3a and the “de novo function” of Dnmt1 are distributive [9]–[12]. However, other groups observe a processive methylation activity for Dnmt3a [13]. In addition, Dnmt activities are modulated by Nuclear protein of 95 kDa (Np95; also known as Uhrf1) and Dnmt3L. Dnmt3L, a cofactor for the de novo methyltransferases, is reported to stimulate Dnmt3a/3b activity, to be needed for de novo establishment for imprint methylation and furthermore to enhance the processive methylation activity of human Dnmt3a [13]–[15]. Np95 is recruiting Dnmt1 to hemimethylated DNA and is interacting with Dnmt3a/3b for gene silencing [16]–[18].
Despite of a lot of in vitro data on Dnmt specificities and interacting partners, relatively little is known about the concerted action in vivo in the genome context and at different types of repetitive elements. Data on ESCs with individual and combined Dnmt knockouts indicated preferences of Dnmts for specific repetitive elements [19]. Different mathematical models were developed to simulate the kinetics of DNA methylation [20]–[25]. However, these calculations were only theoretical or based on only scarce sequencing data. Moreover, most data sets used were based on the bisulfite analysis of only one DNA strand and/or did not discriminate between single Dnmt functions.
In this paper, we present the first comprehensive high resolution methylation analysis for both DNA strands of distinct classes of repetitive elements and four single copy genes known to be methylated in ESCs. Using hairpin linker technology combined with 454 sequencing, we generated individual patterns from embryonic fibroblasts, liver cells, wt ESCs and ESCs depleted for Dnmt1, Dnmt3a, Dnmt3b, Dnmt3a/b, Dnmt3L, Np95 and Suv39h. The Dnmt KO data sets were then used to calculate Dnmt efficiencies with improved hidden Markov models (HMM), extending previous elegant approaches by Sontag et al. and Genereux et al. [20], [25]. The comparative prediction/validation analysis documents a more differentiated view on the relative contributions of individual Dnmts for maintenance and de novo methylation of CpG positions. In addition, the comprehensive hairpin technology allowed us to unambiguously identify the presence and the patterns of non-CpG methylation.
Results
Outline and Quality Monitoring of the Hairpin-Bisulfite Sequencing Strategy
We designed specific hairpin linker protocols to amplify representative fragments of the four major classes of repetitive elements and four single copy genes from bisulfite treated mouse DNA to obtain the methylation pattern of complementary CpGs (CpG dyads) (see for a general scheme Figure S1 and for details see Materials and Methods S1). The repetitive elements selected were i) major Satellites, ii) IAPLTR1, a class of LTR-retrotransposons, iii) the 5′ untranslated region of L1Md_Tf, a long interspersed element (LINE) and iv) B1 elements, representing a class of short interspersed elements (SINEs) (see Figure S2 for locations and Table S1 for references). In this paper, we conveniently refer to the specific repetitive elements as mSat, IAP, L1 and B1, respectively. In addition, we established assays for four single copy genes: alpha feto protein (Afp), testis expressed gene 13 (Tex13), insulin growth factor 2 (Igf2) and Small nuclear ribonucleoprotein-associated protein N (Snrpn). Following amplification, PCR products were sequenced on a 454 GS-FLX sequencer with an average read length of 200–400 bp covering 3 to 12 CpG dyads of the respective amplicons.
The addition of a hairpin linker containing several unmodified cytosines allowed us to directly monitor the bisulfite conversion rates per sequenced molecule. In the linker sequences, the conversion rates ranged from 97,9 to 99,9%, with only B1 showing conversion rates below 98.7%, probably due to the more degenerate sequence composition and occasional back-folding (Table S2).
In contrast to conventional single strand bisulfite sequencing, hairpin bisulfite sequencing allows one to unambiguously distinguish between unmethylated and mutated CpG sites. We identify mutated CpGs in all repetitive elements to various extents (see white positions in Figure 1). A particular abundance of mutated CpGs was found in B1 elements with 44% of CpGs being mutated to TpG (Figure S3a). Previous single strand bisulfite sequencing accounted such TpGs as unmethylated positions estimating the total methylation of B1 elements to be only 10% [26]. When correcting for mutated sites, we find B1 elements to be methylated up to 80% in wt cells (Figure S3b).

Analysis of the Methylation Symmetry at CpG Dyads
Following a precise alignment to reference sequences using BiQAnalyzerHT [27] and the back mapping of complementary CpG positions, we first compared the DNA methylation patterns between mouse wt ESC lines, mouse embryonic liver and cultured mouse embryonic fibroblasts (MEFs) (Figure 1 and Figure 2). In general, mSat, IAPs, B1, Afp and Tex13 are highly methylated in all wt ESC and somatic cells (62–95%), whereas L1 is highly methylated in somatic cells, but only 30 to 52% in ESCs. Igf2 shows in all cell types an intermediate methylation level (Figure S3b).

The hairpin-bisulfite method allows to unambiguously discriminate between unmethylated, hemimethylated and fully methylated CpG dyads. Since it is believed that the maintenance of methylation is very stable and occurs semi-conservative, hemimethylated sites should occur very rarely. The analyses of human DNA showed that hemi-mCpGs occur between 4.8% (sperm) and 20.8% (leukocytes) at human LINE1 elements [24], [28] and 7% hemi-mCpG at human Satellite 2 sequences in different tissues [29].
We found in the analysed mouse cells a range of 1 to 25% of all CpGs in a hemimethylated status across all amplicons (Figure 1 and Figure 2). In differentiated cells, hemimethylated sites occur equally distributed across the analysed sequences - MEFs show the lowest and least variable rate of hemi-mCpG (5,8 to 12% of all methylated CpG dyads) among the analysed elements and in embryonic liver an overall high amount of hemi-mCpGs is detected (16,2 to 30,6% of all methylated CpG dyads) (Figure S3c).
Contrarily, in ESCs the degree of hemimethylation is much more variable at the different types of repetitive elements. While hemimethylation levels are low for IAPs (9,3 to 12,5%) Tex13, Afp and Snrpn (3,8 to 7%), more than 35% of methylated CpGs in L1 and 22% at Igf2 are hemimethylated. Moreover, the extent of hemi-mCpGs at mSat and B1 greatly varied between the three wt ESC lines, but the general tendencies for particular elements are maintained.
The almost exclusive fully or unmethylated patterns of the imprinted gene Snrpn (and H19, data not shown) show the stable maintenance of two non-equilibrium states. The imprinted genes are very important internal controls showing i) that the enzymes responsible for full maintenance are present and fully functional ii) that the occurance of hemimethylated states in other genes/elements is not simple due to an increase of cells analysed in S-phase (i.e. incompleted replications states) in fast dividing ESCs.
Effects of Dnmts Loss on Overall DNA Methylation
Next, we analysed the contributions of Dnmts and cofactors for the maintenance of the methylation pattern by comparing hairpin-bisulfite sequence data of ESCs mutated for Dnmt1, Dnmt3a, Dnmt3b, Dnmt3a and 3b (DKO), Dnmt3L and Np95 (UHRF1), respectively (Figure 2 and Figure 3a).

Deletion of Dnmt1 caused a substantial reduction of DNA methylation in all analysed elements (mSat methylation was reduced by 65%, IAP by 72%, L1 by 76% and B1 by 75%, Tex13 by 82%, Afp by 75%, Igf2 by 82% and Snrpn by 99%, respectively). This tendency was also observed in previous low resolution data obtained for a subset of repetitive sequence elements [19], [30]. Our deep sequencing data however clearly shows that a small subset of sequences maintain a considerable amount of hemi - and fully methylated sites. A triple knockout cell line (TKO, data not shown) does not show any signs of DNA methylation anymore. The effects of Np95 KO at repetitive elements were very similar but not identical to the Dnmt1 KO (see also Bostick et al. [16]) indicating that the major activity of Dnmt1 is indeed mediated by Np95 [16], [17].
In contrast to a general hypomethylation at all elements in Dnmt1/Np95 KOs, the loss of Dnmt3 activities led to element and enzyme specific differences. While methylation at IAP, mSat, Tex13 and Afp did not greatly change in Dnmt3a or Dnmt3b single KOs, the double KO led to a clear decrease of CpG methylation at mSat (24%), IAPs (17%), Tex13 (41%) and Afp (53%). Methylation at L1 and B1 did not change in the Dnmt3b single KO, but was strongly decreased in the Dnmt3a single KO by 64% for L1 and 37% for B1. Igf2 shows decreased level for Dnmt3a and Dnmt3b single KOs (53% for Dnmt3a KO, 34% for Dnmt3b KO). For all three sequences (L1, B1 and Igf2) in the DKO, there is only minor methylation left.
Hence, while either the loss of Dnmt3a or Dnmt3b, respectively, can be compensated by the other enzyme at IAPs, mSat, Afp and Tex13 sequences, the situation is more complex at B1, L1 and Igf2. Finally, Dnmt3L also contributes to maintain a high level of methylation. In the Dnmt3L KO the loss of methylation at all regions is less extensive than in the Dnmt3a/3b DKO, arguing for a stimulatory effect of Dnmt3L on both de novo Dnmts. Note that Dnmt3L KO cells were at passage 15 and underwent already almost twice the amount of replications than the Dnmt3a/b DKO (passage 8).
Loss of Dnmt1 and Np95 KO Leads to an Increase of Hemi-mCpG Sites
For all sequences, we observe a strong increase in the relative amount of hemi-mCpGs (in regard to total methylation) in Dnmt1 KO and Np95 KO (Figure 2, Figure 3b), along with a huge loss of overall methylation. This observation highlights the important role of Dnmt1 in maintaining symmetrical CpG methylation. However, it is very intriguing that in both Dnmt1 and Np95 null backgrounds, we still find a considerable amount of sequences with fully methylated CpG dyads (Figure 2, Figure 3a and 3c).
Chromosomal sequences with hemi-mCpG sites on only the upper or lower strand, respectively, were found frequently, compared to sequences with (dispersed) hemi-mCpG sites on both upper and lower strands. Such dispersed hemimethylation was found in WT ESCs in <2% of mSat, <3,4% of B1, <4,5% of IAP and <7,2% of L1 reads, respectively (see Figure 3c). Note that in Dnmt1KO and/or Np95KO ESCs dispersed hemi-mCpGs were enriched compared to WT.
In contrast to the Dnmt1 KO and Np95 KO, respectively, the abundance of hemimethylated sites does not differ between WT and Dnmt3a/Dnmt3b single KOs and double KO.
CpA Methylation Is Pronounced at Major Satellites in ESCs
The double stranded hairpin sequencing data allowed to unambiguously assign cytosine methylation outside of CpGs. We identified clear non-CpG (mostly CpA) methylation in mSat sequences and the Afp gene in WT ESC lines (Figure 4a and 4b, Figure 5a). This non-CpG methylation is much less pronounced, more sporadic, less position dependent or barely detectable at the other elements (Figure S4). In mSat and Afp amplicons, respectively, cytosines at five non-CpG positions showed a significant methylation (in 6–12% of all reads) clearly above the conversion background of 1.1% (as defined by linker sequence conversion, see above). Most bisulfite unconverted (methylated) positions are found in the CpA sequence context. Interestingly, in most of the sequence reads only one single CpA methylated position was detected; such that 75% for mSat and 55% for Afp of J1 reads had clear single CpA methylation (Figure S5). Finally, our data confirm that methylated cytosines (above technical background) outside of the CpG context are not detectable in differentiated cells (embryonic liver and MEFs (Figure 4b, Figure 5a).
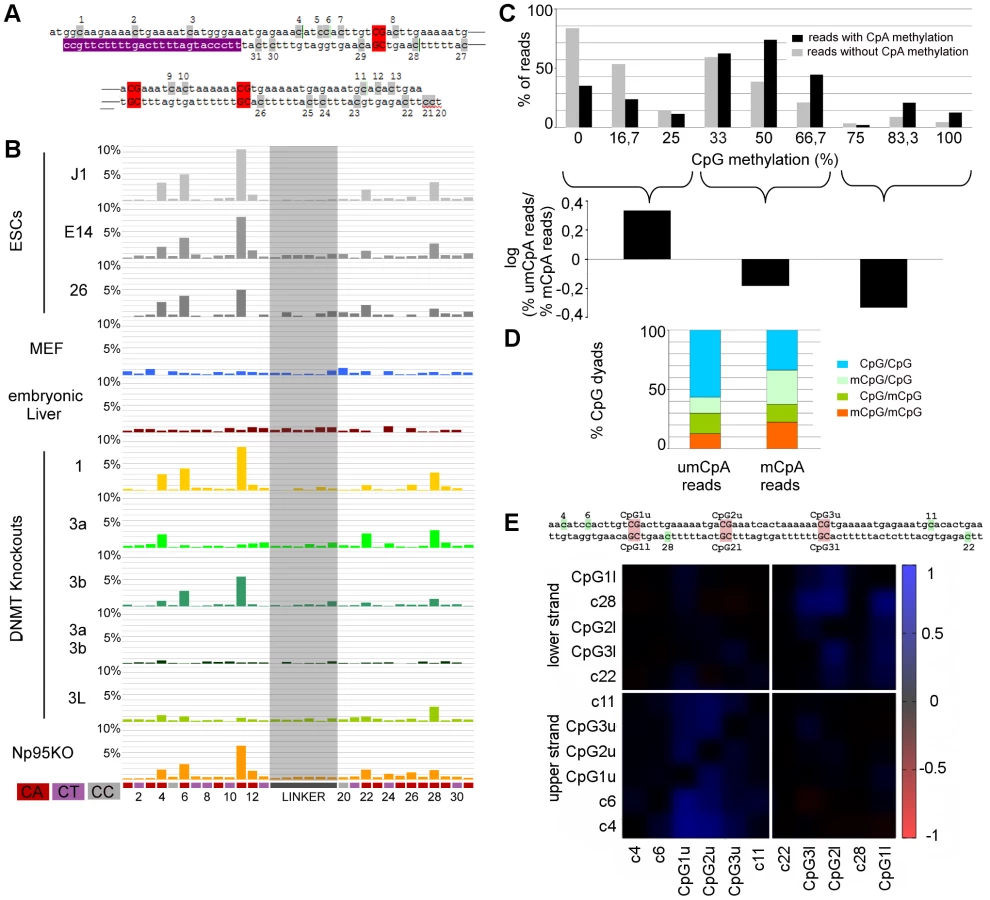

Dnmt3a, 3b Together with 3L Mediate Non-CpG DNA Methylation in Major Satellites
By comparing the presence of methylated cytosines in non-CpG context between wt and the different KO ESC lines (Figure 4b, Figure 5a), we found that in Dnmt1 KO the methylation at all non-CpG positions remained unchanged despite the greatly reduced CpG methylation level. Notably, we found enrichment in CpG methylation at sequences showing non-CpG methylation (Figure 4c, 4d). Moreover, by correlating CpA methylation to CpG methylation in the Dnmt1 KO, we found that CpA methylation is highly linked to neighboured methylated CpG positions at mSat and Afp (Figure 4e and Figure 5b)
In Dnmt3a/3b DKO, CpA methylation above background is completely absent and surprisingly for mSat Dnmt3a and Dnmt3b single KO showed different pattern of CpA methylation. Whereas CpA methylation at position 6 and 11 is greatly reduced in a Dnmt3a KO, the loss of Dnmt3b diminishes CpA methylation at position 4, 22 and 28. Interestingly, the loss of Dnmt3L greatly reduces the methylation at most positions. At the Afp gene, non-CpG methylation also strictly depends on Dnmt3a/3b in combination with Dnmt3L - although here Dnmt3b apparently plays a less important role.
Together these findings clearly point towards a position specific exclusive Dnmt3a and 3b mediated CpA methylation guided by Dnmt3L.
Suv39h KO Decreases CpG Methylation Level at mSat, but Has No Influence on CpA Methylation
The Suv39h1/2 mediated modification of histone H3 at position 9 was reported to influence the targeting of DNA methylation. We therefore included ESCs and MEFs with KO for Suv39h1 and Suv39h2 (Suv39dn) in our analysis for the repetitive elements. Suv39dn ESCs were reported to lack H3K9 trimethylation and Dnmt3b localisation at pericentric heterochromatin. Lehnertz et al. reported reduced DNA methylation (by southern blot) at mSat in Suv39h KO but not at minor Satellites or a C-type retrovirus [31]. Our hairpin bisulfite analysis confirmed this finding on a sequencing basis. DNA methylation at major satellites is reduced by 20% in Suv39dn ESCs, but not at B1, IAP and Line1 elements. Surprisingly, the effect on mSat methylation is almost absent in dnMEFs, which retain 95% of wt methylation (Figure S6a).
Finally, despite of the proposed interaction of Suv39h with Dnmt3b at mSat, we do not observe any influence of the Suv39h absence on CpA methylation, particularly not at the Dnmt3b specific positions 4, 22 and 28 (Figure S6b).
Hidden Markov Model Predicts Methylation Efficiencies of Dnmts
The precise determination of fully methylated, hemimethylated and unmethylated CpG dyads in the comparative data set of some 28.000 sequences (around 146.000 CpG dyads) including wt ESCs and Dnmt KOs allowed us to calculate the element specific methylation efficiencies for the different catalytically active Dnmts in a modified version of the linear HMM proposed by Sontag et al. [20]. We computed maximum likelihood estimates for both methylation efficiencies on unmethylated and hemimethylated CpG dyads separately. As opposed to previous calculations [24], [25], we used the information of Dnmt KOs, to combine these in a single model to obtain Dnmt specific efficiencies at unmethylated and hemimethylated CpG dyads. Furthermore, we did not assume that steady-states are reached in the KO ESC lines. Instead, we estimated the amount of cell generations and inferred parameters during the transient phase of the system, since at least Dnmt3a/3b DKO shows a progressive loss of DNA methylation with increasing passage number [32]. The estimated efficiencies with standard deviations are given in Figure 6a and Table S3. The approximated standard deviations showed that for Dnmt1 efficiencies were accurately estimated for all sequences. For Dnmt3a and Dnmt3b standard deviations are too high for a conclusion at L1, B1 and Afp.

To substantiate the appropriateness of our model and the accuracy of our estimated methylation efficiencies, we predicted the DNA methylation level for the parental wt ESC line (Figure 6b and Table S4). Indeed, we found good predictions for all elements, with maximum error rates of 1.7% (mSat), 4,1% (IAP), 3.9% (Tex13), 2.7% (Afp), 4,1% (L1), 5,9% (B1) and 7,1% (Igf2).
Based on the HMM calculations, we find a high activity of Dnmt1 on hemimethylated CpG dyads. This is 90% or higher for mSat, IAP, Tex13, Afp and B1, but remarkably lower for L1 and Igf2. Furthermore, we found clear evidence for de novo methylation activity of Dnmt1 in vivo. However, it differs for the classes of repetitive elements and single copy genes. While Dnmt1 does not show a remarkable de novo methylation activity (<0.02) for L1, B1 and Igf2, this activity is apparent at IAP, mSat, Tex13 and Afp with calculated efficiencies of 0.36, 0.32, 0.12 and 0.06, respectively (Figure 6a, Table S3). Interestingly, for the de novo methyltransferases Dnmt3a and Dnmt3b, we observe a higher efficiency at hemimethylated sites for some targets, which contrasts in vitro derived data [8], [9].
Discussion
In our work, we present the first high resolution DNA bisulfite methylation analysis of different repetitive elements and selected single copy genes in double stranded DNA. Comparative analysis of wt and KO mouse ES lines revealed detailed insights in the relative contributions of Dnmts to CpG dyad and CpA methylation. In a HMM, we predicted the relative contribution of Dnmts to methylate unmethylated and hemimethylated CpG dyads.
Efficiencies of Dnmts
According to the changed methylation pattern observed in our hairpin-bisulfite analysis and the methylation probabilities of Dnmts estimated with our HMM, the analysed elements can be grouped into three different classes: (i) The imprinted genes (Snrpn and H19 (data not shown)), which exclusively depend on Dnmt1 for maintenance methylation. (ii) IAP, mSat, Tex13 and Afp, which are mainly dependent on Dnmt1 (methylation activity at unmethylated and hemimethylated CpG positions) and show minor effect in the Dnmt3a/3b DKO and (iii) L1, B1 and Igf2, which need Dnmt1 (only methylation activity at hemimethylated CpG positions) and Dnmt3a/3b to work cooperatively. Thus, the HMM model shows that the maintenance contribution for specific genomic regions is clearly distinct from the Dnmt(s) contributions for the de novo acquisition of methylation. Here, the methylation of IAP and L1 is reported to be dependent on either Dnmt3a or Dnmt3b, whereas mSat need Dnmt3b and B1 elements Dnmt3a [33].
Our HMM applied to estimate methylation efficiencies significantly extends previous modelling approaches and allows to draw functional conclusions. First, by separating the effects of all three Dnmts based on KO data, we could estimate probabilities to methylate unmethylated or hemimethylated CpG positions for each enzyme independently. Second, our experimental strategy allowed to precisely assign CpG dyads and to account for experimental measurement errors (bisulfite conversion and mutation errors) as well as the number of cell divisions (passages). Third, we employed numerical techniques to infer optimal methylation efficiencies since analytic solutions of our more complex model are infeasible. By integrating all these parameters, we could functionally extend the previous models developed by Genereux et al. and Sontag et al. [20], [25]. Moreover, beyond prediction, our validations (see Figure 6, Table S4) demonstrate the appropriateness of the model at least for mSat, IAP, Tex13 and Igf2. For B1, L1 and Afp the prediction is very accurate even though the efficiencies of Dnmt3a/3b are difficult to estimate. In contrast to our estimations based on biological Dnmt KO data, a recently published model discriminates between the Dnmts only using theoretical considerations for the Dnmts on WT methylation pattern [34]. However, in this model the authors estimate the processivity of the Dnmts. It will be interesting to adapt their model, using our biological Dnmt KO data.
For Dnmt1, our HMM indicates a significant methylation probability at unmethylated CpG dyads (de novo methylation) in ESCs (up to 22%), depending on the repetitive element/sequence. In vitro experiments analysing the methylation activity of Dnmt1 show 2 to 50 fold higher preference for hemim-CpG dyads, dependent on the substrate or conditions [35]. In vivo, we find 2.5 to 90 fold higher preference for hemimethylated CpGs. Interestingly, pre-existing methylated sites in vitro were shown to enhance the de novo methylation efficiency of Dnmt1 [12],[36]–[38]. Our data corroborate these observations in vivo, linking an increased methylation to a higher de novo methylation activity of Dnmt1 (CpG methylation and methylation activity at unmethylated CpGs is higher at IAP, mSat, Tex13, Afp than both at L1, B1 and Igf2). Differential regulation of the CXXC domain binding capacities at the different sequences could influence the de novo methylation activity of Dnmt1 [39]. The fidelity for Dnmt1 to methylate hemi-mCpG dyads was shown to be 95% to 96% in vitro [12]. Our HMM predicts fidelities of methylating hemi-mCpGs for IAP, mSat, B1, Tex13 and Afp of 90 to 95%, which fits quite well with the in vitro data. However, at L1 and Igf2, the fidelity decreases to less than 80%. This lower fidelity at hemim-CpGs might arise from the presence of 5hmC at L1 elements and Igf2 (see discussion next chapter, reference [40], Figures S7 and S8). 5hmC could hereby not only influence maintenance methylation but also de novo methylation activity, which is enhanced by 5mC content but presumably not 5hmC.
For Dnmt3a and Dnmt3b, we found significant “maintenance” methylation activity. However, the ratio of de novo and maintenance methylation contributions differs across sequence elements. Such context dependent effects were not addressed in former in situ and in vitro modelling studies and may become only evident in the native chromatin context. Since in vitro Dnmt3a and 3b appear to methylate independent of the methylation status of the CpG dyad, the high contribution of Dnmt3a and 3b to maintain full methylation at CpG dyads following replication might be attributed to targeted and enhanced de novo activity stimulated by the presence of CpG methylation density. Some studies show that Dnmt3a/3b can strongly bind to nucleosomes containing methylated DNA [41], [42]. By this Dnmt3a/3b could be triggered to “de novo” methylate hemimethylated sites following replication to maintain full methylation in the absence of Dnmt1.
Distribution and Specificity of CpG Hemimethylation and the Relation to 5hmC
Our experimental approach allowed us to unambiguously assign DNA methylation in total at about 280.000 CpG dyads at 4 repetitive elements and 4 single copy genes on both DNA strands. Besides a general prevalence for symmetrical methylation, we found a substantial portion of hemimethylated CpG dyads in all cell types analysed. The presence of such hemimethylated CpGs can be explained by three different mechanisms: i) the improper recognition of modified cytosines and/or impaired maintenance activity, ii) the selective de novo methylation or iii) active DNA demethylation.
In MEFs and embryonic liver, we find different global tendencies for the occurrence of hemimethylated sites at all analysed elements. However, all three Dnmts are expressed in both cell types (Figure S9). This suggests that in embryonic liver the maintenance fidelity of Dnmts is less pronounced or alternatively DNA demethylation is more pronounced. In ESCs, we observe only at L1 sequences and Igf2 a high level of hemimethylated sites. Apparently, in ESCs, the maintenance methylation machinery is less accurate only at specific sequences and from our data we see that a strong cooperativity of Dnmt1, Dnmt3a and Dnmt3b is needed to maintain methylation at these sequences.
The analyzed cell types show strong cell cycle differences. These might be regarded as reasons for methylation differences. Cultured and fast growing ESCs, for example have been shown to be almost 5 times more likely to be in S-Phase as compared to MEFs [43], [44]. However, in our analysis ESCs show some sequences, which have the same amount of hemimethylated sites as MEFs. Furthermore, in fast growing embryonic liver we observe strong increases of reads showing hemimethylated sites next to fully methylated sites (Figure S10, fraction in dark green). We therefore regard it as unlikely that incomplete methylation can be reduced to the varying number of incomplete S-Phases in the different cell types.
5-hydroxymethylcytosine (5hmC) might contribute to the increase of hemimethylated sites either by impairing maintenance methylation or inducing active DNA demethylation [45]–[48]. 5hmC was reported to be abundant in ESCs, but less so in cultured cells [49]–[51]. Indeed, there is a tendency that 5hmC enrichment is linked to the presence of hemimethylated CpG positions. While Igf2 and L1 regions have an increased level of hemimethylated CpGs and are enriched for 5hmC in ESCs, no ESC specific 5hmC enrichment is found at the Tex13, Afp nor IAP loci, which only show few hemimethylated sites (Figures S7 and S8).
Our HMM calculations indicate that in ESCs sequences enriched for 5hmC do not exhibit de novo methylation activity of Dnmt1 in contrast to 5hmC depleted sequences. Hence, 5hmC might not only impair maintenance methylation but also de novo methylation activity by Dnmt1. However, whether 5hmC indeed blocks Dnmt1 mediated methylation remains to be resolved. Unfortunately, 5hmC profiles cannot be distinguished from 5mC by our bisulfite based sequencing [46], [52], [53] such that the influence of 5hmC on 5mC methylation cannot clearly be assigned. Moreover, the role of 5hmC may not be of importance in some cases, since Np95 apparently recognizes and binds to 5hmC containing DNA and may moderate an effect of 5hmC on Dnmt1 recognition [54]. Finally, a selective conversion of 5mC into 5hmC at individual CpGs could cause mosaic hemimethylated situations by inducing local (hemi-) demethylation. Two general mechanisms, a direct demethylation by further oxidation to carboxylcytosine and subsequent decarboxylation as well as DNA repair coupled processes have been discussed in this respect [46]–[48]. Indeed, the intriguing presence of 5hmC at Line1 and Igf2 regions analyzed might explain the extreme mosaic pictures at these elements. If this is true the estimated de novo methylation rates for both elements may be completely underestimated.
To test if hemimethylated CpG positions are coupled to 5hmC, we analysed the methylation pattern of repetitive elements in the Tet1 KO ESCs. However, the Tet1 KO cells only show a 30% reduction of genome wide 5hmC [55]. We indeed see changed methylation patterns in L1 elements with a slightly increased amount of fully methylated CpG dyads (Figure S11). A combined analysis of Tet KO's (i.e. Tet1+Tet2) might further substantiate the possible link between 5hmC and the increased occurrence of hemimethylated CpG dyads.
Control of CpA Methylation
DNA methylation outside of the CpG context was initially detected in mESCs using nearest neighbour analysis. This analysis revealed a strong prevalence for the CpA context and pointed towards a Dnmt3a dependency [56]. Recent genome wide single stranded bisulfite sequencing using Illumina short reads identified non-CpG methylation at various sequence contexts in human ESCs at rather high rates of 13–25% of all methylated Cs, mainly in CpA context [57]–[59]. In our data set covering a total of 280.000 individual CpG positions and up to 108 bases, we detect non-CpG methylation at specific positions mainly in a CpA context especially confined to mSat sequences and Afp. It is possible that the primer based amplification of our analysis caused some selection against non-CpG methylation and we therefore underestimate the amount of non-CpG methylation. Still, the position dependent non-CpG methylation remains outstanding. We found that non-CpG methylation is exclusively dependent on Dnmts 3a and 3b, in concordance with recent observation in human [59]. However, we can show that both methylate non-CpG positions only in combination with Dnmt3L. Neither the absence of Dnmt1 nor Np95 altered the non-CpG methylation. Moreover, the unchanged non-CpG methylation in Suv39hdn cells reveals that the proposed protective function of H3K9 trimethylation for non-CpG methylation may not be true for mSat [60]. The sequence analysis of Dnmt1 KOs unambiguously shows that non-CpG methylation is linked to Dnmt3a and Dnmt3b mediated CpG methylation. Along this line non-CpG positions are highly co-methylated with some neighbouring CpG positions (Figure 4e, Figure 5b). A recent publication discusses a widespread unspecific non-conserved non-CpG pattern in human pluripotent cells [59]. This contrasts our findings in mouse ESCs, which suggests that non-CpG methylation is mostly locally confined to specific regions such as mSat and Afp and specific non-CpG positions. It will be important to substantiate non-symmetric methylation distribution in human by deep sequencing. Genome wide sequencing approaches at relative low coverage may easily overlook specific patterns as observed in our analysis.
Our data lets us speculate that CpA methylation results from a position specific “side reaction” of Dnmt3a and Dnmt3b stimulated by Dnmt3L. In line with this, Holz-Schietinger et al. show that Dnmt3L increases the processivity of Dnmt3a [13]. Finally, Dnmt3L is much more expressed in ESCs compared to somatic cells, where we do not find any evidence for CpA methylation [61].
Conclusion
Comprehensive hairpin-bisulfite sequencing in Dnmt KO ESCs reveals a complex scenario of sequence, element and cell specific control of DNA methylation pattern at CpG dyads. Based on the sequencing data, we construct a greatly improved HMM, which reveals enzyme, cell type and genome position dependent de novo and maintenance methylation functions for all three Dnmts. This strongly supports previous conclusions by Fatemi et al 2002 and others, that in vivo neither de novo methylation can be exclusively assigned to Dnmt3a/3b nor maintenance methylation exclusively to Dnmt1 [62]. Position dependent non-CpG methylation, mainly in CpA context, occurs at major satellites and the Afp gene exclusively in ESCs. This non-CpG methylation is mediated by Dnmt3a and 3b, depends on the presence of Dnmt3L and is strongly correlated to the methylation of flanking CpG positions.
Materials and Methods
Hairpin-Bisulfite Sequencing
The complete protocol is provided in the SI (Materials and Methods S1). Briefly, genomic DNA was digested with an element specific restriction enzyme and the upper strand and lower strand were linked with a hairpinoligonucleotide. After bisulfite treatment an element specific PCR was performed and the resulting product was sequenced with the 454 sequencing technique.
Estimation of Dnmt Efficiencies
We used CpG dyad methylation data on WT and DnmtKO ESCs in a hidden Markov model to estimate the Dnmt methylation efficiencies by the maximum likelihood method. A detailed description of the model is provided in the SI (Materials and Methods S1).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LiEBestorTHJaenischR 1992 Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69 915 926
2. OkanoMBellDWHaberDALiE 1999 DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99 247 257
3. BirdATaggartMFrommerMMillerOJMacleodD 1985 A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40 91 99
4. EhrlichMGama-SosaMAHuangLHMidgettRMKuoKC 1982 Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10 2709 2721
5. Gama-SosaMAMidgettRMSlagelVAGithensSKuoKC 1983 Tissue-specific differences in DNA methylation in various mammals. Biochim Biophys Acta 740 212 219
6. Bourc'hisDBestorTH 2004 Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431 96 99
7. YoderJAWalshCPBestorTH 1997 Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13 335 340
8. OkanoMXieSLiE 1998 Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19 219 220
9. GowherHJeltschA 2001 Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol 309 1201 1208
10. HermannAGoyalRJeltschA 2004 The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 279 48350 48359
11. GowherHJeltschA 2002 Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J Biol Chem 277 20409 20414
12. VilkaitisGSuetakeIKlimasauskasSTajimaS 2005 Processive methylation of hemimethylated CpG sites by mouse Dnmt1 DNA methyltransferase. J Biol Chem 280 64 72
13. Holz-SchietingerCReichNO 2010 The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J Biol Chem 285 29091 29100
14. GowherHLiebertKHermannAXuGJeltschA 2005 Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem 280 13341 13348
15. Bourc'hisDXuGLLinCSBollmanBBestorTH 2001 Dnmt3L and the establishment of maternal genomic imprints. Science 294 2536 2539
16. BostickMKimJKEstevePOClarkAPradhanS 2007 UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317 1760 1764
17. SharifJMutoMTakebayashiSSuetakeIIwamatsuA 2007 The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450 908 912
18. MeilingerDFellingerKBultmannSRothbauerUBonapaceIM 2009 Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep
19. LiangGChanMFTomigaharaYTsaiYCGonzalesFA 2002 Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol Cell Biol 22 480 491
20. SontagLBLorinczMCGeorg LuebeckE 2006 Dynamics, stability and inheritance of somatic DNA methylation imprints. J Theor Biol 242 890 899
21. LaceyMREhrlichM 2009 Modeling dependence in methylation patterns with application to ovarian carcinomas. Stat Appl Genet Mol Biol 8 Article 40
22. OttoSPWalbotV 1990 DNA methylation in eukaryotes: kinetics of demethylation and de novo methylation during the life cycle. Genetics 124 429 437
23. PfeiferGPSteigerwaldSDHansenRSGartlerSMRiggsAD 1990 Polymerase chain reaction-aided genomic sequencing of an X chromosome-linked CpG island: methylation patterns suggest clonal inheritance, CpG site autonomy, and an explanation of activity state stability. Proc Natl Acad Sci U S A 87 8252 8256
24. LairdCDPleasantNDClarkADSneedenJLHassanKM 2004 Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Natl Acad Sci U S A 101 204 209
25. GenereuxDPMinerBEBergstromCTLairdCD 2005 A population-epigenetic model to infer site-specific methylation rates from double-stranded DNA methylation patterns. Proc Natl Acad Sci U S A 102 5802 5807
26. JeongKSLeeS 2005 Estimating the total mouse DNA methylation according to the B1 repetitive elements. Biochem Biophys Res Commun 335 1211 1216
27. LutsikPFeuerbachLArandJLengauerTWalterJ 2011 BiQ Analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res 39 Suppl 2 W551 556
28. BurdenAFManleyNCClarkADGartlerSMLairdCD 2005 Hemimethylation and non-CpG methylation levels in a promoter region of human LINE-1 (L1) repeated elements. J Biol Chem 280 14413 14419
29. ShaoCLaceyMDubeauLEhrlichM 2009 Hemimethylation footprints of DNA demethylation in cancer. Epigenetics 4 165 175
30. LeiHOhSPOkanoMJuttermannRGossKA 1996 De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 122 3195 3205
31. LehnertzBUedaYDerijckAABraunschweigUPerez-BurgosL 2003 Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13 1192 1200
32. ChenTUedaYDodgeJEWangZLiE 2003 Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23 5594 5605
33. KatoYKanedaMHataKKumakiKHisanoM 2007 Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum Mol Genet 16 2272 2280
34. FuAQGenereuxDPStogerRBurdenAFLairdCD 2012 Statistical inference of in vivo properties of human DNA methyltransferases from double-stranded methylation patterns. PLoS ONE 7 e32225 doi:10.1371/journal.pone.0032225
35. GoyalRReinhardtRJeltschA 2006 Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res 34 1182 1188
36. TollefsbolTOHutchisonCA3rd 1997 Control of methylation spreading in synthetic DNA sequences by the murine DNA methyltransferase. J Mol Biol 269 494 504
37. LorinczMCSchubelerDHutchinsonSRDickersonDRGroudineM 2002 DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol Cell Biol 22 7572 7580
38. BacollaAPradhanSRobertsRJWellsRD 1999 Recombinant human DNA (cytosine-5) methyltransferase. II. Steady-state kinetics reveal allosteric activation by methylated dna. J Biol Chem 274 33011 33019
39. SongJRechkoblitOBestorTHPatelDJ 2011 Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331 1036 1040
40. FiczGBrancoMRSeisenbergerSSantosFKruegerF 2011 Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473 398 402
41. JeongSLiangGSharmaSLinJCChoiSH 2009 Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol Cell Biol 29 5366 5376
42. SharmaSDe CarvalhoDDJeongSJonesPALiangG 2011 Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet 7 e1001286 doi:10.1371/journal.pgen.1001286
43. SavatierPLapillonneHJirmanovaLVitelliLSamarutJ 2002 Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol Biol 185 27 33
44. ElizondoGFernandez-SalgueroPSheikhMSKimGYFornaceAJ 2000 Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol 57 1056 1063
45. ValinluckVSowersLC 2007 Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 67 946 950
46. HeYFLiBZLiZLiuPWangY 2011 Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333 1303 1307
47. ZhangLLuXLuJLiangHDaiQ 2012 Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol
48. ItoSShenLDaiQWuSCCollinsLB 2011 Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science
49. KriaucionisSHeintzN 2009 The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324 929 930
50. KinneySMChinHGVaisvilaRBitinaiteJZhengY 2011 Tissue specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genome. J Biol Chem
51. NestorCEOttavianoRReddingtonJSproulDReinhardtD 2011 Tissue-type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res
52. HayatsuHShiragamiM 1979 Reaction of bisulfite with the 5-hydroxymethyl group in pyrimidines and in phage DNAs. Biochemistry 18 632 637
53. HuangYPastorWAShenYTahilianiMLiuDR 2010 The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 5 e8888 doi:10.1371/journal.pone.0008888
54. FrauerCHoffmannTBultmannSCasaVCardosoMC 2011 Recognition of 5-Hydroxymethylcytosine by the Uhrf1 SRA Domain. PLoS ONE 6 e21306 doi:10.1371/journal.pone.0021306
55. DawlatyMMGanzKPowellBEHuYCMarkoulakiS 2011 Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9 166 175
56. RamsahoyeBHBiniszkiewiczDLykoFClarkVBirdAP 2000 Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A 97 5237 5242
57. ListerRPelizzolaMDowenRHHawkinsRDHonG 2009 Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462 315 322
58. LaurentLWongELiGHuynhTTsirigosA 2010 Dynamic changes in the human methylome during differentiation. Genome Res 20 320 331
59. ZillerMJMullerFLiaoJZhangYGuH 2012 Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet 7 e1002389 doi:10.1371/journal.pgen.1002389
60. ListerRPelizzolaMKidaYSHawkinsRDNeryJR 2011 Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471 68 73
61. SuAIWiltshireTBatalovSLappHChingKA 2004 A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A 101 6062 6067
62. FatemiMHermannAGowherHJeltschA 2002 Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur J Biochem 269 4981 4984
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Rumors of Its Disassembly Have Been Greatly Exaggerated: The Secret Life of the Synaptonemal Complex at the Centromeres
- The NSL Complex Regulates Housekeeping Genes in
- Tipping the Balance in the Powerhouse of the Cell to “Protect” Colorectal Cancer
- Interplay between Synaptonemal Complex, Homologous Recombination, and Centromeres during Mammalian Meiosis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy